

Abstract
Like amyloid beta (Aβ) oligomers, tau aggregates are increasingly recognized as potential key toxic intermediates in Alzheimer's disease (AD) and as therapeutic targets. P‐tau co‐localizes with Aβ in cortical AD synapses and may contribute to synapse dysfunction and loss. Flow cytometry analysis of synaptosomes from AD compared with aged cognitively normal cortex demonstrates increased immunolabeling for three p‐tau antibodies (AT8, PHF‐1 and pS422), indicating phosphorylation at multiple tau epitopes. Sequential extraction experiments show increased soluble p‐tau in AD synapses, but a sizable pool of p‐tau requires detergent solubilization, suggesting endosomal/lysosomal localization. P‐tau is co‐localized with Aβ in individual synaptosomes in dual labeling experiments, and flow cytometry sorting of Aβ‐positive synaptosomes from an AD case reveals a marked enrichment of p‐tau aggregates. The p‐tau enrichment, a 76‐fold increase over the initial homogenate, is consistent with sequestration of p‐tau in internal synaptic compartments. Western analysis of a series of AD and normal cases shows SDS‐stable tau oligomers in the dimer/trimer size range in AD samples. These results indicate that widespread synaptic p‐tau pathology accompanies Aβ accumulations in surviving synaptic terminals, particularly in late‐stage AD.
Keywords: Alzheimer's disease, amyloid beta, flow cytometry, pS422 antibody, synaptosome, tau
INTRODUCTION
A great deal of evidence suggests that early cognitive decline is linked to synaptic dysfunction that precedes the appearance of plaques and tangles, and that amyloid beta (Aβ) may drive tau pathology. We have used flow cytometry analysis of synaptosomes prepared from Alzheimer's disease (AD) brain in order to examine changes in surviving synaptic terminals, and have demonstrated that, in contrast to the discrete spatial locations of amyloid plaques and neurofibrillary tangles, AD synapses accumulate both Aβ and p‐tau 11, 12, 31. This result has been confirmed by others (32) and is consistent with work showing trans‐synaptic induction of tau pathology following injection of Aβ fibrils (10). Co‐localization of Aβ oligomers with early somatodendritic accumulation of tau has also been shown in 3XTg mice (23).
Tau is a soluble protein localized to axons; in tauopathies, it becomes highly phosphorylated, which is thought to contribute to β‐sheet formation, aggregation and eventual assembly into paired helical filaments (PHFs) that form the basis of neurofibrillary tangles. Mislocalization is thought to occur primarily in the somatodendritic compartment when tau levels are increased; however, both pro‐ and anti‐aggregation mouse models with mutated tau protein show synapse loss that appears related to aggregation (6). Thioflavin‐positive tau oligomers, spherical or granular in shape, appear as early as Braak stage I neuropathology, precede filament formation and have recently been suggested as an intermediate of tau filaments 17, 18, 29. Tau fragmentation may also contribute to acceleration of pathology and early neurotoxicity. Aβ‐induced caspase cleavage of tau has been shown to promote tau aggregation 8, 27. Aβ has also been shown to activate calpain, yielding a 17 kDa tau fragment that induced neurodegeneration in cultured hippocampal cells (24). In a study of AD cerebrospinal fluid (CSF), the primary tau peptide was a 26–28 kDa fragment (13).
We have previously demonstrated that p‐tau is co‐localized with Aβ in AD synapses, and that co‐localization is highest in the entorhinal cortex compared with hippocampus and isocortical regions (7). In order to understand the nature and stage of p‐tau pathology in AD synapses, we have examined p‐tau hyperphosphorylation in human AD synaptosomes, and report here that p‐tau phosphorylated at multiple epitopes is elevated in AD synapses, along with SDS‐stable tau oligomers and aggregates.
METHODS
Materials
The monoclonal anti‐Aβ antibody 10G4 has been described previously (19). Polystyrene microsphere size standards were purchased from Polysciences, Inc. (Warrington, PA, USA). Zenon mouse IgG Labeling kits for were purchased from Molecular Probes (Eugene, OR, USA). Anti‐tau pS422 was purchased from Invitrogen (Carlsbad, CA, USA), AT8 from Pierce (Rockford, IL, USA), and Thr231 was a kind gift from Sally Frautschy. Anti‐PHF‐Tau (PHF‐1) was a kind gift from P. Davies.
Human brain specimens
Parietal cortex (A39, A40) samples with a post‐mortem interval ≤12 h were obtained at autopsy from the Alzheimer's Disease Research Centers at University of California, Los Angeles, University of California Irvine, and the University of Southern California. Each diagnosis was established clinically and histopathologically. The mean age for controls was 84.5, and for AD was 86.3; mean post‐mortem delay, 7.56 h.
