

Background: Human SKIL gene encodes for SNON, a negative regulator of the TGF-β/SMAD pathway.
Results: We provide a molecular mechanism of transcriptional regulation of SKIL gene expression by TGF-β/SMADs.
Conclusion: Transcriptional cofactor complex SNON-SMAD4 negatively controls the expression of SKIL gene.
Significance: The formation and function of complex SNON-SMAD4 are impaired in cancer cells lacking SMAD4, which affects TGF β-target gene regulation.
Keywords: Corepressor Transcription, Receptor Serine/Threonine Kinase, SMAD Transcription Factor, Transcription Promoter, Transcription Regulation, Transforming Growth Factor β (TGFβ), SKIL, SNON
Abstract
The human SKI-like (SKIL) gene encodes the SMAD transcriptional corepressor SNON that antagonizes TGF-β signaling. SNON protein levels are tightly regulated by the TGF-β pathway: whereas a short stimulation with TGF-β decreases SNON levels by its degradation via the proteasome, longer TGF-β treatment increases SNON levels by inducing SKIL gene expression. Here, we investigated the molecular mechanisms involved in the self-regulation of SKIL gene expression by SNON. Bioinformatics analysis showed that the human SKIL gene proximal promoter contains a TGF-β response element (TRE) bearing four groups of SMAD-binding elements that are also conserved in mouse. Two regions of 408 and 648 bp of the human SKIL gene (∼2.4 kb upstream of the ATG initiation codon) containing the core promoter, transcription start site, and the TRE were cloned for functional analysis. Binding of SMAD and SNON proteins to the TRE region of the SKIL gene promoter after TGF-β treatment was demonstrated by ChIP and sequential ChIP assays. Interestingly, the SNON-SMAD4 complex negatively regulated basal SKIL gene expression through binding the promoter and recruiting histone deacetylases. In response to TGF-β signal, SNON is removed from the SKIL gene promoter, and then the activated SMAD complexes bind the promoter to induce SKIL gene expression. Subsequently, the up-regulated SNON protein in complex with SMAD4 represses its own expression as part of the negative feedback loop regulating the TGF-β pathway. Accordingly, when the SNON-SMAD4 complex is absent as in some cancer cells lacking SMAD4 the regulation of some TGF-β target genes is modified.
Introduction
TGF-β cytokine regulates several cellular processes such as proliferation, differentiation, and apoptosis mainly through the activation of SMAD transcriptional factors (1–4). Because of the variety of SMAD2 (S2),3 SMAD3 (S3), and Co-SMAD4 (S4) heteromeric complexes that can be generated, the transcription of most TGF-β target genes can be differentially regulated in a cell context-dependent manner (4). In addition, many of the TGF-β/SMAD actions can be antagonized by nuclear SKI and SKI-novel (SNON) proteins, which are two closely related members of the SKI family of oncoproteins that were identified by their homology with the viral transforming protein v-SKI (5–8). SNON and SKI act as SMAD corepressors by interacting with SMAD complexes to inhibit their transcriptional abilities and by recruiting other corepressors and histone deacetylases (HDACs) to diverse TGF-β-responsive gene promoters (5, 9–11). So far, only a few TGF-β target genes have been shown to be directly regulated by SKI and SNON. The SMAD7 gene, a negative regulator of the TGF-β pathway, is likely the best characterized gene negatively regulated by SKI and SNON corepressors (12, 13). In addition, SNON and SKI proteins can also be localized in the cytosol where they might be able to block TGF-β signals by sequestering SMAD proteins and preventing their translocation to the nucleus (5, 14).
TGF-β tightly regulates SNON and SKI protein stability by inducing their degradation via the ubiquitin-proteasome system (UPS) in a SMAD-dependent manner (15–22). Interestingly, the TGF-β/SMAD pathway regulates SNON protein levels in a biphasic manner: it causes a rapid and transient SNON protein degradation via the proteasome followed by an up-regulation of SNON mRNA and protein levels after a longer TGF-β treatment. This newly synthesized SNON protein seems to establish a negative feedback loop to turn off TGF-β signaling; this is an important but poorly understood event (13, 23). The regulation of SNON expression is relevant because SNON has an essential role during embryonic development as well as in homeostasis in the adult organism. SNON is expressed at low levels in embryonic and postnatal tissues, but its expression can be increased in some tissues at specific stages of embryonic development or in different physiological contexts (5, 6). Moreover, SNON protein up-regulation may have a relevant role in regulating the magnitude and duration of TGF-β signaling.
SKIL knock-out causes lethality in mice because Sno gene is required for blastocyst formation (5, 6). Sno+/− mice with very low SNON protein expression are more susceptible to tumorigenesis, suggesting a tumor suppressor role for SNON (5, 6, 24). In contrast, high levels of SNON mRNA and protein have been reported in many cancer cell types, also suggesting an oncogenic role. In fact, the overexpression of SNON seems to contribute to cell resistance to TGF-β-induced growth arrest in some cell types and also induces anchorage-independent growth of chicken and quail embryo fibroblasts (10, 14, 24–27). Therefore, it has been proposed that high levels of SNON protein might lead to tumor growth, whereas low levels may lead to tumor metastasis (26). Interestingly, SNON mRNA and protein levels are up-regulated during liver regeneration, a model of cell proliferation distinct from cancer where SNON probably functions in restraining the mitoinhibitory effect of the TGF-β/SMAD pathway (28); in contrast, low levels of SNON have been observed in renal fibrosis where it might favor TGF-β profibrotic actions (29, 30).
It is clear that a tight regulation of SNON expression is critical for SNON to function adequately in space and time. However, the transcriptional regulation of skil gene is not completely understood. Recently, the mouse Sno (SKIL) gene promoter was cloned and partially characterized in fibroblasts (25). This promoter bears a TGF-β response region with four SMAD-binding element (SBE) groups that bind S2-S4 complexes to activate SKIL gene expression. The promoter also contains a SMAD inhibitory element downstream of the SBE region that binds the inhibitory S3-S4 complex, and that study clearly demonstrated that mouse SKIL gene is a TGF-β/SMAD pathway target. Intriguingly, the authors also observed a prolonged induction of SNON expression by TGF-β, which was shown to have a critical role in fibroblast transformation (25).
Here, we cloned and analyzed a fragment of the human SKIL gene promoter bearing the core and proximal promoter as well as a TGF-β response element (TRE) containing several SBEs. We focused on studying the molecular mechanisms involved in the self-regulation of SKIL gene expression by SNON protein in different cell contexts.
EXPERIMENTAL PROCEDURES
Bioinformatics Analysis
A genomic DNA sequence of ∼5 kb located immediately upstream from the ATG of human SKIL gene was obtained from GenBankTM (accession number AC073288). This sequence was analyzed to predict the putative promoter region, transcription factor binding sites, and TSS of human SKIL gene by using several software tools including GenBank, FPROM, FirstEF, DBTSS, GPminer, and ALGGEN-PROMO (supplemental Table S1 and Fig. S1).
Cloning of the Human SKIL Gene Promoter Bearing TRE and Generation of Plasmid Constructs
Two fragments of the SKIL gene harboring the promoter (408 and 648 bp) were amplified by PCR from human genomic DNA (obtained from freshly isolated human blood leukocytes) using AccuPrime GC-rich DNA polymerase (Invitrogen) and specific primers flanked by KpnI and SacI restriction sites. These SKIL gene fragments are located at positions −3100/−2692 (408 bp) and −3100/−2451 (648 bp) upstream from ATG (+1). The reporter plasmids were constructed by cloning each fragment into KpnI and SacI sites of pGL3-Basic vector (Promega) to obtain the reporter genes skilSBEs(408)-Luc (408-bp fragment) and skilSBEs(648)-Luc (648-bp fragment). The cloned 408- and 648-bp fragments of the SKIL promoter contained all four SBE groups and the TSS (+1). The SKIL promoter region of 408 bp was also cloned with an inverted orientation in the pGL3-Basic vector by subcloning the insert into KpnI and XhoI sites of pcDNA3.1 and then subcloned into HindIII and XhoI sites of pGL3-Basic to obtain the reporter skilSBEs(408, 3′–5′)-Luc. The region of 408 bp was also subcloned into KpnI and SacI sites of pGL3 minimal promoter-E1B-Luc to obtain the reporter skilSBEs(408)-E1B-Luc. All constructs were sequence-verified.
Cell Lines
A549 (human lung carcinoma) cells were maintained in Ham's F-12 medium, and SW480 (human colon carcinoma) cells were cultured in a mixture of Dulbecco's modified Eagle's medium (DMEM)/F-12 medium (1:1), whereas HepG2 (human hepatoma) and AD293 (a clone derived from HEK293 human embryonic kidney cell line) cells were maintained in DMEM. Growth medium was supplemented with 10% fetal bovine serum (FBS) plus antibiotics (penicillin/streptomycin), and cells were maintained under a 5% CO2 atmosphere at 37 °C. AD293 and A549 cells stably expressing pRS/shSnoN (catalog number TR309425 from OriGene), pRetroSuper/shSmad4J hygro (Addgene plasmid 19151), or pBABE/Smad4J Rescue (Addgene plasmid 19153) (31) were maintained in the presence of 10 μg/ml puromycin or 200 μg/ml hygromycin as selection antibiotics.
SNON Site-directed Mutagenesis
DmSNON(ΔS2/S3/S4) and UBmSNON(K437A,K446A) were generated by site-directed mutagenesis on the pCIneo/HA-SnoN (wild type mouse SNON) using specific primers (supplemental Table S3) according to the manufacturer's instructions (Stratagene). All constructs were sequence-verified.
Luciferase Assays
For TGF-β-inducible luciferase assays, A549, HepG2, SW480, and AD293 cells were transiently transfected with the reporter plasmids containing fragments of the SKIL gene promoter and pCMV/β-gal with or without any of the following plasmids: pCMV5/TβRI-HA (wild type (WT)), pCMV5/TβRI-HA (T204D), pCMV5/TβRI-HA (K232R), pCMV5/FLAG-Smad2, pCMV5/FLAG-Smad3, pCMV5/HA-Smad4, pCIneo/HA-Ski, pCIneo/HA-SnoN, pCIneo/HA-DmSnoN, or pCIneo/HA-UBmSnoN. Cells were seeded at 60% confluence in 12-well plates and transiently transfected with 0.5–1 μg of total DNA/well using the Lipofectamine method for A549 cells or calcium phosphate method for SW480 and AD293 cells as described previously (3, 13). 24 h postransfection, cells were treated for 12 h with 100 pm TGF-β1 (PreproTech), then they were lysed, and luciferase activity (Promega) was measured in a luminometer (Turner Biosystems). β-Galactoside activity was used to normalize for transfection efficiency.
RT-PCR Assay
SNON and β-actin mRNA levels were detected by RT-PCR using the primers and conditions described previously (13). In brief, total RNA was isolated using TRIzol (Invitrogen) from cells treated with or without 300 pm TGF-β for the indicated times. Total RNA (2 μg) was used for cDNA synthesis using random hexamers and Moloney murine leukemia virus RT (Invitrogen), and PCR was carried out using Taq PCR Master Mix kit (Qiagen) using specific primers (supplemental Table S2). PCR products were analyzed by electrophoresis on agarose gels.
