

Background: There is little understanding of the mechanisms that explain why testicular germ cell tumors respond so well to chemotherapy.
Results: Noxa is transcriptionally activated by KLF6 and TAp73 and repressed by Sp1 and ΔNp73, and its protein levels correlate with clinical prognosis.
Conclusion: p73 isoforms and Sp1-like factors form a cisplatin-induced transcriptional network that regulate proapoptotic Noxa.
Significance: This might help understand the transcriptional pathways of chemosensitivity.
Keywords: Apoptosis, Bcl-2 Family Proteins, Cancer Biology, Chemoresistance, Transcription Regulation, Noxa, Sp1-like Factors, Embryonal Carcinoma
Abstract
Testicular germ cell tumors (TGCTs) are highly responsive to and curable by cisplatin-based chemotherapy even in advanced stages. We have studied the molecular mechanisms involved in the induction of apoptosis in response to cisplatin, and found that proapoptotic Noxa is transcriptionally up-regulated following cisplatin exposure, even in the absence of p53, in NTERA2 cisplatin-sensitive cells but not in 1411HP-resistant cells. Blockade of Noxa reduced the apoptotic response of embryonal carcinoma (EC) NTERA2 cells to cisplatin. A detailed analysis of the Noxa promoter revealed that p73 and Sp1-like factors, Sp1 and KLF6, played key roles in the transcriptional control of this gene. Overexpression of TAp73 induced Noxa whereas the dominant negative isoform ΔNp73, reduced the levels of Noxa after cisplatin exposure in NTERA2 and 2102EP. Interestingly, down-regulation of Sp1 increased Noxa expression in response to cisplatin. However, blockade of KLF6 decreased cisplatin-induced up-regulation of Noxa in EC cell lines. In addition, tissue microarray analyses of TGCTs revealed that expression of Noxa correlates with good clinical prognosis in patients with embryonal carcinoma. Thus, our data show the transcriptional network that regulates Noxa in EC cells, which is key for their apoptotic response to cisplatin-based chemotherapy, and propose Noxa as a predictive factor of therapeutic response.
Introduction
Although conventional chemotherapy is often regarded as a sub-optimal treatment strategy, with high toxicity and less than curative outcomes in common adult cancers, there are several well documented situations, such as germ-line or pediatric cancers in which these therapies can provide complete cures (1). For example, cisplatin cures most patients with advanced testicular cancer and has been the standard treatment for decades (2, 3). Perhaps because it has been so successful, few studies of this drug response are being conducted. Yet, knowing why this drug and others can produce such efficient cures could illuminate the search for similar opportunities for cancers that are less successfully treated.
Cisplatin is an antineoplastic in the class of alkylating agents that has been shown to induce apoptotic cell death (4). Apoptosis is a tightly regulated and highly efficient cell death system that requires specialized molecular machinery. A key component of this machinery is the Bcl-2 family of apoptosis regulators. Whether a cell dies in response to diverse apoptotic stimuli, including DNA-damaging agents, is determined largely by interactions between antiapoptotic (Bcl-2-like) and proapototic (BH3-only and Bax-like) members of this family (5). A death signal is transmitted through the BH3-only proteins to Bax and Bak which in turn permeabilize the outer mitochondrial membrane allowing the release of apoptogenic factors (6). The tumor suppressor p53 is one of the main transcription factors involved in the regulation of proapoptotic Bcl-2 family members. This protein plays a pivotal role in the decision of whether the outcome of DNA damage will be growth arrest or apoptosis, and this choice is mainly based on its transcriptional activity. Among the BH3-only genes, puma, noxa, and bik have been shown to be induced by the p53 pathway (7–9). Noxa plays a main role in fine-tuning cell death decisions by targeting the prosurvival molecule Mcl-1 for proteasomal degradation. This event appears to be critical for cell death induction in response to factor deprivation or DNA damage (10). Besides p53, Noxa has been shown to be induced by other transcription factors, including HIF1α, E2F1 and p73 (11–13). Although hypoxia-induced expression of Puma is mainly dependent on p53, Noxa has been described as a direct target of HIF1α, and mediates hypoxic cell death in a p53-independent manner in various normal and malignant tissues. Ectopic expression of E2F1 can also trigger apoptosis through direct binding to a consensus E2F1-binding site in the Noxa promoter. In addition, p73 is able to bind to p53 consensus sites, and it promotes the expression of Noxa in different tumor cells (13, 14). Although p53 has been proposed as a mediator of the sensitivity of testicular germ cell tumor (TGCT)2 cells to cisplatin, high levels of p53 does not correlate directly to treatment sensitivity of these tumors, and inactivation of p53 is not a common event in the development of cisplatin resistance (15, 16). Moreover, a subgroup of TGCT patients with embryonal carcinoma and a tumor profile with low p53, showed better survival than that of the overall TGCT patient group (17). These observations support a multifactorial basis for the chemosensitivity of TGCTs (18). Cisplatin has been shown to induce Noxa expression in a number of cell systems including ovarian cancer, neuroectodermal tumors, and TGCTs (19–21), which strongly correlates with an apoptotic response of cancer cells. However, the transcriptional mechanisms involved in the cisplatin-dependent regulation of this proapoptotic gene are still controversial. Here, we have used TGCT cell lines to demonstrate that the key role that Noxa plays in transducing the apoptotic response to cisplatin is controlled by a balance between transcriptional activation mediated by p73 and KLF6 and repression by Sp1. Moreover, by using immunohistochemistry on tissue microarrays, we show for the first time that the expression of Noxa correlates with clinical prognosis in embryonal carcinoma patients.
EXPERIMENTAL PROCEDURES
Cell Lines
The germ cell tumor cell lines with an embryonal carcinoma phenotype, 2102Ep, TERA1, and NTERA2, a yolk sac carcinoma line, 1411HP, and a malignant teratoma cell line, 577MF, were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (Flow Laboratories, Irvine, CA). Drosophila Schneider SL-2 cells were grown in Shield and Sang DES (Drosophila-enriched Schneider) insect medium (Sigma-Aldrich) supplemented with 10% fetal calf serum.
Western Blot Analysis and Immunoprecipitation
Cell extracts (50 μg of protein) were separated on a 8% polyacrylamide gel, and transferred to nitrocellulose. Blots were blocked with 3% BSA and incubated with rabbit antibodies against KLF6, Sp1, Mcl-1, PARP, p53, and p73 (all from Santa Cruz Biotechnology, Santa Cruz, CA), or mouse monoclonal antibodies against Noxa (Millipore), Bad (BD Biosciences, San Diego, CA) and αTubulin, (Santa Cruz Biotechnology), followed by incubation with goat anti-rabbit or anti-mouse antibodies conjugated to alkaline phospatase (Sigma-Aldrich). Bound antibody was detected by a chemiluminescence system (Applied Biosystems, Foster City, CA). For protein immunoprecipitation, NTERA2 cells were lysed in 0.5% Nonidet P-40-containing solution as previously described (22). Cleared lysates were incubated with rabbit anti-Mcl-1 antibodies, and protein A/G conjugated to agarose beads (Santa Cruz Biotechnology). Proteins eluted from the agarose beads were electrophoresed and transferred to nitrocellulose. Membranes were incubated with the indicated antibodies.