P‐2 preparation
Unfixed fresh samples (∼0.3 to 5 g) were minced and slowly frozen on the day of autopsy in 10% DMSO and 0.32 M sucrose (plus chelating agents 0.5 M EDTA and 0.2 M EGTA; phosphatase inhibitors 150 mM NaPP and 1.0 M NaF; protease inhibitor 100 mM PMSF; and buffer 1 M TRIS pH 8.0) and stored at −70°C until homogenization. The P‐2 (crude synaptosome) fraction was prepared as described previously (11) in 0.32 M sucrose solution (plus chelating agents 0.5 M EDTA and 0.2 M EGTA; protease inhibitors 4 µg/mL pepstatin, 5 µg/mL aprotinin, 20 µg/mL trypsin inhibitor, 0.02 M PMSF, 4 µg/mL leu‐peptin, and buffer 1 M TRIS pH 8.0); briefly, the homogenate was first centrifuged at 1000 × g for 10 minutes; the resulting supernatant was centrifuged at 10 000 × g for 20 minutes to obtain the crude synaptosomal pellet. Aliquots of P‐2 were routinely cryopreserved in 0.32 M sucrose and banked at −70°C until the day of the experiment.
Immunolabeling of P‐2 fraction
P‐2 aliquots were immunolabeled for flow cytometry analysis according to a method for staining of intracellular antigens (30). Pellets were fixed in 0.25% buffered paraformaldehyde (1 h, 4°C) and permeabilized in 0.2% Tween20/phosphate‐buffered saline (PBS) (15 minutes, 37°C). Antibodies were labeled directly with Alexa Fluor 488 or 647 reagents according to kit directions. This mixture was added to P‐2 aliquots and incubated at room temperature for 30 minutes. Pellets were washed two times with 1 mL 0.2% Tween20/PBS, resuspended in PBS buffer (0.75 mL) for flow cytometry analysis. The synaptosomal pellet was dispersed for all washes and for incubations with fixative, detergent and antibody, then collected by centrifugation (1310 × g at 4°C).
Flow cytometry
Data were acquired using a BD‐FACSCalibur analytic flow cytometer (Becton‐Dickinson, San Jose, CA, USA) equipped with argon 488 nm, helium‐neon 635 nm and helium‐cadmium 325 nm lasers. Five thousand particles were collected and analyzed for each sample. Debris were excluded by establishing a size threshold set on forward light scatter. Alexa 488 and Alexa 647 fluorochromes were detected by the LSR's FL1, Ssc‐W, photomultiplier tube detectors, respectively. Analysis was performed using FCS Express software (DeNovo Software, ON, Canada).
Synaptosome extracts
Washed P2 fractions were first extracted by sonication in a detergent‐free buffer (10 mM Tris, 1 mM EGTA, 10% sucrose, pH 7.5) and then centrifuged at 25 000 g. The supernatant of this detergent‐free extraction (Sup. A) was used to quantify the levels of aqueous soluble tau species. The remaining pellet was extracted by sonication in the same buffer containing 1% N‐ lauroylsarkosyl (NLS) and centrifuged at 300 000 g. This supernatant was analyzed as the detergent soluble fraction (Sup. NLS).
Luminex assay
The aqueous and detergent soluble fractions were assayed for total tau and p‐tau 181 using specific bead based kits (INNO‐BIA AlzBio3) purchased from Innogenetics (Brussels, Belgium). Assays were performed on a Luminex® instrument using X‐map® Technology (Austin, TX, USA) and xPONENT software. Standard curves were constructed from authentic standards included with the kit. Each extract was analyzed in duplicate for each analyte. Correlation coefficients and Student's t‐tests were calculated using the Vassarstat interactive statistical website (http://www.vassarstats.net; Richard Lowry, Poughkeepsie, NY, USA).
Western blotting
Samples were boiled in Laemmli loading buffer (2%SDS, Invitrogen) and electrophoresed on 4%–20% Tris‐Tricine gradient gels. Gels were stained with Coomasie blue to ensure equal protein loading. Membranes were blocked for 1 h at room temperature in 10% nonfat dried milk in PBS, followed by incubation overnight at 4°C with primary antibodies in PBS containing 0.05% Tween 20 (PBS‐T) and 1.5% (w/v) albumin. After rinsing in PBS‐T, the membranes were incubated with horseradish peroxidase (HRP)‐conjugated anti‐mouse IgG (1:10 000) or anti‐rabbit IgG (1:30 000) in PBS‐T with 1.5% albumin for 1 h. Immunolabeled proteins were visualized by enhanced chemiluminescence (ECL) detection reagents. Resulting films were scanned and quantified using densitometric software (Molecular Analyst II, Bio‐Rad, Hercules, CA, USA).
RESULTS
Multiple soluble p‐tau epitopes are increased in AD synapses, and are concentrated with Aβ in individual terminals
Samples of parietal cortex (Brodmann area 39) from aged cognitively normal controls and from neuropathologically confirmed AD cases were first cryopreserved and later homogenized as described previously to obtain the P‐2 (crude synaptosome) fraction. Synaptosomes are resealed nerve terminals formed during homogenization in isotonic sucrose; synaptosomes typically contain mitochondria, actin, various endosomes and the exocytotic apparatus including vesicles. Abundant synaptic density structure, including adherent postsynaptic elements, is also present. We and others have previously shown tau pathology to be co‐localized with Aβ in AD synapses; the present study was focused on understanding the key phosphorylation epitope(s), peptide species and solubility, and degree of accumulation within Aβ‐positive AD synaptic terminals.