Immunoprecipitation and Western Blot
Cells were lysed with TNTE buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA containing 0.5% Triton X-100 plus a mixture of protease and phosphatase inhibitors) as described previously (13). Proteins were immunoprecipitated with specific anti-SMAD2/SMAD3 (N-20), anti-SMAD4 (C-20), anti-SNON (H-317) polyclonal antibodies (Santa Cruz Biotechnology) or rabbit polyclonal anti-SMAD4 (Upstate/Millipore), and then proteins were separated by SDS-PAGE and detected by immunoblotting with specific primary antibodies and either anti-rabbit or anti-goat secondary HRP-conjugated antibody (Zymed Laboratories Inc.) by using an enhanced chemiluminescence assay (ECL kit from Amersham Biosciences). Phospho-SMAD2 was detected with a specific polyclonal antibody (Chemicon/Millipore).
Chromatin Immunoprecipitation (ChIP) and Sequential ChIP (Re-ChIP) Assays
ChIP assays were carried out as described previously (13) with the following modifications. Cells were treated with 1% formaldehyde at 37 °C for 15 min and then sonicated on ice for 10 cycles of 30 s each with a Fisher Sonic Dismembrator 300. The desired amount of protein-cross-linked DNA extract was precleared in batches, and specific antibodies were used for immunoprecipitation. Oligonucleotide sequences used for PCR were the same used for cloning the human SKIL gene promoter (supplemental Table S2). PCR products were analyzed by electrophoresis on agarose gels. For re-ChIP, the DNA-protein complexes immunoprecipitated with the indicated antibodies were eluted with 10 mm dithiothreitol, diluted 20× in re-ChIP buffer (1% Triton X-100, 2 mm EDTA, 150 mm NaCl, 20 mm Tris-HCl, pH 8.0), and then reimmunoprecipitated with the indicated antibodies (32).
ChIP on Reporter Plasmid
AD293 cells were transiently transfected with 3 μg of skilSBEs(408)-Luc reporter plasmid by the Lipofectamine method. Cells were cross-linked with formaldehyde 48 h post-transfection and harvested, and then a ChIP assay was performed as described previously (33). The obtained DNA fragments were analyzed by PCR with specific primers to amplify the region of SKIL promoter on the reporter (supplemental Table S2).
Northern Blot
Total RNA was purified from either primary cultured mouse hepatocytes or the human HepG2 cell line using TRIzol (Invitrogen). Northern blots were performed as described previously (28).
Wound Healing Assay
Confluent A549 cells were serum-starved for 12 h, and then a wound was generated across the cell monolayer using a 1-mm plastic tip. Cell pictures were taken at different time points (0, 24, 48, and 72 h) after wounding. Data were expressed as a percentage of wound closure.
Statistical Analysis
A Student's t test was used to calculate statistical significance. A p value <0.05 was considered to be significant.
RESULTS
SNON Expression Is Regulated by TGF-β at the Transcriptional Level
SNON is a regulatory protein capable of antagonizing TGF-β/SMAD signaling. Several studies have shown that TGF-β tightly regulates the expression levels of SNON protein and mRNA (13, 15–23, 25, 28, 34, 35). Northern blot analysis using total RNA from HepG2 cells or mouse hepatocytes showed that SNON mRNA was induced after 1-h treatment with 300 pm TGF-β. We detected three main mRNA transcripts for human SKIL gene (∼6.2, 3.5, and 3.0 kb) and for mouse Skil gene (∼6.2, 3.6, and 3.2 kb) (Fig. 1A). The main difference in the size of these transcripts is probably due to different lengths of their 3′-UTRs as has been reported previously (34, 35). In fact, the largest transcript reported in GenBank (accession number NM_005414.4) contains two poly(A) sites in the 3′-UTR (supplemental Fig. S1). Furthermore, the cycloheximide pretreatment induced an accumulation of SNON mRNA transcripts in both HepG2 cells (Fig. 1A) and mouse hepatocytes (data not shown). These results confirm that TGF-β positively regulates SNON expression at the transcriptional level, and it does not require de novo protein synthesis.
FIGURE 1.

TGF-β regulates SNON expression at the transcriptional level. A, TGF-β induces the early expression of three main SNON mRNA transcripts. HepG2 cells and mouse hepatocytes were treated for 1 h with 300 pm TGF-β. HepG2 cells were also pretreated in the absence or presence of 20 μg/ml cycloheximide (CHX) for 20 min. Total RNA was isolated, and Northern blot analysis of SNON mRNA was performed. rRNAs (18 and 28 S) are shown as an RNA loading control. B, to analyze SNON and SNON2 protein expression, whole cell protein extracts from human HepG2 or A549 or mouse C2C12 cells were immunoprecipitated and then detected by WB with specific anti-SNON antibodies (upper panel), or total RNA was obtained from human HepG2 or mouse C2C12 cells treated for 1 h with or without 300 pm TGF-β to analyze SNON and SNON2 mRNA levels by RT-PCR (lower panel). C, the assembled SKIL gene map shows the localization of promoter, TRE, putative TSS, ATG (+1), exon 1 (E1), and exon 2 (E2). D, the pGL3 reporter gene constructs bearing different fragments of the SKIL gene promoter or empty vectors are schematically shown. E, AD293 cells transiently transfected with the indicated reporter plasmids were incubated for 12 h in the absence or presence of 100 pm TGF-β, and cell extracts were analyzed for luciferase activity. F, AD293 cells were transfected with the skilSBEs(408)-E1B-Luc reporter along with plasmids bearing full-length cDNA for S2, S3, or S4, and then cells were incubated for 12 h in the absence or presence of 100 pm TGF-β. Cells were lysed, and samples were analyzed for luciferase activity. Luciferase activity was normalized using β-gal expression and expressed as -fold induction over control. Values are mean ± S.E. (error bars) of three separate experiments in triplicate. *, p < 0.05; **, p < 0.01 compared with control (C).
Interestingly, the analysis of SNON and SNON2 isoform expression showed that human cell lines such as HepG2 and A549 mainly expressed SNON protein isoform, which was also the main isoform induced by TGF-β treatment (Fig. 1B). In contrast, in mouse cell line C2C12 (Fig. 1B) and mouse hepatocytes (data not shown), both SNON and SNON2 protein isoforms were expressed, although SNON2 was the most abundant isoform expressed and the main isoform induced by TGF-β.
To better understand the transcriptional regulation of human SKIL gene, we first performed a bioinformatics analysis of an ∼5-kb genomic DNA sequence from human chromosome 3, which is located immediately upstream of the ATG of SKIL gene (GenBank accession number AC073288). We used the prediction programs GPminer, Genomatix, ALGGEN-PROMO, DBTSS, FPROM, and FirstEF (supplemental Table S1) to determine the promoter region of SKIL gene including the TSS. We also performed an alignment between a human chromosome 3 clone (GenBank accession number AC073288) and the ∼7.2-kb clone of human SNON mRNA (accession number NM_005414.4) using NCBI-BLAST and Aspic. Results revealed that the promoter of SKIL gene including a region with some potential TSSs was located at ∼2.6 kb upstream from the ATG. The analysis showed that the promoter region of SKIL gene has a high G+C content with GC boxes, but neither TATAA nor CAAT boxes were found. Also, we detected a CpG island (∼1.6 kb) spanning the promoter of SKIL gene by using the CpG Island Searcher and EMBOSS programs (supplemental Fig. S1C and Table S1).
Further analysis showed that a ∼450-bp sequence spanning the core promoter of the SKIL gene has four groups of putative binding sites for SMAD transcription factors (SBEs) (supplemental Figs. S1, S2, and S3); this region corresponds to the TRE that is conserved in the mouse Skil gene promoter (supplemental Figs. S1, S2, and S3). These SBEs found in the human SKIL promoter showed high identity to those previously identified on the mouse Skil promoter by footprinting (25). Each one of the four SBEs in the SKIL promoter has one or two consensus sites for SMAD binding that were identified previously as important regulatory motifs for the expression of the mouse Skil gene controlled by TGF-β (25).
On the other hand, the analysis of the 5′-region of SKIL gene structure showed the presence of a small first exon (∼170 bp) followed by the first intron (1933 bp) and part of the second exon (633 kb) (supplemental Fig. S1A). Thus, the SKIL gene contains seven exons, the first exon is noncoding, the ATG is localized at 5′-half of the second exon (supplemental Fig. S1A), and the size of the main SNON mRNA transcript generated is ∼7.2 kb (supplemental Fig. S1B). The position of the putative TSS (+1) of SKIL gene was localized between SBE3 and SBE4 of TRE from data obtained via partial cloning of the 5′-UTR by RT-PCR assay (supplemental Fig. S2, A, B, and C). The 5′-UTR fragment of SNON mRNA (839 bp) was cloned and sequence-verified (supplemental Fig. S2D). This partial nucleotide sequence of the 5′-UTR (from −2738 to −2571 and from −635 to +37 nucleotides) was submitted to GenBank under accession number JX103164 (supplemental Fig. S3).
In addition, we found several consensus sites for different transcription factors on this region by bioinformatics analysis (supplemental Fig. S4). Some of these transcription factors are known SMAD partners such as CCAAT/enhancer-binding proteins, p53, NFκB, STATs, MYOD, and YY1 among others (11). These data support that this region contains a functional gene promoter that is probably regulated by different signaling pathways under specific cellular contexts or in cross-talk with the TGF-β pathway.
The TGF-β-responsive Region of the SKIL Gene Promoter Is Also SMAD-responsive
To study the regulation of the TRE region of the SKIL promoter by TGF-β, we made two main reporter plasmids that include the SBE region and TSS, skilSBEs(408)-Luc and skilSBEs(648)-Luc, each one bearing a fragment of 408 or 648 bp, respectively (Fig. 1, C and D). These reporter gene constructs were transiently transfected into AD293 cells; pGL3-Basic vector was used as a control. As expected, both reporters were clearly responsive to the TGF-β stimulus (Fig. 1E). Thus, our results indicate that the region of the human SKIL gene promoter that contains the SBEs is part of the proximal promoter of SKIL gene and is TGF-β responsive. We also obtained similar results with a reporter plasmid bearing the mouse Skil gene promoter (data not shown).
We also observed that the transcriptional activity of the SKIL gene promoter (skilSBEs(408)-Luc) is specific and orientation-dependent because the same sequence cloned in an inverted orientation into pGL3-Basic (skilSBEs(408, 3′–5′)-Luc) lost TGF-β-induced activity (Fig. 1E). We made another reporter gene named skilSBEs(408)-E1B-Luc, which contains the minimal E1B promoter and the 408-bp fragment of the SKIL promoter; this reporter had a response to TGF-β similar to that of the reporter lacking the E1B promoter (Fig. 1E). These results support the conclusion that the region corresponding to the promoter of SKIL gene is spanned by the TRE.
