

Background: Nucleophosmin leukemia-associated domain binds G-quadruplex DNA.
Results: NMR structural analysis of the 70-residue nucleophosmin C-terminal domain and its interaction with G-quadruplex DNA from the c-MYC promoter was carried out.
Conclusion: The interaction involves helices H1 and H2 of the nucleophosmin terminal three-helix bundle mainly through electrostatic contacts with G-quadruplex phosphates.
Significance: Learning how nucleophosmin interacts with nucleic acids may be crucial in rescuing its function in leukemia.
Keywords: Cell Cycle, Leukemia, Myc, Nuclear Magnetic Resonance, Protein/DNA Interaction, B23, G-quadruplex, Nucleophosmin
Abstract
Nucleophosmin (NPM1) is a nucleocytoplasmic shuttling protein, mainly localized at nucleoli, that plays a key role in several cellular functions, including ribosome maturation and export, centrosome duplication, and response to stress stimuli. More than 50 mutations at the terminal exon of the NPM1 gene have been identified so far in acute myeloid leukemia; the mutated proteins are aberrantly and stably localized in the cytoplasm due to high destabilization of the NPM1 C-terminal domain and the appearance of a new nuclear export signal. We have shown previously that the 70-residue NPM1 C-terminal domain (NPM1-C70) is able to bind with high affinity a specific region at the c-MYC gene promoter characterized by parallel G-quadruplex structure. Here we present the solution structure of the NPM1-C70 domain and NMR analysis of its interaction with a c-MYC-derived G-quadruplex. These data were used to calculate an experimentally restrained molecular docking model for the complex. The NPM1-C70 terminal three-helix bundle binds the G-quadruplex DNA at the interface between helices H1 and H2 through electrostatic interactions with the G-quadruplex phosphate backbone. Furthermore, we show that the 17-residue lysine-rich sequence at the N terminus of the three-helix bundle is disordered and, although necessary, does not participate directly in the contact surface in the complex.
Introduction
Nucleophosmin (also called B23, numatrin, and herein NPM14) is a protein involved in a variety of crucial cellular functions, including ribosome maturation and export, centrosome duplication, and response to stress stimuli (1, 2). NPM1 is a highly mobile protein capable of shuttling between nucleus, nucleoplasm, and cytoplasm, although the bulk of the protein is mainly localized in the nucleoli (3).
The NPM1 gene, overexpressed in a number of solid tumors, has been proposed as a marker for colon, gastric, ovarian, and prostate carcinomas (1). NPM1 is also frequently modified in hematopoietic tumors. For instance, in both lymphoid and myeloid disorders, NPM1 chromosomal translocations lead to the production of several oncogenic fusion proteins (1). Furthermore, NPM1 is the most frequently mutated gene in acute myeloid leukemia, accounting for 35% of all cases (2, 4); over 50 different mutations, always heterozygous and largely associated to a normal karyotype, were discovered (3, 4). Mutations involve duplication or insertion of small base sequences at the last exon of the gene and lead to a C-terminal domain that has acquired four additional residues compared with wild type and a completely different sequence in the last seven. The nucleolar localization signal is compromised with the loss of one or both of the two critical tryptophan residues, and a third nuclear export signal appears. Moreover, the mutated domain is largely destabilized or totally unfolded (5–7). Both the destabilization of NPM1 C-terminal domain and the presence of an additional nuclear export signal contribute to the aberrant and stable localization of the mutated protein in the cytoplasm, which is the salient feature of this type of leukemia (2).
The NPM1 C-terminal domain is known to bind both duplex and single-stranded DNA as well as RNA with no sequence specificity and with a preference for single-stranded nucleic acids (8). This picture was suggestive of a protein playing mainly a chaperone and transport role for preribosomal RNA particles (1, 9). Recently, however, it was shown that NPM1 binds a specific G-rich sequence at the superoxide dismutase 2 (SOD2) gene promoter and participates in the transcriptional activation of this gene (10).
Starting from these premises, we recently showed (11) that (i) NPM1 binds with high affinity DNA sequences that form G-quadruplexes, including the one found at the c-MYC oncogene promoter (see later); (ii) NPM1 is able to induce G-quadruplex formation in G-rich unstructured oligos; and (iii) the region of the SOD2 promoter recognized in vivo by NPM1 is indeed folded as a G-quadruplex in vitro (11). We also investigated the domain boundaries necessary for DNA binding and demonstrated that a 17-residue segment preceding the C-terminal three-helix bundle (5) is necessary for high affinity recognition (11).
G-quadruplex DNA is gaining increasing attention because it is highly represented (especially in selected regions of the genome, including telomeres and gene promoters) and is involved in a number of regulatory processes (12, 13). Interestingly, G-quadruplex regions are frequently found at oncogene promoters, whereas a reduced frequency at tumor suppressor genes is observed (14). For instance, a well characterized G-quadruplex-forming sequence present at the NHEIII region of the c-MYC oncogene promoter (15) is recognized by NPM1 C-terminal domain in vitro (11) and folds as a G-quadruplex both in vitro and in vivo, regulating up to 90% of total c-MYC transcription (15–17). The interaction with proteins that stabilize the G-quadruplex fold, such as nucleolin, causes a marked down-regulation of the gene (18, 19), whereas the opposite happens through the interaction with G-quadruplex-unwinding proteins, such as NMR-H2 (20).
Given its selective localization at oncogene promoters and telomeres, G-quadruplex DNA is an attractive target for tumor treatment, and the structures of several G-quadruplex regions alone and in complex with drugs have been reported (21). Conversely, although the list of proteins that bind G-quadruplex DNA is rapidly increasing and the importance of such interactions for a variety of physiological processes is now clear, very little structural information is available concerning the molecular recognition mechanism of complex formation (22). To the best of our knowledge, only two structures are available: (i) thrombin in complex with a synthetic aptamer that folds as a G-quadruplex (23) and (ii) the Oxytricha nova telomere-binding protein heterodimer bound to its telomeric sequence (24).
