

Background: The nucleosome assembly protein (NAP) family facilitates the dynamic assembly of nucleosomes.
Results: We reveal the large molecular assemblies of NAP-histone complexes.
Conclusion: NAP-histone complexes assemble to form a heterogeneous population of ring-like scaffolds.
Significance: Knowledge of the oligomeric states of NAP-histone complexes is crucial for understanding their diverse functions.
Keywords: Analytical Ultracentrifugation, Biophysics, Electron Microscopy (EM), Histone Chaperone, Nucleosome, X-ray Scattering
Abstract
The nucleosome assembly protein (NAP) family represents a key group of histone chaperones that are essential for cell viability. Several x-ray structures of NAP1 dimers are available; however, there are currently no structures of this ubiquitous chaperone in complex with histones. We have characterized NAP1 from Xenopus laevis and reveal that it forms discrete multimers with histones H2A/H2B and H3/H4 at a stoichiometry of one NAP dimer to one histone fold dimer. These complexes have been characterized by size exclusion chromatography, analytical ultracentrifugation, multiangle laser light scattering, and small-angle x-ray scattering to reveal their oligomeric assembly states in solution. By employing single-particle cryo-electron microscopy, we visualized these complexes for the first time and show that they form heterogeneous ring-like structures, potentially acting as large scaffolds for histone assembly and exchange.
Introduction
Histone chaperones direct the productive assembly and disassembly of nucleosomes by facilitating histone deposition and exchange. They are therefore central to the regulation of multiple cellular processes, such as chromatin remodeling, cell cycle control, DNA replication, and transcription (1). The nucleosome core particle consists of two each of the four core histones H2A, H2B, H3, and H4 and is assembled in a stepwise manner: two H3/H4 heterodimers form a tetramer, which is subsequently flanked by two H2A/H2B heterodimers (2, 3). The extended N-terminal histone tails protrude from the core particle and contain multiple sites for modifications, such as acetylation and methylation, and provide a complex mechanism for controlling compaction and gene expression (4). Histones are predominantly basic proteins but also contain hydrophobic and acidic patches. They repel each other at physiological pH (5) and form non-nucleosomal aggregates with DNA. Histone chaperones prevent these nonspecific interactions and can direct the productive assembly and disassembly of nucleosomes by facilitating histone deposition and exchange (as reviewed in Refs. 6 and 7).
There is a diverse range of histone chaperones, which have a wide range of functions within the cell. Specialized chaperones, such as nucleoplasmin, interact with the H2A/H2B dimer and are involved in histone storage (8), whereas other chaperones, such as ASF1, have a higher preference for H3/H4 and are involved in depositing histones onto DNA for nucleosome formation (9). The Chz1 chaperone shows very high specificity, binding preferentially to the variant H2A.Z/H2B dimer over the canonical H2A/H2B dimer (10). In contrast, chaperones such as NAP1 can bind all of the core histones as well as the linker histone, although with varying affinities (11–13). Following the discovery that NAP1 and ACF can facilitate the assembly of chromatin, organizing nucleosomes in regular spaced arrays (14), NAP1 has become a vital constituent for the generation of in vitro chromatin systems (15) in a range of downstream studies (16–20).
Many binding partners of histone chaperones have been identified, including ATP-dependent remodelers (21), protein kinases and nuclear import and export factors (22), and transcription factors and also other histone chaperones (23). A complex pathway of chaperone activity, dependent upon post-translational modification signals, is becoming more apparent.
The best described NAP1 is from Saccharomyces cerevisiae (yNAP),3 which was shown by analytical ultracentrifugation (AUC) to form a strong dimer in solution (24), even in very high ionic strength buffers (25). In more physiological buffers, further oligomerization of yNAP was observed using size exclusion chromatography (SEC) (12) and AUC (25). Several groups reported that the NAP1 dimer can bind an H3/H4 or H2A/H2B dimer or an H1 monomer (11, 12, 25, 26). Alternative stoichiometries have been suggested, including one NAP1 monomer binding one histone dimer (27, 28), and therefore, the question of the NAP1-histone stoichiometry remains controversial. Recent studies by Noda et al. (29) using a combination of AUC and nondenaturing mass spectrometry reported that complexes of human NAP1 are assembled sequentially from dimers to tetramers, hexamers, and octamers. In contrast, Bowman et al. (30) presented data suggesting that yNAP1 binds histones mainly as a tetrameric assembly.