We used the skilSBEs(408)-Luc reporter construct to characterize the response to TGF-β; thus, different epithelial cell lines were transiently transfected with this reporter, and its activity was measured by a luciferase assay. This reporter was responsive to TGF-β stimulus in all the different cell lines tested such as AD293, A549, and HepG2 (data not shown). We observed that the induction of this reporter was also dependent on TGF-β concentration in all cells tested (data not shown). In addition, the constitutively active form of the TGF-β receptor ALK5 (T204D mutation) was able to induce the SKIL gene promoter, whereas the WT ALK5 and a kinase-deficient ALK5 (K232R mutation) were inactive in AD293 cells (data not shown). Moreover, a pretreatment with the ALK5 inhibitor SB431542 (10 μm) (Tocris), which specifically blocks SMAD2 and SMAD3 phosphorylation, clearly prevented SKIL gene promoter induction by TGF-β in AD293 cells (data not shown).
We then analyzed the activation of SKIL gene promoter by overexpressing different SMAD proteins in AD293 cells. The S2 or S2-S4 overexpression increased SKIL gene promoter expression and also enhanced the induction by TGF-β, whereas S3, S4, S2-S3, S3-S4, or S2-S3-S4 overexpression showed an inhibitory effect (Fig. 1F). Thus, TGF-β positively regulates SKIL promoter activity mainly through specific S2-S4 complexes. Our results agree with data reported previously for the mouse Skil promoter (25).
SMAD and SNON Proteins Bind to and Regulate SKIL Gene Promoter
To examine whether SNON was able to negatively regulate its own expression, we performed luciferase assays by transiently co-transfecting AD293 cells with the skilSBEs(408)-Luc reporter along with plasmids bearing WT HA-Ski or HA-SnoN cDNAs. We observed that SNON and SKI were potent inhibitors of SKIL gene promoter activity (Fig. 2A) and that the SKIL promoter expression inhibited by SNON was dependent on the concentration of the transfected pCIneo/HA-SnoN plasmid (Fig. 2B).
FIGURE 2.
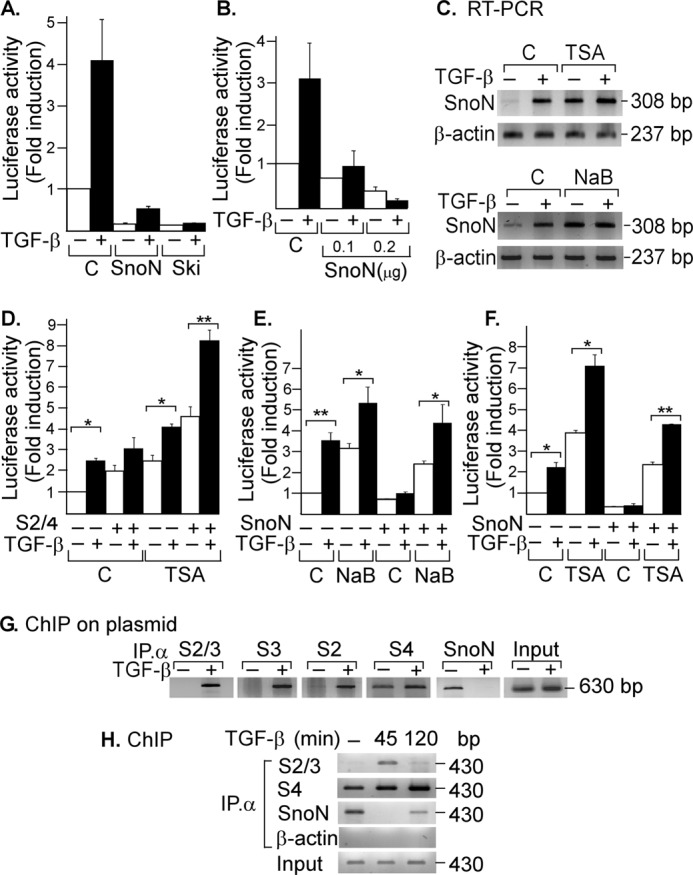
SNON binds to the SKIL promoter and represses its own expression. AD293 cells were transiently transfected with skilSBEs(408)-Luc along with HA-Ski or HA-SnoN cDNAs (A) or with different amounts of pCIneo/HA-SnoN (B). Cells were incubated for 12 h in the absence or presence of 100 pm TGF-β, then lysed, and analyzed for luciferase activity. Luciferase activity was normalized using β-gal expression and is reported as -fold induction over control. Values are expressed as means ± S.E. (error bars) of three separate experiments in triplicate. C, A549 cells were pretreated for 30 min with or without the HDAC inhibitors TSA (100 nm) (upper panel) and NaB (5 mm) (lower panel) and then incubated for 2 h in the absence or presence of 300 pm TGF-β. Total RNA was isolated, and SNON (308-bp) and β-actin (237-bp) mRNAs were amplified by RT-PCR with specific primers (n = 3). D, AD293 cells transfected with skilSBEs(408)-Luc reporter along with plasmids bearing cDNAs for S2 and S4 were pretreated for 30 min with or without 100 nm TSA and then incubated for 12 h in the absence or presence of 100 pm TGF-β. E and F, AD293 cells transfected with skilSBEs(408)-Luc reporter with or without HA-SnoN were pretreated for 30 min with or without 5 mm NaB or 100 nm TSA and then incubated for 12 h in the absence or presence of 100 pm TGF-β, and then cells were lysed and analyzed for luciferase activity. Luciferase activity was normalized using β-gal expression and is reported as -fold induction over control. Values are expressed as means ± S.E. (error bars) of three separate experiments in triplicate. *, p < 0.05; **, p < 0.01 compared with control (C). G, AD293 cells were transiently transfected with skilSBEs(408)-Luc reporter, and 48 h post-transfection, cells were incubated for 45 min in the absence or presence of 500 pm TGF-β (n = 2). H, A549 cells were incubated for 45 min or 2 h in the absence or presence of 500 pm TGF-β (n = 2). ChIP on plasmid (G) or ChIP (H) assays were carried out using anti-S2/S3, anti-S2, anti-S3, anti-S4, or anti-SNON antibody. PCRs were done with primers spanning the SBE region either from the pGL3 vector bearing the promoter fragment (630 bp) or a region from the endogenous SKIL promoter (430 bp).
Several studies have proposed that SNON and SKI can associate with some repressor factors that interact with HDACs to inhibit gene expression (10, 13, 36–39). To understand how SNON inhibits the transcriptional expression of its own gene, we evaluated the effect of HDAC inhibitors trichostatin (TSA) and sodium butyrate (NaB). SNON mRNA levels were analyzed in A549 cells pretreated for 1 h with or without 0.1 μm TSA or 5 mm NaB and then incubated in the presence or absence of 300 pm TGF-β for 2 h. Data showed that HDAC inhibition by TSA or NaB increased basal SNON mRNA levels and facilitated induction of SKIL gene by TGF-β (Fig. 2C). These inhibitors also increased the basal and TGF-β-induced activity of skilSBEs(408)-Luc gene reporter (Fig. 2, D, E, and F). The S2-S4 complexes increased skilSBEs(408)-Luc reporter activity, and this was favored by TSA treatment (Fig. 2D). Interestingly, HDAC inhibitors also blocked the repression of skilSBEs(408)-Luc reporter caused by SNON protein (Fig. 2, E and F). Data showed that SNON self-represses its gene by forming a repressor complex with HDAC activity.
Expression of the SKIL gene is positively regulated by SMADs and negatively regulated by SNON. Thus, we evaluated the binding of endogenous SMAD and SNON proteins to the SKIL gene promoter by ChIP on a plasmid assay. We first analyzed the binding of endogenous S2, S3, and S4 proteins on skilSBEs(408)-Luc reporter plasmid previously transfected along with HA-SnoN cDNA in AD293 cells. Cell extracts were immunoprecipitated with specific antibodies for anti-S2/S3, -S2, -S3, -S4, or -SNON, whereas anti-β-actin antibody was used as a control, and the DNA that co-immunoprecipitated with these proteins was used to amplify the region of the pGL3 vector bearing the SKIL promoter fragment (630 bp) by PCR. It was observed that endogenous S2 and S3 proteins interacted with the SBE region only in response to TGF-β, whereas SNON was bound to that region only at the basal state. Intriguingly, S4 was bound to the SKIL promoter in basal conditions, and the binding was further increased by TGF-β stimulation (Fig. 2G). These results demonstrate that SNON and SMAD proteins bind differentially to the human SKIL gene promoter and indicate that the repression and activation of the SKIL gene promoter occur at different time points during TGF-β signaling.
We further evaluated the dynamic of endogenous SMAD and SNON binding to the SKIL gene promoter in response to TGF-β, considering that TGF-β exerts a fine-tuned time-dependent regulation of SNON levels. We performed ChIP assays using A549 cells treated with 500 pm TGF-β for different time points (0, 45, and 120 min). Immunoprecipitations were carried out with anti-S2/S3 and anti-SNON specific antibodies, and then the co-immunoprecipitated DNA was amplified by PCR with specific primers for the SKIL gene promoter (430 bp). Data showed that the endogenous activated S2 and S3 were transiently bound to the SKIL gene promoter after TGF-β treatment (45 min), and then their binding decreased at 120 min (Fig. 2H). In contrast, we observed that endogenous SNON protein was associated with the SKIL gene promoter at the basal level, but this association decreased shortly after TGF-β treatment, which coincides with SNON down-regulation induced by TGF-β. Interestingly, after a longer TGF-β treatment (>2 h) when SNON protein levels were up-regulated, the SNON protein was observed to bind back to the SKIL promoter to repress it probably as part of a negative feedback loop generated by TGF-β itself (Fig. 2H). This result demonstrates that SNON and SMADs bind to the SKIL gene promoter and also provides a molecular mechanism for the temporal repression and activation of SKIL gene promoter during TGF-β/SMAD signaling.
The Expression of SNON Is Regulated at Different Levels
SNON expression is regulated at multiple levels such as gene transcription, mRNA stability, and translation as well as at the level of protein stability. For this reason, it is difficult to find the correlation between SNON mRNA and protein levels at specific time points after TGF-β stimulus. Therefore, to analyze the time course of induction of SNON mRNA in response to TGF-β, A549 cells were incubated for different times with or without 300 pm TGF-β. Total RNA was then isolated, and a fragment (308 bp) of SNON mRNA and a fragment (317 bp) of SMAD7 mRNA were amplified by RT-PCR with specific primers (Fig. 3, A and B). Analyzing the time course of induction of SNON, we observed that SNON mRNA levels were increased 2 h after stimulus and remained elevated until 4 h later (Fig. 3A). We observed that 2 h after TGF-β stimulation the SNON-S4 repressor complex was again positioned on the SKIL promoter instead of the S2-S4 activator complex; thus, under this scenario, we investigated why the levels of SNON mRNA remained elevated for so long.
FIGURE 3.