For initial studies, flow cytometry analysis of synaptosomes was used in order to examine a large and highly pure population (5000/sample) of human synaptic terminals on a terminal‐by‐terminal basis (11). Figure 1 illustrates the flow cytometry analysis; a positive control sample immunolabeled for the presynaptic docking protein SNAP‐25 (Figure 1A) demonstrates the purity of synaptosomes (93.74%) analyzed within the size gate drawn on forward scatter, which is proportional to particle size. The size gate is drawn based on size standards and includes only particles between 0.75 and 1.5 microns (31). Figure 1B shows background labeling in a representative sample. Representative synaptic p‐tau immunolabeling with the AT8 antibody (directed against Ser 202, Thr 205) is shown for an aged cognitively normal case (11.26%; Figure 1C) and for an AD case (55.32%; Figure 1D). Synaptic levels for additional p‐tau epitopes in a representative AD sample are shown below; PHF‐1 (directed against phosphorylation at Ser 396 and Ser 404) labeled 42.74% (Figure 1E), and the pS422 antibody, which detects tau phosphorylated at Ser 422, labeled 20.34% (Figure 1F).
Figure 1.
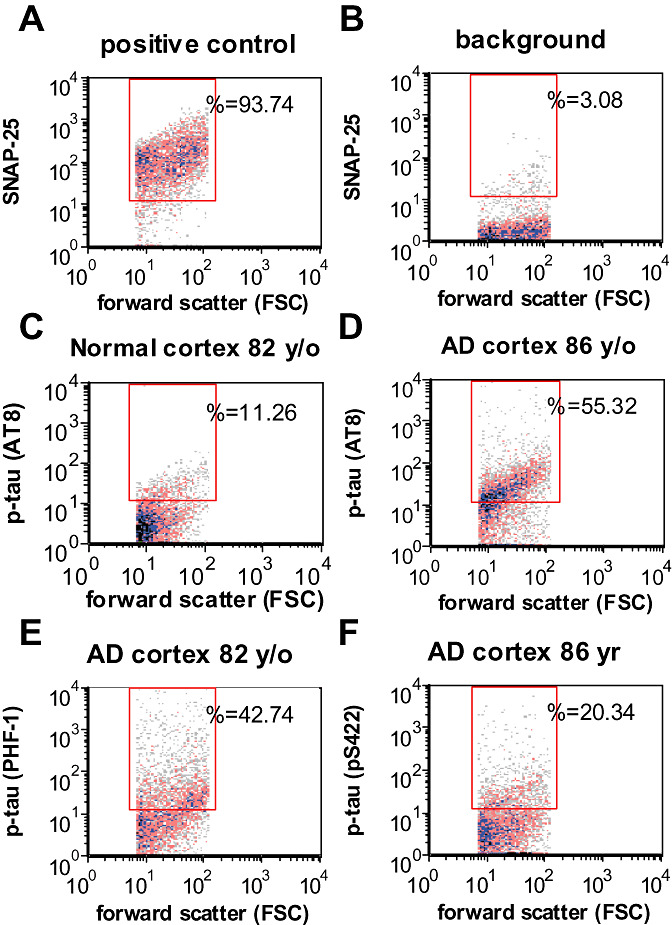
Flow cytometry and biochemical analysis of p‐tau in AD synaptosomes: Representative samples showing positive control labeled with SNAP‐25, a presynaptic marker (A), unstained background labeling (B), and synaptic p‐tau labeling in AD (C) and normal parietal cortex (D) for the p‐tau epitope detected by AT8. Representative samples are also shown for p‐tau epitopes detected by PHF‐1 (E) and pS422 (F). AD = Alzheimer's disease.
The aggregate data for synaptic p‐tau immunolabeling in flow cytometry experiments are shown in Figure 2A. Flow cytometry allows quantification of the brightness of fluorescence in relative fluorescence units (RFU) for individual synaptosomes within each sample; the level of p‐tau immunolabeling by the antibodies AT8, PHF‐1 and pS422 is shown. With each antibody, p‐tau was elevated in AD compared with cognitively normal samples (P < 0.05).
Figure 2.
