

Background: P-glycoprotein is an ABC transporter that confers multidrug resistance.
Results: Linkage of the nucleotide-binding domains with short but not long cross-linkers highly activated ATPase activity.
Conclusion: Drug substrates activate P-gp ATPase activity by promoting close association of the nucleotide-binding domains.
Significance: Understanding the mechanism of P-glycoprotein will aid in the development of inhibitors to overcome multidrug resistance.
Keywords: ABC Transporter, ATPases, Cysteine-mediated Cross-linking, Drug Resistance, Membrane Proteins, P-glycoprotein
Abstract
The P-glycoprotein (P-gp, ABCB1) drug pump protects us from toxic compounds and confers multidrug resistance. Each of the homologous halves of P-gp is composed of a transmembrane domain (TMD) with 6 TM segments followed by a nucleotide-binding domain (NBD). The predicted drug- and ATP-binding sites reside at the interface between the TMDs and NBDs, respectively. Crystal structures and EM projection images suggest that the two halves of P-gp are separated by a central cavity that closes upon binding of nucleotide. Binding of drug substrates may induce further structural rearrangements because they stimulate ATPase activity. Here, we used disulfide cross-linking with short (8 Å) or long (22 Å) cross-linkers to identify domain-domain interactions that activate ATPase activity. It was found that cross-linking of cysteines that lie close to the LSGGQ (P517C) and Walker A (I1050C) sites of NBD1 and NBD2, respectively, as well as the cytoplasmic extensions of TM segments 3 (D177C or L175C) and 9 (N820C) with a short cross-linker activated ATPase activity over 10-fold. A pyrylium compound that inhibits ATPase activity blocked cross-linking at these sites. Cross-linking between the NBDs was not inhibited by tariquidar, a drug transport inhibitor that stimulates P-gp ATPase activity but is not transported. Cross-linking between extracellular cysteines (T333C/L975C) predicted to lock P-gp into a conformation that prevents close NBD association inhibited ATPase activity. The results suggest that trapping P-gp in a conformation in which the NBDs are closely associated likely mimics the structural rearrangements caused by binding of drug substrates that stimulate ATPase activity.
Introduction
ATP-binding cassette (ABC)2 membrane transport proteins couple ATP hydrolysis to the movement of a diverse range of substrates across cell membranes (1). A distinguishing feature of ABC proteins is that they contain at least two transmembrane domains (TMDs) with at least 6 transmembrane (TM) segments each and two nucleotide-binding domains (NBDs) each of which contains a “Walker A” and a LSGGQ (signature) sequence.
The P-glycoprotein drug pump (P-gp, ABCB1) was the first human ABC protein to be discovered during efforts to determine how cancer cells developed multidrug resistance (2). P-gp mediates the ATP-dependent efflux of a wide range of hydrophobic compounds (such as anticancer drugs, hydrophobic drugs, steroids, peptides, detergents, and lipids) that enter cells by diffusion through the plasma membrane (3–5). P-gp is expressed in the epithelium of liver, kidney, and gastrointestinal tract and at the blood-brain or blood-testes barrier. Its location likely contributes to multidrug resistance in diseases such as cancer and AIDS (4).
The 1280 amino acids of human P-gp (6) are organized as two tandem repeats of 610 amino acids that are joined by a linker region. Each repeat consists of an NH2-terminal TMD containing 6 TM segments followed by a NBD. Drug substrates appear to bind at multiple drug-binding sites within a cavity at the interface between the TMDs (7–10). Mg.ATP binds at the interface between the NBDs. In this nucleotide-sandwich conformation, each nucleotide would interact with the phosphate-binding loop (P-loop) of one NBD (located at N-terminal end) and the ABC signature motif (LSGGQ) of the other NBD (located in the central region). ATP hydrolysis likely occurs by an alternating site mechanism as inhibition of ATPase activity at either site causes a complete loss of ATPase activity (11–15).
The mechanism of how ATP hydrolysis is coupled to drug transport is unknown. It has been proposed that P-gp can exist in at least two major conformations during the catalytic cycle, an inward-facing (open) conformation with separated NBDs and a drug-binding pocket that is exposed to the cytoplasm and an outward-facing (closed) conformation with close association of the NBDs and exposure of the drug-binding pocket to the extracellular surface (16). Drug substrates and ATP binding were predicted to induce a closed conformation. Hydrolysis of the first ATP would promote drug transport and hydrolysis of the second ATP would convert the protein back to an open conformation.
The crystal structure of a mouse P-gp (mdr1a) showed that P-gp in the absence of ATP was in an open conformation with the opposing Walker A and LSGGQ segments of the NBDs far apart and the drug-binding pocket open to the cytoplasm (7). The presence of drug substrates (inhibitors) in the predicted drug-binding pocket caused little change in the structure.
Structural changes between the NBDs have been investigated by cysteine cross-linking. It was found that cross-linking could be observed between naturally occurring cysteines in the P-loop or cysteines introduced into the signature motifs (17–19). Drug substrates that activated P-gp ATPase activity were observed to promote cross-linking between the NBDs (19). A potential criticism of these initial studies was that cross-linking inactivated the transporter and may not represent conformations normally found during the catalytic cycle.
In a recent study (20), it was observed that spontaneous cross-linking between cysteines located at the C-terminal ends of the NBDs yielded mouse Mdr3 P-gps that retained drug-stimulated ATPase activity. In the mouse inward-facing mouse P-gp crystal structure, the cross-linkable cysteines would be predicted to be in close proximity (segments of the C-terminal ends were not resolved in the crystal structure) while the N-terminal and central regions that form the nucleotide-binding sandwich structure would be far apart. These results showed that the C-terminal ends of the NBDs were not required to undergo significant separation during the catalytic cycle. Movement between the NBDs during the catalytic cycle may only involve the central or N-terminal regions closer to the Walker A and LSGGQ sequences.
A large cavity was also observed in electron microscopic projection structures of two-dimensional crystals of human P-gp reconstituted into a lipid bilayer (21). In this study however, addition of nucleotides resulted in the disappearance of the central cavity and a sideways motion of the two halves of the transporter. These results suggested that binding of nucleotide converted P-gp from the open conformation to a closed conformation.
In the absence of drug substrates, P-gp exhibits basal ATPase activity, which may allow it to cycle between these conformations. Drug substrates like verapamil and rhodamine appear to induce further conformational changes as they can stimulate ATPase activity over 10-fold. Indeed, P-gp ATPase activity can be constitutively activated to high levels upon covalent attachment of thiol-reactive verapamil or rhodamine derivatives to cysteines within the drug-binding pocket in the TMDs (22–24). It is possible that covalent attachment of drug substrates activates P-gp ATPase activity by promoting formation of the closed conformation that keeps the NBD N-terminal and central regions in close proximity. If this was the case, then it should be possible to mimic activation of P-gp ATPase activity by covalently linking the NBDs in close proximity at sites close to the opposing Walker A (located at N-terminal region) and LSGGQ (located in central region) sites. On the other hand, covalent linkage of the NBD N-terminal and central regions in close proximity may inhibit P-gp ATPase activity because separation of the NBDs during formation of the open conformation may be an essential step in the catalytic cycle.
To address whether covalent linkage of the NBD N-terminal and central regions in close proximity would activate or inhibit ATPase activity, we introduced cysteines close to the LSGGQ site in NBD1 (central region) and the opposing Walker A site in NBD2 (N-terminal region). The introduced cysteines are predicted to lie in close proximity in the closed conformation and far apart in the open conformation. Cross-linking with a short but not a long cross-linker was found to highly activate ATPase activity. These results indicate that maintaining the NBD N-terminal and central regions in close proximity mimics activation of ATPase activity by covalent attachment of drug substrates to the drug-binding domain. We also report that a pyrylium compound (P10) that inhibits ATPase activity blocked cross-linking between the NBDs while an inhibitor (tariquidar) that is not transported (25) but stimulates ATPase activity did not inhibit cross-linking.