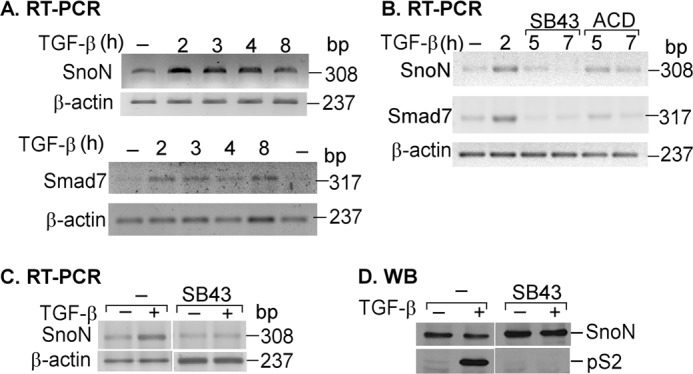
SNON mRNA and protein levels are regulated at different levels after TGF-β stimulus. A, A549 cells were incubated for different times with or without 300 pm TGF-β. Total RNA was isolated, and then SNON (308-bp), SMAD7 (317-bp), and β-actin (237-bp) mRNAs were amplified by RT-PCR with specific primers (n = 3). B, A549 cells were incubated for different times in the presence or absence of 300 pm TGF-β, and in the indicated cases, 5 μg/ml actinomycin D (ACD) or 10 μm SB431542 (SB43) was added 2 h post-treatment with TGF-β. Total RNA was isolated, and RT-PCR was performed with specific primers for SNON (308 bp), SMAD7 (317 bp), and β-actin (237 bp) (n = 2). C and D, A549 cells were preincubated for 30 min in the presence or absence of 10 μm SB431542 and then incubated for 1 h with or without 300 pm TGF-β. Total RNA was isolated, and RT-PCR was performed with specific primers for SNON and β-actin (n = 2) (C), or whole cell protein extracts were immunoprecipitated with anti-SNON or anti-S2/S3 antibody and then detected by WB with anti-SNON or anti-phospho-S2 antibody (n = 3) (D).
To evaluate whether the levels of SNON mRNA remained elevated due to an increase in its stability or to continuous SKIL gene transcription, we used actinomycin D to inhibit transcription and SB431542 to inhibit SMAD activation. A549 cells were incubated for 2 h in the presence or absence of 300 pm TGF-β, and then 5 μg/ml actinomycin D or 10 μm SB431542 was added for different times (Fig. 3B). Total RNA was isolated, and RT-PCR was performed with specific primers for SNON. We observed that SB431542 induced a faster decrease of SNON mRNA levels than actinomycin D, suggesting that activated SMADs may play a role not only in inducing SKIL gene transcription but also in controlling SNON mRNA stability (Fig. 3B). Similar results were obtained when SMAD7 mRNA levels were analyzed (Fig. 3, A and B); SMAD7 was identified previously as a SNON target gene (13).
Because multiple factors are involved in controlling SNON expression, it has been difficult to observe a correlation between SNON mRNA and protein levels. Thus, we studied the effect of SB431542 on SNON expression. Serum-starved A549 cells were preincubated for 30 min in the absence or presence of 10 μm SB431542 and then incubated for 2 h with or without 300 pm TGF-β. Total RNA was isolated, and RT-PCR was performed with specific primers for SNON (Fig. 3C), or whole cell protein extracts were immunoprecipitated with anti-SNON or anti-S2/S3 antibody and then detected by immunoblot with anti-SNON or anti-phospho-S2 (Fig. 3D). Data showed that SB431542 treatment decreased SNON mRNA levels but increased SNON protein levels at the basal conditions. Thus, the inactivation of SMADs seems to promote low levels of SNON mRNA and high levels of SNON protein, suggesting a role for SMADs in controlling both SNON mRNA and protein stability.
SNON Is Removed from SKIL Gene Promoter upon TGF-β Stimulation Independently of Its Degradation
It has been proposed that SNON and SKI corepressors maintain some TGF-β target genes repressed in the absence of ligand; however, only a few SKI and SNON target genes have been identified so far. Upon TGF-β stimulation, S2 and S3 translocate into the nucleus and induce a rapid degradation of SNON and SKI proteins via the proteasome. Thus, it has been argued that SNON and SKI degradation induced by TGF-β is necessary to allow the activation of different TGF-β target genes, but the exact underlying mechanisms have not been clearly determined. TGF-β/SMAD signaling induces SNON protein degradation via the UPS involving E3 ubiquitin ligases such as Arkadia, SMURF2, or anaphase-promoting complex; activated R-SMADs participate as adapters that recruit the E3 ubiquitin ligases required for the polyubiquitination of SKI and SNON (15–22). Previously, we reported that the antibiotic anisomycin (ANISO) can also down-regulate SNON and SKI proteins via the proteasome but through a new mechanism that is independent of SMAD proteins and from its known ribotoxic effects, and now anisomycin has become a useful tool to down-regulate SKI and SNON levels in specific cell types (16, 40).
We set up an assay to evaluate whether SNON degradation induced by TGF-β was required to induce SKIL gene expression. A549 cells were preincubated for 2 h with or without 50 μm MG132, a specific proteasomal inhibitor, and then cells were incubated for 45 min in the absence or presence of 300 pm TGF-β or 10 μm ANISO. TGF-β and ANISO decreased SNON protein levels in the absence of a proteasome inhibitor, whereas treatment with MG132 prevented SNON down-regulation but not S2 phosphorylation (Fig. 4A). We also observed that SNON protein levels were increased over basal in A549 cells preincubated with MG132 likely because of greater protein stability, whereas the basal mRNA levels did not change significantly. However, SNON mRNA levels were induced by TGF-β with or without MG132 pretreatment, but the induction was lower in cells pretreated with MG132 (Fig. 4B). These data indicated that TGF-β and ANISO reduce SNON protein levels by the UPS as shown previously (13, 16).
FIGURE 4.
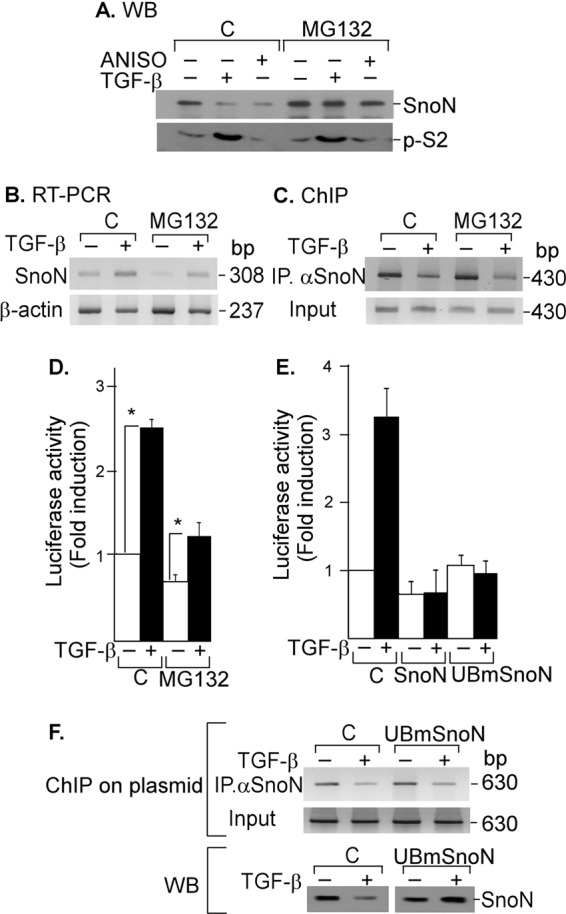
TGF-β signal removes SNON from SKIL gene promoter independently of its degradation. A, A549 cells were pretreated for 2 h without or with 50 μm MG132 and then incubated for 45 min in the absence or presence of 300 pm TGF-β or 10 μm ANISO. Proteins were immunoprecipitated with anti-SNON or anti-SMAD2 antibody followed by WB (n = 2) (A), or total RNA was isolated, and SNON (308-bp) and β-actin (237-bp) mRNAs were amplified by RT-PCR with specific primers (n = 2) (B), or ChIP assays were carried out using anti-SNON antibody, and PCRs were done with primers spanning SKIL SBE region (430 bp) (n = 3) (C). D, A549 cells transfected with the skilSBEs(408)-Luc reporter were pretreated for 2 h without or with 50 μm MG132 and then incubated for 12 h with or without 100 pm TGF-β. Luciferase activity was evaluated and normalized using β-gal expression and is reported as -fold induction over control. Values are mean ± S.E. (error bars) of three separate experiments in triplicate. *, p < 0.05 compared with control (C). E, to further analyze whether SNON degradation was required to regulate SKIL gene, we used UBmutSNON, which is unable to be ubiquitinated or degraded. AD293 cells were transiently transfected with skilSBEs(408)-Luc reporter along with WT SNON or UBmSNON, and 24 h post-transfection, cells were incubated for 12 h with or without 100 pm TGF-β. Luciferase activity was evaluated and normalized using β-gal expression and is reported as -fold induction over control. Values are mean ± S.E. (error bars) of three separate experiments in triplicate. F, AD293 cells were transiently transfected with the skilSBEs(408)-Luc reporter with or without UBmSNON. Cells were incubated for 45 min with or without 500 pm TGF-β, and a ChIP on plasmid assay was carried out using anti-SNON antibody for IP. PCRs were done with primers spanning the SBE region cloned into pGL3 vector (630 bp) (upper panel). Endogenous SNON and UBmSNON protein levels were detected by Western blot (lower panel).
MG132 pretreatment showed a slight inhibitory effect on TGF-β actions such as the increase of SNON mRNA levels (Fig. 4B), SNON binding to SKIL gene promoter (Fig. 4C), and activation of SKIL gene promoter (Fig. 4D). In the latter case, MG132 pretreatment decreased basal skilSBEs-Luc reporter gene expression, whereas the TGF-β-induced expression of the SKIL promoter was only slightly affected (Fig. 4D). These data suggest that TGF-β promotes SNON protein down-regulation to fully induce SKIL gene expression. To test this, we used a SNON mutant (UBmSNON) unable to be polyubiquitinated and degraded (17). We observed that UBmSNON repressed skilSBEs-Luc reporter activity in a similar way as WT SNON (Fig. 4E). We also performed ChIP on a reporter plasmid assay using skilSBEs-Luc and UBmSNON. We observed that UBmSNON was recruited to the SKIL gene reporter similarly to WT HA-SNON, and it is very interesting that TGF-β was able to remove the UBmSNON protein from the SKIL promoter even though TGF-β did not cause its degradation (Fig. 4F). Thus, we demonstrated that SNON protein degradation is not a prerequisite for SNON to be removed from its target gene promoters. However, we also showed that it is important for TGF-β signaling that activated SMAD complexes remove SNON from the promoter to bind the promoter and fully induce SKIL gene expression as it has been suggested previously (17).