The first crystal structure of a NAP1 protein was from S. cerevisiae (31). High conservation is seen throughout the NAP1 homologs, exemplified by the distant relatives S. cerevisiae and Xenopus laevis sharing 33% sequence identity (supplemental Fig. S1). Xenopus NAP (xNAP) is far closer to the human homolog, with 89% identity. Subsequent crystal structures of the Plasmodium falciparum NAP family (NAPS and NAPL) illustrated the structural similarity between the P. falciparum and S. cerevisiae homologs and led to the determination of a putative histone-binding surface (32). Despite numerous attempts by various groups, an x-ray structure of a NAP1-histone complex remains elusive, and conflicting reports on the stoichiometry and oligomerization states of NAP1-histone complexes persist. These details are vital if we are to address and understand the mechanisms of NAP1-mediated function. Here, we report the analysis of the histone chaperone NAP1 from X. laevis. A soluble proteolytic fragment lacking 49 amino acids from the unstructured N terminus is referred to hereafter as xNAP. We employed a combination of biophysical techniques to reveal both the stoichiometry and high order assemblies of complexes that xNAP forms with X. laevis histone heterodimers H2A/H2B and H3/H4. These results, together with single-particle cryo-electron microscopy, demonstrate specific stoichiometric interactions between xNAP and histones and reveal how they combine to form large dynamic ring-like assemblies.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The hexahistidine-tagged xNAP construct in the pET28b vector was transformed into Escherichia coli BL21(DE3)pLysS cells and expressed as described previously (33). The soluble fraction was isolated, and the recombinant xNAP protein was purified using nickel affinity followed by Mono Q anion exchange chromatography. The X. laevis histone constructs contained within the pET3a and pET3d vectors were transformed into E. coli BL21(DE3)pLysS cells and expressed as described previously (3). The insoluble fraction (containing the inclusion bodies) was solubilized as described previously by Luger et al. (3), and DNA contamination was removed by incubation with 1 g of hydroxylapatite resin. The samples were buffer-exchanged into 4 m urea, 20 mm sodium acetate (pH 5.2), 2 mm Tris-HCl, 200 mm NaCl, 2 mm β-mercaptoethanol, and 0.1 mm EDTA. The histone proteins were purified by cation exchange chromatography using a HiTrap SP HP chromatography column (GE Healthcare).
Size Exclusion Chromatography
xNAP and xNAP-histone complexes were analyzed using a Superose 6 10/300 column equilibrated in 10 mm Tris-HCl (pH 7.5), 3 mm MgCl2, and (unless indicated otherwise) 150 mm NaCl. SEC with multiangle laser light scattering (MALLS) was performed using a Superose 6 column in the same buffer. The scattered light intensity and protein concentration of the column eluate were recorded using a DAWN HELEOS laser photometer and an Optilab rEX differential refractometer, respectively. The weight average molecular mass of the sample peaks in the eluate was determined from the combined data from both detectors using ASTRA software (Version 5.1, Wyatt Technology) with a value of dn/dc = 0.186 ml/g.
Analytical Ultracentrifugation
A Beckman Optima XL-A analytical ultracentrifuge equipped with an An-50 Ti analytical rotor was used. Partial specific volumes and buffer densities were calculated using the program SEDNTERP (34) and corrected for temperature.
Sedimentation Velocity
400 μl of sample (either xNAP or xNAP-histone complexes) and 425 μl of SEC buffer were loaded into the corresponding sectors of a double-sector cell with a 12-mm optical path length. The rotor was accelerated to 15,000 or 40,000 rpm, and readings of absorbance at 276 nm versus radial distance were taken in step mode with a radial step size of 0.005 cm every 10 min at 10 °C. The raw data were analyzed in terms of the size distribution function (c(s)) using the program SEDFIT (35).
Sedimentation Equilibrium
90 μl of the xNAP-histone complexes at 0.2, 0.4, and 0.8 mg/ml were loaded into a six-channel cell with a 12-mm optical path length. 100 μl of SEC buffer was loaded into the three corresponding reference channels. Absorbance scans at 276 nm were collected at 3000, 5000, and 7000 rpm in step mode with a radial step size of 0.001 cm. At each spin speed, samples were left for 18 h to equilibrate prior to data collection. The rotor was then accelerated to 40,000 rpm to determine any residual absorbance. Data analysis was performed using the program Origin 6.0 (MicroCal, Inc.) with a specially designed AUC package developed by Beckman Coulter. A weighted fit was applied to a single-species model to analyze the equilibrium curves individually and then globally.
Small-angle X-ray Scattering (SAXS)
Data from xNAP and the xNAP-histone complexes were collected from the Diamond Light Source on beamline I22 and the European Synchrotron Radiation Facility on beamlines ID14-3 and ID02. Beamline parameters are detailed in supplemental Table S1. The data were normalized to the intensity of the incident beam, and scattering of the buffer was subtracted using in-house programs (Diamond Light Source) or PRIMUS43 (36) (European Synchrotron Radiation Facility). The averaged curves were processed using PRIMUS43 and GNOM44 (37) to obtain the radius of gyration and to generate a p(r) plot. Three-dimensional ab initio models were calculated using DAMMIF, averaged by DAMAVER, refined with DAMFILT (38, 39), and rendered using PyMOL (Schrödinger, LLC).
Cryo-electron Microscopy
3.5 μl of purified xNAP2-H3/H4 or xNAP2-H2A/H2B protein complex at 0.75 mg/ml in SEC buffer were applied to a 200 mesh glow-discharged C-flat 2/2 type copper grid (2-μm hole size; Protochips Inc.). The grid was blotted at room temperature and 100% humidity inside an FEI Vitrobot Mark IV device using a 3-s blotting time. Subsequently, it was plunged into liquid ethane for vitrification. The vitrified grid was loaded on a liquid nitrogen precooled Gatan 626 cryo-holder for examination under the electron beam.
Single-particle data collection was performed stroboscopically (40) on an FEI Tecnai F20 transmission electron microscope equipped with a field emission gun operating at 200 kV. The images were recorded under low dose settings with an incident flux density of 5 e− Å−2 s−1 on a 4k × 4k Gatan UltraScan 4000 slow-scan CCD on-axis camera and hardware binned providing 2k × 2k images with a pixel size of 4.6 Å2. The magnification at the detector plane was ×65,500. At each position, three images were recorded at a requested defocus of −6 μm. In total, 347 image series were acquired. Within each series, images were aligned and averaged computationally; these averages were used for further analysis.