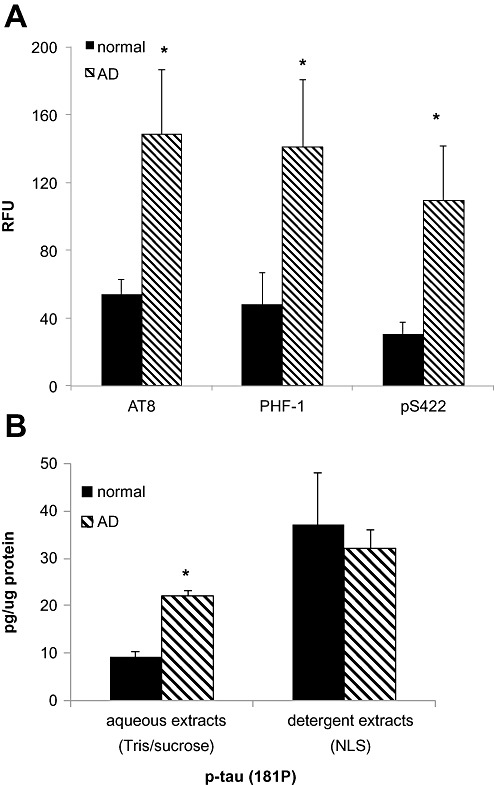
Flow cytometry and immunoassay of p‐tau in AD synaptosomes and detergent extracts. A. Size of positive synaptosome fraction for the p‐tau epitopes detected by the antibodies AT8, PHF‐1 and pS422, in cognitively normal aged (n = 3) and late stage (Braak VI) AD cases (n = 4); data were collected from 5000 terminals for each sample, *P < 0.05. B. Bead‐based immunoassay of p‐tau 181 in aqueous and detergent (1% N‐laurylsarcosyl) extracts of synaptosome‐enriched fractions from normal (n = 2) and AD cases (n = 7), *P < 0.002. AD = Alzheimer's disease.
To confirm and extend flow cytometry observations, sequential extractions were prepared from AD and normal synaptosome‐enriched fractions. These fractions (ie, washed P‐2 or crude synaptosome pellet) are enriched for presynaptic and postsynaptic proteins, approximately twofold above the initial homogenate, and were used in order to obtain sufficient volume for the analyses (31). A bead‐based immunoassay was used to measure p‐tau phosphyorylated at Thr 181 (p‐181 tau). For the initial soluble extract, washed P‐2 pellets were sonicated in detergent‐free buffer; this pellet was then sonicated in buffer plus detergent (1% N‐laurylsarcosyl). In aqueous extracts, p‐tau was elevated in AD samples (21.93 pg/µg protein ± 1.37) compared with aged normal control samples (9.16 ± 1.05; P < 0.002; Figure 2B). P‐tau levels were higher in detergent extracts compared with aqueous extracts for both normal and AD cases; however, the detergent extract p‐tau level was not different for AD (32.02 ± 3.91) compared with normal cases (37.11 ± 11.05). These results confirm flow cytometry observations of widespread synaptic p‐tau pathology, and indicate AD‐specific accumulation of p‐tau within a soluble pool. Detergent extract data demonstrate that a sizable but less soluble pool of p‐tau accumulates in aged normal as well as AD synapses.
Flow cytometry analysis of synaptosomes dual labeled for Aβ (with the 10G4 antibody) and for p‐tau (with the pS422 antibody; Figure 3A,B) confirmed that approximately 50% of Aβ‐positive terminals contain p‐tau in parietal cortex. We have previously demonstrated that the N‐terminal antibodies 10G4 and 6E10 label a distinct population of synaptosomes compared with APP antibodies (31). Dual labeling in a 24‐month‐old 3XTg mouse showed levels of co‐localization (Figure 3C) similar to that observed in AD cases.
Figure 3.
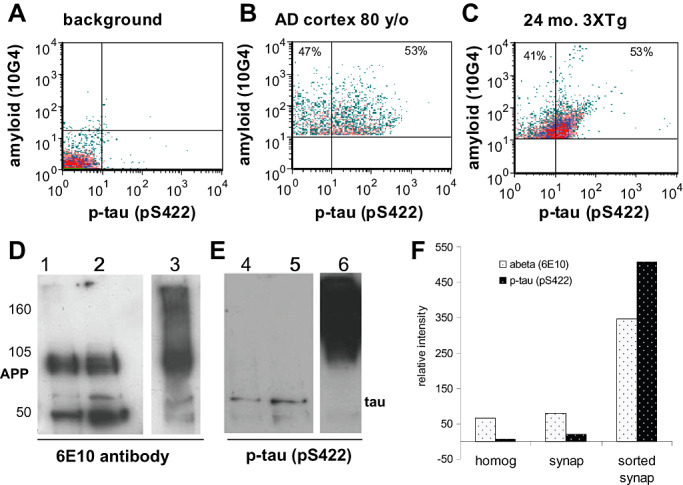
Co‐localization of Aβ and p‐tau within individual synapses. Flow cytometry of dual labeled synaptosomes; representative samples show unstained background labeling (A); 53% of Aβ‐positives are positive for p‐tau with the pS422 antibody in human AD cortex (B, upper right quadrant) as well as 24‐month‐old 3XTg mouse cortex (C, upper right quadrant). Flow sorting was used to collect 108 million Aβ‐positive synaptosomes. The sorted fraction (lanes 3,6) was compared with the initial homogenate (lanes 1, 4) and to total gradient purified synaptosomes (lanes 2,5); Western blots are shown for APP and Aβ (D; 6E10 antibody) and for p‐tau (E; pS422). (F) Western blots were quantified for relative intensity of bands detected by Aβ (6e10) and p‐tau (pS422). AD = Alzheimer's disease.