EXPERIMENTAL PROCEDURES
Construction of Mutants
The seven endogenous cysteines at positions 137, 431, 717, 956, 1074, 1125, and 1227 were replaced with alanine to create a Cys-less P-gp (26). The Cys-less P-gp cDNA was modified to contain a 10-histidine tag at the COOH-terminal end to facilitate purification of the expressed protein by nickel-chelate chromatography (11). Pairs of cysteines were introduced into various domains of Cys-less P-gp to test for the effects of cross-linking as described previously (27). Mutant P517C(NBD1)/I1050C(NBD2) was constructed to test the effects of cross-linking between the NBDs while mutant D177C(ICL1)/N820C(ICL3) was constructed to examine the effects of cross-linking between the intracellular loops (ICL) of TMD1 and TMD2. Mutants C137(TM2 of TMD1)/A935C(TM11 of TMD2) and T333C(TM6 of TMD1)/L975C(TM12 of TMD2) were constructed to examine the effects of cross-linking between the TM segments of the TMDs at locations predicted to lie close to the intracellular and extracellular membrane surfaces, respectively. The L443C(NBD1)/S909C(TMD2) mutant was constructed to examine the effects of cross-linking between NBD1 and the fourth intracellular loop (ICL4) in TMD2.
Disulfide Cross-linking Analysis
The double cysteine mutants were transiently expressed in HEK293 cells. Whole cells expressing mutant T333C(TM6)/L975C(TM12) were treated with the cross-linker bis(maleimido)ethane (BMOE) because Cys-333 and Cys-975 are predicted to be accessible from the extracellular surface. BMOE is a short (8 Å) homobifunctional, maleimide cross-linker that would not be sensitive to thiol-reducing agents that would be released when whole cells are lysed. Transfected cells were suspended in TBS, pH 7.4 (1 × 106 cells per ml) and treated at 20 °C for 15 min with 0.5 mm BMOE. Reactions were stopped by addition of 2× SDS sample buffer (125 mm Tris-HCl, pH 6.8, 20% (v/v) glycerol, and 4% (w/v) SDS) containing 50 mm EDTA, and 2% β-mercaptoethanol). Samples (1 × 104 cells) were subjected to immunoblot analysis. Intramolecular disulfide cross-linking between P-gp domains can be detected because the cross-linked product migrates with a slower mobility on SDS-PAGE gels (28).
Mutants containing cysteines predicted to reside on the intracellular surface of the cell were cross-linked with methanethiosulfonate (MTS) cross-linkers because these agents are more reactive than maleimide-type cross-linkers. Membranes were prepared as described previously (19) and suspended in TBS, pH 7.4. Samples of the membranes were then treated with 0.05 mm of the homobifunctional MTS cross-linkers 1,4-butanediyl bismethanethiosulfonate (M4M) or 3,6,9,12,15-pentaoxaheptane-1,17-diyl bismethanethiosulfonate (M17M) for 15 min at 20 °C. M4M and M17M (Toronto Research Chemicals, Toronto, ON) could span maximum distances of about 7.8 or 22 Å (29, 30), respectively. Mutants 175C(ICL1)/N820C(ICL3) and D177C(ICL1)/N820C(ICL3) were also treated with 0.02 mm of the short (4 Å) cross-linker 1,1-methanediyl bismethanethiosulfonate (M1M) for 15 min at 20 °C. The reactions were performed using a protein concentration of 0.4 mg/ml. Bismaleimidoethane (BMOE) cross-linker (Fisher Scientific, Ottawa, ON) with a spacer arm of about 8 Å was used to cross-link cysteine mutants in whole cells. The reactions were stopped by addition of 2× SDS sample buffer (125 mm Tris-HCl, pH 6.8, 20% (v/v) glycerol, and 4% (w/v) SDS) containing 50 mm EDTA and no reducing agent. The reaction mixtures (1 μg protein) were then subjected to SDS-PAGE (6.5% (w/v) polyacrylamide gels; 1.5 mm 15-slot minigels) and immunoblot analysis with a rabbit polyclonal antibody against P-gp. The gel lanes were scanned and the amount of cross-linked product relative to total P-gp (cross-linked plus 170 kDa protein) was analyzed using the NIH Image program and an Apple computer.
Purification of P-gp and Measurement of ATPase Activity
Histidine-tagged P-gps were expressed in HEK 293 cells and then isolated by nickel-chelate chromatography as described previously (11). Recovery of P-gp was monitored by immunoblot analysis with rabbit anti-P-gp polyclonal antibody (31). A sample of the isolated histidine-tagged P-gp was mixed with an equal volume of 10 mg/ml sheep brain phosphatidylethanolamine (Type II-S, Sigma) that had been washed and suspended in TBS. ATPase activity was measured in the presence of various concentrations of tariquidar (MedKoo Biosciences, Inc. Chapel Hill, NC; or the pyrylium compounds P10 or P12 (32).
Homology Models of P-gp
The model of human P-gp in the open conformation was generated by Bikardi et al. (33) and was based on that of mouse P-gp (7). The closed conformation was generated by Globisch et al. (34) based on the structure of Sav 1866 (35). Pymol (36) was used to view the structures.
RESULTS
Cross-linking of the NBDs Activates ATPase Activity
Fig. 1A shows a model of human P-gp in the open conformation (33) while Fig. 1B shows the predicted structure of human P-gp in the closed conformation (34). In the open conformation (Fig. 1A) the NBDs are widely separated but are in close proximity in the closed conformation (Fig. 1B). To address whether trapping the NBDs in close proximity (closed conformation) would activate or inhibit ATPase activity, cysteines were introduced at locations in the central part of NBD1 (P517C) and the N-terminal region of NBD2 (I1050C) of Cys-less P-gp predicted to be outside the catalytic sites but close to the LSGGQ (residues 531–535) and Walker A sites (residues 1070–1077) of NBD1 and NBD2, respectively. The opposing LSGGQ and Walker A sites are predicted to form an ATP-binding site between the NBDs to yield a nucleotide-binding sandwich upon binding of ATP. In the open conformation P517C and I1050C are predicted to be far apart (22.9 Å; measured from the α carbons) but close (7.8 Å) in the closed conformation.
FIGURE 1.

Schematic models of P-gp in the inward- and outward-facing conformations. The predicted models of human P-gp in the open (A) (33) and closed (B) (34) conformations are shown. The branched lines on the extracellular loop connecting TM1 and TM2 represent the glycosylated sites. The colors highlight the TMD1 (green), NBD1 (blue), TMD2 (black), and NBD2 (red) domains. The location of residues that were mutated to cysteine to test for the effect of cross-linking between NBD1 and NBD2 (P517C/I1050C), NBD1 and TMD2 (L443C/S909C), ICL1 and ICL3 (D177C/N820C), TM segments 2 and 11 (C137/A935C) or 6 and 12 (T333C/L975C) are shown.
The P517C/I1050C mutant was expressed in HEK 293 cells. Membranes prepared from the transfected cells were treated with or without a short (M4M, maximum cross-linking span of 7.8 Å) or long (M17M, maximum cross-linking span of 22 Å) thiol reactive cross-linker. Samples of the cross-linked membranes were subjected to immunoblot analysis. It was found that the mutant was efficiently cross-linked (>90%) with both M4M and M17M (Fig. 2A).
FIGURE 2.

Cross-linking of the NBDs with M4M highly stimulates ATPase activity. A, membranes prepared from HEK 293 cells expressing the NBD1/NBD2 mutant P517C/I1050C were treated without (Control) or with cross-linkers M4M or M17M. The reactions were stopped by addition of SDS sample buffer containing no thiol-reducing agent and samples subjected to immunoblot analysis. The positions of the cross-linked (X-link) and mature (170 kDa) P-gps are indicated. B, membranes expressing mutant P517C/I1050C or Cys-less P-gp were treated with or without (None) cross-linker and histidine-tagged P-gp isolated by nickel-chelate chromatography. The isolated P-gps were mixed with lipid and ATPase activities were measured in the absence (−) or presence (+) of 0.3 mm verapamil. In some cases the samples were treated with 10 mm dithiothreitol (+DTT) before assay. Each value is the mean ± S.D. (n = 3).
To test the effect of covalently linking NBD1 and NBD2 on catalytic activity, ATPase activity of mutant P517C/NI1050C was measured in the presence or absence of verapamil before and after cross-linking with the short M4M cross-linker. Verapamil was used because it is transported by P-gp (37) and it highly stimulates (over 10-fold) the ATPase activity of Cys-less human P-gp (19). Prior to cross-linking (Fig. 2B), the ATPase activity mutant P517C/I1050C was highly activated (11.5-fold) by verapamil and exhibited about 80% (1.5 + 0.2 μmol Pi/min/mg P-gp) of the verapamil-stimulated ATPase activity of Cys-less P-gp (1.9 + 0.3 μmol Pi/min/mg P-gp). Cross-linking of P517C/I1050C with M4M increased its basal ATPase activity (14-fold higher than the basal ATPase activity of untreated P-gp) (Fig. 2B). The activity of the M4M cross-linked mutant was not further activated by verapamil. The enhanced activity of M4M cross-linked P517C/I1050C was not due to carry over of M4M during purification because M4M had little effect on Cys-less P-gp ATPase activity (Fig. 2B). Reduction of M4M cross-linked P517C/I1050C with dithiothreitol decreased its activity back to basal levels (Fig. 2B).