Here we present the high resolution NMR structure of the NPM1 DNA-binding domain and analyze its interaction with the G-quadruplex DNA from the NHEIII region of the c-MYC promoter. We show (i) that the contact surface involves largely amino acids belonging to helices H1 and H2 of the terminal three-helix bundle and (ii) that a well defined G-quadruplex region is recognized through several electrostatic interactions with the phosphate backbone. The N-terminal lysine-rich region of the NPM1 C-terminal domain, which we show to be unstructured, does not participate directly in the interacting surface although it proved necessary to increase affinity (11). The data presented below unveil the interaction surface between G-quadruplex DNA and NPM1 and may inspire the search for small molecules or aptamers aimed at restoring a native-like fold in NPM1 leukemic mutants.
EXPERIMENTAL PROCEDURES
Oligonucleotides
The oligonucleotides used in this study are Pu27 of sequence 5′-TGGGGAGGGTGGGGAGGGTGGGGAAGG-3′ and Pu24I of sequence 5′-TGAGGGTGGIGAGGGTGGGGAAGG-3′. Pu27 and Pu24I were purchased from Primm (Milan, Italy) and Integrated DNA Technologies, Inc. (Coralville, IA), respectively, and were both HPLC-purified. Lyophilized oligos were dissolved in 20 mm phosphate buffer pH 7.0, 100 mm KCl and annealed. For annealing, oligos were heated to 95 °C for 15 min and then allowed to gently cool down to room temperature overnight. After annealing, the parallel G-quadruplex assembly of both oligos was assessed by inspecting CD spectra collected with a Jasco J-710 spectropolarimeter.
Protein Sample Preparation
A DNA construct for residues 225–294 of human NPM1 was cloned into pET28+(a) vector and transformed into Escherichia coli BL21(DE3) cells. For isotope enrichment, cells were grown in a minimal medium with (15NH4)2SO4 and [13C]glucose. Protein expression was induced with 1.0 mm isopropyl β-d-thiogalactopyranoside at 20 °C, and cells were harvested after 16 h. NPM1-C70 was purified as reported previously (11). The hexahistidine tag at the N terminus of the protein was thrombin-cleaved and removed by nickel-nitrilotriacetic acid affinity column.
Structure Calculations of the Free Protein
The 1H, 13C, and 15N resonance frequencies of NPM1-C70 were assigned using all classical NMR experiments. NMR experiments used for resonance assignment and structure calculations were performed on 13C,15N-labeled NPM1-C70 sample containing 10% D2O. NMR spectra were collected at 298 K, processed using standard Bruker software (TOPSPIN 2.1), and analyzed with CARA (25).
Structure calculations were performed with the software package UNIO using as input the amino acid sequence; the chemical shift lists; and three 1H-1H NOE experiments, two-dimensional NOESY, three-dimensional 13C-edited NOESY, and three-dimensional 15N-edited NOESY, recorded at 900 MHz with a mixing time of 100 ms. The standard protocol included in UNIO with seven cycles of peak picking using ATNOS (26), NOE assignment with CANDID (27), and structure calculation with CYANA-2.1 (28) was used. ϕ and ψ dihedral angles were obtained from the chemical shift analysis using TALOS+ software (29–31). In each ATNOS/CANDID cycle, the angle constraints were combined with the updated NOE upper distance constraints in the input for subsequent CYANA-2.1 structure calculation cycle.
The 20 conformers with the lowest target function values were subjected to restrained energy minimization in explicit water with AMBER 11.0 (32, 33). NOE and torsion angle constraints were used. The quality of the structures was evaluated using the programs PROCHECK, PROCHECK-NMR (34), and WHAT IF (35). Statistics about the energy-minimized family of conformers are reported in Table 1. The atomic coordinates and structural restraints for NPM1-C70 have been deposited in the Protein Data Bank with accession code 2llh. Resonance assignments are also available at BioMagResBank (accession number 18048).
TABLE 1.
Statistical analysis of the energy-minimized family of conformers of NPM1-C70
r.m.s., root mean square; r.m.s.d., root mean square deviation; rad, radian; BB, backbone; HA, heavy atoms.
| AMBER 10.0a (20 structures) | |
|---|---|
| r.m.s. deviations per meaningful distance constraint (Å)b | |
| Intraresidue (189) | 0.0065 ± 0.0063 |
| Sequential (279) | 0.0208 ± 0.0062 |
| Medium range (220)c | 0.0198 ± 0.0031 |
| Long range (40) | 0.0106 ± 0.0122 |
| Total (728) | 0.0178 ± 0.0015 |
| r.m.s. violations per meaningful dihedral angle constraints (°)b | |
| φ (42) | 1.20 ± 1.01 |
| ψ (42) | 6.17 ± 4.19 |
| Average no. of NOE violations larger than 0.3 Å | 0.05 ± 0.22 |
| Average NOE deviation (Å2)d | 0.01 ± 0.03 |
| Average angle deviation (rad2)d | 0.66 ± 0.10 |
| r.m.s.d. to the mean structure (Å) (BB)e | 0.80 ± 0.21 |
| r.m.s.d. to the mean structure (Å) (HA)e | 1.40 ± 0.15 |
| Structural analysis | |
| Residues in most favorable regions (%)f,g | 96.5 |
| Residues in allowed regions (%)f,g | 3.3 |
| Residues in generously allowed regions (%)f,g | 0.1 |
| Residues in disallowed regions (%)f,g | 0.0 |
| G-factorf,g | 0.03 |
| Structure Z-scoresg,h | |
| First generation packing quality | 1.27 |
| Second generation packing quality | 4.51 |
| Ramachandran plot appearance | −1.50 |
| χ1/χ2 rotamer normality | −3.04 |
| Backbone conformation | −0.50 |
a AMBER indicates the energy-minimized family of 20 structures.
b The number of meaningful constraints for each class is reported in parentheses.
c Medium range distance constraints are those between residues (i, i + 2), (i, i + 3), (i, i + 4), and (i, i + 5).
d NOE and torsion angle constraints were applied with force constants of 20 kcal mol−1 Å−2 and 20 kcal mol−1 rad−2, respectively.
e The r.m.s.d. to the mean structure is reported considering the segment 293–291.
f As it results from the Ramachandran plot analysis. For the PROCHECK statistics, an overall G-factor larger than −0.5 is expected for a good quality structure.
g The statistical analysis is reported considering the segment 293–291.
h Values based on WHAT IF output. A Z-score is defined as the deviation from the average value for this indicator observed in a database of high resolution crystal structures expressed in units of the standard deviation of this database-derived average. Typically, Z-scores below a value of −3 are considered poor, and those below −4 are considered bad.