All image processing was done with the XMIPP package (41) except for particle picking, which was done using the e2boxer program from EMAN2 (42). Particles were boxed out from averaged images and converted to XMIPP image format. The Fourier space reference-free maximum likelihood two-dimensional alignment and classification (MLF2D) were carried out with a high resolution cutoff of 30 Å to classify particles into 50 different two-dimensional classes.
GraFix and Negative Stain Electron Microscopy
To improve the quality of the samples and the imaging, we employed the GraFix method of sample preparation (43, 44). Briefly, samples of xNAP2-H3/H4 were applied to a gradient of 5–35% sucrose containing 0.2% glutaraldehyde and centrifuged at 41,000 rpm in an SW 41 swing-out rotor for 24 h at 4 °C. The gradients were fractionated in 100 mm Tris buffer.
The particles were allowed to bind to a freshly glow-discharged continuous carbon layer supported by a 200 mesh copper grid. The adhered sample was washed three times with buffer, followed by two washes with a heavy atom stain of 5% ammonium molybdate. As a final staining step, the same heavy atom solution was applied for 30 s. The carbon sandwich method was used by floating a second carbon layer onto the grid (45). This method resulted in a relative uniform background staining. Transmission electron microscopy data were collected on an FEI Tecnai 12 microscope with a BioTWIN lens operated at 80 kV and equipped with a LaB6 filament and an FEI Eagle 4k × 4k CCD camera. The data were recorded at a magnification at the detector plane of ×68,000 on hardware binned images with a requested underfocus of 1.5 μm. Particles were selected using EMAN2 (42) and classified using XMIPP (41).
RESULTS
Characterization of xNAP
To investigate the oligomeric assembly states of xNAP, we expressed and purified the main structural and functional domain as identified previously by limited proteolysis (46). This domain comprises 342 amino acids from the native coding sequence, corresponding closely to the folded region observed in the homologous yNAP x-ray crystal structure (31), and includes the extended β-hairpin that was shown to be required for oligomerization (supplemental Fig. S1) (47).
Studies with yNAP have demonstrated that the oligomeric state is strongly dependent on the ionic strength of the buffer (25, 31, 48). However, in view of the fact that there is only 33% sequence identity between the yeast and Xenopus proteins, xNAP was characterized under a range of ionic conditions using SEC-MALLS, AUC, and SAXS (see supplemental “Experimental Procedures”). At high ionic strength (500 mm NaCl), the free xNAP protein was also principally a dimer in solution with an overall shape consistent with the yNAP crystal structure (supplemental Figs. S2 and S3).
Lowering the ionic strength of the buffer had a striking effect on the oligomerization state of the xNAP protein. In SEC experiments at 50 mm NaCl, the dimer peak observed under high salt conditions was completely absent. Instead, a broad peak corresponding to multiple larger species (>400 kDa) was seen (supplemental Fig. S2A). At an ionic strength approaching physiological conditions (150 mm NaCl), we observed an intermediate situation with multiple broad elution peaks (supplemental Fig. S2B), indicating a complex equilibrium of high order oligomeric species. These were further characterized by SEC-MALLS (supplemental Fig. S2B) and AUC (supplemental Fig. S2, D and E), revealing species ranging from 8 to 18 S.
xNAP Forms Specific Stoichiometric Complexes with Histones H2A/H2B and H3/H4
Following the characterization of xNAP alone, we used similar hydrodynamic techniques to examine the interactions with histones. We purified recombinant X. laevis xNAP and histones and reconstituted high purity H2A/H2B and H3/H4 complexes. The H2A/H2B and H3/H4 complexes were individually added to a fixed quantity of xNAP and analyzed by SEC and SEC-MALLS. To explore the controversial binding stoichiometry of the xNAP-histone complexes, three ratios were analyzed, including those ratios published previously in the literature. The xNAP dimer (xNAP2) was mixed with the H2A/H2B or H3/H4 heterodimer at a molar ratio of 2:1, 1:1, or 1:2 xNAP dimer to histone dimer. Samples were then dialyzed in 150 mm NaCl buffer overnight and run individually on a Superose 6 column. For all three mixtures of xNAP2 + H2A/H2B examined, a characteristic elution peak at 11.5 ml was observed (supplemental Fig. S4, A–C). This high molecular mass peak was discrete from those resulting from the individual donor components, free xNAP2 and free histones. A shoulder to the right of the main peak was observed in the 2:1 ratio profile (supplemental Fig. S4A), and this was shown to be unbound xNAP by SDS-PAGE analysis. This shoulder was replaced by a more defined peak in the 1:1 and 1:2 ratios, which was shown to be the xNAP-histone complex by SDS-PAGE examination. These data demonstrate that all of the xNAP2 was bound to histones at the 1:1 ratio. Similarly for xNAP2 + H3/H4, a main elution peak at 11.5 ml was observed for all of the ratios examined. Unbound xNAP was again observed in the 2:1 ratio profile as a shoulder on the right-hand side of the main peak (supplemental Fig. S4D). Excess histones, as produced in the 1:2 ratio experiments, were not observed as separate peaks by SEC and were later found to form nonspecific interactions with the Superose resin. In a separate experiment, it was shown that these immobilized histones could be recovered as intact complexes by flowing free xNAP through the same column (data not shown). Neither excess histones nor excess xNAP2 was detected in the 1:1 ratio, confirming this as the probable stoichiometry.