Chromatin Immunoprecipitation (ChIP)
Purification of sonicated nuclear lysates and immunoprecipitation were performed as described elsewhere (23). Precipitates using anti-Sp1, anti-KLF6, anti-p73 (Santa Cruz Biotechnology), or irrelevant IgG were heated at 65 °C for at least 4 h to reverse the formaldehyde cross-linking. Proteins were digested in the presence of 20 μg/μl Proteinase K, and then DNA fragments were purified with a QIAquick Spin Kit (Qiagen, Valencia, CA). Quantitative PCR was performed using primers 5′-ACGTCCAGCGTTTGCAGATG-3′ and 5′-GACGTAGGGAAACTAGACGCC-3′ that generate a 258-bp fragment containing the Sp1 and p53 sites, and primers 5′-GGGATTACAGGCGTGAGAGA-3′ and 5′-GTGCTGTAGATCGATTGTGTTTG-3′ that generate a 127-bp fragment, 3 kb further upstream, used as a negative control of immunoprecipitation.
Electrophoretic Mobility Shift Assay (EMSA)
Cells were lysed and nuclear fractions were resuspended in 20 mm HEPES pH 7.9, 420 mm NaCl, 1 mm EDTA, 1 mm EGTA, and 20% glycerol. Nuclear extracts (10 μg of total protein) were incubated with a 32P-labeled double stranded DNA probe corresponding to a sequence from the Noxa promoter that contained the Sp1 binding site (5′-GCGGGGCGGGGACAGGGGCGGGGACAGGGGCGGGCC-3′). Samples were run on a 5% non-denaturing polyacrylamide gel in 200 mm Tris borate, 2 mm EDTA. Gels were dried and visualized by autoradiography. Supershifts were performed using antibodies against Sp1 and GATA1 (used here as an irrelevant antibody) (both from Santa Cruz Biotechnology).
Analyses of mRNA Expression
Total RNA from germ cell tumor cell lines was extracted using the RNeasy mini kit (Qiagen). To assess the expression of individual genes, a cDNA was generated and amplified by using primers for human noxa (5′-ATGCTGCGTTTCACCAGGG-3′) and (5′-TCCATGCTACTTGCACTTGTTCCT-3′), bim (5′-CCAGCACCCATGAGTTGTGAC-3′, and 5′-TGGTCTTCGTGCTGCTTGG-3′), and β-actin (5′-GCGGGAAATCGTGCGTGACATT-3′, and 5′-GATGGAGTTGAAGGTAGTTTCGTG-3′). Quantitative real-time PCR was performed in a 7000 Sequence Detection System (Applied Biosystems).
Cell Viability Assays
Viability in response to cisplatin was analyzed by using the WST-1 reagent (Roche Applied Science, Indianapolis, IN) that is cleaved to formazan dye by mitochondrial dehydrogenases, which directly correlates to the number of metabolically active cells in the culture. Apoptosis was determined with a DeadEnd fluorometric TUNEL system (Promega Corp., Madison, WI) following the manufacturer's recommendations.
Transfection Experiments and Gene Reporter Assays
A genomic PCR fragment from the promoter region of Noxa, containing the sequence from −1292 to +85 bp relative to the transcription start site, was cloned into KpnI and XhoI sites of the pGL2-basic luciferase reporter vector (Promega). The authenticity of the construct was confirmed by sequencing. Serial deletions were obtained by digestion with NsiI (cuts at position −892), StuI (cuts at position −423) or by PCR with the following primers that contained a KpnI sequence at the 5′ end, 5′-GAGGTACCAAGTAATTTCGGGGCCGAGC-3′ (for a fragment starting at −256), 5′-GAGGTACCTCGCTGCTCAGCGGGGTACTT-3′ (for a fragment starting at −177), 5′-GAGGTACCTCCTGCTCCCATAACGCCGTCT-3′ (for a fragment starting at −139), 5′-GAGGTACCTAGTTTCCCTACGTCACCAG-3′ (for a fragment starting at −75), 5′-GAGGTACCCTGGACAAAAGCGTGGTCTC-3′ (for a fragment starting at −44). In all PCRs, the same antisense primer (5′-GACTCGAGTGAACACGAACAGTCCTGCA-3′) with a XhoI site at the 5′-end was used. Cell lines were co-transfected with 1 μg of pGL2 vector containing the different promoter fragments and 50 ng of pRSV-β-gal by lipofection using Superfect (Qiagen). When indicated, cells were co-transfected with 1 μg of pGL2-Noxa-promoter and 1 μg of expression plasmids containing TAp73, ΔNp73 (24), KLF6, or KLF6 Sv2 (25). 24 h post-transfection, cell extracts were prepared and analyzed for the relative luciferase activity by a dual-light reporter gene assay system (Applied Biosystems). Results were normalized for transfection efficiency with values obtained with pRSV-β-gal. Site-directed mutagenesis was carried out by using the QuickChange Site-directed mutagenesis kit (Stratagene, La Jolla, CA) with the following primers to mutate Egr1 (5′-CCGAGCTGCTCTGCTCCAAACGCCGCGGGTCG-3′), p53 (5′-CGCCGCGGGTCGGTGCTAGGGTCCGGGCAGGTCG-3′) and Sp1 (5′-CGGGATGGGGACAGGGATGGGGACAGGGATGGGCCGGG-3′) sites (changes are underlined). In all cases, DNA inserts were sequenced to verify the mutation.
Gene Silencing
Cells were cultured at a density of 1 × 106 cells/ml and transfected with shRNA plasmids specific for Noxa, p53 and Sp1, or control scrambled shRNA containing plasmid (MISSION shRNA library, Sigma-Aldrich) using Lipofectamine or nucleofection (Amaxa, Cologne, Germany) for 24 h.
Prognostic Groups of Patients
A retrospective cohort of 75 consecutive male patients with metastatic germ cell tumors treated at the Catalan Institute of Oncology from August 1989 to November 2004 were included in this study. Clinical prognosis was classified according to the International Germ Cell Cancer Collaborative Group (26) as good prognosis (60%), defined by complete or partial response by imaging procedures (computed tomography-scan) and negative serum tumor markers; bad prognosis (21%), defined by partial response or stabilization of disease and positive tumor markers (this group includes 41.6% of resistant cases); and intermediate prognosis (19%). All patients were treated with cisplatin-based chemotherapy following the Spanish Germ Cell Group protocol, and they were considered resistant when progression or relapse occurred despite adequate initial treatment.
Tissue Microarray
Formalin-fixed paraffin-embedded tissue samples of representative tumor regions from primary tumors and/or resected metastasis were collected for the preparation of eight tissue microarrays. Briefly, three tissue cylinders with a diameter of 1.0 mm were punched from morphologically representative areas of each donor tissue block and brought into a recipient paraffin block using a manual tissue arrayer. For mixed tumors, three cylinders of each component were included.