Structure Calculations of the Complex
To identify intermolecular NOEs in the NPM1-C70·Pu24I complex, a ω1-13C-edited, ω2-13C-filtered NOESY experiment was recorded in a two-dimensional plane (1H-1H plane) on 13C,15N-labeled NPM1-C70·unlabeled Pu24I (36). The selected temperature was 290 K, and the mixing time used was 120 ms. 1H, 13C, and 15N backbone resonances of NPM1-C70 in the complex were assigned by performing all the typical experiments for backbone assignment.
To calculate a structural model for the interaction between NPM1-C70 and Pu24I, a data-driven molecular docking was performed using the HADDOCK protocol. HADDOCK comprises a series of Python scripts that run on top of the structure determination programs ARIA and CNS (37–41). The method relies on the definition of ambiguous interaction restraints (AIRs) derived from experimental data.
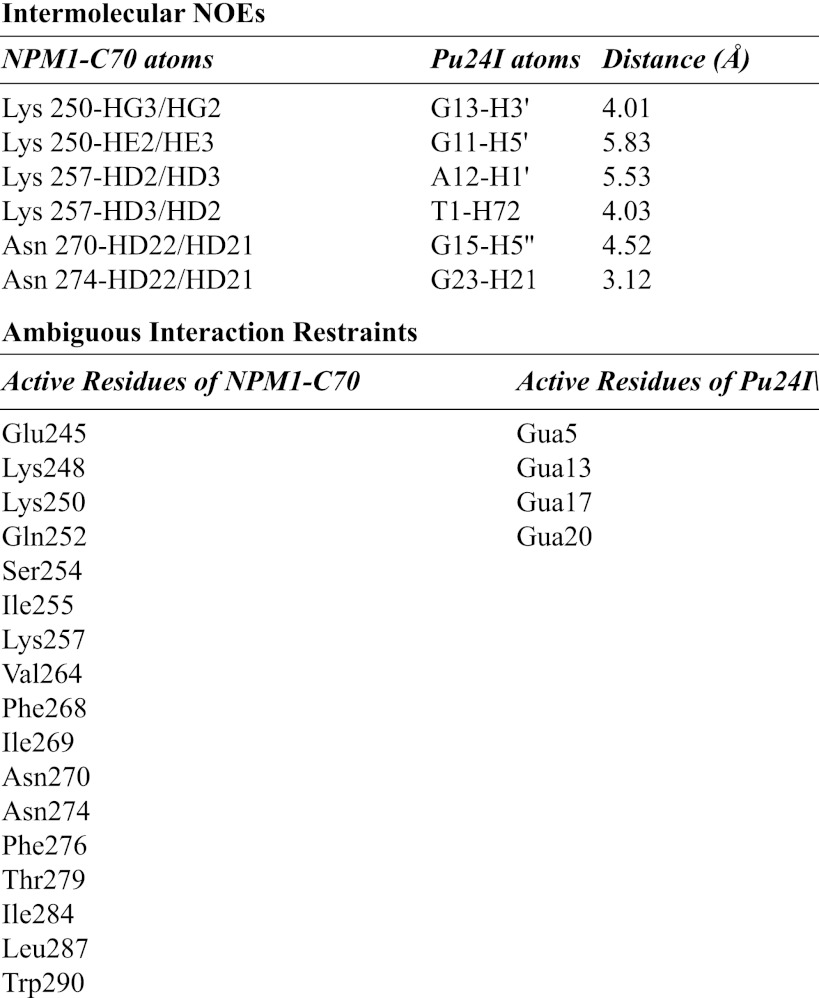
For our docking calculations, we defined as unambiguous restraints residues resulting from intermolecular NOE cross-peaks on both protein and DNA. Furthermore, we defined as AIRs (i) residues experiencing chemical shift variations above the average variations plus one standard deviation (δHN = 0.043 ± 0.025 ppm; Fig. 3B, black line); (ii) residues whose signals broadened their line width (Glu245, Lys248, and Phe268) and/or disappeared during the titration (Phe276); and (iii) DNA atoms interacting with the protein, also identified by chemical shift perturbation, as detected in homonuclear experiments (one-dimensional and two-dimensional). Residues used as AIRs are listed in Table 2.
FIGURE 3.

Interaction of NPM1-C70 with the Pu24I G-quadruplex. A, 15N HSQC spectra of the protein before (black) and after addition of stoichiometric amounts of unlabeled Pu24I (red). Representative chemical shift variations are labeled, indicating relevant residues. B, chemical shift variations cluster in the three-helix bundle, whereas they are not found at the N-terminal 225–242 segment. The horizontal black line indicates the average chemical shift variation plus one standard deviation upon Pu24I addition. C, residues experiencing chemical shift variations higher than the average plus one standard deviation are highlighted on the structure of the protein. Residues belonging to helices H1 and H2 are solvent-exposed. A few hydrophobic residues belonging to helix H3 are also affected, indicating coupling between the helices upon Pu24I binding. Heteronuclear 15N-1H R1 (D) and heteronuclear 15N-1H R2 values (E) for NPM1-C70 in complex with Pu24I indicate that the N-terminal region flanking the three-helix bundle remains unstructured after Pu24I binding. F, the increase of heteronuclear 15N{1H} NOE values in the three-helix bundle upon Pu24I binding (see Fig. 1D for comparison) suggests increased rigidity.
TABLE 2.
Intermolecular NOEs and AIRs used for HADDOCK docking calculations
Gua, guanine.
The HADDOCK docking protocol consists of (i) randomization of orientation and rigid body minimization, (ii) semirigid simulated annealing in torsion angle space, and (iii) final refinement in Cartesian space with explicit solvent. The rigid body docking step was performed five times with 1000 structures generated at each stage, the best 200 of which were refined in the semiflexible stage and subsequently in explicit water. Electrostatic and van der Waals terms were calculated with an 8.5-Å distance cutoff (37).