SNON Protein Negatively Self-regulates Its Expression at Transcriptional Level
To demonstrate that SNON can regulate its own expression at the transcriptional level, we used two different strategies to down-regulate SNON protein levels and evaluated their effect on SKIL gene expression. First, A549 cells were pretreated with or without 10 μm ANISO for 1 h and then incubated for 45 min in the absence or presence of 300 pm TGF-β, and SNON protein levels were evaluated by immunoprecipitation (IP)/Western blot (WB) (Fig. 5A). These data showed that SNON protein was degraded after TGF-β or ANISO treatment as we reported previously (13, 16). Thus, we evaluated how SNON protein down-regulation induced by ANISO contributes to the regulation of SKIL gene expression. First, SNON mRNA levels were analyzed by RT-PCR in A549 cells pretreated for 20 min with 20 μg/ml cycloheximide to inhibit protein synthesis and then incubated for 2 h in the absence or presence of TGF-β or ANISO. β-Actin mRNA (237 bp) was amplified as an RNA loading control. These data showed that TGF-β induced an increase of SNON mRNA levels, whereas ANISO increased basal and TGF-β-induced SNON mRNA levels (Fig. 5B). It is possible that SNON protein down-regulation induced by ANISO caused a derepression of SKIL gene. This was supported by the observation that endogenous SNON was not positioned on the SKIL promoter when A549 cells were pretreated with ANISO (Fig. 5C). These data suggest that SNON protein down-regulation by ANISO may relieve the basal repression of SKIL gene and may facilitate the TGF-β effect on SKIL gene induction.
FIGURE 5.
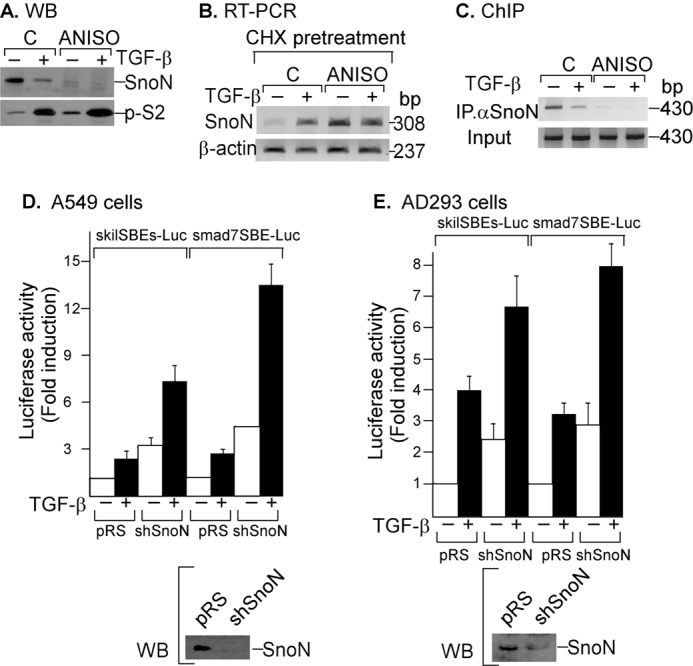
SNON negatively self-regulates its expression at the transcriptional level. A, A549 cells were preincubated for 1 h in the absence or presence of 10 μm ANISO and then incubated for 45 min with or without 300 pm TGF-β. Whole cell protein extracts were immunoprecipitated with specific anti-SNON and anti-S2/S3 antibodies and then analyzed by immunoblotting with specific anti-SNON and anti-phospho-SMAD2 (p-S2) antibodies. B, A549 pretreated with cycloheximide (CHX) for 20 min were then preincubated for 1 h in the absence or presence of 10 μm ANISO and then incubated for 2 h with or without 300 pm TGF-β (n = 2). Total RNA was isolated, and RT-PCR was performed with specific primers for SNON and β-actin. C, A549 cells were pretreated for 1 h without or with 10 μm ANISO and then treated for 45 min with or without 500 pm TGF-β, and a ChIP assay was carried out using anti-SNON antibody for IP (n = 3). PCRs were done with primers spanning the SKIL SBE region. A549 (D) or AD293 (E) cells stably expressing an shRNA to knock down SNON were co-transfected with the skilSBEs(408)-Luc or smad7SBE-Luc reporter plasmid, and 24 h post-transfection, cells were incubated for 12 h in the absence or presence of 100 pm TGF-β. Luciferase activity was normalized using β-gal expression and is reported as -fold induction over control (C). Values are mean ± S.E. (error bars) of three separate experiments in triplicate (upper panels). Endogenous SNON protein levels were detected in each cell type by Western blot (lower panels).
To validate the hypothesis that SNON was involved in basal repression of SKIL gene promoter, the endogenous SNON protein expression was knocked down using shRNA in AD293 and A549 cells. SNON knockdown cells (shSnoN) with low SNON protein levels (Fig. 5, D and E, lower panels) showed higher skilSBEs(408)-Luc reporter basal expression as well as higher TGF-β-induced activity (Fig. 5, D and E, upper panels). Because the levels of SNON expression could affect the expression of several TGF-β and SNON target genes, we also investigated the SMAD7 gene expression. We observed that basal and TGF-β-induced activity of smad7SBE-Luc was also increased in SNON knockdown cells (Fig. 5, D and E, upper panels). Therefore, it is very likely that the levels of SNON protein may affect the basal expression of most of its target genes.
SMAD4 Protein Is Required to Induce SKIL Gene Expression by TGF-β and to Bind and Repress SKIL Gene Promoter by SNON
We evaluated the role of S4 in recruiting R-SMAD and SNON proteins to the SKIL promoter. SNON and SKI do not possess DNA binding ability, and they seem to be recruited to TGF-β-responsive gene promoters through their interaction with SMAD proteins, mainly S4 (41, 42). Moreover, the SKIL gene seems to be induced by TGF-β in either an S4-dependent or S4-independent manner (43, 44).
To study the participation of S4 in the regulation of the human SKIL promoter by TGF-β/SMADs and SNON, we used the AD293 cell line stably expressing pRetroSuper/shS4J hygro (shS4; cells with low levels of SMAD4 protein by RNAi) or pRetroSuper/shS4J hygro plus pBABE/Smad4J Rescue (S4R; cells that overexpress SMAD4 protein because S4 mRNA cannot be degraded) (Fig. 6A, upper panel). TGF-β transiently increased SNON mRNA levels in AD293 cells, and the highest levels were observed between 2 and 4 h after treatment (Fig. 6A, middle panel). We also observed that TGF-β increased SNON mRNA levels at 2 h in control and S4R cells but not in shS4 cells where the induction of SKIL gene by TGF-β was reduced (Fig. 6A, lower panel). Interestingly, the basal expression of SNON mRNA was increased in shS4 cells with respect to control and S4R cells (Fig. 6A, lower panel). Our data indicate that S4 is necessary not only for repressing SKIL gene expression at the basal level but also for inducing its expression by TGF-β in these cells.
FIGURE 6.
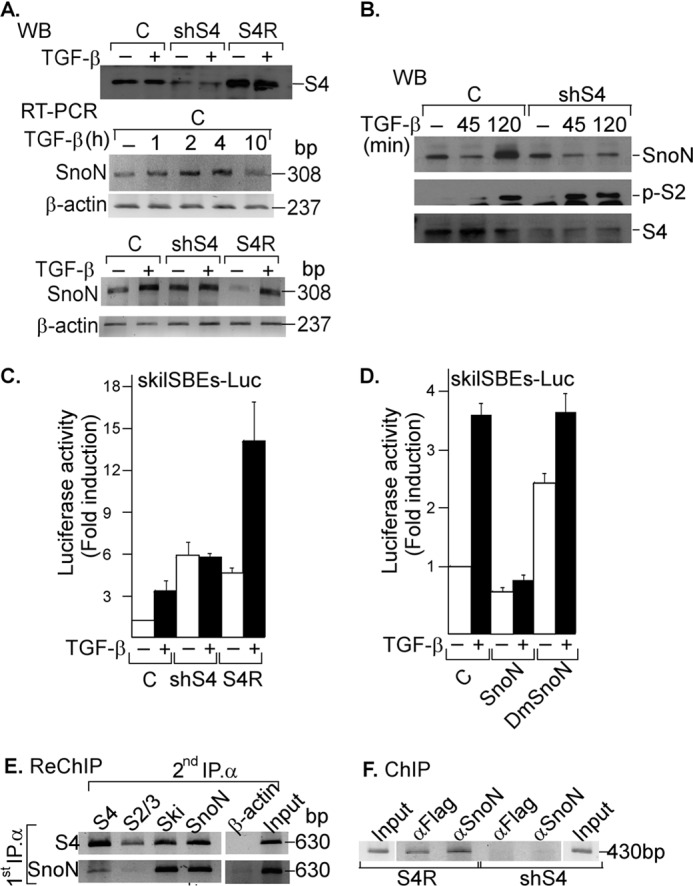
SNON depends on SMAD4 protein for SKIL promoter binding and repression. A, control AD293 cells (C) or cells stably expressing shSmad4 (shS4) or shSmad4 plus rescue S4 cDNA (S4R) were incubated for 2 h with or without 300 pm TGF-β. Whole cell protein extracts were subjected to IP with anti-S4 specific antibody followed by immunoblotting (upper panel). Total RNA was also isolated from control AD293 cells (upper and lower panels) or from shS4 or S4R AD293 cells (lower panel) incubated with or without 300 pm TGF-β for different times, and SNON (308-bp) and β-actin (237-bp) mRNAs were amplified by RT-PCR with specific primers (n = 2). B, control AD293 cells or stably expressing shSmad4 (shS4) AD293 cells were incubated for different times with 300 pm TGF-β. Whole cell protein extracts were subjected to IP with anti-SNON, anti-S2/S3, or anti-S4 antibody followed by WB with anti-SNON, anti-phospho-S2 (p-S2), or anti-S4 antibody (n = 2). Control or shS4 AD293 cells were transiently transfected with skilSBEs(408)-Luc reporter plasmid with or without plasmids bearing cDNA for SMAD4 rescue (S4R) (C), and AD293 cells were transiently transfected with skilSBEs(408)-Luc reporter along with plasmids bearing cDNA for WT SNON or DmSNON (D). Twenty-four hours post-transfection, cells were incubated for 12 h in the absence or presence of 100 pm TGF-β. Luciferase activity was normalized using β-gal expression and is reported as -fold induction over control. Values are mean ± S.E. (error bars) (n = 6). AD293 cells were transiently transfected with skilSBEs-Luc reporter, and 48 h post-transfection, a re-ChIP assay was carried out (n = 2) (E), or a ChIP assay was carried out in shS4 and S4R AD293 cells (F); both assays were performed using anti-S4, anti-FLAG, or anti-SNON antibody for the first IP; anti-S4, anti-S2/S3, anti-SNON, or anti-SKI antibody for the second IP; and anti-β-actin antibody as a control. PCRs were performed with primers for pGL3 plasmid bearing SKIL promoter (630 bp) or for SKIL promoter (430 bp).