The co‐accumulation of p‐tau and Aβ within a large population of individual cortical synaptosomes was validated further using gradient‐purified synaptosomes prepared from an 80‐year‐old female AD case (case 102) and labeled for Aβ with the 10G4 antibody. A flow‐sorting experiment collected 108 million Aβ‐positive, size‐gated synaptosomes in a 12‐h experiment. The sorted samples were separated by SDS gel electrophoresis; Western blots (Figure 3D, lanes 1–3) labeled with the 6E10 antibody against Aβ demonstrate that the sorted synaptosomes (lane 3) show a marked increase (∼fivefold) in APP and Aβ aggregates compared with the initial homogenate and purified synaptosomes (lanes 1 and 2, respectively). Two prominent Aβ oligomer bands between 50 and 75 kDa were also observed in the sorted sample. The same membrane was then probed for p‐tau with the pS422 antibody. Interestingly, the sorted synaptosomes show decreased tau monomer and a massive enrichment (∼76‐fold; Figure 3E, lane 6) of p‐tau aggregates in the sample sorted for Aβ‐positives, highlighting the degree of tau pathology that accompanies Aβ accumulation in surviving synapses. A minor band at ∼70 kDa is labeled for both Aβ and p‐tau and likely represents a complex that contains both proteins. Western blot quantification is shown in Figure 3F. In sorted synaptosomes, the disproportionate increase in p‐tau compared with Aβ indicates a marked concentration of p‐tau aggregates in Aβ‐bearing synapses, possibly within interior endosomal compartments that are not accessible to antibodies in intact synaptosomes analyzed by flow cytometry.
Tau oligomers and fragments are prominent in synaptosome‐enriched fractions from AD brain
The size of synaptic tau peptides was examined in Western blots using synaptosome‐enriched fractions from a series of AD (n = 7) and aged normal control (n = 4) parietal cortex samples. The series also included three non‐AD comparison cases, two with Parkinson's disease (PD), and a tauopathy case without diffuse or neuritic plaques that showed striking tau‐immunoreactive neurofibrillary tangles and neuropil threads on neuropathological examination. The neurologic controls were included to determine the disease specificity of synaptic pathology in AD, and to include mixed pathology. For example, one of the PD cases displayed mixed pathology with a 2‐year history of dementia; a Lewy body AD diagnosis was considered for this case. We have performed a detailed examination of Aβ peptide species in the same set of cases (30); case information and lane number for Figure 4 are indicated in Table 1.
Figure 4.
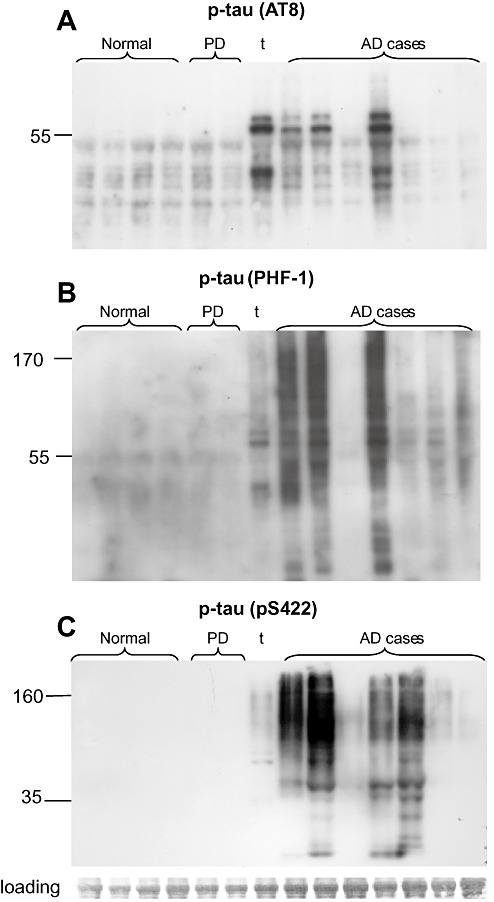
P‐tau Western blots in synaptosome‐enriched fractions. Western blots are shown for a series of cognitively normal (n = 4) and AD cases (n = 7); neurological controls include two PD cases and one tauopathy case; the right PD case (lane 6) had a 2‐year history of dementia. Western blots are shown for the same cases for the p‐tau antibodies AT8 (A), PHF‐1 (B) and pS422 (C). AD = Alzheimer's disease; PD = Parkinson's disease.
Table 1.
