

Background: The transcription factor Bcl11b plays essential roles during T-cell development.
Results: Bcl11b activity in thymocytes is regulated by MAPK-mediated phosphorylation and subsequent sumoylation and ubiquitination.
Conclusion: A regulatory pathway links thymocyte stimulation, MAPK activation, and Bcl11b-dependent regulation of gene expression during late T-cell development.
Significance: This work has implications for the role of Bcl11b in T-cell development and leukemogenesis.
Keywords: Phosphorylation, Post-translational Modification, Signal Transduction, Sumoylation, Transcription Regulation, Bcl11b, Ctip2, Thymocytes
Abstract
The transcriptional regulatory protein Bcl11b is essential for T-cell development. We have discovered a dynamic, MAPK-regulated pathway involving sequential, linked, and reversible post-translational modifications of Bcl11b in thymocytes. MAPK-mediated phosphorylation of Bcl11b was coupled to its rapid desumoylation, which was followed by a subsequent cycle of dephosphorylation and resumoylation. Additionally and notably, we report the first instance of direct identification by mass spectrometry of a site of small ubiquitin-like modifier (SUMO) adduction, Lys-679 of Bcl11b, in a protein isolated from a native, mammalian cell. Sumoylation of Bcl11b resulted in recruitment of the transcriptional co-activator p300 to a Bcl11b-repressed promoter with subsequent induction of transcription. Prolonged treatment of native thymocytes with phorbol 12,13-dibutyrate together with the calcium ionophore A23187 also promoted ubiquitination and proteasomal degradation of Bcl11b, providing a mechanism for signal termination. A Bcl11b phospho-deSUMO switch was identified, the basis of which was phosphorylation-dependent recruitment of the SUMO hydrolase SENP1 to phospho-Bcl11b, coupled to hydrolysis of SUMO-Bcl11b. These results define a regulatory pathway in thymocytes that includes the MAPK pathways and upstream signaling components, Bcl11b and the associated nucleosome remodeling and deacetylation (NuRD) complex, SENP proteins, the Bcl11b protein phosphatase 6, the sumoylation machinery, the histone acetyltransferase p300, and downstream transcriptional machinery. This pathway appears to facilitate derepression of repressed Bcl11b target genes as immature thymocytes initiate differentiation programs, biochemically linking MAPK signaling with the latter stages of T-cell development.
Introduction
Post-translational modification (PTM)6 of proteins plays an essential role in creating and maintaining a highly dynamic system that integrates external signals from the cellular microenvironment into contextually appropriate responses (1). Relatively transient PTMs, such as phosphorylation, acetylation, ubiquitination, and sumoylation, are utilized in signal transduction relays. Control over the lifetime of a particular PTM on a given protein is governed by regulation of an array of opposing enzymes, e.g. kinases and phosphatases, acetyltransferases and deacetylases, SUMO ligases and SUMO proteases (SENPs), ubiquitin ligases and deubiquitinating enzymes (DUBs).
PTMs play particularly important roles in modulating the activity of transcriptional regulatory proteins. The addition of a phosphoryl group may alter protein conformation or create a physical docking site for interaction with other proteins containing phosphoamino acid binding motifs, and both may impact the activity of transcriptional regulatory proteins (2, 3). Covalent attachment of the small ubiquitin-like modifier (SUMO) protein to acceptor lysine residues in target proteins has emerged as a key PTM in the regulatory control of many proteins, most notably transcription factors (4, 5). Protein sumoylation may alter the intracellular localization, stability, and/or protein-protein interactions of target proteins (4, 6). The SUMO motif can serve as a docking site to recruit co-regulatory proteins harboring the canonical SUMO-interacting motif (7).
A compelling amount of data has demonstrated the critical importance of cross-talk between different classes of PTMs. Phosphorylation can either enhance or inhibit sumoylation within the same protein. A phosphorylation-dependent sumoylation motif in target proteins has been identified that when phosphorylated enhances substrate sumoylation (8). This motif is found in a number of transcription factors, such as GATA-1, MEF2A, estrogen-related receptor-α and -γ, and HSF1 (8–10). Conversely, sumoylation of transcription factors, such as AIB1, Elk1, and KAP1, is negatively regulated by the phosphorylation status of these factors (11–13). A complex interplay also exists between sumoylation and ubiquitination of target proteins. Initially, the SUMO and ubiquitin pathways were viewed as having a largely antagonistic relationship by competing for overlapping target site residues (14, 15). However, recent studies reveal that sumoylated substrates can be targeted to the ubiquitin system by a family of RING finger ubiquitin ligases, known as SUMO-targeted ubiquitin ligases (STUbL proteins), promoting target protein degradation by the 26 S proteasome (16, 17).