SNON protein levels were also evaluated in control and shS4 AD293 cells. We observed that its levels decreased at 45 min and then increased after 2 h of TGF-β treatment in control cells (Fig. 6B), whereas TGF-β was unable to induce SNON expression at 2 h in the shS4 cells (Fig. 6B). Phospho-SMAD2 levels were increased after TGF-β treatment in both control and shS4 AD293 cells (Fig. 6B). Furthermore, we also observed that SKIL gene promoter activity induced by TGF-β was dependent on S4 expression because this effect only occurred in control and S4R cells (Fig. 6C). TGF-β was also able to active 3TP-Lux in a SMAD4-dependent manner (data not shown) as reported previously (43, 44). Intriguingly, SNON could not repress its expression when SMAD4 was absent, and as a consequence, the levels of SNON mRNA as well as the activity of SKIL promoter were increased (Fig. 6, A and C). Our results show that S4 protein is needed to induce SKIL gene expression by TGF-β and for the self-repression caused by SNON in AD293 cells.
To further evaluate the relevance of SNON-SMAD interaction in the regulation of SKIL gene expression, we constructed a double mutant of SNON (DmSNON) unable to associate with R-SMAD and SMAD4 (41). We observed that, in contrast to WT SNON, the DmSNON was unable to repress both basal and TGF-β-induced activity of skilSBEs(408)-Luc reporter (Fig. 6D). Our data indicated that SNON requires interaction with SMADs to repress the expression of SKIL gene, and previous studies have shown that SKI requires S4 to inhibit the SMAD7 gene promoter (12, 42). To evaluate whether the regulation of SKIL gene by SNON also requires its association with SMAD4, a re-ChIP assay on reporter plasmid was carried out in AD293 cells expressing skilSBEs-Luc using anti-SNON and anti-S4 antibodies for first and second IPs. Our results showed that endogenous SNON and SKI associate with endogenous S4, and this association appears to mediate the binding of SNON and SKI proteins to the SKIL promoter (Fig. 6E). We also observed by endogenous ChIP assays in S4R and shS4 AD293 cells that SNON can bind the SKIL promoter only when S4 is expressed (Fig. 6F). It is possible that SKI and SNON may also depend on S4 to bind to other TGF-β target gene promoters as occurs with the SKIL gene. We also found that SNON interacts with S4 to negatively regulate its own basal and TGF-β-induced expression.
The Absence of SNON-SMAD4 Complex Affects the Biological Outcome of TGF-β Signaling
In the epithelial AD293 cell line, we observed that TGF-β requires S4 to induce SKIL gene expression, whereas SNON depends on S4 protein for binding and repression of SKIL gene promoter. Because the response of cells to TGF-β depends on the cell context, we evaluated the effect of TGF-β on SKIL gene expression in two different cell types: A549 cells that undergo epithelial-mesenchymal transition in response to TGF-β and exhibit a high level of SNON protein and a low level of SKI protein and the colon cancer cell line SW480 that lacks S4, has a mesenchymal phenotype, and contains very high levels of both SNON and SKI proteins that are not down-regulated in response to TGF-β. We first examined the SNON expression levels in these cells by WB. The levels of SNON protein were very high in SW480 cells compared with AD293 and A549 cells (Fig. 7A). Thus, we decided to study the role played by SNON and S4 expression levels in the formation of SNON-S4 complex as well as the role of this complex in the regulation of human SKIL gene by TGF-β in different cellular contexts.
FIGURE 7.
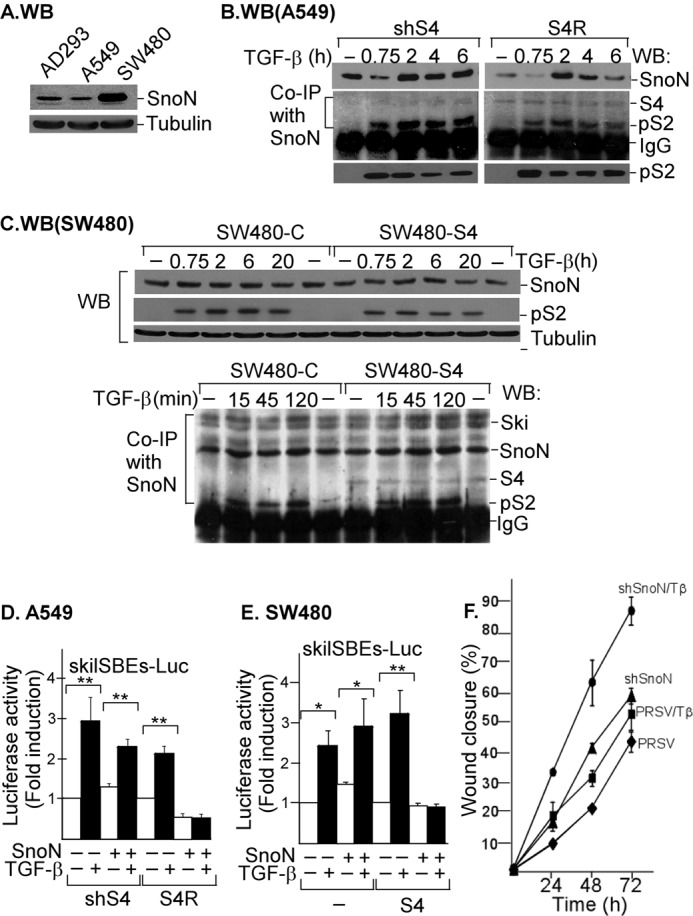
The absence of SNON-SMAD4 complex affects TGF-β signaling outcome. A, the levels of SNON protein were detected by WB in total protein extracts from AD293, A549, and SW480 cells. The levels of tubulin were used as a loading control (n = 3). For IP/WB assays, the following cell types were incubated for different times with or without 300 pm TGF-β: A549 cells stably expressing shSmad4 (shS4) or expressing shSmad4 plus rescue S4 cDNA (S4R) (n = 2) (B) and total cell lysates from control or transiently expressing S4 SW480 cells were subjected to WB (upper panel) or IP with anti-SNON or anti-SMAD2/3 specific antibody (lower panel) followed by WB with anti-SNON, anti-SKI, anti-S4, or anti-phospho-S2 (pS2) antibody (n = 2) (C). For reporter gene assays, the following cell types were transiently transfected with skilSBEs(408)-E1B-Luc: A549 cells stably expressing shSmad4 (shS4) or expressing shSmad4 plus rescue S4 cDNA (S4R) (D) and control (−) or transiently expressing S4 or SNON SW480 cells (E). Transfected cells were then incubated for 12 h with or without 100 pm TGF-β, and then luciferase activity was measured and normalized using β-gal expression. Data are reported as -fold induction over control. Values are mean ± S.E. (error bars) of three separate experiments in triplicate (upper panels). *, p < 0.05; **, p < 0.01 compared with control (C). F, confluent and serum-starved A549 cells stably expressing empty vector pRSV (♦ and ■) or shSnoN (▴ and ●) were wounded, and then the wound closure was monitored for different times in the absence or presence of 50 pm TGF-β (Tβ) (■ and ●). The graph shows a representative experiment, and data are shown as percentage of wound closure (mean ± S.E. (error bars), n = 3).
We evaluated SNON protein levels after TGF-β treatment for different times in shS4 and S4R A549 cells (Fig. 7B), SW480 cells lacking endogenous S4 expression, and SW480 cells transiently expressing S4 (Fig. 7C). We observed that SNON protein levels decreased at 45 min and then increased at 2 h in both shS4 and S4R A549 cells; however, we also observed a higher basal level of SNON protein expression as well as high levels of SNON protein after 4 h or longer times of TGF-β treatment in shS4 cells than in S4R cells (Fig. 7B).
In the case of the SW480 cell line, both control or overexpressing S4, SNON protein levels were just slightly increased in response to TGF-β even though the phospho-S2 levels were highly increased (Fig. 7C). Interestingly, TGF-β was unable to cause SNON degradation at any time point even though the phospho-S2 levels remained elevated until 20 h after TGF-β treatment (Fig. 7C, upper panel). Also, we observed a strong interaction among SNON, SKI, and phospho-S2 in control SW480 cells after TGF-β treatment (Fig. 7C, lower panel). However, we observed an interaction between SNON and S4 only in SW480 cells transiently expressing S4. Furthermore, TSA was unable to increase SNON mRNA levels in SW480 cells (data not shown), supporting the idea that in these cells the basal SNON-S4 repressor complex is absent.
Interestingly, we observed that the induction of SKIL gene reporter by TGF-β was independent of S4 expression in A549 cells because this effect occurred in both shS4 and S4R A549 cells (Fig. 7D). Similar data were obtained in SW480 cells (Fig. 7E). Additionally, the analysis of smad7-Luc reporter showed a similar regulation (data not shown). Intriguingly, when SMAD4 was absent or its levels were low, SNON could not repress its own expression, and as a consequence, the activity of SKIL promoter was increased (Fig. 7, D and E). Our results showed that S4 protein is always indispensable for the SKIL gene repression caused by SNON.
To further study the relevance of SNON levels in regulating the response of the cell to TGF-β, we evaluated the migration of A549 cells by using a wound healing assay. Confluent and serum-starved A549 cells stably expressing empty vector (pRSV) or shSnoN were wounded, and the wound closure was monitored for different times in the presence or absence of TGF-β (Fig. 7F). We observed that the knockdown of SNON increased the migratory phenotype of A549 cells and promoted the TGF-β effect. These data show the relevance of SNON levels to control TGF-β signals.
DISCUSSION
SNON is a negative regulator of SMAD transcriptional factors that may control many actions of TGF-β (1–9, 41, 42). Changes in SNON levels have been associated with diverse physiological processes such as embryonic development, hepatic regeneration, and muscular differentiation as well as with some diseases such as fibrosis and cancer, which are also related to altered TGF-β signaling (26–30, 45). Much evidence suggests that the regulation of SNON expression is essential in homeostasis and very complex. So far, TGF-β and hepatocyte growth factor are the main known signals that can induce SNON expression (25, 46). Because SNON expression might be critical in diverse physiological and pathological processes, here we aimed to describe the molecular mechanisms controlling human SKIL gene expression by the SNON-SMAD4 complex and TGF-β signaling.
Our bioinformatics analysis showed that the TRE spans the SKIL gene promoter. Also, the TSS of SKIL gene was located between SBE3 and SBE4 of TRE by partial cloning of the 5′-UTR of SNON mRNA by RT-PCR assay. A previous report showed by ChIP assay that TGF-β induced RNA polymerase II recruitment to the TRE of SKIL gene (47), which supports our finding that the TRE region spans the core promoter of SKIL gene. Intriguingly, TGF-β-responsive SKIL gene promoter is spanned by four groups of SBEs (supplemental Fig. S1). The affinity of SMAD proteins for one SBE is very low, but the presence of multiple SBEs probably increases the SMAD binding affinity to improve transcriptional activation through cooperative interactions between multiple SMAD-SBE contacts by the activated SMAD complex as suggested previously (1, 2, 4, 11–13, 48, 49).
Our analysis also showed that the ATG is located in the 5′-half of the second exon of the SKIL gene, whereas the first exon is small, GC-rich, and non-coding, and the first intron is very large. Interestingly, these particular characteristics of SKIL gene show that the first exon and first intron contain the previously identified regulatory sequences, the SMAD inhibitory element and hepatocyte growth factor-responsive element, which were identified as relevant elements for the regulation of SKIL gene expression by TGF-β and hepatocyte growth factor, respectively (25, 46, 50). We also found that the SKIL promoter is a TATA-less core promoter, and it is spanned by a large CpG island (supplemental Fig. S1) (51).