The large increase in basal ATPase activity observed when mutant P517C/I1050C was cross-linked with the short M4M cross-linker appeared to be caused by trapping of the NBDs in close proximity rather than nonspecific cross-linking effects because treatment with the long M17M cross-linker caused a much smaller increase (about 2-fold) in basal ATPase activity (Fig. 2B). The reduced ATPase activity observed after cross-linking with M17M did not appear to be due to a general inhibition of ATPase activity by M17M because addition of verapamil further increased the ATPase activity of the M17M cross-linked mutant by over 6-fold (from 0.3 + 0.2 to 1.9 + 0.3 μmol Pi/min/mg P-gp).
The cross-linking results observed with mutant P517C/I1050C suggest that covalently linking NBD1 and NBD2 in close proximity with M4M mimics activation of ATPase activity with compounds that highly activate P-gp ATPase activity.
Cross-linking of ICL1 (TMD1) and ICL3 (TMD2) in Close Proximity Also Activates P-gp ATPase Activity
To test if cross-linking of other segments predicted to undergo large conformational changes during the reaction cycle would also activate P-gp ATPase activity, we performed cross-linking studies on mutant D177C/N820C. Residue Asp-177 is located in ICL1(TMD1) while Asn-820 is located in ICL3(TMD2) (Fig. 1). Asp-177 and Asn-820 are predicted to be outside the drug- or ATP-binding sites. In the open conformation (Fig. 1A) Asp-177 and Asn-820 are predicted to be far apart (23.7 Å; measured from the α carbons) while in the closed conformation, residues Asp-177 and Asn-820 are predicted to lie in close proximity (about 7.4 Å apart) (Figs. 1B and 3A).
FIGURE 3.

Cross-linking of ICL1 and ICL3 with M4M highly stimulates ATPase activity. A, model showing predicted orientations of Leu175, Asp177, and Asn820 in the closed conformation. B, membranes prepared from HEK 293 cells expressing the ICL1/ICL3 mutants D177C/N820C or L175C/N820C were treated with cross-linkers M1M, M4M, or M17M or no cross-linker (Control). The reactions were stopped by addition of SDS sample buffer containing no thiol-reducing agent and samples subjected to immunoblot analysis. The positions of the cross-linked (X-link) and mature (170 kDa) P-gps are indicated. C, membranes expressing mutants D177C/N820C or L175C/N820C were treated with or without (None) cross-linker and histidine-tagged P-gp isolated by nickel-chelate chromatography. The isolated P-gps were mixed with lipid and ATPase activities were measured in the absence (−) or presence (+) of 0.3 mm verapamil. In some cases the samples were treated with 10 mm dithiothreitol (+DTT) before assay. Each value is the mean ± S.D. (n = 3).
Membranes prepared from cells expressing mutant D177C/N820C were treated with M4M and M17M. Fig. 3B shows the mutant was efficiently cross-linked (>90%) with both cross-linkers.
The cross-linked P-gps were then isolated by nickel-chelate chromatography and their ATPase activities compared with the uncross-linked parent. It was found that the effect of cross-linking on ATPase of mutant D177C/N820C (Fig. 3C) was very similar to those observed for mutant P517C/I1050C (Fig. 2B). Prior to cross-linking, mutant D177C/N820C showed verapamil-stimulated ATPase activity that was similar to the Cys-less parent. The basal ATPase activity of the M4M cross-linked D177C/N820C mutant was similar to that of uncross-linked mutant assayed in the presence of verapamil (Fig. 3B). No further stimulation of ATPase activity was observed when the M4M cross-linked D177C/N820C was assayed in the presence of verapamil. Reduction of the cross-link with dithiothreitol restored the basal ATPase activity to levels similar to the uncross-linked mutant. Cross-linking with M17M caused about a 2-fold increase in basal ATPase activity. The ATPase activity of the M17M cross-linked mutant was further activated by verapamil.
In a previous study (38), we observed that mutant L175C(ICL1)/N820C(ICL3) but not D177C(ICL1)/N820C(ICL3) could be cross-linked at 0 °C with the short (4 Å) M1M cross-linker. In this study, we also observed that the majority of mutant L175C/N820C was cross-linked when the mutant was treated with 0.02 mm M1M at 20 °C while only a low level of cross-linking (about 25%) was observed when D177C/N820C was treated under the same conditions. Since D177C is predicted to have a more favorable orientation toward N820C compared with L175C (Fig. 3A), it would be expected that cross-linking of D177C/N820C would be more efficient than L175C/N820C. One possibility is that a separate M1M molecule preferentially modifies each cysteine of mutant D177C/N820C as it was found that cross-linking was reduced when the concentration of M1M was increased (data not shown). We previously observed that mutant L175C/N820 only showed efficient cross-linking with a narrow concentration range of M1M (about 10–20 μm). Another possibility is that in human P-gp the orientation of TM3 and its cytoplasmic extension (ICL1) may be slightly different than that found in the crystal structures of Sav 1866 and mouse P-gp. For example, the orientation of TM3 in the mouse crystal structure was not compatible with an arginine-scanning mutagenesis study performed on human P-gp (10). Both mutants however, could be efficiently cross-linked with M4M and M17M (Fig. 3B).
We tested the effects of M4M and M17M cross-linking on the ATPase activity of mutant L175C/N820C and found that the effects were similar to those observed with mutant D177C/N820C (Fig. 3C). Cross-linking of L175C/N820C with M4M highly activated the basal ATPase activity of the mutant while cross-linking with M17M caused little activation. The ATPase activity of M17M cross-linked L175C/N820C could still be highly activated when assayed in the presence of verapamil. These results show that covalently linking ICL1 and ICL3 in close proximity highly activates its ATPase activity.
ATPase Activity Is Not Activated when the Homologous Halves Are Cross-linked at Locations Predicted Not to Undergo Large Distance Changes during the Open to Closed Conformational Change
Mutant C137/A935C contains natural Cys-137 in TM segment 2 (TMD1) and the A935C mutation in TM segment 11 (TMD2). Cysteines at positions 137 and 935 are predicted to be in close proximity in both the open (5.0 Å; measured from α carbons) (Fig. 1A) and closed (6.6 Å) (Fig. 1B) conformations. Mutant L443C/S909C contains a cysteine (L443) in NBD1 and a cysteine (S909C) in the ICL4 loop that connects the cytoplasmic extensions of TM segments 10 and 11. Cysteines at positions 443 and 909 are predicted to show only small changes in proximity between the open (8.3 Å) (Fig. 1A) and closed (12.0 Å) (Fig. 1B) conformations. In previous studies we showed that mutants C137/A935C (39) and L443C/S909C (27) had verapamil-stimulated ATPase activities similar to Cys-less P-gp.
Membranes prepared from cells expressing mutants C137/A935C or L443C/S909C were treated with M4M cross-linker. Both mutants were efficiently cross-linked (>90%) (Fig. 4A). The histidine-tagged mutants were then isolated and assayed for verapamil-stimulated ATPase activity. It was found that cross-linking did not cause any significant alteration in their basal or verapamil-stimulated ATPase activities (Fig. 4B). These results indicate that stimulation of ATPase activity observed in M4M cross-linked mutants P517C/I1050C (Fig. 2B), D177C/N820C, or L175C/N820C (Fig. 3C) was likely a specific effect of trapping the protein in the closed conformation.
FIGURE 4.