Bcl11b (also known as Ctip2) is a transcriptional regulatory protein that harbors multiple C2H2 zinc fingers and is highly expressed in the CNS, thymus, and epithelial tissues (18, 19). Disruption of the Bcl11b locus in mice results in defective neuronal (20, 21), skin (22), craniofacial (23), and T-cell (20, 24–26) development. Disruption of the human BCL11B locus has been associated with T-cell malignancies (27–29), suggesting that the protein acts as a tumor suppressor in T-cells (30). Bcl11b is essential to at least three critical checkpoints within the T-cell developmental program: 1) the commitment step of T-cell development at which CD4−CD8− (double negative (DN)) progenitors at the DN2 stage (CD25+CD44+) down-regulate CD44 expression to become DN3 (CD25+CD44−) cells (25); 2) the DN3 → DN4 transition, also known as β selection (31), and 3) differentiation of CD4+CD8+ (double positive; DP) cells into either CD4+ or CD8+ single positive (SP) cells, a process known as positive selection. Mice conditionally null for Bcl11b in DP cells exhibit defects in positive selection (24, 32) likely due to large scale dysregulation of the expression of genes key to both SP differentiation programs (24).
Bcl11b appears to function predominantly, but not exclusively, as a repressor of transcription in thymocytes and neuroblastoma cells. Bcl11b interacts indirectly with histone deacetylases (HDACs) within the context of the nucleosome remodeling and deacetylation (NuRD) (33, 34) or SIRT1 (35) complexes, both of which are recruited to target promoters by interaction with Bcl11b. Bcl11b also acts as a transcriptional activator in a promoter-dependent manner (22, 24, 36). However, the mechanistic basis for control of the transcriptional regulatory activity of Bcl11b is largely unknown.
Signaling cascades initiated by T-cell receptor (TCR) activation direct the reprogramming of the gene expression network in progenitor cells that drives T-cell differentiation and maturation toward the mature, SP phenotypes (37). The MAP kinase pathway in thymocytes, which is downstream of TCR activation, is implicated in both early (DN3 → DN4 transition (38–40)) and late (DP → SP transition) selection events in T-cell development (41–43). Upon activation, MAP kinases phosphorylate a number of transcription factors, leading to alterations in the transcriptional regulatory activity of these factors on target promoters (37). Mice lacking upstream proteins in the MAP kinase pathway, such as Ras, RasGRP1, Raf, and phosphatidylinositol 3-kinase as well as MEK and Erk1/2, exhibit a severe block in positive selection (43) similar to the phenotype for mice lacking Bcl11b in DP cells (24, 32), suggesting that Bcl11b may be a required effector downstream of TCR/MAP kinase signaling in thymocyte development.
We hypothesized that Bcl11b may be regulated by PTMs in the context of T-cell signaling events. To test this hypothesis, we adapted a previously described in vitro system that was shown to serve as a surrogate for TCR activation (44, 45). Data presented herein suggest that the MAP kinase pathways dynamically regulate the PTM status of Bcl11b; treatment with a phorbol ester and a calcium ionophore resulted in the sequential and rapid phosphorylation, desumoylation, dephosphorylation, resumoylation, ubiquitination, and ultimately degradation of Bcl11b. Furthermore and importantly, these PTMs modified the transcriptional regulatory activity of Bcl11b on a natural transcriptional target, the mouse Id2 promoter. Our findings strongly suggest that Bcl11b is a highly regulated node in MAP kinase pathways, linking TCR activation to altered gene expression programs that are required to initiate the SP differentiation program in DP thymocytes. This study provides a better understanding of the mechanistic basis for and the molecular consequences of cell signaling-dependent modification of Bcl11b by PTMs.
EXPERIMENTAL PROCEDURES
Chemicals and Antibodies
Phorbol 12,13-dibutyrate (P1269) PDBu and anti-FLAG antibody (F4042) were purchased from Sigma. A23187, U0126, and SB202190 were purchased from EMD Biosciences (Gibbstown, NJ). Calyculin A was purchased from Cell Signaling Technology (Danvers, MA). Antibodies to Bcl11b (ab18465), SUMO1 (ab32058), and SUMO2/3 (ab3742) were purchased from Abcam (Cambridge, MA). Antibodies to phosphothreonine (9386), phospho-Erk1/2 (4370), and phospho-p38 (9211) were purchased from Cell Signaling Technology. Antibodies to MTA2 (A300-395A), HDAC1 (A300-713A), HDAC2 (A300-705A), and p300 (A300-358A; used for immunoprecipitation (IP) studies) were purchased from Bethyl Laboratories (Montgomery, TX). A second anti-p300 antibody was purchased from Santa Cruz Biotechnology (N-15, sc584) for chromatin immunoprecipitation (ChIP) experiments. The anti-HA antibody was purchased from Aves Labs, Inc. (Tigard, Oregon), and the anti-ubiquitin antibody was purchased from Enzo Life Sciences (Plymouth Meeting, PA). Cell culture medium was purchased from VWR, and fetal bovine serum was from Hyclone.