For comparison, Fig. 1A shows three overlaid profiles of H2A/H2B, xNAP, and the resulting complex from xNAP2 + H2A/H2B at a 1:1 ratio. SDS-PAGE verified that both xNAP and H2A/H2B were present in the characteristic complex peak at 11.5 ml (Fig. 1B). SEC-MALLS analysis revealed that the elution peak was composed of oligomers with a wide range of molecular masses (Fig. 1A). A similar result was obtained with the complex of xNAP2 + H3/H4 (1:1 ratio), again with clear evidence of a mixture of oligomeric states (Fig. 1, C and D). To confirm the stoichiometry, SDS-PAGE densitometry analysis was performed. The xNAP-histone complexes were made by adding the individual components at a 1:1 stoichiometry, followed by SEC to ensure removal of any excess free components. Three concentrations of the individual proteins were analyzed to determine the staining efficiency and to allow for normalization (supplemental Table S2). The peak fraction of the complex (at three concentrations) was loaded on the same gel as the individual components for direct comparison (Fig. 1). H2A and H2B have the same electrophoretic mobility, resulting in a single band. After normalizing for the difference in staining efficiency, the ratio of xNAP2 to H2A/H2B was shown to be 0.95:1.00. This provides further evidence that the stoichiometry of binding was one xNAP dimer to one H2A/H2B heterodimer (xNAP2-H2A/H2B). Histones H3 and H4 have different electrophoretic mobilities; therefore, the summation of the two bands can be compared directly with the band corresponding to xNAP (Fig. 1D). After normalizing for staining efficiency, the stoichiometry observed for xNAP to H3 to H4 was 2.0:1.1:1.0, demonstrating that one xNAP dimer bound one H3/H4 dimer (xNAP2-H3/H4). No significant concentration dependence was observed for either complex in the range of 0.5–2.0 mg/ml (supplemental Fig. S5). Together, these data confirm that the stoichiometric unit within these large oligomers is one xNAP dimer to one histone dimer.
FIGURE 1.

xNAP2-histone complex formation. Analytical SEC was used to monitor complex formation of the xNAP2-histone complexes at 150 mm NaCl. Elution profiles were overlaid to allow for direct comparison of the individual components and their resulting complexes: xNAP2-H2A/H2B (A) and xNAP2-H3/H4 (C). SEC-MALLS was performed on each xNAP2-histone complex, and the corresponding molar mass curves are shown in red. Staining efficiencies of xNAP2 and histones were measured by performing densitometry on SDS-PAGE bands at three different concentrations as indicated in B and D. To determine the stoichiometry of the eluting xNAP2-histone complexes, the peak fraction (green asterisk or number sign for each complex, respectively) was analyzed by SDS-PAGE at three concentrations as indicated in B and D. The total integrated intensity is shown above each band in red. mAU, milli-absorbance units.
xNAP2-Histone Complexes Exist as Large Heterogeneous Oligomers in Equilibrium
Although several structures have been reported for the NAP1 dimer from various species, structural information for the NAP1-histone complexes remains elusive. We therefore employed AUC together with solution scattering techniques to reveal the structural parameters of the complexes formed between xNAP and histones H2A/H2B or H3/H4 under physiological ionic conditions (150 mm NaCl). The purified complexes were analyzed by sedimentation velocity and sedimentation equilibrium (Fig. 2 and supplemental Figs. S6 and S7).
FIGURE 2.

AUC of xNAP2-histone complexes. Sedimentation velocity was performed on the xNAP2-histone complexes at 15,000 rpm. The velocity scans and fit lines and the resultant c(s) plots are shown for xNAP2-H2A/H2B (A and B) and xNAP2-H3/H4 (C and D). mAU, milli-absorbance units; RMSD, root mean square deviation.
Analysis of velocity data for the xNAP2-H2A/H2B complex at 0.2 mg/ml (data not shown) and 0.8 mg/ml (Fig. 2) resulted in an excellent fit to the Lamm equation with a low root mean square deviation of 0.005 (Fig. 2A). The c(s) plot shows a large broad peak, indicating the presence of multiple species, together with a minor peak at 6 S (Fig. 2B). The frictional ratio (f/f0) of 2.3, together with the broad distribution, indicates sample heterogeneity and that the species present were significantly non-spherical in shape and/or highly hydrated. To estimate the molecular masses, a continuous c(M) distribution plot was derived for the two concentrations of xNAP2-H2A/H2B that were measured. The estimated molecular masses from c(M) plots using the average f/f0 were 912 and 984 kDa, respectively.
The xNAP2-H3/H4 data also fit well to the Lamm equation with a root mean square deviation of 0.005 (Fig. 2C). The frictional ratio obtained (f/f0 = 2.4) suggested that the xNAP2-H3/H4 complex was also distinctly non-spherical in shape. The c(s) plots reveal a heterogeneous sample with two peaks at 9.6 and 14 S (Fig. 2D). The molecular masses, obtained from analysis using a continuous c(M) distribution model, were 605 and 1060 kDa, respectively. For both complexes, the values are based on a single f/f0 and therefore represent an estimate because there are at least two distinct species that may have different shapes and/or unusually high hydrated volumes. In view of this, samples were also analyzed by sedimentation equilibrium.