Immunohistochemistry and Scoring
Paraffin-embeded sections were deparaffinized in xylene and rehydrated in downgraded alcohols and distilled water. Then, tissue microarrays were incubated with anti-Noxa antibodies (OP180, Millipore), followed by a specific secondary antibody using the DAB Map detection kit (Ventana Medical Systems, Tucson, AR). Sections were counterstained with hematoxylin and analyzed by light microscopy. For immunoreactivity, a previously reported 0–3 semi-quantitative scoring system for both the intensity of stain and the percentage of positive cells (labeling frequency percentage) was used (27). For intensity of stain, each case was visually compared with both positive and negative controls. The grading scale ranged from no detectable signal (0 points), to strong signal seen at low power (3 points). Labeling frequency was scored as 0 (0%), 1 (1 to 33%), 2 (34 to 66%), or 3 (67–100%). The multiplicative index of intensity and labeling was considered for analysis. Each image was interpreted by three independent observers (T.H., S.F., and E.de A.).
Statistical Analysis
All statistics were calculated with the SPSS statistical package (version 13.0). The Student's t test was used to compare continuous variables between two groups. The significance level was set at p < 0.05.
RESULTS
Noxa Expression and Resistance to Cisplatin in Germ Cell Tumor Cell Lines
We first studied the expression levels of Noxa in TGCT cell lines. As shown in Fig. 1A, 577ML4, NTERA2, TERA1, and 2102Ep cells expressed the Noxa protein, although the levels varied significantly between the different cell lines. Interestingly, 1411HP cells which are known to be resistant to cisplatin (28) expressed the lowest levels of Noxa. This expression pattern was also evident at the level of mRNA (Fig. 1B) suggesting a correlation between the transcriptional control of this proapoptotic gene and the cellular response to cisplatin. Another BH3-only gene, Bim, did not show expression differences in 1411HP cells with respect to other cell lines (Fig. 1B). The best known function of Noxa is to bind to and neutralize the antiapoptotic activity of Mcl-1. We verified the presence of this protein complex in our cell system by immunoprecipitation experiments. An antibody against Mcl-1 was able to co-immunoprecipitate Noxa, but not the BH3-only protein Bad (Fig. 1C). Of note, the Mcl-1 protein was absent or expressed at very low levels in 1411HP cell line (Fig. 1A). Next, we determined whether the expression of Noxa correlated with the apoptotic response of cells to cisplatin. A TUNEL assay showed that the proportion of apoptotic cells was higher than 50% in both NTERA2 and 2102Ep cell lines but less than 9% in 1411HP after 24h of treatment with cisplatin (Fig. 2A). This result was confirmed by analyzing the digestion of PARP, a substrate of caspases. Following cisplatin exposure, the cleavage product of PARP was readily detected in NTERA2 and 2102Ep cell lines. However, only a small proportion of PARP was digested even after 48 h of incubation of 1411HP cells in the presence of cisplatin (Fig. 2B). These data correlated with the induction (about 3-fold) of Noxa in NTERA2 cells in response to cisplatin, in contrast to the low levels of Noxa protein in treated 1411HP cells, which were even lower than those in untreated NTERA2 (Fig. 2C). In order to verify that the expression of Noxa in response to cisplatin was transcriptionally regulated, NTERA2 and 1411HP cells were transfected with a luciferase reporter driven by the noxa promoter and then treated with cisplatin. After 3 h of exposure to the chemotherapeutic drug, the luciferase activity increased about 10 times in NTERA2 (Fig. 2D). However, no variations were observed in 1411HP under the same experimental conditions.
FIGURE 1.

Noxa expression in germ cell tumor cell lines. The expression levels of the indicated Bcl-2 family members were determined by A, Western blot analysis and B, quantitative RT-PCR. C. Cell extracts from NTERA2 were analyzed by immunoprecipitation (IP) with antibodies against Mcl-1 at two different concentrations, and by subsequent blotting with antibodies specific for Mcl-1, Noxa, and Bad. Histograms represent the mean ± S.D. of three independent experiments.
FIGURE 2.

1411HP cells are resistant to cisplatin. A, TGCT cell lines were treated with 12 μm cisplatin for 24 h and then cell death was determined by cytometry using TUNEL assay. Percentages of apoptosis are indicated. B, cisplatin-induced apoptosis was also determined by Western blot analysis of PARP cleavage at 24 h or at the indicated time points. C, NTERA2 and 1411HP cells were treated with cisplatin and the protein expression of Noxa was analyzed by Western blot. The levels of α-tubulin were also analyzed to assure equal loading. D, NTERA2 and 1411HP were transfected with a luciferase reporter plasmid containing the Noxa promoter. Following treatment with cisplatin, cell extracts were analyzed for the relative luciferase activity. Histograms represent the mean ± S.D. of three independent experiments.
Down-regulation of Noxa Reverts Resistance to Cisplatin
To determine the functional relevance of Noxa in the apoptotic response of TGCT cells to cisplatin, we blocked the expression of Noxa in NTERA2 cells by an interference RNA strategy. Two out of five Noxa-targeted shRNAs promoted a significant reduction of Noxa protein levels. One of these, sh2-Noxa, almost abrogated the expression of the BH3-only protein (Fig. 3A). Cells transfected with sh2-Noxa were more resistant (about two times more viable cells) to cisplatin than control transfected cells, reducing the cell viability to almost 20% (Fig. 3B). Similar but less pronounced effects in cell viability were observed when cells were transfected with sh3-Noxa, and no significant difference with respect to controls were detected in cells transfected with the other three shRNAs (data not shown). These results indicate that Noxa contributes to the sensitivity of TGCT cells to cisplatin and provides a molecular rationale to explain the good therapeutic response of testicular germ cell tumors.
FIGURE 3.
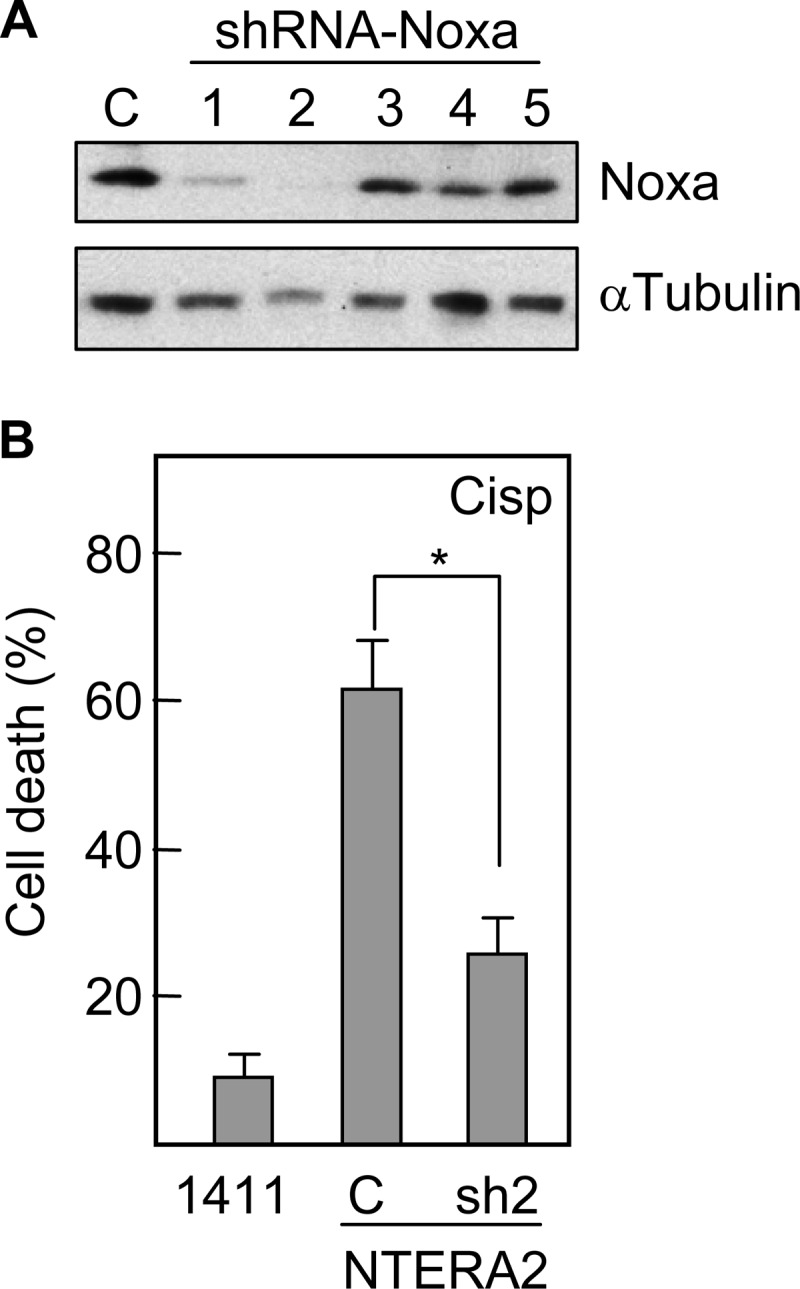
Down-regulation of Noxa reduces the sensitivity of NTERA2 cells to cisplatin. A, NTERA2 cells were transfected with different shRNA-containing plasmids specific for Noxa and the levels of the targeted protein were analyzed by Western blot. B, participation of Noxa in TGCT cell chemosensitivity was determined by comparing viabilities of Noxa knockdown cells (sh2) and mock-transfected cells (C) after treatment with cisplatin. 1411HP cells were included as a control of chemoresistance. Cell viability was analyzed by using the WST-1 reagent. Asterisk represents significant differences (p < 0.001). Histograms represent the mean ± S.D. of three independent experiments.