For the docking procedure, the structures of NPM1-C70 (here determined) and Pu24I (Protein Data Bank code 2A5P) were used as starting points. The coordinates for the lowest energy NPM1-C70·Pu24I structural model are included as supplemental material.
Relaxation Data
Heteronuclear relaxation experiments were performed on 15N-labeled samples of NPM1-C70 at 700 MHz. The 15N backbone longitudinal (R1) and transverse (R2) relaxation rates as well as heteronuclear 15N{1H} NOEs were measured using a standard protocol (42, 43).
RESULTS
The Structure of the NPM1-C70 Domain
The short C-terminal domain of NPM1 (residues 242–294; hereafter NPM1-C53) was shown by Grummitt et al. (5) to fold as a three-helix bundle. However, NPM1-C53 is poorly competent for DNA binding, and the 17-residue lysine-rich region at its N terminus (residues 225–241) is necessary for high affinity (11). Therefore, we decided to determine by NMR the structure of this longer construct encompassing the last 70 residues of the NPM1 sequence (residues 225–294; hereafter NPM1-C70). Statistics about structure determination are shown in Table 1.
The structure depicted in Fig. 1A comprises a well defined three-helix bundle, similar to the NPM1-C53 construct (5), in terms of length, relative orientation, and hydrophobic interactions between all the paired helices (Fig. 1A). On the other hand, the lysine-rich region (residues 225–241) that enhances DNA binding is unstructured as indicated by high values for 15N-1H R1 (Fig. 1B) and low values for 15N-1H R2 (Fig. 1C) and heteronuclear NOEs (Fig. 1D) with no secondary structure elements nor any propensity to fractionally take up secondary structures as clearly shown from the chemical shift index analysis (Fig. 1, B and C). Conversely, the folded part of the NPM1-C70 construct shows 15N-1H R1 and 15N-1H R2 values typical of an 8-kDa protein (Fig. 1, B and C) and positive values for heteronuclear 15N{1H} NOEs values (Fig. 1D), although the latter are lower than those of a rigid protein, thus indicating the occurrence of fast internal motions.
FIGURE 1.

NPM1-C70 quadruplex-binding C-terminal domain encompassing residues 225–294. A, NMR solution structure of NPM1-C70 showing the 20 lowest energy structures. A lysine-rich natively unstructured segment (amino acids 225–242) precedes the terminal three-helix bundle. B, 15N-1H R1 NOE values. C, 15N-1H R2 NOE values. Low R1 and high R2 values for segment 225–242 are consistent with the N-terminal tail being unstructured. D, heteronuclear 15N{1H} NOEs are positive but smaller than expected for an 8-kDa protein, indicating fast internal motion in the three-helix bundle. Red lines indicate average values for the 225–242 and the 243–294 segments, respectively.
The Complex of NPM1-C70 with c-MYC G-quadruplex DNA
NPM1-C70 binds a DNA oligonucleotide resembling a specific sequence found at the NHEIII region of the c-MYC promoter (11). This 27-mer region (called Pu27) of sequence 5′-TGGGGAGGGTGGGGAGGGTGGGGAAGG-3′ is known to form a parallel G-quadruplex structure in the presence of K+ at physiological concentration (100–150 mm) (11, 18). Pu27 contains five runs of three or more consecutive guanines and therefore can, in principle, populate several G-quadruplex structures with different topologies (13). These multiple conformations are indeed observed in the free state as monitored by multiple sets of NMR signals in slow exchange in the one-dimensional 1H NMR spectra (Fig. 2A). Moreover, Pu27 maintains different quadruplex topologies also when bound to NPM1-C70 (Fig. 2A). The structural heterogeneity of the Pu27 sample complicates the analysis of its interaction with NPM1-C70. However, recently, Patel and coworkers (44) showed that a c-MYC-derived shorter oligonucleotide of 24 residues containing a guanine to inosine substitution in one of the guanine runs populates only one of the possible G-quadruplex conformations (called Pu24I; 5′-TGAGGGTGGIGAGGGTGGGGAAGG-3′). Therefore, we decided to use this oligonucleotide for further studies. The assignment of the 1H nuclei of Pu24I was provided by courtesy of Anh Tuan Phan and Vitaly Kuryavyi (44). First, by comparing the one-dimensional 1H NMR spectra of Pu24I in the free state and bound to NPM1-C70, we confirmed that Pu24I displays only a single G-quadruplex topology that is retained after NPM1-C70 binding (Fig. 2B). Then, to assess whether Pu24I undergoes major conformational changes upon NPM1-C70 binding, we performed intramolecular NOE experiments for the bound state of Pu24I and compared it with its free state. To characterize the bound state, a ω1-13C-filtered, ω2-13C-filtered NOESY experiment was recorded in a two-dimensional plane (1H-1H plane), whereas, in the case of the Pu24I free state, a standard two-dimensional NOESY was performed. As shown in Fig. 2C, no major variations are visible in the superimposition of the two spectra, indicating that the G-quadruplex structure of Pu24I is maintaining its conformation when bound to NPM1-C70.
FIGURE 2.

Analysis of Pu27 and Pu24I G-quadruplex conformations. A, superimposition of the one-dimensional 1H NMR spectra of Pu27 in the free state (blue) and bound (red) to NPM1-C70. Both spectra were acquired at 700 MHz and at 290 K. B, superimposition of the one-dimensional 1H NMR spectra of Pu24I in the free state (blue) and bound (red) to NPM1-C70. Both spectra were acquired at 700 MHz and 290 K. C, details of the superimposition between ω1-13C-filtered, ω2-13C-filtered NOESY experiments in a two-dimensional plane (1H-1H plane) of Pu24I·NPM1-C70 complex (black) and a classical two-dimensional NOESY of Pu24I in its free state (red). Both spectra were acquired at 700 MHz and 290 K.