Case information for synaptosome samples. Abbreviations: AD = Alzheimer's disease; PD = Parkinson's disease; PMI = Post‐mortem interval.
| Lane | Case No. | Sex | Age | PMI (h) | Braak & Braak score |
|---|---|---|---|---|---|
| Normal cases | |||||
| 1 | 726 | F | 97 | 5.5 | — |
| 2 | 758 | M | 93 | 8.5 | — |
| 3 | 1509 | M | 82 | 14.9 | — |
| 4 | 789 | F | 105 | 9 | — |
| PD cases | |||||
| 5 | 711 | M | 74 | 7.8 | PD |
| 6 | 720 | M | 63 | 3.5 | PD vs. Lewy body AD; 2‐year dementia |
| AD cases | |||||
| 7 | 024 tauopathy | F | 72 | 5 | Dementia |
| 8 | 782 | F | 89 | 12 | IV–V; 10‐year dementia |
| 9 | 731 | F | 87 | 10 | V–VI; 7‐year dementia |
| 10 | 102 | F | 80 | 11 | VI; 7‐year dementia |
| 11 | 716 | F | 86 | 5 | VI; 11‐year dementia |
| 12 | 163 | F | 76 | 3.6 | VI; 26‐year dementia |
| 13 | 718 | M | 83 | 7 | V; 2‐year dementia |
| 14 | 776 | F | 99 | 11.5 | V; 2‐year dementia |
The same cases were used in the same order for the Western blots in Figure 4; strong tau oligomer bands were observed in 3–4 of the seven AD cases with the p‐tau antibodies PHF‐1 and pS422 (Figure 4B,C). As previously reported for brain homogenates (18), the oligomer bands in synaptosome‐enriched fractions have a smeared appearance; particularly with pS422, with major bands in the size range of tau dimer to trimer. In contrast, the AT8 antibody does not label oligomeric assemblies but is clearly increased in three AD cases (Figure 4A). An antibody directed against phosphorylation at Thr231 also shows smeared tau oligomers in some AD cases (data not shown). In general, p‐tau pathology, particularly synapse‐associated oligomers, is more prominent in Braak stage VI cases and is associated with a longer history of dementia. Interestingly, only the phosphorylation epitope detected by the AT8 antibody (Ser 202; Thr 205) is prominent in the tauopathy case. In cases with tau pathology, all three p‐tau antibodies detect a ladder of fragments between 20 and 55 kDa, indicating a heterogenous mix of monomeric plus fragmented tau. PD cases displayed little to no tau pathology, a result in contrast to Aβ oligomer pathology previously observed in the same two PD cases (31).
DISCUSSION
Tau protein is required for the full expression of AD pathology 26, 28, but little is known about prefibrillar aggregation intermediates leading to neurofibrillary tangles. Therefore, tau pathology in AD synaptic terminals may be an important component of synapse loss, and inhibition of tau aggregation should be considered along with Aβ aggregation as a viable therapeutic strategy for early cognitive deficits (3). The present experiments demonstrate that extensive tau pathology accompanies cortical Aβ pathology; tau fragments as well as tau phosphorylated at multiple epitopes accumulates and assembles into oligomers in extant synaptic terminals. Flow‐sorting and detergent‐extraction experiments confirm that p‐tau pathology occurs within individual synapses also positive for Aβ, and suggest sequestration or endosomal/lysosome localization of some p‐tau within the synapse.
Tau is generally considered to be a soluble protein; however, total tau was evenly distributed between the two fractions, with 45% in the aqueous and 55% in the detergent‐soluble fractions. Taken together with the flow results for p‐tau, these data indicate that in aged normal control and AD synapses, a significant fraction of p‐tau requires detergent solubilization, consistent with release from interior endosomal/lysosomal compartments, which are abundant within synaptosomes and upregulated in AD brain 2, 4, 21, 22.
Neurofibrillary tangle (NFTs) development follows a well‐established regional progression, often used as the basis for neuropathological staging, beginning in transentorhinal regions and ending in neocortical regions. Lesions have been described as evolving in a stepwise manner, beginning with a pre‐NFT phase, followed by an intraneuronal NFT phase and ending with a mature extracellular NFT (1), but the early stages of tau processing and phosphorylation have been difficult to resolve, and have not previously associated with the synaptic compartment. In contrast to the present results, pS422 phosphorylation was not increased in a study of medial temporal cortex homogenates from AD cases (34); our results may indicate that pS422 phosphorylation is more prominent in synaptic terminals or in the later‐affected parietal cortex used in the present study.