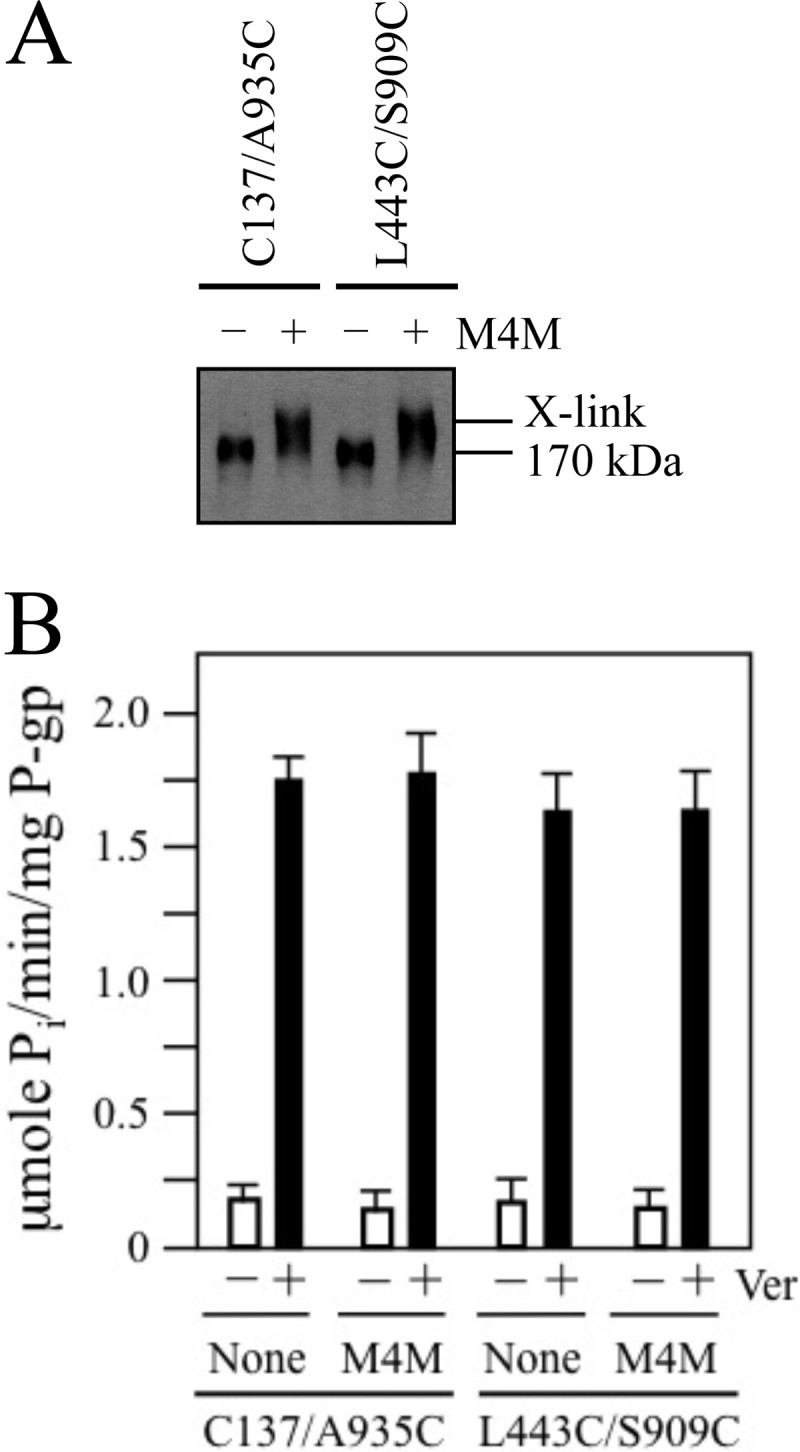
Cross-linking between NBD1 and TMD2 or TM segments 2 and 11 with M4M does not activate or inhibit ATPase activity. A, membranes prepared from HEK 293 cells expressing the NBD1/TMD2 mutant L443C/S909C or the TM2/TM11 mutant C137/A935C were treated with (+) or without (−) the M4M cross-linker. The reactions were stopped by addition of SDS sample buffer containing no thiol-reducing agent and samples subjected to immunoblot analysis. The positions of the cross-linked (X-link) and mature (170 kDa) P-gps are indicated. B, membranes expressing mutants L443C/S909C or C137/A935C were treated with or without (None) M4M cross-linker and histidine-tagged P-gp isolated by nickel-chelate chromatography. The isolated P-gps were mixed with lipid and ATPase activities were measured in the absence (−) or presence (+) of 0.3 mm verapamil. Each value is the mean ± S.D. (n = 3).
Would an Inhibitor of ATPase Activity Affect Cross-linking Between the NBDs?
To address this question, we first examined interactions of human P-gp with tariquidar, a potent inhibitor of rodent P-gp drug transport and ATPase activity (25). For example, 100 nm tariquidar was found to decrease the resistance of P-gp-expressing cells to doxorubicin by 30-fold (40). In patients, a single dose was shown to inhibit P-gp transport activity for 48 h (41).
Tariquidar differs from most other P-gp substrates because it was found to inhibit ATPase activity of hamster P-gp (25). It has been reported however, that the substrate specificity of human P-gp can differ from rodent P-gps (42). To test if tariquidar also inhibited the ATPase activity of human P-gp, histidine-tagged human P-gp was isolated by nickel-chelate chromatography, and ATPase activity was measured in the presence of various concentrations of tariquidar. It was observed that human P-gp was different from hamster P-gp as tariquidar highly stimulated ATPase activity by 10-fold (Fig. 5A). The level of stimulation of ATPase activity by tariquidar was comparable to that of verapamil (both about 10-fold) but the concentration required for half-maximal activation was about 75-fold lower for tariquidar (0.33 μm for tariquidar compared with 25 μm for verapamil). Since tariquidar is not transported by human P-gp (40), the results suggest that its effect on P-gp may mimic those of cross-linked NBDs (Fig. 2B) with both causing activation of ATPase activity.
FIGURE 5.

Pyrylium compound P10 inhibits NBD cross-linking, ATPase activity and P-gp-mediated drug resistance. A, ATPase activity of isolated histidine-tagged Cys-less P-gp was determined in the presence of various concentrations of tariquidar (structure shown in inset). B, structures of pyrylium compounds P10 and P12. C, Cys-less P-gp ATPase activity was determined in the presence of various concentrations of P10 or P12. Inhibition of verapamil-stimulated ATPase activity was performed in the presence of 0.1 mm verapamil and increasing concentrations of P10. D, membranes prepared from HEK 293 cells expressing mutants P517C(NBD1)/I1050C(NBD2) or D177C(ICL1)/N820C(ICL3) were pretreated in the absence (None) or presence of 0.1 mm P10 or P12. The membranes were then treated with (+) or without (−) 0.05 mm M4M cross-linker and samples subjected to immunoblot analysis. The positions of cross-linked (X-link) and mature (170 kDa) forms of P-gp are indicated. E, percent of cross-linked product relative to total P-gp (cross-linked plus 170 kDa P-gp) was determined. Each value is the mean ± S.D. (n = 3). F, BHK cells (Control) or BHK cells expressing wild-type P-gp were incubated in the presence of various levels of colchicine in the presence (+) or absence (−) of 5 μm P10 or 100 nm tariquidar and the concentration of colchicine required to reduce cell viability by 50% (LD50) was determined. The results represent the average LD50 obtained from analysis of three independent assays ± S.D.
We tested whether another inhibitor of ATPase activity (the pyrylium compound P10) (Fig. 5B) would block cross-linking between the NBDs because tariquidar did not inhibit human P-gp ATPase activity. P10 inhibits basal and verapamil-stimulated P-gp ATPase activity (half-maximal inhibition of verapamil-stimulated ATPase activity with 32 μm P10) (Fig. 5C). The pyrylium compound P12 was used as a control compound because it activates Cys-less P-gp ATPase activity 11.5-fold (half-maximal activation observed with 18 μm P12) (Fig. 5C). Mutants that showed activation of ATPase after cross-linking with M4M (P517C/I1050C and D177C/N820C) were tested for inhibition of cross-linking. It was observed that the ATPase inhibitor P10 inhibited cross-linking of both mutants whereas the P12 ATPase activator did not (Fig. 5, D and E).
We then tested the effects of P10 and tariquidar on P-gp mediated resistance to the cytotoxic P-gp substrate colchicine. It was found that both P10 and tariquidar reduced P-gp-mediated colchicine resistance by about 7- and 23-fold, respectively (Fig. 5F).
Trapping P-gp in the Predicted Open Conformation Inhibits ATPase Activity
The P10 compound may inhibit ATPase activity by binding to P-gp to favor an inward-facing conformation and reduce the probability of generating an ATP-bound sandwich configuration. Examination of the open conformation shown in Fig. 1A suggests that a potential way to trap P-gp in this conformation would be to cross-link cysteines placed in the extracellular ends of TM segments in each half of P-gp. For example, the extracellular ends of TM segments 6 (TMD1) and 12 (TMD2) would be predicted to be about 10 Å apart (measured from the α carbons) in the open conformation (Fig. 1A) and about 20 Å apart in the closed conformation (Fig. 1B). Accordingly, we introduced cysteines in regions of TM segments 6 (T333C) and 12 (L975C) predicted to lie at or close to the extracellular surface of the cell (Fig. 1). The T333C and L975C mutations were selected because we previously showed that they had little effect on P-gp activity and treatment of the single cysteine mutants with a thiol-reactive derivative of verapamil had little effect on activity (43).