To further investigate the interaction of NPM1-C70 with Pu24I, we titrated 15N-labeled NPM1-C70 with increasing amounts of unlabeled Pu24I. We observed both the appearance of new peaks and the disappearance of others, indicating the formation of a new species that exchanges slowly with the free protein on the NMR time scale, i.e. < 10−2 s−1 (Fig. 3A). The ratio of the intensity of the signals for a given resonance in the two species changes linearly with the amount of Pu24I and reaches a plateau upon addition of stoichiometric amounts of Pu24I, consistent with a 1:1 protein·Pu24I complex. NPM1-C70 interacts with Pu24I mainly via hydrophilic residues located in helices H1 and H2 and exposed on the same side of the protein (Fig. 3, B and C). Interestingly, a few residues located on helix Η3 also display chemical shift variations (Fig. 3, B and C). Because the latter are buried and interact with helices Η1 and Η2, we conclude that a strong coupling is present among the three helices.
In the protein·Pu24I complex, no chemical shift variations were detected for residues belonging to the NPM1-C70 N-terminal unstructured segment, indicating that this tail is not directly involved in the complex (Fig. 3B). This was confirmed by analysis of the 15N heteronuclear relaxation data in the complex where the N-terminal tail is still characterized by high values for heteronuclear 15N-1H R1 (Fig. 3D) and low values for heteronuclear 15N-1H R2 (Fig. 3E) and heteronuclear NOEs (Fig. 3F), consistent with a natively unstructured state. Conversely, the folded C-terminal region of NPM1-C70 shows 15N-1H R1 and 15N-1H R2 values typical of a complex of 16-kDa molecular mass (in agreement with the sum of the molecular weights of NPM1-C70 and Pu24I) and positive values for heteronuclear 15N{1H} NOE values (Fig. 3, D–F). Both results suggest that the complex between NPM1-C70 and Pu24I is stable, consistent with the high affinity observed previously for the Pu27 oligonucleotide (11). The relaxation data analysis also pointed out that the heteronuclear 15N{1H} NOE values increase in the folded region of NPM1-C70, indicating an increasing rigidity for the three-helix bundle of NPM1-C70 upon binding of Pu24I (Fig. 3F).
Experimentally Restrained Molecular Docking of the NPM1-C70·Pu24I complex
To gain additional information on the structure of the complex, 13C-filtered NOESY experiments were performed. Six intermolecular NOEs were detected between NPM1-C70 and Pu24I in the complex (Table 2). An example of assignment of NOE cross-peaks is shown in Fig. 4A. They involve Lys and Asn residues of the protein (whose side chains are free to rotate during the subsequent docking calculation) and sugar backbone protons on Pu24I. These distance restraints, together with the chemical shift perturbations defined as AIRs (also listed in Table 2), were used to calculate a structural model of the NPM1-C70·Pu24I complex within the data-driven HADDOCK docking program (see “Experimental “Procedures” for details about the protocol used). Importantly, HADDOCK calculations identified a single cluster of docking poses. In particular, 184 final complex structures were obtained at the end of the procedure with an root mean square deviation from the lowest energy solution of 1.5 ± 0.9 Å. As an example, the 20 lowest energy docking poses are reported in Fig. 4B.
FIGURE 4.

Intermolecular NOEs and HADDOCK calculations. A, an example of NOE cross-peak assignment from ω1-13C-edited, ω2-13C-filtered NOESY experiments in a two-dimensional plane (1H-1H plane) of the NPM1-C70·Pu24I complex acquired on a 13C,15N-labeled NPM1-C70·unlabeled Pu24I sample. B, the 20 lowest energy complex structures obtained by NMR data-restrained molecular docking calculations using the HADDOCK protocol. NPM1-C70 is represented in blue, and Pu24I is represented in green.
As shown in Fig. 5A, the interaction surface involves one side of the three-helix bundle, the interacting residues all being located on helices H1 and H2, with a buried area of 1358.6 ± 92.2 Å2. Based on the assignment of the intermolecular NOEs (Fig. 4A and Table 2), the interaction involves side chains of NPM1-C70 residues Lys250, Lys257, Asn270, and Asn274. On the Pu24I side, intermolecular NOEs indicate the involvement of protons of the backbone (mainly the deoxyribose ring) of nucleotides T1, G11, A12, G13, G15, and G23. From the structure model analysis, a linear stretch of nucleotides (from G11 to G16) located on one side of the G-quadruplex scaffold (Fig. 5A) intercalates into a groove formed by helices H1 and H2 (Fig. 5B). Interestingly, this stretch contributes to the formation of each of the three stacked guanine tetrads in the G-quadruplex scaffold. By analyzing the 20 lowest energy docking solutions, it appears that residues Lys250, Lys257, Asn270, and Asn274 always participate in the formation of salt bridges or hydrogen bonds with backbone phosphates belonging to the G11–G16 linear stretch. In addition, residues Lys267 and Cys275 are always found at the interface where they contact phosphate groups in the G11–G16 stretch in all 20 lowest energy docking poses.
FIGURE 5.

Structural model of the NPM1-C70·Pu24I complex. A, ribbon representation of the lowest energy model showing helices H1 and H2 of NPM1-C70 approaching the G-quadruplex “laterally” and interacting with a specific segment of the backbone (in orange). B, NPM1-C70 is represented with its electrostatic surface (blue for positive and red for negative), whereas Pu24I is shown in ribbon representation. The Pu24I structure is shown in transparency to highlight the small positively charged groove in between helices H1 and H2 that accommodates a stretch of Pu24I nucleotides (G11–G16; colored in orange). The long unstructured tail is also positively charged and may play a role in long range electrostatic interactions with the approaching oligonucleotide.
DISCUSSION
The propensity of NPM1 to interact with nucleic acids appears to be crucial for several functions that this protein plays both in the nucleoli and in the cytoplasm where a small fraction of the protein is always present due to continuous shuttling back and forth from the nucleus. For instance, a great deal of data show that NPM1 controls both ribosome assembly and transport (9). Recently, it was also shown that NPM1 is selectively deposited on the mRNA body during polyadenylation, suggesting a putative role in a variety of post-transcriptional events, including splicing (45). A role of NPM1 in the control of gene transcription has been also suggested either through its association with several transcription factors at gene promoters (9) or through its direct interaction with the G-rich sequence found at the SOD2 gene promoter (10). We further showed that the latter region is folded as a G-quadruplex at least in vitro and that G-quadruplexes are bound by NPM1-C70 with high affinity (11).