Analysis of Regulatory Elements in the Noxa Promoter
Based on our previous findings, we looked for transcription factor binding sites in the noxa promoter that were functional in TGCT cells. We found consensus binding sequences for HIF1α, p53, Egr1, Sp1, and E2F1 transcription factors within 1.3 kb upstream from the transcription start site (Fig. 4A). Restriction fragments of the promoter were sequentially obtained and cloned into a luciferase reporter construct. Luciferase assays demonstrated that the promoter activity was located in a fragment of 423 bp that contained Egr1, p53, Sp1, and E2F1 sites (Fig. 4B). Loss of a sequence from −423 to −177 caused a 5-fold reduction in luciferase activity indicating the relevance of Egr1 and/or p53 sites to the expression of Noxa in NTERA2 cells. Further deletion to position −75 which eliminated the Sp1 site but maintained the E2F1 sequence, completely abrogated the activity of the promoter. To further narrow down the sequence containing the active binding sites, PCR fragments from positions −256 and −139 were generated and the corresponding luciferase reporter constructs were transfected into NTERA2 and 2102Ep cells. The biggest fragment retained the activity of the original 1.3 kb promoter sequence, and this activity was drastically reduced when the promoter contained a deletion up to position −139 (Fig. 4C). Again, luciferase activity was abolished in both cell lines transfected with a construct containing the first 75 nucleotides of the promoter. These data confirmed that Egr1 and/or p53, and Sp1 sites were important for the activity of Noxa promoter. To identify the contribution of each one of these three sites, we eliminated their binding capacity by changing key nucleotides from their consensus sequences. Mutation of Egr1 did not modify the levels of luciferase activity (Fig. 4D). However, the capacity of the promoter to induce luciferase was significantly reduced following mutation of p53 (about 12-fold reduction) and Sp1 (about 3-fold reduction) binding sites.
FIGURE 4.

Sp1 and p53 sites in the Noxa promoter are necessary for transactivation in TGCT cells. A, scheme of the Noxa promoter showing the consensus sites for different transcription factors. Numbering starts at the first nucleotide upstream from the transcription start site (TSS). B and C, NTERA2 and 2102Ep cells were transfected with a luciferase reporter vector containing sequential deletion fragments of the Noxa promoter. Following 24 h of transfection, cell extracts were prepared and analyzed for the relative luciferase activity. D, reporter vector containing mutant Egr1, p53, or Sp1 site was introduced into NTERA2 cells and luciferase activity was determined. Results were normalized for transfection efficiency with values obtained with pRSV-β-gal. Histograms represent the mean ± S.D. of three independent experiments.
p73 and Sp1 Play Key Roles in the Cisplatin-induced Up-regulation of Noxa
To further explore the contribution of each transcription factor to the induction of Noxa under apoptosis-inducing conditions, we inhibited the expression of p53 or Sp1 in NTERA2 cells by using interference RNAs and determined the expression of Noxa protein in response to cisplatin. p53, which has been shown to promote the expression of Noxa in different cell systems (10), was clearly induced in control cells but not in cells transfected with a p53-specific shRNA (Fig. 5A). Surprisingly, the levels of Noxa were very similar in both cell populations following exposure to the drug. However, consistent with the apoptotic role of p53, cells defective in this transcription factor were less responsive to cisplatin (cell death decreased from 61% in control cells to 37% in p53-defective cells) (Fig. 5B), which suggests that p53 may contribute to apoptotic cell death through Noxa-independent mechanisms. Another p53 family member, p73, shares a remarkable homology in the sequence-specific DNA binding domain with p53 (29) and is able to bind to p53 consensus sites and to activate p53 target genes. ChIP analyses demonstrated the binding of p73 to the corresponding site in the Noxa promoter, which was further enhanced (about 3-fold increase) in the presence of cisplatin (Fig. 5C). The protein levels of both, transcriptionally active (TA) and dominant negative (ΔN) isoforms of p73 were either unchanged or slightly down-regulated following treatment with cisplatin (Fig. 5A). To study the contribution of p73 in this transcriptional system, cells were transfected with TAp73 or the truncated isoform, ΔNp73. Overexpression of TAp73 induced Noxa protein, whereas functional blockade of p73 reduced the expression of Noxa (Fig. 5D). Moreover, TAp73 promoted an increase (about 2-fold) in luciferase activity when this factor was cotransfected with a Noxa promoter-luciferase reporter construct into p53-defective NTERA2 cells. On the contrary, ΔNp73 caused a 4-fold reduction in luciferase activity (Fig. 5E). Consistent with these results, ΔNp73 reduced cisplatin-induced cell death from 42% in control cells to 17% in ΔNp73-tranfected cells (Fig. 5F), and decreased the upregulatioin of Noxa mRNA levels in response to cisplatin (Fig. 5G). In order to confirm these data in another embryonal carcinoma cell line, we transfected 2102Ep cells with ΔNp73 and found that these cells also had a reduced capacity to up-regulate Noxa (Fig. 5H) and to undergo apoptosis (data not shown) following treatment with cisplatin. These data indicate a direct participation of p73 in the expression of Noxa and therefore, in the response of NTERA2 cells to cisplatin.
FIGURE 5.