Since the T333C and L975C mutations were predicted to reside at or close to the extracellular surface of the cell, we performed cross-linking analysis on intact cells expressing the mutant. The maleimide cross-linker BMOE (a flexible cross-linker that can span an S-S distance of 6.3–10.5 Å) (44) was used instead of M4M. The rationale for using BMOE is that cross-links formed by BMOE but not M4M are resistant to reducing compounds such as glutathione that are released during cell lysis.
Fig. 6A shows that P-gp mutant T333C/L975C was efficiently cross-linked (> 90% efficiency) when intact cells were treated with BMOE. To test for the effect of cross-linking on activity, histidine-tagged T333C/L975C P-gp was isolated by nickel-chelate chromatography before and after cross-linking with BMOE. It was found that cross-linking reduced verapamil-stimulated ATPase activity by over 10-fold (Fig. 6B). By contrast, treatment of Cys-less P-gp with BMOE had no significant effect on activity (Fig. 6B).
FIGURE 6.

Cross-linking of cysteines on the extracellular surface inhibits ATPase activity. A, cells expressing mutant T333C(TM6)/L975C(TM12) were treated with (+) or without (−) 0.5 mm BMOE. Samples were subjected to immunoblot analysis. B, cells expressing Cys-less P-gp or mutant T333C(TM6)/L975C(TM12) were treated with 0.5 mm BMOE. P-gp was then isolated by nickel-chelate chromatography and ATPase activity determined in the presence (+) or absence (−) of 0.3 mm verapamil. Each value is the mean ± S.D. (n = 3). C, cells expressing mutant T333C/L975C were pretreated with 0.1 mm P10, 0.1 mm P12, 0.03 mm tariquidar (Tar), or no compound (None) and then incubated in the absence (−) or presence (+) of 0.5 mm BMOE. Samples were subjected to immunoblot analysis. D, membranes prepared from cells expressing mutant P517C/I1050C were pretreated without (None) or with 0.03 mm tariquidar (Tar) followed by incubation in the absence (−) or presence (+) of 0.05 mm M4M cross-linker. Samples were then subjected to immunoblot analysis. The positions of mature (170 kDa) and cross-linked (X-link) P-gps are indicated.
We then tested the effect of the stimulators (tariquidar, P12) and inhibitor (P10) of ATPase activity on cross-linking of mutant T333C/L975C. It was observed that the stimulators inhibited cross-linking whereas P10 did not (Fig. 6C). The results suggest that the stimulators may promote the closed conformation where the T333C and L975C residues are far apart. By contrast, tariquidar did not inhibit cross-linking of P517C(NBD1)/I1050C(NBD2) with M4M cross-linker (Fig. 6D).
DISCUSSION
Transport by ABC transporters is predicted to occur by an alternating access mechanism driven by ATP binding and hydrolysis at the NBDs that switches access to a pocket formed within the TMDs from one side of the membrane to the other. Based on the crystal structure of mouse P-gp (7), it was proposed that the NBDs may have to come far apart to accommodate bulky drug molecules. This proposal was tested in a recent cross-linking study with mouse P-gp. Verhalen and Wilkens (20) introduced cysteines at the C-terminal ends of the NBDs because they are predicted to be close together in the mouse crystal structure while the N-terminal and central regions (containing the Walker A and LSGGQ sites) are far apart. It was observed that the cysteines spontaneously cross-linked yielding a molecule that retained 30–68% drug-stimulated ATPase activity. Cross-linking had little effect on basal ATPase activity. The results of the mouse cross-linking study showed that further separation of the NBDs was not an essential step in the catalytic cycle. Similar results were observed with the maltose transporter as a cross-linking study (45) showed that the C-terminal ends of the NBDs (MalK) stayed together during the entire transport cycle. By contrast the opposing Walker A (N-terminal ends) and LSGGQ (central region) sites in MalK are widely separated in the inward-facing conformation but in close proximity in the outward-facing conformation (46).
In contrast to the mouse cross-linking study, we observed that cross-linking of the NBDs of human P-gp highly activated basal ATPase activity to levels normally observed with verapamil, one of the most potent activators of ATPase activity. An explanation for the increased activity is that P517C and I1050C are located at the N-terminal end and central segments of the NBDs close to the LSGGQ and Walker A sites of NBD1 and NBD2, respectively. Cross-linking P517C/I1050C (or D177C/N820C) with M4M would hold the Walker A and LSGGQ sites in close proximity (Fig. 7A) to increase the probability of generating the ATP-bound sandwich conformation and therefore increase the basal ATPase activity. The longer cross-linker M17M may not have highly activated ATPase activity because it could have still allowed the cross-linked protein to adopt an open conformation with separation of the opposing LSGGQ and Walker A segments (Fig. 7B).
FIGURE 7.

Models based on open and closed crystal structures showing relative locations of NBD1 LSGGQ and NBD2 Walker A sites. A, model of P-gp in the closed conformation showing the N-terminal LSGGQ sequence (residues 531–535) to be in close proximity to the C-terminal Walker A sequence (residues 1070–1077) if the P517C/I1050C or D177C/N820C pairs of cysteines were cross-linked with M4M. B, P-gp in the open conformation in which these residues are far apart.
Mutagenesis (47) and nucleotide trapping experiments (12, 13, 48) suggests that ATP hydrolysis occurs in an alternating fashion with the NBDs staying associated for at least two hydrolysis steps. The observation that cross-linked mutants P517C/I1050C, D177C/N820C, and L175C/N820C showed high levels of ATPase activity indicates that the protein does not have to adopt an inward-facing conformation with separation of the NBDs for ATP hydrolysis to continue after the first two hydrolysis steps.
Fluorescence resonance energy transfer (FRET) spectroscopy studies showed that vanadate trapping of nucleotide, ATP, and the drug substrates could modulate proximity of the NBDs (49). Pairs of cysteines were introduced into NBD1 and NBD2 for ensemble and single molecule FRET spectroscopy after labeling with fluorescent dyes. In ensemble FRET, it was found that addition of drug and ATP promoted association of the NBDs, while vanadate trapping of nucleotide further decreased the donor to acceptor ratio. Single-molecule FRET spectroscopy showed that a drug substrate that highly stimulated ATPase activity (verapamil) displayed a higher FRET efficiency compared with a drug substrate that caused a smaller stimulation of ATPase activity (cyclosporine). These results suggested that a substrate that promotes ATPase activity enhances the ability of P-gp to adopt the ATP-bound sandwich conformation.
In the model of the P-gp reaction cycle (49), it was postulated that binding of drugs and 2 ATP molecules causes the protein to adopt an inward-facing conformation that differs from the apo form in which the TMDs and NBDs are more closely associated (49). ATP hydrolysis at the first site would convert the high affinity drug-binding site to low affinity site by lateral movements or rotation of the helices and exposure to the extracellular surface in an outward-facing conformation would cause release of the drug. Evidence that ATP hydrolysis appears to cause lateral movement or rotation of the helices were the observations that ATP hydrolysis was required for cross-linking of mutant L332C(TM6)/L975C (50) and ATP hydrolysis shifted cross-linking of V982C in TM12 from L339C to F343C in TM6 (51). ATP then replaces ADP at the first site and closing of the first site induces tightly bound ATP at the second site to enter a transition state. Hydrolysis of a second ATP has been postulated to reset P-gp to the ground state with separation of the NBDs (52, 53).
Our results suggest that separation of the NBDs after one NBD catalytic cycle (hydrolysis of a pair of ATP molecules) is not required to begin another NBD catalytic cycle. Why then does P-gp need to hydrolyze ATP at two different sites? One possibility is that P-gp contains multiple drug-binding sites (9, 23, 54) and that hydrolysis at each ATP-binding site only disrupts a subset of the drug-binding sites. In support of this prediction, it has been reported that hydrolysis of ATP at the different ATP-binding sites causes different conformational changes (55, 56). It was proposed that each ATP hydrolysis event might be linked to different conformational changes (55).