Tau oligomers have been previously observed in homogenates and have been suggested to be intermediates of tau filaments and an early sign of AD. (14) The present results extend this observation to the synaptic compartment, which is shared with Aβ oligomers 17, 31. In vitro observations suggest that small tau oligomers form early in tau aggregation and self‐associate to create larger oligomers (25). Interestingly, recent work suggests that toxic oligomeric assemblies are not limited to Aβ and tau; α‐synuclein oligomers may contribute to toxicity in PD and dementia with Lewy bodies (16). In some studies, specific tau phosphorylation sites have been associated with pathology stage; for example, serine 422 has been associated with pretangle neuropil threads in some studies (15) and with intraneuronal tangles in others (1). However, reported results vary widely, and other studies report that tau phosphorylation in the neocortex does not follow the stereotypical patterns seen in limbic cortex (20). In the present experiments, all of the AD cases were late stage (Braak V–VI); therefore, it is not possible to draw conclusions about the sequence of Aβ and p‐tau pathology progression. However, it is also possible that pathology in surviving terminals, even from later stage cases, may represent earlier events in the sequence of synapse dysfunction and loss. Ongoing studies in early AD cases will clarify whether synaptic tau oligomers appear in earlier disease stages.
Like oligomerization, tau fragmentation may contribute to acceleration of pathology and synaptic dysfunction and loss. Aβ‐induced caspase cleavage of tau has been shown to promote tau aggregation (8) and has been suggested as an early event in tangle formation (26). A caspase‐truncated (Asp 421) tau fragment has been observed in NFTs and dystrophic neurites in AD brain (8). More recently, the same caspase cleavage event was observed to occur just before tangle formation in vivo, and subsequently led to intracellular aggregates, phosphorylation and recruitment of full length tau into aggregated assemblies (5). A calpain‐generated 17–20 kDa fragment has also been hypothesized to contribute to tau‐mediated neurotoxicity (23), although recent work suggests a larger size and lack of toxicity for this particular fragment (9).
The smeared appearance of tau oligomers in our Western blots has been observed by others (18), and likely indicates extensive aggregation of multiple p‐tau fragments along with full‐length p‐tau. The multiple fragments and smeared appearance of synaptic tau oligomers suggest that aggregation and fragmentation processes occur together in synapses; this combination of p‐tau peptides may be particularly toxic (33). Given that inhibition of tau aggregation is a therapeutic target, and that evidence also supports anti‐tau oligomer vaccination as a potential treatment, the present results highlight a need for a more complete understanding of pretangle pathology progression in the AD brain.
ACKNOWLEDGMENTS
This work was supported by NIH AG27465 to KHG, by NIA AG18879 to CAM. HVV is supported by the Daljit S and Elaine Sarkaria Chair in Diagnostic Medicine. Tissue was obtained from the Alzheimer's Disease Research Center Neuropathology Cores of USC (NIA 050 AG05142), UCLA (NIA P50 AG 16970) and UC Irvine (NIA P50 AG016573). Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility supported by NIH CA16042 and AI 28697, and by the JCCC, the UCLA AIDS Institute, the David Geffen School of Medicine and the Chancellor's Office at UCLA.
Funding: This work was supported by NIH 5AG016570 to KHG, by NIA AG18879 to CAM, and by NIH CA 16042 and AI 28697 to the Jonsson Cancer Center at UCLA.
REFERENCES
- 1. Augustinack JC, Schneider A, Mandelkow EM, Hyman BT (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol 103:26–35. [DOI] [PubMed] [Google Scholar]
- 2. Bernstein HG (1997) Tissue kallikrein‐like immunoreactive material decorates neuritic plaques of Alzheimer patients. Clin Neuropathol 16:69–71. [PubMed] [Google Scholar]
- 3. Bulic B, Pickhardt M, Mandelkow EM, Mandelkow E (2010) Tau protein and tau aggregation inhibitors. Neuropharmacology 59:276–289. [DOI] [PubMed] [Google Scholar]
- 4. Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S et al (1995) Gene expression and cellular content of cathepsin D in Alzheimer's disease brain: evidence for early up‐regulation of the endosomal‐lysosomal system. Neuron 14:671–680. [DOI] [PubMed] [Google Scholar]
- 5. de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires‐Jones TL, Hyman BT (2010) Caspase activation precedes and leads to tangles. Nature 464:1201–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eckermann K, Mocanu MM, Khlistunova I, Biernat J, Nissen A, Hofmann A et al (2007) The beta‐propensity of Tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J Biol Chem 282:31755–31765. [DOI] [PubMed] [Google Scholar]
- 7. Fein JA, Sokolow S, Miller CA, Vinters HV, Yang F, Cole GM, Gylys KH (2008) Co‐localization of amyloid beta and tau pathology in Alzheimer's disease synaptosomes. Am J Pathol 172:1683–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL et al (2003) Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci U S A 100:10032–10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garg S, Timm T, Mandelkow EM, Mandelkow E, Wang Y (2011) Cleavage of Tau by calpain in Alzheimer's disease: the quest for the toxic 17 kD fragment. Neurobiol Aging 32:1–14. [DOI] [PubMed] [Google Scholar]