Cisplatin-induced expression of Noxa is dependent on p73 but not p53 in embryonal carcinoma cells. NTERA2 cells transfected with a vector expressing a shRNA targeted against p53 were treated with cisplatin and A, the expression levels of Noxa, p53 and p73 isoforms were determined by Western blot analysis, and B, cell death was determined following 24 h of treatment. C, ChIP analysis of the association of p73 with the Noxa promoter in the presence or in the absence of cisplatin. Immunoprecipitates were analyzed by quantitative PCR using primers specific to the target site or a 3 kb upstream region. Fold enrichment is expressed relative to IgG immunoprecipitates. D, protein levels of Noxa were analyzed in NTERA2 cells overexpressing an active (TA) or a dominant negative form (ΔN) of p73. Samples were run on the same gel but not in consecutive lanes. The levels of α-tubulin were also analyzed to assure equal loading. E, p53 knockdown cells were transfected with TAp73 or ΔNp73 and a luciferase-Noxa promoter vector and then analyzed for luciferase activity. F, p53 knockdown cells transfected with ΔNp73 were treated with cisplatin and 24 h later cell death was quantified. G, p53 knockdown NTERA, and H, 2102EP cells were transfected with ΔNp73 and treated with cisplatin for 24 h. The mRNA levels of Noxa were determined by quantitative RT-PCR. *, p < 0.01; **, p < 0.001. Histograms represent the mean ± S.D. of three independent experiments.
Next, we knocked down Sp1 transcription factor (Fig. 6A). Specific shRNAs virtually eliminated the expression of Sp1 but not of other member of the same protein family, KLF6. Unexpectedly, Noxa protein levels were increased. Consistent with this result, Sp1 deficiency caused a 5-fold increase in luciferase activity in cells cotransfected with the Noxa promoter-reporter construct (Fig. 6B), and induced a higher cell death response to cisplatin (mean of 80%) as compared with control cells (mean of 56%) (Fig. 6C). Moreover, the binding of Sp1 to its corresponding site in the Noxa promoter was reduced following cisplatin exposure as determined by EMSA (Fig. 6D), which correlates with a decrease in the protein levels of Sp1, but not of KLF6 (Fig. 6E). ChIP analyses of NTERA2 cells also revealed a decrease of Sp1 binding to the corresponding site in the Noxa promoter following treatment with cisplatin (Fig. 6F). Interestingly, KLF6, that has been reported as a candidate tumor suppressor gene in different cell systems, binds to the same site in the Noxa promoter and this binding increased in response to cisplatin (Fig. 6F). These data suggest a competition between Sp1 and KLF6 for binding to Noxa promoter.
FIGURE 6.

Sp1 is blocking the expression of Noxa. A, NTERA2 cells were transfected with shRNAs specific for Sp1 and the protein levels of Noxa, Sp1 and KLF6 were determined by Western blot. The levels of α-tubulin were also analyzed to assure equal loading. B, NTERA2 cells lacking Sp1 were transfected with a Noxa promoter-luciferase reporter vector. Following 24 h of transfection, cell extracts were analyzed for the relative luciferase activity. C, Sp1-deficient NTERA2 cells were treated with cisplatin and cell death was quantified 24 h later. C, cells transfected with an irrelevant shRNA-containing plasmid. *, p = 0.02; **, p < 0.001. D, NTERA2 cells were treated with cisplatin and the formation of protein-DNA binding complexes was analyzed by EMSA. Antibodies against Sp1 and irrelevant GATA1 were used for supershift. Arrowheads indicate the specific bands. E, proteins levels of Noxa, Sp1, and KLF6, with or without cisplatin exposure were determined by Western blotting. F, a ChIP assay was performed to study the in vivo binding of Sp1 and KLF6 to the promoter of Noxa in response to cisplatin. Cross-linked chromatin was incubated with the indicated antibodies. Immunoprecipitates were analyzed by quantitative PCR using primers specific to the target site or a 3 kb upstream region. Fold enrichment is expressed relative to IgG immunoprecipitates. Histograms represent the mean ± S.D. of three independent experiments. *, p < 0.05.
The Balance between KLF6 and Sp1 Determines the Transcriptional Control of Noxa
Sp1 belongs to a complex family of transcriptional regulators that include more than 20 Sp1-like proteins and Kruppel-like factors (KLF), and competition for DNA binding has been shown for different family members (30). Protein analysis showed that exogenous KLF6 failed to modify Noxa levels in wild type NTERA2 cells. However, in the absence of Sp1, KLF6 increased the expression of Noxa (Fig. 7A). The capacity of KLF6 to transactivate the Noxa promoter was first analyzed in Drosophila Schneider cells, which lack Sp1 and KLF6 (25). In these cells, exogenous KLF6 was able to induce expression of a luciferase gene driven by the Noxa promoter, in a dose-dependent manner (Fig. 7B). Moreover, KLF6 overexpression induced a slight increase (about 1.5-fold) in the activity of the Noxa promoter-luciferase construct in NTERA2 cells, but promoted a stronger activation of luciferase (about 4-fold) in cells lacking Sp1. However, overexpression of a transactivation defective variant of KLF6, Sv2, did not have any significant effect on the Noxa promoter activity (Fig. 7C). To study whether KLF6 was mediating at least in part, the transactivation of the Noxa promoter in response to cisplatin, we took advantage of the capacity of Sv2 to inhibit the binding of functionally competent KLF6. Luciferase assays showed that the alternatively spliced variant of KLF6 induced an average of 40% reduction in the activity of the Noxa promoter following treatment with cisplatin (Fig. 7D), and consistently Sv2 decreased the cisplatin-induced up-regulation of Noxa mRNA levels in both NTERA2 (Fig. 7E) and 2102EP (Fig. 7F) cell lines. Thus, KLF6 promotes the transcriptional activation of Noxa in response to chemotherapy in embryonal carcinoma cells, and its relative contribution depends on the endogenous levels of Sp1, which is acting as a transcriptional repressor of Noxa in these tumor cells.
FIGURE 7.

KLF6 is a transcriptional activator of Noxa in NTERA2 cells. A, wild type and Sp1-deficient cells were transfected with KLF6 and the levels of Noxa protein were analyzed. The expression of α-tubulin was also analyzed to assure equal loading. B, Drosophila Schneider cells were cotransfected with the Noxa promoter-luciferase construct and increasing amounts of a KLF6-containing plasmid and then, luciferase activity was determined. C, NTERA2 cells were transfected with the Noxa promoter-luciferase construct and the indicated plasmids, and the promoter activation was determined by analyzing luciferase activity. D, NTERA2 cells cotransfected with the Noxa promoter and Sv2 in the presence or in the absence of cisplatin. After 3 h of treatment luciferase activity was analyzed. E, NTERA2 and F, 2102Ep cells were cotransfected with the Noxa promoter and Sv2. The levels of Noxa mRNA in response to cisplatin were determined by quantitative RT-PCR. Asterisk, significant differences (p < 0.01). Histograms represent the mean ± S.D. of three independent experiments.