The results of this study suggests that P-gp may be trapped in a futile ATP hydrolysis cycle if rotation of the helices does not cause release of a tightly bound drug substrate after hydrolysis of the first ATP molecule. Evidence that P-gp can be trapped in a futile reaction cycle if a drug substrate cannot be released were the observation that covalent attachment of thiol-reactive substrates to cysteines introduced into the drug-binding pocket will highly activate ATPase activity. For example, covalent labeling of F728C (TM7) (22), L65C (TM1) (57), or I306C (TM5) (24) with a thiol-reactive derivative of verapamil increased basal ATPase activity of P-gp by 7–12-fold. Covalent labeling of T199C (TM3) with a thiol-reactive derivative of rhodamine increased basal activity 7-fold (58). Since covalent attachment of the drug substrates to the drug-binding domain prevents drug release, P-gp may be locked in a futile ATPase cycle where the N-terminal and central regions of the NBDs remain closely associated.
The working model in Fig. 8 incorporates the alternating site models proposed previously (12, 13, 15, 49) and includes the finding that P-gp can be trapped in an outward-facing conformation with highly activated ATPase activity. The model predicts that hydrolysis at each ATP-binding site causes altered conformational changes in the TMDs. In support of this prediction, we observed that E556Q and E1201Q mutations to the catalytic carboxylates had different effects on cross-linking between TM segments 6 and 12 (59). We predict that covalent attachment of thiol-reactive drug substrates to cysteines in the TMDs (or M4M cross-linking of mutants P517C/I1050C, D177C/N820C, or L175C/N820C) traps P-gp in states IV-VI (Fig. 8) that show a high rate of ATP hydrolysis.
FIGURE 8.

Models of P-gp catalytic cycle and constitutive activation of ATPase activity. For clarity, only TM6 and TM12 of TMD1 and TMD2 are shown since we previously showed that hydrolysis of ATP or binding of drug substrate can promote rotation or conformational changes with these TM segments (19, 51). The cycle starts with P-gp in an open conformation (I). Although binding of drug substrate is shown as the first step for discussion, binding of drug substrate and ATP can bind in a random order. Binding of drug substrate can alter packing of the TM segments by an induced-fit mechanism (II) (19). Binding of ATP has been shown to cause repacking of the TM segments (III) (66) and cause a decrease in affinity for some drug substrates (67). Binding of ATP also promotes formation of the nucleotide-sandwich conformation and an outward-facing conformation (IV). ATP hydrolysis at one site causes rotation or movement of the TM segments (51). If drug is released, then hydrolysis of the second ATP would reset P-gp to a resting state. If the drug substrate is not released, then hydrolysis of an ATP molecule at the second site causes additional conformational changes to promote drug release (V). If the substrate is not released (example would be covalent attachment of a thiol-reactive drug substrate to a cysteine in the TM segments) (VI) or the NBDs are held in close proximity by cross-linking then P-gp will continue to hydrolyze ATP at a high rate.
Could noncovalent attachment of a drug substrate lock P-gp into a conformation that highly activates ATP hydrolysis without drug release? Tariquidar appears to be a candidate since it is not transported by P-gp (25, 40), yet it highly activates human P-gp ATPase activity (this study). Kannan et al. (40) reported that P-gp expressing cells actually accumulated 2-fold more [3H]tariquidar than parental cells not expressing P-gp. It was postulated that P-gp expressing cells accumulated [3H]tariquidar due to its binding to P-gp in the drug resistant KB cell line. By contrast tariquidar was found to be a substrate of the breast cancer resistance protein (BCRP/ABCG2) as it stimulated its ATPase activity and BCRP transported [3H]tariquidar (40).
Tariquidar has been reported to act as a noncompetitive inhibitor of P-gp (25). Tariquidar may resemble modulators such as flupentixols that appear to modulate ATPase activity but bind outside of the drug-binding sites (60, 61). Perhaps binding of tariquidar traps P-gp in an outward-facing conformation that mimics cross-linking (states IV-VI, Fig. 8).
Inhibitory antibodies have also been identified that bind to the TMDs outside the drug-binding sites but activate ATPase activity (62). It was found that both antibodies MRK16 and UIC2 block drug efflux without inhibiting ATP hydrolysis after binding to epitopes on the extracellular surface. Binding of MRK16 activated ATPase activity 2-fold. It was proposed that binding of the antibodies allowed the NBDs to cycle through the conformations required for ATP hydrolysis but the antibodies sterically blocked drug release.
By contrast we found that cross-linking between the extracellular loops inhibits ATPase activity and might lock P-gp in an open conformation such that the NBDs are too far apart to carry out efficient ATP hydrolysis. It cannot be ruled out however, that cross-linking between TM segments 6 and 12 indirectly blocks ATP hydrolysis by inhibiting rotation of one or both of these segments. We previously showed that ATP hydrolysis appeared to cause rotation of TM segments 6 and 12 because of the way it altered the cross-linking pattern between the helices (51). The effects may be transient as no crystal structure of a drug transporter to date has shown rotation of a TM segment. Other evidence to support the prediction that TM6 and TM12 (as well as TM11) can rotate was the observation that different drug substrates altered cross-linking between these TM segments (proposed induced fit mechanism for drug recognition) (63).
The requirement for rotation or motion of the TM segments during the catalytic cycle may explain why direct cross-linking of the cysteines in mutant C137/A935C (TM2/TM11) (39) inhibited activity whereas cross-linking with M4M did not (this study). M4M is a flexible cross-linker that can span distances from about 5 to 8 Å. Restriction of protein motion by cross-linking may also explain why we only observed a small activation of ATPase activity when mutant L175C(ICL1)/N820C(ICL3) was cross-linked with M1M (spans about 4 Å with low flexibility) (38) while it was highly activated when cross-linked with M4M (Fig. 3C).
The effects on ATPase activity of cross-linking mutant L443C(NBD1)/S909C(TMD2) were very similar to those observed with mutant C137(TM2)/A935C(TM11). In both cases, cross-linking with the flexible M4M cross-linker had little effect on basal or verapamil-stimulated ATPase activities (Fig. 4), whereas direct cross-linking of the introduced cysteines using oxidant (copper phenanthroline) inhibited activity (27, 39). One explanation is that some movement is required at the C137/A935C and L443C/S909C interfaces during the catalytic cycle. For example, residues Cys137 and Ala935 are predicted to be 6.55 Å apart in the closed conformation (Fig. 7A) but 5.02 Å apart in the open conformation (Fig. 7B). Residues Leu443 and Ser909 are predicted to be 8.34 Å apart in the closed conformation (Fig. 7A) but 12.04 Å apart in the open conformation (Fig. 7B). Direct cross-linking of cysteines in the mutants would trap C137/A935C or L443C/S909C at a distance of about 5.6 Å, the average distance between α carbons in a disulfide bond (64).
A similar scenario was observed in a cross-linking study of the lactose transporter (65). It was found that cross-linking between cysteines in helices with short cross-linkers inhibited activity, whereas full or partial activity was observed when the mutants were cross-linked with long, flexible cross-linkers.
Another plausible explanation as to why direct cross-linking of mutants C137/A935C or L443C/S909C inhibited activity is that cross-linking may have trapped the protein in an inactive conformation with the sites closer than normal. In the closed conformation, residues Cys137 and Ala935 or Leu443 and Ser909 are predicted to be 6.55 Å or 8.34 Å apart, respectively. Direct cross-linking would reduce the distance to about 5.6 Å.
In summary, our findings show that cross-linking to bring the central region of NBD1 (close to the LSGGQ site) close to the N-terminal region of NBD2 (close to the Walker A site) highly activates ATPase activity and suggests that these regions undergo significant motion with respect to one another during the catalytic cycle. By contrast, a cross-linking study of the C-terminal ends of P-gp suggests that the protein resembles the maltose transporter (MalK subunit) where the C-terminal ends stay together during the entire reaction cycle. Our findings also suggest that separation of the N-terminal and central regions of the NBDs as observed in the mouse crystal structure (Fig. 1A) is not required to reset P-gp after one cycle of ATP hydrolysis (hydrolysis of one ATP molecule at each NBD). Cross-linking may increase the probability of generating an ATP-bound sandwich conformation to highly activate ATPase activity, an effect that may mimic stimulation of ATPase activity by potent stimulators such as verapamil and tariquidar but reduced by inhibitors such as the P10 pyrylium compound.
This work was supported, in whole or in part, by National Institutes of Health Grant GM-94367 (to M. R. D.) and by grants from the Canadian Cancer Society (19074) (to D. M. C.), the Canadian Institutes of Health Research (25043) (to D. M. C.).