Based on these premises, we analyzed the three-dimensional structure of NPM1-C70 bound to G-quadruplexes. Among several oligonucleotides tested, we focused our attention on the interaction of NPM1-C70 with the G-quadruplex at the NHEIII region of the c-MYC promoter because this is the DNA sequence bound with the highest affinity (11) whose structure is known (40).
The structure of NPM1-C70 alone and bound to the DNA fragment was investigated by a combination of NMR data and docking calculations guided by experimental restraints. As shown in Fig. 5, NPM1-C70 binds the Pu24I G-quadruplex through a specific surface at the interface between helices H1 and H2. Several positively charged and polar residues establish interactions mainly with phosphate groups of a linear stretch of nucleotides that fits the small groove at the H1-H2 interface. Interestingly, this stretch contributes to the formation of the main G-quadruplex scaffold, whereas the interactions with nucleotides belonging to the connecting loops appear marginal. This may explain why NPM1 recognizes with comparable affinity several G-quadruplexes that differ in loop length and distribution (11).
Among the NPM1-C70 residues found at the interface with the Pu24I G-quadruplex, Lys257 and Lys267 are acetylated in vivo by p300 and deacetylated by SIRT1 (46). NPM1 acetylation results in dislocation of the protein from the nucleoli to the nucleoplasm where NPM1 interacts with transcriptionally active RNA polymerase II. Our finding that these two residues are at the interface with Pu24I DNA suggests that loss of nucleolar localization may be due to impaired DNA binding at the nucleoli coupled to acetylation.
Interestingly, we also found from our docking simulations that Cys275 is always located at the center of the surface buried by Pu24I. Cys275 is targeted and alkylated by the natural antitumoral compound (+)-avrainvillamide (47). Treatment of LNCaP or T-47D cells with (+)-avrainvillamide leads to an increase in cellular p53 concentrations and promotes apoptosis. It is therefore conceivable that these effects may be linked to (+)-avrainvillamide-mediated impairment of NPM1 nucleic acid binding efficiency.
The N-terminal tail of the NPM1-C70 construct, although necessary for high affinity binding of any DNA tested (11), is natively unstructured both in the isolated domain and in the complex with Pu24I. This finding parallels previous observations in other systems. For instance, the affinity of the cAMP-responsive element-binding protein KID domain for the KIX domain of the cAMP-responsive element-binding protein-binding protein is significantly reduced when an unstructured portion of the domain that does not participate directly to the complex contact surface is deleted (48). Similarly, the interaction of splicing factor 1 with the large subunit of the U2 small nuclear RNA auxiliary factor (U2AF65) is affected by flanking unstructured regions that do not physically contact the partner (49). This phenomenon, which is more frequent than previously anticipated, has been termed “flanking fuzziness” (50). Furthermore, it was shown recently that a large fraction of transcription factors are characterized by the presence of unstructured regions that flank the DNA-binding domain at one or both ends and that these regions may impact the affinity for specific or nonspecific DNA sequences (51).
What could the role of this unstructured N-terminal tail of NPM1-C70 be? Although this issue is still under investigation, it is conceivable that the presence of an unstructured segment adjacent to the interacting domain may provide a larger platform for long range electrostatic interactions or even transient physical contacts that facilitate the fine tuning of binding (see Fig. 5B) (50). This is supported by the observation that two concomitant Lys to Ala substitutions (Lys229-Lys230) in the unstructured segment of NPM1-C70 result in a dramatic decrease of global affinity (11).
Furthermore, exposed unstructured regions may be modified by post-translational modifications, driving regulatory changes. Several residues in the unstructured segment may be modified, such as Lys229 and Lys230 that are among the lysines acetylated by p300 and deacetylated by SIRT1 (46). A number of putative phosphorylation sites are also present in the tail, including Ser227, Thr234, Thr237, and Ser242; among these, the phosphorylations of Ser227 by PKC and of Thr234-Thr237 by the Cdk1·cyclinB complex were experimentally validated (52, 53). Both acetylation and phosphorylation may therefore interfere with NPM1 nucleic acid binding and play a role in NPM1 activity and trafficking throughout the cell cycle.
Leukemia-associated NPM1 mutations cause dramatic destabilization up to total unfolding of the terminal three-helix bundle that is responsible for the aberrant cytosolic translocation of the protein. Scaloni et al. (6, 7) showed that a malleable “native-like” structure that accelerates folding is retained in the denatured state of the wild-type NPM1-C53 three-helix bundle and involves helices H2 and H3 (H3 is also the site of leukemic mutations), whereas helix H1 is totally unfolded in the denatured state. A possible strategy to rationally target NPM1 for the treatment of this type of leukemia might be that of developing a drug able to stabilize in the leukemic variant a native-like structure by altering through binding the folding-unfolding equilibrium in favor of the native state. Our folding studies suggested that such a drug should preferentially target helix H1. Because we have shown here that G-quadruplex DNA specifically binds a region in between helices H1 and H2 of NPM1-C70, we may attempt to rationally design aptamers or other smaller molecules that by mimicking the binding properties of G-quadruplexes to NPM1 C-terminal domain might stabilize a native-like state in the leukemic variant.
Acknowledgments
We sincerely thank Prof. Brunangelo Falini (University of Perugia, Italy) for the introduction to nucleophosmin and precious collaboration. The WeNMR project (European FP7 e-Infrastructure grant, Contract Number 261572, supported by the national GRID Initiatives of Belgium, Italy, Germany, the Netherlands (via the Dutch BiG Grid project), Portugal, UK, South Africa, Taiwan, and the Latin America GRID infrastructure via the Gisela project) is acknowledged for the use of web portals and computing and storage facilities.
This work was supported by Associazione Italiana Ricerca sul Cancro Grants IG2007-4798 (to M. B.) and IG2011-11712 (to L. F.) and by Ministero dell'Istruzione, Università e Ricerca of Italy Grants RBRN07BMCT_007 (to M. B.), RBRN07BMCT (to L. B. and I. B.), and PRIN2009FAKHZT_001 (to L. B. and I. B.).