In the TGF-β pathway, SKI and SNON are two important SMAD corepressors (5, 6). SNON can form homodimers or heterodimers with SKI, and they play a crucial role in cellular transformation and transcriptional repression (7, 8). Interestingly, SNON and SKI protein levels can be regulated by TGF-β because the UPS rapidly degrades them upon ligand stimulus (15–22). In addition, TGF-β induces an increase of SNON mRNA and protein levels at treatments longer than 1 h (13, 23). Our work focused on demonstrating that SNON protein negatively regulates the basal expression of SKIL gene and its induction by the TGF-β/SMAD pathway; we also demonstrated that this last event is part of a negative feedback loop generated by TGF-β itself (Figs. 3 and 5). Our data support the idea that the transcriptional regulation of SKIL gene by SKI and SNON could be considered as a general mechanism to control other TGF-β target genes because the SMAD7 gene appears to be similarly regulated by SKI and SNON (Fig. 3) (12, 13). Therefore, it is clear that the transcriptional self-regulation of SNON could potentially affect the expression of many of its target genes, which can be analyzed when more SNON target genes are identified.
Because SNON and SKI cannot bind directly to DNA, we explored how these corepressors bind to the SKIL promoter. It has been reported that SKI binds DNA through S4 to repress the basal activity of SMAD7 gene independently of R-SMADs (12, 13, 52). Here, we demonstrated that SNON negatively self-regulates its expression because SNON binds to the SKIL promoter and recruits a repressor complex that also contains SKI, SMAD4, and proteins with HDAC activity. We also provide evidence that activated R-SMADs promote SNON protein removal from SKIL promoter independently of inducing its degradation. However, SNON degradation via the UPS is important to decrease the availability of SNON protein that may compete with activated R-SMAD complexes for TGF-β target gene promoters.
SMAD4 is not always required in SMAD transcriptional complexes to activate TGF-β target genes, and some TGF-β target genes can be differentially regulated by S4 and R-SMADs (43, 44, 53, 54). In fact, tumor cells deficient in S4 or expressing mutant S4 or cells with low levels of S4 due to shRNAs display a differential regulation of some but not all TGF-β target genes (43, 55, 56). Interestingly, SKIL was identified previously as a TGF-β target gene that may be regulated via an S4-dependent or S4-independent manner (43, 57). Here, we showed that the induction of SKIL gene by TGF-β effectively may rely on S4, but it depends on the cell type, which is similar to previous reports (43, 57).
Notably, the self-repression mediated by SNON is clearly S4-dependent because SNON was unable to inhibit the basal and TGF-β-induced SKIL gene expression when S4 protein levels were decreased in all cell types tested. After restoring S4 expression or when S4 was overexpressed, it was possible to rescue the interaction of SNON with S4 and the recruitment of S4-SNON complex to SKIL promoter and as a result to decrease SNON mRNA levels. We confirmed that SNON associates with activated R-SMADs, S4, and SKI. Thus, when SNON does not bind S4, then it cannot repress the SKIL promoter. Using a re-ChIP assay, we confirmed that SNON and SKI proteins in combination with S4 bind and repress the SKIL promoter basal expression. Furthermore, we demonstrated that the absence of the SNON-S4 repressor complex affects the regulation of SKIL gene expression by TGF-β. Because S4 is a functional partner for SNON and SKI, it is very likely that S4 is also required for transcriptional repression of many other TGF-β target genes.
It is possible that the self-regulation of SNON expression could be affected in some cellular contexts. It was reported previously that a prolonged induction of SNON expression by TGF-β in fibroblast plays a critical role for cell oncogenic transformation; intriguingly, in these cells, the negative feedback loop seems to be absent (25). Nevertheless, we have demonstrated that when high levels of SNON mRNA are observed in cells where SMAD proteins remain activated for a long time, it is possible that the negative feedback loop is functioning to stop the gene transcription, but at the same time, a mechanism controlling the stability of mRNAs might be also participating.
The absence of this self-regulation of SNON could be a new mechanism causing up-regulation of TGF-β inhibitors such as SMAD7 and SKIL (Sno) genes in some diseases such as cancer, and it may favor TGF-β resistance in cancer cells by affecting the regulation of TGF-β target genes. This new mechanism could also be evident mainly in cancer cells lacking SMAD4 such as those from colon and pancreas (58) where the absence of the SNON-SMAD4 repressor complex might be responsible for the changes observed in cell phenotype (25, 26). Furthermore, it is highly probable that in those cancerous cells lacking SMAD4 the functions of both corepressors SNON and SKI are impaired.
In summary, SNON and SKI are bound to the SKIL gene promoter to repress its basal expression in a manner similar to that for SMAD7 gene regulation. The SKIL gene is a target of SKI and SNON corepressors, and both seem to be involved in maintaining the SKIL promoter in a repressed state in the absence of TGF-β signaling. That this effect depends on the association with SMAD4 is noteworthy. In contrast, upon TGF-β stimulus, SNON and SKI are removed from the SKIL promoter and replaced by the activated SMAD complex. Thus, TGF-β positively regulates the human SKIL gene expression through the S2-S4 complex. Later, after longer TGF-β treatment, SNON protein levels are increased, and SNON binds back to the SKIL promoter. Thus, SNON functions as a negative feedback control regulating SKIL (Sno) expression (supplemental Fig. S5). This regulation of SNON levels could be critical for appropriate control of the TGF-β signal and for cell homeostasis. Thus, any deregulation of this negative feedback loop might be involved in the development of diverse diseases such as fibrosis and cancer.
Acknowledgments
We thank Drs. Claudia González-Espinosa, Marco A. Briones-Orta, and Jacqueline Hernández-Damián for helpful discussions. We appreciate the kind gifts of plasmids from Drs. J. L. Wrana (Mount Sinai Hospital, Toronto, Ontario, Canada), H. F. Lodish (Whitehead Institute, Cambridge, MA), and M. Klüppel (Northwestern University, Chicago, IL). We thank Drs. Rosa Navarro and Jorge Ramírez-Salcedo (Instituto de Fisiología Celular, Universidad Nacional Autónoma de México), and José Vázquez-Prado (Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional) for reagents. We also thank Dr. L. Ongay and members of Unidad de Biología Molecular (Instituto de Fisiología Celular, Universidad Nacional Autónoma de México).
This work was supported in part by Universidad Nacional Autónoma de México/Dirección General Asuntos del Personal Académico/Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica Program Grants IN222909 and IN206012 and Consejo Nacional de Ciencia y Tecnología (CONACyT) Grants 49493-Q and 101826.

This article contains supplemental Figs. S1–S5 and Tables S1–S3.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) JX103164.
- S2
- SMAD2
- IP
- immunoprecipitation
- S3
- SMAD3
- S4
- SMAD4
- SBE
- SMAD-binding element
- SNON
- SKI-novel
- TRE
- TGF-β response element
- UPS
- ubiquitin-proteasome system
- WB
- Western blot
- SKIL
- SKI-like
- TSS
- transcription start site
- HDAC
- histone deacetylase
- re-ChIP
- sequential ChIP
- TSA
- trichostatin
- NaB
- sodium butyrate
- ANISO
- anisomycin
- R-SMAD
- receptor-regulated SMAD.
REFERENCES
- 1. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 2. Attisano L., Wrana J. L. (2002) Signal transduction by the TGF-β superfamily. Science 296, 1646–1647 [DOI] [PubMed] [Google Scholar]
- 3. Macías-Silva M., Abdollah S., Hoodless P. A., Pirone R., Attisano L., Wrana J. L. (1996) MADR2 is a substrate of the TGFβ receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell 87, 1215–1224 [DOI] [PubMed] [Google Scholar]
- 4. Massagué J., Seoane J., Wotton D. (2005) Smad transcription factors. Genes Dev. 19, 2783–2810 [DOI] [PubMed] [Google Scholar]
- 5. Luo K. (2004) Ski and SnoN: negative regulators of TGF-β signaling. Curr. Opin. Genet. Dev. 14, 65–70 [DOI] [PubMed] [Google Scholar]
- 6. Liu X., Sun Y., Weinberg R. A., Lodish H. F. (2001) Ski/Sno and TGF-β signaling. Cytokine Growth Factor Rev. 12, 1–8 [DOI] [PubMed] [Google Scholar]
- 7. Cohen S. B., Zheng G., Heyman H. C., Stavnezer E. (1999) Heterodimers of the SnoN and Ski oncoproteins form preferentially over homodimers and are more potent transforming agents. Nucleic Acids Res. 27, 1006–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heyman H. C., Stavnezer E. (1994) A carboxyl-terminal region of the ski oncoprotein mediates homodimerization as well as heterodimerization with the related protein SnoN. J. Biol. Chem. 269, 26996–27003 [PubMed] [Google Scholar]
- 9. Sun Y., Liu X., Eaton E. N., Lane W. S., Lodish H. F., Weinberg R. A. (1999) Interaction of the Ski oncoprotein with Smad3 regulates TGF-β signaling. Mol. Cell 4, 499–509 [DOI] [PubMed] [Google Scholar]
- 10. Shinagawa T., Dong H. D., Xu M., Maekawa T., Ishii S. (2000) The sno gene, which encodes a component of the histone deacetylase complex, acts as a tumor suppressor in mice. EMBO J. 19, 2280–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin X., Chen Y.-G., Feng X.-H. (2008) in The TGF-β Family (Derynck R., Miyazono K., eds) pp. 287–332, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 12. Denissova N. G., Liu F. (2004) Repression of endogenous Smad7 by Ski. J. Biol. Chem. 279, 28143–28148 [DOI] [PubMed] [Google Scholar]
- 13. Briones-Orta M. A., Sosa-Garrocho M., Moreno-Alvarez P., Fonseca-Sánchez M. A., Macías-Silva M. (2006) SnoN co-repressor binds and represses smad7 gene promoter. Biochem. Biophys. Res. Commun. 341, 889–894 [DOI] [PubMed] [Google Scholar]
- 14. Krakowski A. R., Laboureau J., Mauviel A., Bissell M. J., Luo K. (2005) Cytoplasmic SnoN in normal tissues and nonmalignant cells antagonizes TGF-β signaling by sequestration of the Smad proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 12437–12442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun Y., Liu X., Ng-Eaton E., Lodish H. F., Weinberg R. A. (1999) SnoN and Ski protooncoproteins are rapidly degraded in response to transforming growth factor β signaling. Proc. Natl. Acad. Sci. U.S.A. 96, 12442–12447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vázquez-Macías A., Ruíz-Mendoza A. B., Fonseca-Sánchez M. A., Briones-Orta M. A., Macías-Silva M. (2005) Downregulation of Ski and SnoN co-repressors by anisomycin. FEBS Lett. 579, 3701–3706 [DOI] [PubMed] [Google Scholar]