Noxa Protein Levels Correlate with Clinical Prognosis
To translate our previous data to a clinically relevant setting, we analyzed the expression levels of Noxa in 436 tissue sections from tumors of different histological types, including seminoma (18.2%), embryonal carcinoma (36%), teratoma (17.2%), yolk sac tumor (15.3%), and choriocarcinoma (13.3%) in tissue microarrays. We considered good, intermediate and bad prognostic groups, as classified by the International Germ Cell Cancer Collaborative Group (26). We also included a group of patients refractory to cisplatin-based chemotherapy, which have a poor prognosis. Clearly visible differences in Noxa expression were observed between tissue sections within the same histological type (Fig. 8, A and B). Notably, significant differences in the protein levels of Noxa between the prognostic groups were only observed in embryonal carcinoma, the same histological type as NTERA2. In this tumor, we found higher levels of Noxa in disease with good prognostic criteria (81 tissue sections) as compared with intermediate (p < 0.0001) and bad (p = 0.001) prognosis disease (16 and 25 tissue sections analyzed, respectively) (Fig. 8C). Comparison between the good prognostic group and the group of patients with refractory disease (35 sections) showed no significant difference (Fig. 8C), suggesting that alternative apoptosis regulatory pathways might be active in tumor cells refractory to chemotherapy. Overall, the extent and intensity of embryonal carcinoma tissue staining with Noxa was statistically decreased in cases with intermediate or low response to cisplatin-based chemotherapy.
FIGURE 8.

Noxa expression correlates with response to cisplatin-based chemotherapy in embryonal carcinoma. A and B, representative examples of negative and intensively positive staining for Noxa in embryonal carcinoma components of TGCT patients on a tissue microarray. C, quantification of Noxa levels in tumor tissue sections from patients with good (GP), intermediate (IP), or bad (BP) prognosis. RF, patients refractory to chemotherapy.
DISCUSSION
It is well established that certain cancers, including testicular carcinoma, Wilms tumor, and pediatric Hodgkin disease, are chemosensitive (31–33), even with drugs that show low efficacy in other tumors. However, there is little understanding of the underlying mechanisms that might explain why these cancers respond so well to chemotherapy. The National Cancer Institute has identified this issue as one of the provocative questions that need to be addressed to drive progress against cancer (34). Thus, unraveling the properties of cancers that render them susceptible to eradication by chemotherapy, might help understand how certain therapies work, or develop new approaches to treat chemoresistant tumors. We have focused on TGCTs as a model of chemosensitivity to cisplatin-based therapies. Cisplatin is a genotoxic agent and the p53 pathway is known to participate in regulating the cellular response to this drug. Using TGCT cell lines, we found a much higher basal expression of Noxa in cisplatin-sensitive cells (NTERA2, 2102Ep) than in chemoresistant cells (1411HP). Furthermore, cisplatin-induced apoptosis increased the levels of Noxa in chemosensitive cell lines, which is consistent with the strong up-regulation of Noxa by platinum compounds described in other cell models (35). Down-regulation of Noxa by interference strategies reverted the sensitivity to cisplatin indicating that this proapoptotic BH3-only protein plays a key role in the cellular response to cisplatin in TGCT cells. Although cisplatin is widely used to treat different tumors, induction of Noxa by this drug has not been observed in other tumor models such as non-small cell lung cancer cells (36). Other proapoptotic members of the BH3-only family, including Puma and Bim, have been associated with the apoptotic response to cisplatin (37, 38), which exemplifies the redundancy of this family of apoptosis regulators. To further understand the mechanisms that control the expression levels of Noxa in TGCT cells, we analyzed in detail the promoter of this gene and found that p53 and Sp1 sites but not HIF1α, Egr1, or E2F1 are essential for the transcriptional activity of the promoter. Interestingly, induction of Noxa was dependent on p73 but not p53, which is consistent with the observation that Noxa induction is key in determining apoptosis of different tumor cells in a p53-independent manner (20, 39). Although recent data suggested that p53 plays a major role in Noxa expression and the chemotherapeutic response of TGCT cell lines (21), other authors have showed that the unique sensitivity of TGCT cells to cisplatin could be attributed to p53-dependent and p53-independent pathways (40, 41). Even though, it is likely that the cellular context or clonal differences in the expression levels or activity of p53 and p73, which bind to the same response elements, may determine the contribution of each one to the activation of a target gene. In line with this, it has been proposed that regulation of p53 family protein activity depends on the relative expression of transcriptionally active proteins and inactive isoforms (42). An alternative explanation may reside in clonal differences in the expression of genes that could promote some degree of spontaneous differentiation (43), as short-term differentiation of NTERA2 cells causes a loss in p53-dependent hypersensitivity (21). In addition to p73, we found that the Sp1 site in the Noxa promoter participates in the transcriptional control of this gene. It has been shown that the apoptotic activity of butyrate in colon cancer cells requires a reduction of the Sp1 binding to its consensus site in the Bak promoter in favor of Sp3 binding (44). In NTERA2 cells, we found that knockdown of Sp1 correlates with up-regulation of Noxa and a stronger response to cisplatin, and that another member of the Sp1-like family, KLF6, is able to transactivate Noxa in competition with Sp1. Consistent with this, competition for DNA binding has been shown for some members of the family; for example, Sp1 competes with KLF4, KLF9, and KLF13 (30). Moreover, Sp1 and p73 appears to cooperatively activate apoptotic genes such as Puma, and caspase-3 (45, 46). However, we demonstrate that Sp1 may exert a transcriptional suppression of a gene involved in the apoptotic response to chemotherapeutic drugs. Accumulating evidence suggests that the tumor suppressor gene KLF6 and its dominant-negative splicing variants play important roles in the development and/or progression of cancer (47). A recent work described that a splice variant of KLF6, KLF6-SV1, targets Noxa for degradation in cancer cell lines (48). SV1 antagonizes wild type KLF6 function and may be acting as a tumor promoter gene (49). Therefore, we provide the first description of a competitive transcription factor binding mechanism formed by Sp1 and KLF6 that control a proapoptotic member of the Bcl-2 family. Finally, we showed for the first time that Noxa positivity of embryonal carcinoma components in TGCT tissue specimens correlated with a good response to cisplatin-containing chemotherapy. This result is consistent with our in vitro data in which Noxa induction plays a key role in determining the apoptotic response to cisplatin. However, no correlation was observed in a group of patients refractory to treatment, most likely due to activation of alternative pathways regulating apoptosis. In line with this, it has been recently described that cisplatin-resistance in testicular embryonal carcinoma cells correlates with high amounts of cytoplasmic p21, resulting in CDK2 inhibition and reduced levels of cisplatin-induced apoptosis (50). In conclusion, we demonstrate the relevance of a transcriptional network formed by p73 isoforms, Sp1, and KLF6 in the regulation of Noxa levels in TGCT cells, which mediates their apoptotic response to cisplatin. Moreover, immunoreactivity for Noxa in the embryonal carcinoma component was more often observed in good prognostic patients, thus providing a predictive marker of therapeutic response.
This work was supported by Instituto de Salud Carlos III Grants RD06/0020/0074 (to J. L. F.-L.), RD06/0020/0059 (to E. A.), RD06/0020/0017 (to M. D. D.) (Red Tematica de Investigacion Cooperativa en Cancer), PI11/00397 (to M. D. D.), and PI081491 (to X. G.); and a grant from Instituto de Formacion e Investigación Marqués de Valdecilla (IFIMAV), API2011-04 (to J. L. F.-L.).
- TGCT
- testicular germ cell tumor
- EC
- embryonal carcinoma
- KLF
- Kruppel-like factor.