- ABC
- ATP-binding cassette
- P-gp
- P-glycoprotein
- NBD
- nucleotide-binding domain
- HEK
- human embryonic kidney
- TM
- transmembrane
- TMD
- transmembrane domain
- ICL
- intracellular loop
- MTS
- methanethiosulfonate
- M1M
- 1,1-methanediyl bismethanethiosulfonate
- M4M
- 1,4-butanediyl bismethanethiosulfonate
- M17M
- 3,6,9,12,15-pentaoxaheptane-1,17-diyl bismethanethiosulfonate
- BMOE
- bis(maleimido)ethane.
REFERENCES
- 1. Rees D. C., Johnson E., Lewinson O. (2009) ABC transporters: the power to change. Nat. Rev. Mol. Cell Biol. 10, 218–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Juliano R. L., Ling V. (1976) A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 455, 152–162 [DOI] [PubMed] [Google Scholar]
- 3. Sharom F. J. (2006) Shedding light on drug transport: structure and function of the P-glycoprotein multidrug transporter (ABCB1). Biochem. Cell Biol. 84, 979–992 [DOI] [PubMed] [Google Scholar]
- 4. Ambudkar S. V., Dey S., Hrycyna C. A., Ramachandra M., Pastan I., Gottesman M. M. (1999) Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 39, 361–398 [DOI] [PubMed] [Google Scholar]
- 5. Eckford P. D., Sharom F. J. (2009) ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 109, 2989–3011 [DOI] [PubMed] [Google Scholar]
- 6. Chen C. J., Chin J. E., Ueda K., Clark D. P., Pastan I., Gottesman M. M., Roninson I. B. (1986) Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 47, 381–389 [DOI] [PubMed] [Google Scholar]
- 7. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., Chang G. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dey S., Ramachandra M., Pastan I., Gottesman M. M., Ambudkar S. V. (1997) Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 94, 10594–10599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lugo M. R., Sharom F. J. (2005) Interaction of LDS-751 with P-glycoprotein and mapping of the location of the R drug binding site. Biochemistry 44, 643–655 [DOI] [PubMed] [Google Scholar]
- 10. Loo T. W., Bartlett M. C., Clarke D. M. (2009) Identification of residues in the drug translocation pathway of the human multidrug resistance P-glycoprotein by arginine mutagenesis. J. Biol. Chem. 284, 24074–24087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Loo T. W., Clarke D. M. (1995) Covalent modification of human P-glycoprotein mutants containing a single cysteine in either nucleotide-binding fold abolishes drug-stimulated ATPase activity. J. Biol. Chem. 270, 22957–22961 [DOI] [PubMed] [Google Scholar]
- 12. Sauna Z. E., Kim I. W., Nandigama K., Kopp S., Chiba P., Ambudkar S. V. (2007) Catalytic cycle of ATP hydrolysis by P-glycoprotein: evidence for formation of the E.S reaction intermediate with ATP-γS, a nonhydrolyzable analogue of ATP. Biochemistry 46, 13787–13799 [DOI] [PubMed] [Google Scholar]
- 13. Siarheyeva A., Liu R., Sharom F. J. (2010) Characterization of an asymmetric occluded state of P-glycoprotein with two bound nucleotides: implications for catalysis. J. Biol. Chem. 285, 7575–7586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Delannoy S., Urbatsch I. L., Tombline G., Senior A. E., Vogel P. D. (2005) Nucleotide binding to the multidrug resistance P-glycoprotein as studied by ESR spectroscopy. Biochemistry 44, 14010–14019 [DOI] [PubMed] [Google Scholar]
- 15. Senior A. E., al-Shawi M. K., Urbatsch I. L. (1995) The catalytic cycle of P-glycoprotein. FEBS Lett. 377, 285–289 [DOI] [PubMed] [Google Scholar]
- 16. Gutmann D. A., Ward A., Urbatsch I. L., Chang G., van Veen H. W. (2010) Understanding polyspecificity of multidrug ABC transporters: closing in on the gaps in ABCB1. Trends Biochem. Sci. 35, 36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Urbatsch I. L., Gimi K., Wilke-Mounts S., Lerner-Marmarosh N., Rousseau M. E., Gros P., Senior A. E. (2001) Cysteines 431 and 1074 are responsible for inhibitory disulfide cross-linking between the two nucleotide-binding sites in human P-glycoprotein. J. Biol. Chem. 276, 26980–26987 [DOI] [PubMed] [Google Scholar]
- 18. Loo T. W., Bartlett M. C., Clarke D. M. (2002) The “LSGGQ” motif in each nucleotide-binding domain of human P-glycoprotein is adjacent to the opposing walker A sequence. J. Biol. Chem. 277, 41303–41306 [DOI] [PubMed] [Google Scholar]
- 19. Loo T. W., Bartlett M. C., Clarke D. M. (2003) Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains. J. Biol. Chem. 278, 1575–1578 [DOI] [PubMed] [Google Scholar]
- 20. Verhalen B., Wilkens S. (2011) P-glycoprotein retains drug-stimulated ATPase activity upon covalent linkage of the two nucleotide binding domains at their C-terminal ends. J. Biol. Chem. 286, 10476–10482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee J. Y., Urbatsch I. L., Senior A. E., Wilkens S. (2008) Nucleotide-induced structural changes in P-glycoprotein observed by electron microscopy. J. Biol. Chem. 283, 5769–5779 [DOI] [PubMed] [Google Scholar]
- 22. Loo T. W., Bartlett M. C., Clarke D. M. (2006) Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket. Biochem. J. 399, 351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loo T. W., Bartlett M. C., Clarke D. M. (2003) Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J. Biol. Chem. 278, 39706–39710 [DOI] [PubMed] [Google Scholar]
- 24. Loo T. W., Bartlett M. C., Clarke D. M. (2003) Permanent activation of the human P-glycoprotein by covalent modification of a residue in the drug-binding site. J. Biol. Chem. 278, 20449–20452 [DOI] [PubMed] [Google Scholar]
- 25. Martin C., Berridge G., Mistry P., Higgins C., Charlton P., Callaghan R. (1999) The molecular interaction of the high affinity reversal agent XR9576 with P-glycoprotein. Br. J. Pharmacol. 128, 403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loo T. W., Clarke D. M. (1995) Membrane topology of a cysteine-less mutant of human P-glycoprotein. J. Biol. Chem. 270, 843–848 [DOI] [PubMed] [Google Scholar]
- 27. Loo T. W., Bartlett M. C., Clarke D. M. (2008) Processing mutations disrupt interactions between the nucleotide binding and transmembrane domains of P-glycoprotein and the cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 283, 28190–28197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Loo T. W., Clarke D. M. (1999) The human multidrug resistance P-glycoprotein is inactive when its maturation is inhibited: potential for a role in cancer chemotherapy. Faseb J. 13, 1724–1732 [DOI] [PubMed] [Google Scholar]
- 29. Loo T. W., Clarke D. M. (2001) Determining the dimensions of the drug-binding domain of human P-glycoprotein using thiol cross-linking compounds as molecular rulers. J. Biol. Chem. 276, 36877–36880 [DOI] [PubMed] [Google Scholar]
- 30. Loo T. W., Clarke D. M. (2002) Vanadate trapping of nucleotide at the ATP-binding sites of human multidrug resistance P-glycoprotein exposes different residues to the drug-binding site. Proc. Natl. Acad. Sci. U.S.A. 99, 3511–3516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loo T. W., Clarke D. M. (1995) P-glycoprotein. Associations between domains and between domains and molecular chaperones. J. Biol. Chem. 270, 21839–21844 [DOI] [PubMed] [Google Scholar]
- 32. Ebert S. P., Wetzel B., Myette R. L., Conseil G., Cole S. P., Sawada G. A., Loo T. W., Bartlett M. C., Clarke D. M., Detty M. R. (2012) Chalcogenopyrylium compounds as modulators of the ATP-binding cassette transporters P-glycoprotein (P-gp/ABCB1) and multidrug resistance protein 1 (MRP1/ABCC1). J. Med. Chem. 55, 4683–4699 [DOI] [PubMed] [Google Scholar]