Sedimentation equilibrium was performed at three concentrations and three rotor speeds for each xNAP2-histone complex (supplemental Figs. S6 and S7). Because analysis of the individual species could not easily be deconstructed from these data, a global analysis using the single-species model was employed. This gave a weighted average of the species in solution and is consistent with the velocity measurements, providing molecular masses in the region of 1.07 and 1.09 MDa for the xNAP2-H2A/H2B and xNAP2-H3/H4 complexes, respectively.
The xNAP2-H2A/H2B and xNAP2-H3/H4 complexes were further analyzed using SAXS to obtain additional shape information. Complexes were purified using SEC immediately prior to analysis to remove nonspecific aggregation and uncomplexed components. Guinier analysis of the resulting scattering curve from xNAP-H2A/H2B resulted in an RG of 80.6 ± 1.0 Å with an excellent fit. Fourier transformation of the scattering data to obtain the distance distribution function (p(r)) indicated that the Dmax is 250 Å (Fig. 3A). The xNAP2-H3/H4 assemblies were of a similar magnitude, with an RG of 85.1 ± 2.8 Å and a Dmax of 275 Å (Fig. 3C). In both cases, back-transformation of the distance distribution plot gave a good fit to the experimental data (Fig. 3, B and D). Analysis of the scattering curves using the program DAMMIN consistently produced oblate ring-like envelopes with approximate dimensions of 250 × 110 Å for xNAP2-H3/H4, although a number of different symmetries were equally consistent with the scattering data (Fig. 4, A–D), possibly reflecting the heterogeneous nature of the samples as indicated by AUC and SEC-MALLS. The data from the xNAP2-H2A/H2B preparations were noisier compared with the data from xNAP2-H3/H4 and are likely to reflect a more heterogeneous sample preparation. The relative increase in the vertical dimensions of these side projections (Fig. 4, I–L, upper row) may simply be an averaging feature of a wider population of conformations or less stable complexes.
FIGURE 3.

SAXS of xNAP2-H2A/H2B (beamline I22, Diamond Light Source) and xNAP2-H3/H4 (beamline ID02, European Synchrotron Radiation Facility). X-ray scattering was measured at a protein concentration of 1.3 mg/ml in 150 mm NaCl, and the buffer scattering was subtracted. The p(r) plots, together with the Dmax and estimated RG, are shown for xNAP2-H2A/H2B (A) and xNAP2-H3/H4 (C). The original scattering data compared with the back-transformed curve are also shown for xNAP2-H2A/H2B (B) and xNAP2-H3/H4 (D).
FIGURE 4.

Cryo-electron microscopy of xNAP2-H3/H4 and xNAP2-H2A/H2B complexes. Two orientations of ab initio DAMAVER envelope models based on the SAXS data (Fig. 3) are shown with P5, P6, P7, or P8 symmetry imposed for xNAP2-H3/H4 (A–D) and xNAP2-H2A/H2B (I–L). Ten DAMMIF input models were used in each case. E–H, cryo-EM images showing reference-free class averages of the xNAP2-H3/H4 complex in different oligomeric states. M–P, corresponding reference-free class averages of xNAP2-H2A/H2B.
Cryo-electron Microscopy Reveals Multiple Ring-like Assemblies of xNAP-Histone Complexes
Further structural characterization of the xNAP2-H3/H4 and xNAP2-H2A/H2B complexes was carried out using single-particle cryo-electron microscopy. The purified samples showed clear heterogeneity, as demonstrated in Fig. 4 (E–H and M–P), where a selection of two-dimensional class averages of the particles is shown. Ring-like assemblies could be identified in many classes, and it is tempting to suggest that the individual dark areas of electron density may correspond to the individual stoichiometric units of xNAP-histone complexes. Given that one stoichiometric unit corresponds to a molecular mass of ∼110 kDa, the molecular masses of the particles shown are estimated to be in the range 0.5–1.0 MDa. The particles appear to have a preferred orientation within the vitrified ice layer and are seen lying on their flat face, with respect to the grid surface, most often. The approximate diameters of the xNAP2-H3/H4 complexes range from 220 Å (Fig. 4E; class has 290 members) up to 270 Å (Fig. 4H; class has 177 members). There is evidence of significant density occupying the central core of each of these class average structures, although this density is often weaker than that of the surrounding ring. It is possible that this represents an additional stoichiometric unit that is less positionally defined and therefore becomes smeared during the averaging process. If this is the case, the molecular mass of the largest particle shown would be on the order of 1 MDa, corresponding to the observed molecular masses observed in AUC and SEC-MALLS experiments. The maximum dimensions are in agreement with those obtained by SAXS experiments, suggesting that these ring-like structures are present in solution. Although the xNAP2-H2A/H2B complexes appear rather less defined, the same general characteristics are clearly visible, including the overall dimensions and heterogeneity.