- 10. Gotz J, Chen F, van Dorpe J, Nitsch RM (2001) Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293:1491–1495. [DOI] [PubMed] [Google Scholar]
- 11. Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM (2004) Synaptic changes in Alzheimer's disease: increased amyloid‐beta and gliosis in surviving terminals is accompanied by decreased PSD‐95 fluorescence. Am J Pathol 165:1809–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gylys KH, Fein JA, Yang F, Miller CA, Cole GM (2007) Increased cholesterol in Abeta‐positive nerve terminals from Alzheimer's disease cortex. Neurobiol Aging 28:8–17. [DOI] [PubMed] [Google Scholar]
- 13. Johnson GV, Seubert P, Cox TM, Motter R, Brown JP, Galasko D (1997) The tau protein in human cerebrospinal fluid in Alzheimer's disease consists of proteolytically derived fragments. J Neurochem 68:430–433. [DOI] [PubMed] [Google Scholar]
- 14. Kayed R (2010) Anti‐tau oligomers passive vaccination for the treatment of Alzheimer disease. Hum Vaccin 6:931–935. [DOI] [PubMed] [Google Scholar]
- 15. Kimura T, Ono T, Takamatsu J, Yamamoto H, Ikegami K, Kondo A et al (1996) Sequential changes of tau‐site‐specific phosphorylation during development of paired helical filaments. Dementia 7:177–181. [DOI] [PubMed] [Google Scholar]
- 16. Lasagna‐Reeves CA, Castillo‐Carranza DL, Guerrero‐Muoz MJ, Jackson GR, Kayed R (2010) Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49:10039–10041. [DOI] [PubMed] [Google Scholar]
- 17. Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A (2006) Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci Res 54:197–201. [DOI] [PubMed] [Google Scholar]
- 18. Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H et al (2007) Granular tau oligomers as intermediates of tau filaments. Biochemistry 46:3856–3861. [DOI] [PubMed] [Google Scholar]
- 19. Mak K, Yang F, Vinters HV, Frautschy SA, Cole GM (1994) Polyclonals to beta‐amyloid(1‐42) identify most plaque and vascular deposits in Alzheimer cortex, but not striatum. Brain Res 667:138–142. [DOI] [PubMed] [Google Scholar]
- 20. Nakano H, Kobayashi K, Sugimori K, Shimazaki M, Miyazu K, Hayashi M, Furuta H (2004) Regional analysis of differently phosphorylated tau proteins in brains from patients with Alzheimer's disease. Dement Geriatr Cogn Disord 17:122–131. [DOI] [PubMed] [Google Scholar]
- 21. Nixon RA (2000) A “protease activation cascade” in the pathogenesis of Alzheimer's disease. Ann N Y Acad Sci 924:117–131. [PubMed] [Google Scholar]
- 22. Nixon RA (2005) Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol Aging 26:373–382. [DOI] [PubMed] [Google Scholar]
- 23. Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM (2006) Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem 281:39413–39423. [DOI] [PubMed] [Google Scholar]
- 24. Park SY, Ferreira A (2005) The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta‐amyloid‐induced neurodegeneration. J Neurosci 25:5365–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L et al (2011) Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem 286:23063–23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rapoport SI (2002) Hydrogen magnetic resonance spectroscopy in Alzheimer's disease. Lancet Neurol 1:82. [DOI] [PubMed] [Google Scholar]
- 27. Rissman RA, Poon WW, Blurton‐Jones M, Oddo S, Torp R, Vitek MP et al (2004) Caspase‐cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest 114:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roberson ED, Scearce‐Levie K, Palop JJ, Yan F, Cheng IH, Wu T et al (2007) Reducing endogenous tau ameliorates amyloid beta‐induced deficits in an Alzheimer's disease mouse model. Science 316:750–754. [DOI] [PubMed] [Google Scholar]
- 29. Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen SH, Takashima A (2007) Assembly of two distinct dimers and higher‐order oligomers from full‐length tau. Eur J Neurosci 25:3020–3029. [DOI] [PubMed] [Google Scholar]
- 30. Schmid I, Uittenbogaart CH, Giorgi JV (1991) A gentle fixation and permeabilization method for combined cell surface and intracellular staining with improved precision in DNA quantification. Cytometry 12:279–285. [DOI] [PubMed] [Google Scholar]
- 31. Sokolow S, Henkins KM, Bilousova T, Miller CA, Vinters HV, Poon W et al (2011) AD synapses contain abundant Abeta monomer and multiple soluble oligomers, including a 56‐kDa assembly. Neurobiol Aging (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takahashi RH, Capetillo‐Zarate E, Lin MT, Milner TA, Gouras GK (2010) Co‐occurrence of Alzheimer's disease β‐amyloid and tau pathologies at synapses. Neurobiol Aging 31:1145–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang YP, Biernat J, Pickhardt M, Mandelkow E, Mandelkow EM (2007) Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer‐like aggregation of full‐length tau in a neuronal cell model. Proc Natl Acad Sci U S A 104:10252–10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou XW, Li X, Bjorkdahl C, Sjogren MJ, Alafuzoff I, Soininen H et al (2006) Assessments of the accumulation severities of amyloid beta‐protein and hyperphosphorylated tau in the medial temporal cortex of control and Alzheimer's brains. Neurobiol Dis 22:657–668. [DOI] [PubMed] [Google Scholar]