REFERENCES
- 1. Guminski A. D., Harnett P. R., deFazio A. (2002) Scientists and clinicians test their metal-back to the future with platinum compounds. Lancet Oncol. 3, 312–318 [DOI] [PubMed] [Google Scholar]
- 2. Feldman D. R., Bosl G. J., Sheinfeld J., Motzer R. J. (2008) Medical treatment of advanced testicular cancer. JAMA 299, 672–684 [DOI] [PubMed] [Google Scholar]
- 3. de Wit R., Fizazi K. (2006) Controversies in the management of clinical stage I testis cancer. J. Clin. Oncol. 24, 5482–5492 [DOI] [PubMed] [Google Scholar]
- 4. Wang D. (2005) Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 4, 307–320 [DOI] [PubMed] [Google Scholar]
- 5. Yip K. W., Reed J. C. (2008) Bcl-2 family proteins and cancer. Oncogene 27, 6398–6406 [DOI] [PubMed] [Google Scholar]
- 6. Fernandez-Luna J. (2008) Regulation of pro-apoptotic BH3-only proteins and its contribution to cancer progression and chemoresistance. Cell Signal. 20, 1921–1926 [DOI] [PubMed] [Google Scholar]
- 7. Oda E., Ohki R., Murasawa H., Nemoto J., Shibue T., Yamashita T., Tokino T., Taniguchi T., Tanaka N. (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058 [DOI] [PubMed] [Google Scholar]
- 8. Nakano K., Vousden K. H. (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7, 683–694 [DOI] [PubMed] [Google Scholar]
- 9. Mathai J. P., Germain M., Marcellus R. C., Shore G. C. (2002) Induction and endoplasmic reticulum location of BIK/NBK in response to apoptotic signaling by E1A and p53. Oncogene 21, 2534–2544 [DOI] [PubMed] [Google Scholar]
- 10. Ploner C., Kofler R., Villunger A. (2008) Noxa: at the tip of the balance between life and death. Oncogene 27, S84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim J. Y., Ahn H. J., Ryu J. H., Suk K., Park J. H. (2004) BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1α. J. Exp. Med. 199, 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hershko T., Ginsberg D. (2004) Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J. Biol. Chem. 279, 8627–8634 [DOI] [PubMed] [Google Scholar]
- 13. Rocco J. W., Leong C. O., Kuperwasser N., DeYoung M. P., Ellisen L. W. (2006) p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 9, 45–56 [DOI] [PubMed] [Google Scholar]
- 14. Ruhul Amin A. R., Thakur V. S., Gupta K., Agarwal M. K., Wald D. N., Shin D. M., Agarwal M. L. (2012) N-(phosphonacetyl)-l-Aspartate induces TAp73-dependent apoptosis by modulating multiple Bcl-2 proteins: potential for cancer therapy. Oncogene doi: 10.1038/onc.2012.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kersemaekers A. M., Mayer F., Molier M., van Weeren P. C., Oosterhuis J. W., Bokemeyer C., Looijenga L. H. (2002) Role of P53 and MDM2 in treatment response of human germ cell tumors. J. Clin. Oncol. 20, 1551–1561 [DOI] [PubMed] [Google Scholar]
- 16. García-Velasco A., Durán I., García E., Tarón M., Ballestín C., Castellanos D., Cortés-Funés H., Paz-Ares L. (2012) Biological markers of cisplatin resistance in advanced testicular germ cell tumours. Clin. Transl. Oncol. 14, 452–457 [DOI] [PubMed] [Google Scholar]
- 17. Mazumdar M., Bacik J., Tickoo S. K., Dobrzynski D., Donadio A., Bajorin D., Motzer R., Reuter V., Bosl G. J. (2003) Cluster analysis of p53 and Ki67 expression, apoptosis, alpha-fetoprotein, and human chorionic gonadotrophin indicates a favorable prognostic subgroup within the embryonal carcinoma germ cell tumor. J. Clin. Oncol. 21, 2679–2688 [DOI] [PubMed] [Google Scholar]
- 18. Mayer F., Stoop H., Scheffer G. L., Scheper R., Oosterhuis J. W., Looijenga L. H., Bokemeyer C. (2003) Molecular determinants of treatment response in human germ cell tumors. Clin. Cancer Res. 9, 767–773 [PubMed] [Google Scholar]
- 19. Al-Bahlani S., Fraser M., Wong A. Y., Sayan B. S., Bergeron R., Melino G., Tsang B. K. (2011) P73 regulates cisplatin-induced apoptosis in ovarian cancer cells via a calcium/calpain-dependent mechanism. Oncogene 30, 4219–4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Armstrong J. L., Veal G. J., Redfern C. P., Lovat P. E. (2007) Role of Noxa in p53-independent fenretinide-induced apoptosis of neuroectodermal tumours. Apoptosis 12, 613–622 [DOI] [PubMed] [Google Scholar]
- 21. Gutekunst M., Oren M., Weilbacher A., Dengler M. A., Markwardt C., Thomale J., Aulitzky W. E., van der Kuip H. (2011) p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cisplatin. PLoS One 6, e19198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horita M., Andreu E. J., Benito A., Arbona C., Sanz C., Benet I., Prosper F., Fernandez-Luna J. L. (2000) Blockade of the Bcr-Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5-dependent expression of Bcl-xL. J. Exp. Med. 191, 977–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shang Y., Hu X., DiRenzo J., Lazar M. A., Brown M. (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103, 843–852 [DOI] [PubMed] [Google Scholar]
- 24. Fernandez-Garcia B., Vaqué J. P., Herreros-Villanueva M., Marques-Garcia F., Castrillo F., Fernandez-Medarde A., León J., Marín M. (2007) p73 cooperates with Ras in the activation of MAP kinase signaling cascade. Cell Death Differ. 14, 254–265 [DOI] [PubMed] [Google Scholar]
- 25. Botella L. M., Sanz-Rodriguez, Komi Y., Fernandez-L A., Varela E., Garrido-Martin E. M., Narla G., Friedman S. L., Kojima S. (2009) TGF-β regulates the expression of transcription factor KLF6 and its splice variants and promotes co-operative transactivation of common target genes through a Smad3-Sp1-KLF6 interaction. Biochem. J. 419, 485–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmoll H. J., Souchon R., Krege S., Albers P., Beyer J., Kollmannsberger C., Fossa S. D., Skakkebaek N. E., de Wit R., Fizazi K., Droz J. P., Pizzocaro G., Daugaard G., de Mulder P. H., Horwich A., Oliver T., Huddart R., Rosti G., Paz Ares L., Pont O., Hartmann J. T., Aass N., Algaba F., Bamberg M., Bodrogi I., Bokemeyer C., Classen J., Clemm S., Culine S., de Wit M., Derigs H. G., Dieckmann K. P., Flasshove M., Garcia del Muro X., Gerl A., Germa-Lluch J. R., Hartmann M., Heidenreich A., Hoeltl W., Joffe J., Jones W., Kaiser G., Klepp O., Kliesch S., Kisbenedek L., Koehrmann K. U., Kuczyk M., Laguna M. P., Leiva O., Loy V., Mason M. D., Mead G. M., Mueller R. P., Nicolai N., Oosterhof G. O., Pottek T., Rick O., Schmidberger H., Sedlmayer F., Siegert W., Studer U., Tjulandin S., von der Maase H., Walz P., Weinknecht S., Weissbach L., Winter E., Wittekind C. (2004) European consensus on diagnosis and treatment of germ cell cancer: a report of the European Germ Cell Cancer Consensus Group (EGCCCG). Ann. Oncol. 15, 1377–1399 [DOI] [PubMed] [Google Scholar]