Negative Stain Electron Microscopy Reveals Alternative Conformations in High Contrast
The more stable xNAP2-H3/H4 complex was selected for higher contrast negative stain electron microscopy studies employing the sandwich method (45). Individual complexes were stabilized using the GraFix preparation method (44) utilizing a sucrose and cross-linking gradient centrifugation step. The GraFix procedure yielded four peaks, with peaks 3 and 4 showing the largest particles. We employed different negative stains (including uranyl acetate, uranyl formate, and methylamine tungstate) and found that ammonium molybdate yielded the best structural preservation of the complex. Peak fractions 3 and 4 show mainly wheel-like structures, where the outer ring contains six blobs. The outer ring shows no perfect 6-fold symmetry but seems to be distorted to different extents among the different classes found. Clear density can be seen enclosed by the outer ring, although the number of blobs (typically four) and their relative orientation vary. Fig. 5 (A–C) shows class average top views of the particles; Fig. 5 (D–F) shows side views. The latter reveals that the complex is not a flat disc structure but rather shows a variant three-dimensional stacking. The overall shape and dimensions of these particles are consistent with the corresponding ab initio SAXS envelopes for these complexes (Fig. 4, A–D).
FIGURE 5.
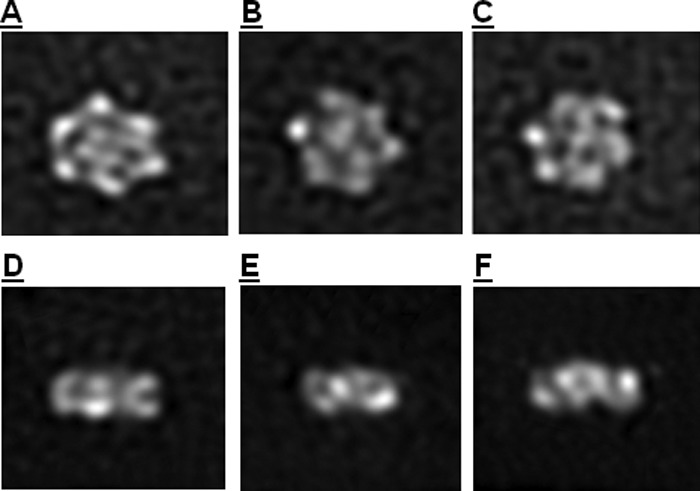
Negative stain electron microscopy of GraFix-prepared xNAP2-H3/H4 complex. Over 10,000 negatively stained particles were imaged and classified using XMIPP. Class averages show variable conformations of similarly sized particles. A–C, three class averages of top views; D–F, class averages of side views.
DISCUSSION
The current x-ray crystal structures of NAP1 reveal a dimeric organization, although there is evidence from crystal packing in the yNAP structure (Protein Data Bank code 2Z2R) that the extended β-strands may form the basis for higher oligomerization (47). Here, we have shown that although free xNAP is predominantly a dimer under high salt conditions, it forms multiple large oligomers in physiological ionic strength buffers. Despite numerous studies, much controversy surrounds the binding stoichiometry and affinities for NAP1-histone complexes from a variety of species. Previous studies have focused on S. cerevisiae and P. falciparum NAP interacting with either Xenopus or yeast histones. AUC experiments have suggested that one dimer of S. cerevisiae NAP associates with one dimer of X. laevis histones, forming a complex with a sedimentation coefficient of 5.5 S (25). Under 100 mm KCl conditions, Tóth et al. (25) observed these complexes associating into higher order assemblies estimated to be (NAP2-H2A/H2B)4 and (NAP2-H3/H4)4, both with a sedimentation coefficient of 16.6 S. Our studies focused on the interactions between NAP and histones from the higher eukaryote X. laevis, and we found that under physiological ionic conditions, xNAP forms stoichiometric complexes with histones at a ratio of one xNAP dimer to one histone heterodimer (either H2A/H2B or H3/H4). Our study provides a definitive answer to the issue of stoichiometry in the X. laevis system and is in accordance with the findings of McBryant et al. (12) and Tóth et al. (25) with yNAP. Moreover, it provides for the first time direct visualization of the architecture and multiple symmetries found in NAP-histone assemblies from any species.
One would expect that the large ring-like structures observed in our cryo-electron microscopy studies could form the basis for a scaffold that allows the controlled assembly and exchange of histones in chromatin. Recent electron microscopy studies have shown how nucleoplasmin can act as a platform for binding and transporting histones. It is notable that the xNAP-histone complexes are significantly larger than the nucleoplasmin-histone complexes and are in fact over double the diameter of a fully folded nucleosome. It could be envisaged that to maintain a stable environment while exchanging highly charged histone proteins, a large scaffold could generate a protective neutralizing local environment. Further high resolution electron microscopy studies are currently under way to gain additional insight into the structural organization of these complexes.
Acknowledgments
We thank Andrew McCarthy (European Molecular Biology Laboratory, Grenoble, France) for help and access to laboratory facilities, John Baldwin (Liverpool John Moores University, Liverpool, United Kingdom) for provision of native chicken histones, and Colyn Crane-Robinson and Alan Thorne (University of Portsmouth) for helpful discussions. We thank all of the beamline scientists from central facilities, including Phil Callow (Institut Laue-Langevin, Grenoble), Shirley Callow and Adam Round (European Synchrotron Radiation Facility, Grenoble), and Mark Malfois (Diamond Light Source, Oxfordshire, United Kingdom).
This work was supported by a Research Councils UK fellowship (to J. E. M.), a University of Portsmouth Institute of Biomedical and Biomolecular Science bursary (to E. R. N.), and Netherlands Organisation for Scientific Research Project 016.072.321 (to R. B. G. R., M. K., and F. K. N.).

This article contains supplemental “Experimental Procedures,” Figs. S1–S7, and Tables S1 and S2.