This article contains a supplemental file showing the coordinates for the lowest energy NPM1-C70·Pu24I structural model.
The atomic coordinates and structure factors (code 2llh) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- NPM1
- nucleophosmin
- NPM1-C70
- 70-residue NPM1 C-terminal domain
- SOD2
- superoxide dismutase 2
- AIRs
- ambiguous interaction restraints
- NHE III
- nuclease hypersensitive element III.
REFERENCES
- 1. Grisendi S., Mecucci C., Falini B., Pandolfi P. P. (2006) Nucleophosmin and cancer. Nat. Rev. Cancer 6, 493–505 [DOI] [PubMed] [Google Scholar]
- 2. Falini B., Sportoletti P., Martelli M. P. (2009) Acute myeloid leukemia with mutated NPM1: diagnosis, prognosis and therapeutic perspectives. Curr. Opin. Oncol. 21, 573–581 [DOI] [PubMed] [Google Scholar]
- 3. Falini B., Gionfriddo I., Cecchetti F., Ballanti S., Pettirossi V., Martelli M. P. (2011) Acute myeloid leukemia with mutated nucleophosmin (NPM1): any hope for a targeted therapy? Blood Rev. 25, 247–254 [DOI] [PubMed] [Google Scholar]
- 4. Falini B., Mecucci C., Tiacci E., Alcalay M., Rosati R., Pasqualucci L., La Starza R., Diverio D., Colombo E., Santucci A., Bigerna B., Pacini R., Pucciarini A., Liso A., Vignetti M., Fazi P., Meani N., Pettirossi V., Saglio G., Mandelli F., Lo-Coco F., Pelicci P. G., Martelli M. F.; GIMEMA Acute Leukemia Working Party (2005) Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 352, 254–266 [DOI] [PubMed] [Google Scholar]
- 5. Grummitt C. G., Townsley F. M., Johnson C. M., Warren A. J., Bycroft M. (2008) Structural consequences of nucleophosmin mutations in acute myeloid leukemia. J. Biol. Chem. 283, 23326–23332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scaloni F., Gianni S., Federici L., Falini B., Brunori M. (2009) Folding mechanism of the C-terminal domain of nucleophosmin: residual structure in the denatured state and its pathophysiological significance. FASEB J. 23, 2360–2365 [DOI] [PubMed] [Google Scholar]
- 7. Scaloni F., Federici L., Brunori M., Gianni S. (2010) Deciphering the folding transition state structure and denatured state properties of nucleophosmin C-terminal domain. Proc. Natl. Acad. Sci. U.S.A. 107, 5447–5452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hingorani K., Szebeni A., Olson M. O. (2000) Mapping the functional domains of nucleolar protein B23. J. Biol. Chem. 275, 24451–24457 [DOI] [PubMed] [Google Scholar]
- 9. Lindström M. S. (2011) NPM1/B23: a multifunctional chaperone in ribosome biogenesis and chromatin remodeling. Biochem. Res. Int. 2011, 195209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu Y., Fang F., Dhar S. K., St Clair W. H., Kasarskis E. J., St Clair D. K. (2007) The role of a single-stranded nucleotide loop in transcriptional regulation of the human sod2 gene. J. Biol. Chem. 282, 15981–15994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Federici L., Arcovito A., Scaglione G. L., Scaloni F., Lo Sterzo C., Di Matteo A., Falini B., Giardina B., Brunori M. (2010) Nucleophosmin C-terminal leukemia-associated domain interacts with G-rich quadruplex forming DNA. J. Biol. Chem. 285, 37138–37149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huppert J. L., Balasubramanian S. (2005) Prevalence of quadruplexes at the human genome. Nucleic Acids Res. 33, 2908–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huppert J. L. (2008) Four-stranded nucleic acids: structure, function and targeting of G-quadruplexes. Chem. Soc. Rev. 37, 1375–1384 [DOI] [PubMed] [Google Scholar]
- 14. Qin Y., Hurley L. H. (2008) Structures, folding patterns, and functions of intramolecular DNA G-quadruplexes found in eukaryotic promoter regions. Biochimie 90, 1149–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Siddiqui-Jain A., Grand C. L., Bearss D. J., Hurley L. H. (2002) Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. U.S.A. 99, 11593–11598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grand C. L., Han H., Muñoz R.M., Weitman S., Von Hoff D. D., Hurley L. H., Bearss D. J. (2002) The cationic porphyrin TMPyP4 down-regulates c-MYC and human telomerase reverse transcriptase expression and inhibits tumor growth in vivo. Mol. Cancer Ther. 1, 565–573 [PubMed] [Google Scholar]
- 17. Brown R. V., Danford F. L., Gokhale V., Hurley L. H., Brooks T. A. (2011) Demonstration that drug-targeted downregulation of MYC in non-Hodgkins lymphoma is directly mediated through the promoter G-quadruplex. J. Biol. Chem. 286, 41018–41027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. González V., Guo K., Hurley L., Sun D. (2009) Identification and characterization of nucleolin as a c-myc G-quadruplex-binding protein. J. Biol. Chem. 284, 23622–23635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. González V., Hurley L. H. (2010) The cMYC NHEIII (1): function and regulation. Annu. Rev. Pharmacol. Toxicol. 50, 111–129 [DOI] [PubMed] [Google Scholar]
- 20. Thakur R. K., Kumar P., Halder K., Verma A., Kar A., Parent J. L., Basundra R., Kumar A., Chowdhury S. (2009) Metastases suppressor NM23-H2 interaction with G-quadruplex DNA within c-MYC promoter nuclease hypersensitive element induces c-MYC expression. Nucleic Acids Res. 37, 172–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neidle S. (2009) The structures of quadruplex nucleic acids and their drug complexes. Curr. Opin. Struct. Biol. 19, 239–250 [DOI] [PubMed] [Google Scholar]
- 22. Sissi C., Gatto B., Palumbo M. (2011) The evolving world of protein-G-quadruplex recognition: a medicinal chemist's perspective. Biochimie 93, 1219–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Padmanabhan K., Padmanabhan K. P., Ferrara J. D., Sadler J. E., Tulinsky A. (1993) The structure of α-thrombin inhibited by a 15-mer single-stranded DNA aptamer. J. Biol. Chem. 268, 17651–17654 [DOI] [PubMed] [Google Scholar]