- 17. Stroschein S. L., Bonni S., Wrana J. L., Luo K. (2001) Smad3 recruits the anaphase-promoting complex for ubiquitination and degradation of SnoN. Genes Dev. 15, 2822–2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Izzi L., Attisano L. (2004) Regulation of the TGFβ signalling pathway by ubiquitin-mediated degradation. Oncogene 23, 2071–2078 [DOI] [PubMed] [Google Scholar]
- 19. Wan Y., Liu X., Kirschner M. W. (2001) The anaphase-promoting complex mediates TGF-β signaling by targeting SnoN for destruction. Mol. Cell 8, 1027–1039 [DOI] [PubMed] [Google Scholar]
- 20. Bonni S., Wang H. R., Causing C. G., Kavsak P., Stroschein S. L., Luo K., Wrana J. L. (2001) TGF-β induces assembly of a Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat. Cell Biol. 3, 587–595 [DOI] [PubMed] [Google Scholar]
- 21. Levy L., Howell M., Das D., Harkin S., Episkopou V., Hill C. S. (2007) Arkadia activates Smad3/Smad4-dependent transcription by triggering signal-induced SnoN degradation. Mol. Cell. Biol. 27, 6068–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagano Y., Mavrakis K. J., Lee K. L., Fujii T., Koinuma D., Sase H., Yuki K., Isogaya K., Saitoh M., Imamura T., Episkopou V., Miyazono K., Miyazawa K. (2007) Arkadia induces degradation of SnoN and c-Ski to enhance transforming growth factor-β signaling. J. Biol. Chem. 282, 20492–20501 [DOI] [PubMed] [Google Scholar]
- 23. Stroschein S. L., Wang W., Zhou S., Zhou Q., Luo K. (1999) Negative feedback regulation of TGF-β signaling by the SnoN oncoprotein. Science 286, 771–774 [DOI] [PubMed] [Google Scholar]
- 24. Pearson-White S., McDuffie M. (2003) Defective T-cell activation is associated with augmented transforming growth factor β sensitivity in mice with mutations in the Sno gene. Mol. Cell. Biol. 23, 5446–5459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu Q., Pearson-White S., Luo K. (2005) Requirement for the SnoN oncoprotein in transforming growth factor β-induced oncogenic transformation of fibroblast cells. Mol. Cell. Biol. 25, 10731–10744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu Q., Krakowski A. R., Dunham E. E., Wang L., Bandyopadhyay A., Berdeaux R., Martin G. S., Sun L., Luo K. (2007) Dual role of SnoN in mammalian tumorigenesis. Mol. Cell. Biol. 27, 324–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Edmiston J. S., Yeudall W. A., Chung T. D., Lebman D. A. (2005) Inability of transforming growth factor-β to cause SnoN degradation leads to resistance to transforming growth factor-β-induced growth arrest in esophageal cancer cells. Cancer Res. 65, 4782–4788 [DOI] [PubMed] [Google Scholar]
- 28. Macias-Silva M., Li W., Leu J. I., Crissey M. A., Taub R. (2002) Up-regulated transcriptional repressors SnoN and Ski bind Smad proteins to antagonize transforming growth factor-β signals during liver regeneration. J. Biol. Chem. 277, 28483–28490 [DOI] [PubMed] [Google Scholar]
- 29. Tan R., Zhang J., Tan X., Zhang X., Yang J., Liu Y. (2006) Downregulation of SnoN expression in obstructive nephropathy is mediated by an enhanced ubiquitin-dependent degradation. J. Am. Soc. Nephrol. 17, 2781–2791 [DOI] [PubMed] [Google Scholar]
- 30. Yang J., Zhang X., Li Y., Liu Y. (2003) Downregulation of Smad transcriptional corepressors SnoN and Ski in the fibrotic kidney: an amplification mechanism for TGF-β1 signaling. J. Am. Soc. Nephrol. 14, 3167–3177 [DOI] [PubMed] [Google Scholar]
- 31. Padua D., Zhang X. H., Wang Q., Nadal C., Gerald W. L., Gomis R. R., Massagué J. (2008) TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 133, 66–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Furlan-Magaril M., Rincón-Arano H., Recillas-Targa F. (2009) in Methods in Molecular Biology: DNA-Protein Interactions (Moss T., Leblanc B., eds) Vol. 543, pp. 253–266, Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 33. Izzi L., Silvestri C., von Both I., Labbé E., Zakin L., Wrana J. L., Attisano L. (2007) Foxh1 recruits Gsc to negatively regulate Mixl1 expression during early mouse development. EMBO J. 26, 3132–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nomura N., Sasamoto S., Ishii S., Date T., Matsui M., Ishizaki R. (1989) Isolation of human cDNA clones of ski and the ski-related gene, sno. Nucleic Acids Res. 17, 5489–5500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pearson-White S., Crittenden R. (1997) Proto-oncogene Sno expression, alternative isoforms and immediate early serum response. Nucleic Acids Res. 25, 2930–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nomura T., Khan M. M., Kaul S. C., Dong H. D., Wadhwa R., Colmenares C., Kohno I., Ishii S. (1999) Ski is a component of the histone deacetylase complex required for transcriptional repression by Mad and thyroid hormone receptor. Genes Dev. 13, 412–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kokura K., Kaul S. C., Wadhwa R., Nomura T., Khan M. M., Shinagawa T., Yasukawa T., Colmenares C., Ishii S. (2001) The Ski protein family is required for MeCP2-mediated transcriptional repression. J. Biol. Chem. 276, 34115–34121 [DOI] [PubMed] [Google Scholar]
- 38. Nagase T., Nomura N., Ishii S. (1993) Complex formation between proteins encoded by the ski gene family. J. Biol. Chem. 268, 13710–13716 [PubMed] [Google Scholar]
- 39. Wilkinson D. S., Ogden S. K., Stratton S. A., Piechan J. L., Nguyen T. T., Smulian G. A., Barton M. C. (2005) A direct intersection between p53 and transforming growth factor β pathways targets chromatin modification and transcription repression of the α-fetoprotein gene. Mol. Cell. Biol. 25, 1200–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Macías-Silva M., Vázquez-Victorio G., Hernández-Damián J. (2010) Anisomycin is a multifunctional drug: More than just a protein synthesis inhibitor. Curr. Chem. Biol. 4, 124–132 [Google Scholar]
- 41. He J., Tegen S. B., Krawitz A. R., Martin G. S., Luo K. (2003) The transforming activity of Ski and SnoN is dependent on their ability to repress the activity of Smad proteins. J. Biol. Chem. 278, 30540–30547 [DOI] [PubMed] [Google Scholar]
- 42. Wu J. W., Krawitz A. R., Chai J., Li W., Zhang F., Luo K., Shi Y. (2002) Structural mechanism of Smad4 recognition by the nuclear oncoprotein Ski: insights on Ski-mediated repression of TGF-β signaling. Cell 111, 357–367 [DOI] [PubMed] [Google Scholar]
- 43. Levy L., Hill C. S. (2005) Smad4 dependency defines two classes of transforming growth factor β (TGF-β) target genes and distinguishes TGF-β-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol. Cell. Biol. 25, 8108–8125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koinuma D., Tsutsumi S., Kamimura N., Imamura T., Aburatani H., Miyazono K. (2009) Promoter-wide analysis of Smad4 binding sites in human epithelial cells. Cancer Sci. 100, 2133–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Imoto I., Pimkhaokham A., Fukuda Y., Yang Z. Q., Shimada Y., Nomura N., Hirai H., Imamura M., Inazawa J. (2001) SNO is a probable target for gene amplification at 3q26 in squamous-cell carcinomas of the esophagus. Biochem. Biophys. Res. Commun. 286, 559–565 [DOI] [PubMed] [Google Scholar]
- 46. Tan R., Zhang X., Yang J., Li Y., Liu Y. (2007) Molecular basis for the cell type specific induction of SnoN expression by hepatocyte growth factor. J. Am. Soc. Nephrol. 18, 2340–2349 [DOI] [PubMed] [Google Scholar]
- 47. Xi Q., He W., Zhang X. H., Le H. V., Massagué J. (2008) Genome-wide impact of the BRG1 SWI/SNF chromatin remodeler on the transforming growth factor β transcriptional program. J. Biol. Chem. 283, 1146–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Benchabane H., Wrana J. L. (2003) GATA- and Smad1-dependent enhancers in the Smad7 gene differentially interpret bone morphogenetic protein concentrations. Mol. Cell. Biol. 23, 6646–6661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Briones-Orta M. A., Tecalco-Cruz A. C., Sosa-Garrocho M., Caligaris C., Macías-Silva M. (2011) Inhibitory Smad7: emerging roles in health and disease. Curr. Mol. Pharmacol. 4, 141–153 [PubMed] [Google Scholar]
- 50. Kalari K. R., Casavant M., Bair T. B., Keen H. L., Comeron J. M., Casavant T. L., Scheetz T. E. (2006) First exons and introns—a survey of GC content and gene structure in the human genome. In Silico Biol. 6, 237–242 [PubMed] [Google Scholar]
- 51. Sandelin A., Carninci P., Lenhard B., Ponjavic J., Hayashizaki Y., Hume D. A. (2007) Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nat. Rev. Genet. 8, 424–436 [DOI] [PubMed] [Google Scholar]
- 52. Tabata T., Kokura K., Ten, Dijke P., Ishii S. (2009) Ski co-repressor complexes maintain the basal repressed state of the TGF-beta target gene, SMAD7, via HDAC3 and PRMT5. Genes Cells 14, 17–28 [DOI] [PubMed] [Google Scholar]
- 53. Ikushima H., Miyazono K. (2010) Cellular context-dependent “colors” of transforming growth factor-β signaling. Cancer Sci. 101, 306–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ijichi H., Otsuka M., Tateishi K., Ikenoue T., Kawakami T., Kanai F., Arakawa Y., Seki N., Shimizu K., Miyazono K., Kawabe T., Omata M. (2004) Smad4-independent regulation of p21/WAF1 by transforming growth factor-β. Oncogene 23, 1043–1051 [DOI] [PubMed] [Google Scholar]
- 55. Zhao S., Ammanamanchi S., Brattain M., Cao L., Thangasamy A., Wang J., Freeman J. W. (2008) Smad4-dependent TGF-β signaling suppresses RON receptor tyrosine kinase-dependent motility and invasion of pancreatic cancer cells. J. Biol. Chem. 283, 11293–11301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Müller N., Reinacher-Schick A., Baldus S., van Hengel J., Berx G., Baar A., van Roy F., Schmiegel W., Schwarte-Waldhoff I. (2002) Smad4 induces the tumor suppressor E-cadherin and P-cadherin in colon carcinoma cells. Oncogene 21, 6049–6058 [DOI] [PubMed] [Google Scholar]
- 57. Hesling C., Fattet L., Teyre G., Jury D., Gonzalo P., Lopez J., Vanbelle C., Morel A. P., Gillet G., Mikaelian I., Rimokh R. (2011) Antagonistic regulation of EMT by TIF1γ and Smad4 in mammary epithelial cells. EMBO Rep. 12, 665–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang G., Yang X. (2010) Smad4-mediated TGF-β signaling in tumorigenesis. Int. J. Biol. Sci. 6, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]