- yNAP
- yeast nucleosome assembly protein
- xNAP
- Xenopus NAP
- AUC
- analytical ultracentrifugation
- SEC
- size exclusion chromatography
- MALLS
- multiangle laser light scattering
- SAXS
- small-angle x-ray scattering.
REFERENCES
- 1. Akey C. W., Luger K. (2003) Histone chaperones and nucleosome assembly. Curr. Opin. Struct. Biol. 13, 6–14 [DOI] [PubMed] [Google Scholar]
- 2. Kleinschmidt J. A., Seiter A., Zentgraf H. (1990) Nucleosome assembly in vitro: separate histone transfer and synergistic interaction of native histone complexes purified from nuclei of Xenopus laevis oocytes. EMBO J. 9, 1309–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 4. Tse C., Sera T., Wolffe A. P., Hansen J. C. (1998) Disruption of higher order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol. Cell. Biol. 18, 4629–4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karantza V., Freire E., Moudrianakis E. N. (1996) Thermodynamic studies of the core histones: pH and ionic strength effects on the stability of the (H3-H4)/(H3-H4)2 system. Biochemistry 35, 2037–2046 [DOI] [PubMed] [Google Scholar]
- 6. Hondele M., Ladurner A. G. (2011) The chaperone-histone partnership: for the greater good of histone traffic and chromatin plasticity. Curr. Opin. Struct. Biol. 21, 698–708 [DOI] [PubMed] [Google Scholar]
- 7. Tyler J. K. (2002) Chromatin assembly. Cooperation between histone chaperones and ATP-dependent nucleosome remodeling machines. Eur. J. Biochem. 269, 2268–2274 [DOI] [PubMed] [Google Scholar]
- 8. Taneva S. G., Muñoz I. G., Franco G., Falces J., Arregi I., Muga A., Montoya G., Urbaneja M. A., Bañuelos S. (2008) Activation of nucleoplasmin, an oligomeric histone chaperone, challenges its stability. Biochemistry 47, 13897–13906 [DOI] [PubMed] [Google Scholar]
- 9. Natsume R., Eitoku M., Akai Y., Sano N., Horikoshi M., Senda T. (2007) Structure and function of the histone chaperone CIA/ASF1 complexed with histones H3 and H4. Nature 446, 338–341 [DOI] [PubMed] [Google Scholar]
- 10. Zhou Z., Feng H., Hansen D. F., Kato H., Luk E., Freedberg D. I., Kay L. E., Wu C., Bai Y. (2008) NMR structure of chaperone Chz1 complexed with histones H2A.Z-H2B. Nat. Struct. Mol. Biol. 15, 868–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kepert J. F., Mazurkiewicz J., Heuvelman G. L., Tóth K. F., Rippe K. (2005) NAP1 modulates binding of linker histone H1 to chromatin and induces an extended chromatin fiber conformation. J. Biol. Chem. 280, 34063–34072 [DOI] [PubMed] [Google Scholar]
- 12. McBryant S. J., Park Y. J., Abernathy S. M., Laybourn P. J., Nyborg J. K., Luger K. (2003) Preferential binding of the histone (H3-H4)2 tetramer by NAP1 is mediated by the amino-terminal histone tails. J. Biol. Chem. 278, 44574–44583 [DOI] [PubMed] [Google Scholar]
- 13. Park Y. J., Chodaparambil J. V., Bao Y., McBryant S. J., Luger K. (2005) Nucleosome assembly protein 1 exchanges histone H2A-H2B dimers and assists nucleosome sliding. J. Biol. Chem. 280, 1817–1825 [DOI] [PubMed] [Google Scholar]
- 14. Ito T., Bulger M., Kobayashi R., Kadonaga J. T. (1996) Drosophila NAP1 is a core histone chaperone that functions in ATP-facilitated assembly of regularly spaced nucleosomal arrays. Mol. Cell. Biol. 16, 3112–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fyodorov D. V., Kadonaga J. T. (2003) Chromatin assembly in vitro with purified recombinant ACF and NAP1. Methods Enzymol. 371, 499–515 [DOI] [PubMed] [Google Scholar]
- 16. Lorch Y., Maier-Davis B., Kornberg R. D. (2006) Chromatin remodeling by nucleosome disassembly in vitro. Proc. Natl. Acad. Sci. U.S.A. 103, 3090–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Santos S. F., Zanette D., Fischer H., Itri R. (2003) A systematic study of bovine serum albumin (BSA) and sodium dodecyl sulfate (SDS) interactions by surface tension and small-angle x-ray scattering. J. Colloid Interface Sci. 262, 400–408 [DOI] [PubMed] [Google Scholar]
- 18. Rattner B. P., Yusufzai T., Kadonaga J. T. (2009) HMGN proteins act in opposition to ATP-dependent chromatin-remodeling factors to restrict nucleosome mobility. Mol. Cell 34, 620–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y., Moqtaderi Z., Rattner B. P., Euskirchen G., Snyder M., Kadonaga J. T., Liu X. S., Struhl K. (2009) Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat. Struct. Mol. Biol. 16, 847–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Torigoe S. E., Urwin D. L., Ishii H., Smith D. E., Kadonaga J. T. (2011) Identification of a rapidly formed non-nucleosomal histone-DNA intermediate that is converted into chromatin by ACF. Mol. Cell 43, 638–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krogan N. J., Hughes T. R. (2006) Signals and systems. Genome Biol. 7, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Altman R., Kellogg D. (1997) Control of mitotic events by Nap1 and the Gin4 kinase. J. Cell Biol. 138, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Koning L., Corpet A., Haber J. E., Almouzni G. (2007) Histone chaperones: an escort network regulating histone traffic. Nat. Struct. Mol. Biol. 14, 997–1007 [DOI] [PubMed] [Google Scholar]