- 33. Bikadi Z., Hazai I., Malik D., Jemnitz K., Veres Z., Hari P., Ni Z., Loo T. W., Clarke D. M., Hazai E., Mao Q. (2011) Predicting P-glycoprotein-mediated drug transport based on support vector machine and three-dimensional crystal structure of P-glycoprotein. PLoS One 6, e25815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Globisch C., Pajeva I. K., Wiese M. (2008) Identification of putative binding sites of P-glycoprotein based on its homology model. ChemMedChem. 3, 280–295 [DOI] [PubMed] [Google Scholar]
- 35. Dawson R. J., Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 36. DeLano W. L. (2002) DeLano Scientific, Palo Alto, CA. U.S.A., www.pymol.org
- 37. Omote H., Al-Shawi M. K. (2002) A novel electron paramagnetic resonance approach to determine the mechanism of drug transport by P-glycoprotein. J. Biol. Chem. 277, 45688–45694 [DOI] [PubMed] [Google Scholar]
- 38. Loo T. W., Bartlett M. C., Clarke D. M. (2010) Human P-glycoprotein is active when the two halves are clamped together in the closed conformation. Biochem. Biophys. Res. Commun. 395, 436–440 [DOI] [PubMed] [Google Scholar]
- 39. Loo T. W., Bartlett M. C., Clarke D. M. (2004) Val-133 and Cys-137 in transmembrane segment 2 are close to Arg-935 and Gly-939 in transmembrane segment 11 of human P-glycoprotein. J. Biol. Chem. 279, 18232–18238 [DOI] [PubMed] [Google Scholar]
- 40. Kannan P., Telu S., Shukla S., Ambudkar S. V., Pike V. W., Halldin C., Gottesman M. M., Innis R. B., Hall M. D. (2011) The “Specific” P-glycoprotein inhibitor tariquidar is also a substrate and an inhibitor for breast cancer resistance protein (BCRP/ABCG2). ACS Chem. Neurosci. 2, 82–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fox E., Bates S. E. (2007) Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev. Anticancer Ther. 7, 447–459 [DOI] [PubMed] [Google Scholar]
- 42. Tang-Wai D. F., Kajiji S., DiCapua F., de Graaf D., Roninson I. B., Gros P. (1995) Human (MDR1) and mouse (mdr1, mdr3) P-glycoproteins can be distinguished by their respective drug resistance profiles and sensitivity to modulators. Biochemistry 34, 32–39 [DOI] [PubMed] [Google Scholar]
- 43. Loo T. W., Clarke D. M. (2001) Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J. Biol. Chem. 276, 14972–14979 [DOI] [PubMed] [Google Scholar]
- 44. Green N. S., Reisler E., Houk K. N. (2001) Quantitative evaluation of the lengths of homobifunctional protein cross-linking reagents used as molecular rulers. Protein. Sci. 10, 1293–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Samanta S., Ayvaz T., Reyes M., Shuman H. A., Chen J., Davidson A. L. (2003) Disulfide cross-linking reveals a site of stable interaction between C-terminal regulatory domains of the two MalK subunits in the maltose transport complex. J. Biol. Chem. 278, 35265–35271 [DOI] [PubMed] [Google Scholar]
- 46. Khare D., Oldham M. L., Orelle C., Davidson A. L., Chen J. (2009) Alternating access in maltose transporter mediated by rigid-body rotations. Mol. Cell 33, 528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tombline G., Bartholomew L. A., Tyndall G. A., Gimi K., Urbatsch I. L., Senior A. E. (2004) Properties of P-glycoprotein with mutations in the “catalytic carboxylate” glutamate residues. J. Biol. Chem. 279, 46518–46526 [DOI] [PubMed] [Google Scholar]
- 48. Urbatsch I. L., Sankaran B., Weber J., Senior A. E. (1995) P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 270, 19383–19390 [DOI] [PubMed] [Google Scholar]
- 49. Verhalen B., Ernst S., Börsch M., Wilkens S. (2012) Dynamic ligand-induced conformational rearrangements in P-glycoprotein as probed by fluorescence resonance energy transfer spectroscopy. J. Biol. Chem. 287, 1112–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Loo T. W., Clarke D. M. (1997) Drug-stimulated ATPase activity of human P-glycoprotein requires movement between transmembrane segments 6 and 12. J. Biol. Chem. 272, 20986–20989 [DOI] [PubMed] [Google Scholar]
- 51. Loo T. W., Clarke D. M. (2001) Cross-linking of human multidrug resistance P-glycoprotein by the substrate, Tris-(2-maleimidoethyl)amine, is altered by ATP hydrolysis. Evidence for rotation of a transmembrane helix. J. Biol. Chem. 276, 31800–31805 [DOI] [PubMed] [Google Scholar]
- 52. Jones P. M., O'Mara M. L., George A. M. (2009) ABC transporters: a riddle wrapped in a mystery inside an enigma. Trends Biochem. Sci. 34, 520–531 [DOI] [PubMed] [Google Scholar]
- 53. Sauna Z. E., Ambudkar S. V. (2001) Characterization of the catalytic cycle of ATP hydrolysis by human P-glycoprotein. The two ATP hydrolysis events in a single catalytic cycle are kinetically similar but affect different functional outcomes. J. Biol. Chem. 276, 11653–11661 [DOI] [PubMed] [Google Scholar]
- 54. Shapiro A. B., Ling V. (1997) Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem. 250, 130–137 [DOI] [PubMed] [Google Scholar]
- 55. Hrycyna C. A., Ramachandra M., Germann U. A., Cheng P. W., Pastan I., Gottesman M. M. (1999) Both ATP sites of human P-glycoprotein are essential but not symmetric. Biochemistry 38, 13887–13899 [DOI] [PubMed] [Google Scholar]
- 56. Vigano C., Julien M., Carrier I., Gros P., Ruysschaert J. M. (2002) Structural and functional asymmetry of the nucleotide-binding domains of P-glycoprotein investigated by attenuated total reflection Fourier transform infrared spectroscopy. J. Biol. Chem. 277, 5008–5016 [DOI] [PubMed] [Google Scholar]
- 57. Loo T. W., Bartlett M. C., Clarke D. M. (2006) Transmembrane segment 1 of human P-glycoprotein contributes to the drug-binding pocket. Biochem. J. 396, 537–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Loo T. W., Bartlett M. C., Clarke D. M. (2007) Suppressor mutations in the transmembrane segments of P-glycoprotein promote maturation of processing mutants and disrupt a subset of drug-binding sites. J. Biol. Chem. 282, 32043–32052 [DOI] [PubMed] [Google Scholar]
- 59. Loo T. W., Bartlett M. C., Clarke D. M. (2007) Nucleotide binding, ATP hydrolysis, and mutation of the catalytic carboxylates of human P-glycoprotein cause distinct conformational changes in the transmembrane segments. Biochemistry 46, 9328–9336 [DOI] [PubMed] [Google Scholar]
- 60. Mandal D., Moitra K., Ghosh D., Xia D., Dey S. (2012) Evidence for modulatory sites at the lipid-protein interface of the human multidrug transporter P-glycoprotein. Biochemistry 51, 2852–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ghosh P., Moitra K., Maki N., Dey S. (2006) Allosteric modulation of the human P-glycoprotein involves conformational changes mimicking catalytic transition intermediates. Arch. Biochem. Biophys. 450, 100–112 [DOI] [PubMed] [Google Scholar]
- 62. Ritchie T. K., Kwon H., Atkins W. M. (2011) Conformational analysis of human ATP-binding cassette transporter ABCB1 in lipid nanodiscs and inhibition by the antibodies MRK16 and UIC2. J. Biol. Chem. 286, 39489–39496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Loo T. W., Bartlett M. C., Clarke D. M. (2003) Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J. Biol. Chem. 278, 13603–13606 [DOI] [PubMed] [Google Scholar]
- 64. Schmidt B., Ho L., Hogg P. J. (2006) Allosteric disulfide bonds. Biochemistry 45, 7429–7433 [DOI] [PubMed] [Google Scholar]
- 65. Zhou Y., Guan L., Freites J. A., Kaback H. R. (2008) Opening and closing of the periplasmic gate in lactose permease. Proc. Natl. Acad. Sci. U.S.A. 105, 3774–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rosenberg M. F., Velarde G., Ford R. C., Martin C., Berridge G., Kerr I. D., Callaghan R., Schmidlin A., Wooding C., Linton K. J., Higgins C. F. (2001) Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. EMBO J. 20, 5615–5625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Martin C., Berridge G., Mistry P., Higgins C., Charlton P., Callaghan R. (2000) Drug binding sites on P-glycoprotein are altered by ATP binding prior to nucleotide hydrolysis. Biochemistry 39, 11901–11906 [DOI] [PubMed] [Google Scholar]