- 27. Mora J., Rodríguez E., de Torres C., Cardesa T., Ríos J., Hernández T., Cardesa A., de Alava E. (2012) Activated growth signaling pathway expression in Ewing sarcoma and clinical outcome. Pediatr. Blood Cancer 58, 532–538 [DOI] [PubMed] [Google Scholar]
- 28. Mueller T., Voigt W., Simon H., Fruehauf A., Bulankin A., Grothey A., Schmoll H. J. (2003) Failure of activation of caspase-9 induces a higher threshold for apoptosis and cisplatin resistance in testicular cancer. Cancer Res. 63, 513–521 [PubMed] [Google Scholar]
- 29. Kaghad M., Bonnet H., Yang A., Creancier L., Biscan J. C., Valent A., Minty A., Chalon P., Lelias J. M., Dumont X., Ferrara P., McKeon F., Caput D. (1997) Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90, 809–819 [DOI] [PubMed] [Google Scholar]
- 30. Kaczynski J., Cook T., Urrutia R. (2003) Sp1- and Kruppel-like transcription factors. Genome Biol. 4, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hussain S. A., Ma Y. T., Palmer D. H., Hutton P., Cullen M. H. (2008) Biology of testicular germ cell tumors. Expert Rev. Anticancer Ther 8, 1659–1673 [DOI] [PubMed] [Google Scholar]
- 32. Davidoff A. M. (2009) Wilms' tumor. Curr. Opin. Pediatr 21, 357–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schellong G., Pötter R., Brämswig J., Wagner W., Prott F. J., Dörffel W., Körholz D., Mann G., Rath B., Reiter A., Weissbach G., Riepenhausen M., Thiemann M., Schwarze E. W. (1999) High cure rates and reduced long-term toxicity in pediatric Hodgkin's disease: the German-Austrian multicenter trial DAL-HD-90. The German-Austrian Pediatric Hodgkin's Disease Study Group. J. Clin. Oncol. 17, 3736–3744 [DOI] [PubMed] [Google Scholar]
- 34. Printz C. (2011) NCI explores “provocative questions”: seeks to address the less researched aspects of cancer. Cancer 117, 4337–4338 [DOI] [PubMed] [Google Scholar]
- 35. Sheridan C., Brumatti G., Elgendy M., Brunet M., Martin S. J. (2010) An ERK-dependent pathway to Noxa expression regulates apoptosis by platinum-based chemotherapeutic drugs. Oncogene 29, 6428–6441 [DOI] [PubMed] [Google Scholar]
- 36. Voortman J., Checinska A., Giaccone G., Rodriguez J. A., Kruyt F. A. (2007) Bortezomib, but not cisplatin, induces mitochondria-dependent apoptosis accompanied by up-regulation of noxa in the non-small cell lung cancer cell line NCI-H460. Mol. Cancer Ther. 6, 1046–1053 [DOI] [PubMed] [Google Scholar]
- 37. Wang J., Zhou J. Y., Wu G. S. (2011) Bim protein degradation contributes to cisplatin resistance. J. Biol. Chem. 286, 22384–22392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang M., Wei Q., Wang J., Du Q., Yu J., Zhang L., Dong Z. (2006) Regulation of PUMA-α by p53 in cisplatin-induced renal cell apoptosis. Oncogene 25, 4056–4066 [DOI] [PubMed] [Google Scholar]
- 39. Qin J. Z., Stennett L., Bacon P., Bodner B., Hendrix M. J., Seftor R. E., Seftor E. A., Margaryan N. V., Pollock P. M., Curtis A., Trent J. M., Bennett F., Miele L., Nickoloff B. J. (2004) p53-independent NOXA induction overcomes apoptotic resistance of malignant melanomas. Mol. Cancer Ther. 3, 895–902 [PubMed] [Google Scholar]
- 40. Schweyer S., Soruri A., Meschter O., Heintze A., Zschunke F., Miosge N., Thelen P., Schlott T., Radzun H. J., Fayyazi A. (2004) Cisplatin-induced apoptosis in human malignant testicular germ cell lines depends on MEK/ERK activation. Br. J. Cancer 91, 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burger H., Nooter K., Boersma A. W., van Wingerden K. E., Looijenga L. H., Jochemsen A. G., Stoter G. (1999) Distinct p53-independent apoptotic cell death signalling pathways in testicular germ cell tumour cell lines. Int. J. Cancer 81, 620–628 [DOI] [PubMed] [Google Scholar]
- 42. Malaguarnera R., Vella V., Vigneri R., Frasca F. (2007) p53 family proteins in thyroid cancer. Endocr Relat Cancer 14, 43–60 [DOI] [PubMed] [Google Scholar]
- 43. Bahrami A. R., Matin M. M., Andrews P. W. (2005) The CDK inhibitor p27 enhances neural differentiation in pluripotent NTERA2 human EC cells, but does not permit differentiation of 2102Ep nullipotent human EC cells. Mech Dev. 122, 1034–1042 [DOI] [PubMed] [Google Scholar]
- 44. Chirakkal H., Leech S. H., Brookes K. E., Prais A. L., Waby J. S., Corfe B. M. (2006) Upregulation of BAK by butyrate in the colon is associated with increased Sp3 binding. Oncogene 25, 7192–7200 [DOI] [PubMed] [Google Scholar]
- 45. Ming L., Sakaida T., Yue W., Jha A., Zhang L., Yu J. (2008) Sp1 and p73 activate PUMA following serum starvation. Carcinogenesis 29, 1878–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sudhakar C., Jain N., Swarup G. (2008) Sp1-like sequences mediate human caspase-3 promoter activation by p73 and cisplatin. FEBS J. 275, 2200–2213 [DOI] [PubMed] [Google Scholar]
- 47. Huh S. J., Chen Y. L., Friedman S. L., Liao J., Huang H. J., Cavenee W. K., Robertson G. P. (2010) KLF6 Gene and early melanoma development in a collagen I-rich extracellular environment. J. Natl. Cancer Inst. 102, 1131–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Difeo A., Huang F., Sangodkar J., Terzo E. A., Leake D., Narla G., Martignetti J. A. (2009) KLF6-SV1 is a novel antiapoptotic protein that targets the BH3-only protein NOXA for degradation and whose inhibition extends survival in an ovarian cancer model. Cancer Res. 69, 4733–4741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Narla G., DiFeo A., Yao S., Banno A., Hod E., Reeves H. L., Qiao R. F., Camacho-Vanegas O., Levine A., Kirschenbaum A., Chan A. M., Friedman S. L., Martignetti J. A. (2005) Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 65, 5761–5768 [DOI] [PubMed] [Google Scholar]
- 50. Koster R., di Pietro A., Timmer-Bosscha H., Gibcus J. H., van den Berg A., Suurmeijer A. J., Bischoff R., Gietema J. A., de Jong S. (2010) Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J. Clin. Invest. 120, 3594–3605 [DOI] [PMC free article] [PubMed] [Google Scholar]