- 24. Ishimi Y., Yasuda H., Hirosumi J., Hanaoka F., Yamada M. (1983) A protein which facilitates assembly of nucleosome-like structures in vitro in mammalian cells. J. Biochem. 94, 735–744 [DOI] [PubMed] [Google Scholar]
- 25. Tóth K. F., Mazurkiewicz J., Rippe K. (2005) Association states of nucleosome assembly protein 1 and its complexes with histones. J. Biol. Chem. 280, 15690–15699 [DOI] [PubMed] [Google Scholar]
- 26. Zlatanova J., Seebart C., Tomschik M. (2007) Nap1: taking a closer look at a juggler protein of extraordinary skills. FASEB J. 21, 1294–1310 [DOI] [PubMed] [Google Scholar]
- 27. Andrews A. J., Downing G., Brown K., Park Y. J., Luger K. (2008) A thermodynamic model for Nap1-histone interactions. J. Biol. Chem. 283, 32412–32418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McQuibban G. A., Commisso-Cappelli C. N., Lewis P. N. (1998) Assembly, remodeling, and histone binding capabilities of yeast nucleosome assembly protein 1. J. Biol. Chem. 273, 6582–6590 [DOI] [PubMed] [Google Scholar]
- 29. Noda M., Uchiyama S., McKay A. R., Morimoto A., Misawa S., Yoshida A., Shimahara H., Takinowaki H., Nakamura S., Kobayashi Y., Matsunaga S., Ohkubo T., Robinson C. V., Fukui K. (2011) Assembly states of the nucleosome assembly protein 1 (NAP1) revealed by sedimentation velocity and nondenaturing MS. Biochem. J. 436, 101–112 [DOI] [PubMed] [Google Scholar]
- 30. Bowman A., Ward R., Wiechens N., Singh V., El-Mkami H., Norman D. G., Owen-Hughes T. (2011) The histone chaperones Nap1 and Vps75 bind histones H3 and H4 in a tetrameric conformation. Mol. Cell 41, 398–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park Y. J., Luger K. (2006) The structure of nucleosome assembly protein 1. Proc. Natl. Acad. Sci. U.S.A. 103, 1248–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gill J., Yogavel M., Kumar A., Belrhali H., Jain S. K., Rug M., Brown M., Maier A. G., Sharma A. (2009) Crystal structure of malaria parasite nucleosome assembly protein: distinct modes of protein localization and histone recognition. J. Biol. Chem. 284, 10076–10087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Friedeberg C., Scarlett G., McGeehan J., Abu-Daya A., Guille M., Kneale G. (2006) Identification of a structural and functional domain in xNAP1 involved in protein-protein interactions. Nucleic Acids Res. 34, 4893–4899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Philo J. S. (2006) Improved methods for fitting sedimentation coefficient distributions derived by time-derivative techniques. Anal. Biochem. 354, 238–246 [DOI] [PubMed] [Google Scholar]
- 35. Schuck P. (2000) Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 37. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 38. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karuppasamy M., Karimi Nejadasl F., Vulovic M., Koster A. J., Ravelli R. B. (2011) Radiation damage in single-particle cryo-electron microscopy: effects of dose and dose rate. J. Synchrotron Radiat. 18, 398–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scheres S. H., Núñez-Ramírez R., Sorzano C. O., Carazo J. M., Marabini R. (2008) Image processing for electron microscopy single-particle analysis using XMIPP. Nat. Protoc. 3, 977–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J. (2007) EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 [DOI] [PubMed] [Google Scholar]
- 43. Kastner B., Fischer N., Golas M. M., Sander B., Dube P., Boehringer D., Hartmuth K., Deckert J., Hauer F., Wolf E., Uchtenhagen H., Urlaub H., Herzog F., Peters J. M., Poerschke D., Lührmann R., Stark H. (2008) GraFix: sample preparation for single-particle electron cryomicroscopy. Nat. Methods 5, 53–55 [DOI] [PubMed] [Google Scholar]
- 44. Stark H. (2010) GraFix: stabilization of fragile macromolecular complexes for single-particle cryo-EM. Methods Enzymol. 481, 109–126 [DOI] [PubMed] [Google Scholar]
- 45. De Carlo S., Stark H. (2010) Cryonegative staining of macromolecular assemblies. Methods Enzymol. 481, 127–145 [DOI] [PubMed] [Google Scholar]
- 46. Friedeberg C. (2005) Structural and Functional Characterization of the Nucleosome Assembly Protein, xNAP1. Ph.D. thesis, The University of Portsmouth [Google Scholar]
- 47. Park Y. J., McBryant S. J., Luger K. (2008) A β-hairpin comprising the nuclear localization sequence sustains the self-associated states of nucleosome assembly protein 1. J. Mol. Biol. 375, 1076–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McBryant S. J., Peersen O. B. (2004) Self-association of the yeast nucleosome assembly protein 1. Biochemistry 43, 10592–10599 [DOI] [PubMed] [Google Scholar]