- 24. Horvath M. P., Schultz S. C. (2001) DNA G-quartets in a 1.86 Å resolution structure of an Oxytricha nova telomeric protein-DNA complex. J. Mol. Biol. 310, 367–377 [DOI] [PubMed] [Google Scholar]
- 25. Keller R. (2004) The Computer Aided Resonance Assignment Tutorial, CANTINA Verlag, Goldau, Switzerland [Google Scholar]
- 26. Herrmann T., Güntert P., Wüthrich K. (2002) Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR 24, 171–189 [DOI] [PubMed] [Google Scholar]
- 27. Herrmann T., Güntert P., Wüthrich K. (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 [DOI] [PubMed] [Google Scholar]
- 28. Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 29. Wishart D. S., Sykes B. D. (1994) The 13C chemical shift index: a simple method for the identification of protein secondary structure using 13C chemical shift data. J. Biomol. NMR 4, 171–180 [DOI] [PubMed] [Google Scholar]
- 30. Eghbalnia H. R., Wang L., Bahrami A., Assadi A., Markley J. L. (2005) Protein energetic conformational analysis from NMR chemical shifts (PECAN) and its use in determining secondary structural elements. J. Biomol. NMR 32, 71–81 [DOI] [PubMed] [Google Scholar]
- 31. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bertini I., Case D. A., Ferella L., Giachetti A., Rosato A. (2011) A grid-enabled web portal for NMR structure refinement with AMBER. Bioinformatics 27, 2384–2390 [DOI] [PubMed] [Google Scholar]
- 33. Case D. A., Darden T. A., Cheatham T. E., 3rd, Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Hayik S., Roitberg A., Seabra G., Swails J., Goetz A. W., Kolossvai I., Wong K. F., Paesani F., Vanicek J., Wolf R. M., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M. J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Salomon-Ferrer R., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A., Kollman P. A. (2010) AMBER 11, University of California, San Francisco [Google Scholar]
- 34. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 35. Vriend G. (1990) WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8, 52–56, 29 [DOI] [PubMed] [Google Scholar]
- 36. Zwahlen C., Legault P., Vincent S. J., Greenblatt J., Konrat R., Kay L. E. (1997) Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: application to a bacteriophage N-peptide/boxB RNA complex. J. Am. Chem. Soc. 119, 6711–6721 [Google Scholar]
- 37. Dominguez C., Boelens R., Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 38. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR System: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 39. Linge J. P., O'Donoghue S. I., Nilges M. (2001) Automated assignment of ambiguous nuclear Overhauser effects with ARIA. Methods Enzymol. 339, 71–90 [DOI] [PubMed] [Google Scholar]
- 40. Linge J. P., Habeck M., Rieping W., Nilges M. (2003) ARIA: automated NOE assignment and NMR structure calculation. Bioinformatics 19, 315–316 [DOI] [PubMed] [Google Scholar]
- 41. de Vries S. J., van Dijk M., Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 42. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., Kay L. E. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 [DOI] [PubMed] [Google Scholar]
- 43. Orekhov V. Yu., Pervushin K. V., Arseniev A. S. (1994) Backbone dynamics of (1–71) bacterioopsin studied by two-dimensional 1H-15N NMR spectroscopy. Eur. J. Biochem. 219, 887–896 [DOI] [PubMed] [Google Scholar]
- 44. Phan A. T., Kuryavyi V., Gaw H. Y., Patel D. J. (2005) Small-molecule interaction with a five-guanine-tract G-quadruplex structure from the human MYC promoter. Nat. Chem. Biol. 1, 167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Palaniswamy V., Moraes K. C., Wilusz C. J., Wilusz J. (2006) Nucleophosmin is selectively deposited on mRNA during polyadenylation. Nat. Struct. Mol. Biol. 13, 429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shandilya J., Swaminathan V., Gadad S. S., Choudhari R., Kodaganur G. S., Kundu T. K. (2009) Acetylated NPM1 localizes in the nucleoplasm and regulates transcriptional activation of genes implicated in oral cancer manifestation. Mol. Cell. Biol. 29, 5115–5127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wulff J. E., Siegrist R., Myers A. G. (2007) The natural product avrainvillamide binds to the oncoprotein nucleophosmin. J. Am. Chem. Soc. 129, 14444–14451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zor T., Mayr B. M., Dyson H. J., Montminy M. R., Wright P. E. (2002) Roles of phosphorylation and helix propensity in the binding of the KIX domain of CREB-binding protein by constitutive (c-Myb) and inducible (CREB) activators. J. Biol. Chem. 277, 42241–42248 [DOI] [PubMed] [Google Scholar]
- 49. Selenko P., Gregorovic G., Sprangers R., Stier G., Rhani Z., Krämer A., Sattler M. (2003) Structural basis for the molecular recognition between human splicing factors U2AF65 and SF1/mBBP. Mol. Cell 11, 965–976 [DOI] [PubMed] [Google Scholar]
- 50. Tompa P., Fuxreiter M. (2008) Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 33, 2–8 [DOI] [PubMed] [Google Scholar]
- 51. Guo X., Bulyk M. L., Hartemink A. J. (2012) Intrinsic disorder within and flanking the DNA-binding domains of human transcription factors. Pac. Symp. Biocomput. 2012, 104–115 [PMC free article] [PubMed] [Google Scholar]
- 52. Beckmann R., Buchner K., Jungblut P. R., Eckerskorn C., Weise C., Hilbert R., Hucho F. (1992) Nuclear substrates of protein kinase C. Eur. J. Biochem. 210, 45–51 [DOI] [PubMed] [Google Scholar]
- 53. Okuwaki M., Tsujimoto M., Nagata K. (2002) The RNA binding activity of a ribosome biogenesis factor, nucleophosmin/B23, is modulated by phosphorylation with a cell cycle-dependent kinase and by association with its subtype. Mol. Biol. Cell 13, 2016–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]