Cell Culture
Primary thymocytes were isolated from 4–8-week-old wild-type mice and cultured at 37 °C in RPMI 1640 medium plus 5% fetal bovine serum (FBS) and penicillin/streptomycin for 4 h prior to drug treatment. All procedures involving mice were conducted with approval of the Oregon State University Institutional Animal Care and Use Committee. HEK293T cells were cultured as described (33).
Drug Treatments
Primary thymocytes were cultured for 4 h before treating with vehicle (DMSO) or a mixture consisting of PDBu (100 nm) and A23187 (500 nm) (P/A). In some studies, cells were pretreated with the indicated kinase inhibitors for 30 min prior to P/A treatment.
Sample Preparation for Mass Spectrometry
Natively expressed mouse thymic Bcl11b was prepared for mass spectrometry analysis from immunoprecipitates after electrophoretic purification by 4–12% SDS-PAGE (BisTris/MES; Bio-Rad). Gel slices were identified after staining with colloidal Coomassie G-250 (Biosafe Coomassie, Bio-Rad) and subsequently excised; destained in 25 mm ammonium bicarbonate, 50% acetonitrile; and dehydrated in 100% acetonitrile. Peptides were generated from tryptic and combined cyanogen bromide/tryptic digests. For in-gel cyanogen bromide digests, dried gel slices were rehydrated in a solution of 200 mm cyanogen bromide in 70% trifluoroacetic acid and incubated at room temperature for 12–14 h while protected from light. Volatile reactants were subsequently removed by two cycles of water addition and centrifugal vacuum concentration, each at 10-fold the original reaction volume. This was followed by two cycles of 25 mm ammonium bicarbonate, 50% acetonitrile; dehydration in 100% acetonitrile; and drying by vacuum centrifugation. Prior to tryptic digestion, the dried gel slices were rehydrated and reduced in 25 mm dithiothreitol, 50 mm ammonium bicarbonate for 30 min a 50 °C; alkylated in 50 mm 2-chloroacetamide, 50 mm ammonium bicarbonate (46) for 30 min at room temperature; and dehydrated again in 100% acetonitrile. In-gel tryptic digests were conducted by rehydrating gel slices in 12.5 ng/ml trypsin (Trypsin Gold, mass spectrometry grade, Promega) in 0.025% ProteaseMax (Promega), 50 mm ammonium bicarbonate and incubated at 37 °C for 3 h. Product peptides were twice extracted from gel slices in 0.01% ProteaseMax, 50 mm ammonium bicarbonate; acidified with trifluoroacetic acid; and concentrated by vacuum centrifugation.
Mass Spectrometry and Data Analysis
Mass spectrometry analyses were conducted on a nanoAcuity UPLC (Waters, Milford, MA)-coupled hybrid linear ion trap/Fourier transform-ion cyclotron resonance mass spectrometer (7-tesla LTQ-FTMS Ultra, Thermo Fisher, San Jose, CA). Data-dependent analysis used both instrument mass analyzers. The Fourier transform-ion cyclotron mass analyzer (FT-ICR) was used to record high resolution spectra of the parent peptide ions and to select them for further analysis, and the linear ion trap mass analyzer (LTQ) was used to record the tandem mass spectra of these selected parent ions. Peptide samples were concentrated on a C18 trap column and from there resolved on a 100-μm × 150-mm Waters BEH130 1.7-μm C18 column using a two-segment acetonitrile gradient, 3–28% in 60 min followed by 28–76% between 60 and 90 min, in 0.1% formic acid at a flow rate of 230 nl/min. Mass spectra were collected for 70 min after a 1-min delay from the start of the gradient. The electrospray ionization source (Michrom Advance captive spray source, Michrom Bioresources, Auburn, CA) was operated with a spray voltage of 1.7–2 kV, a capillary voltage of 24–48 V, a capillary temperature of 150 °C, and a tube lens offset of 135–210 V (the specific setting varied within the given ranges). Full-scan spectra (400–1800 m/z for phosphopeptides and 350–2000 m/z for sumoylation analysis) were recorded in the FT-ICR cell at a resolution of 100,000 (m/z 400) with a target value of 1 × 106 charges accumulated for a maximum of 500 ms. Charge state screening was used to reject singly charged peptides, and a threshold of 1000 charge counts was required to trigger tandem mass analysis. The five most intense ions meeting these criteria were selected with an isolation width of 2 thomsons for collision-induced dissociation tandem mass acquisition in the linear ion trap and then dynamically excluded from reanalysis for 45 s. The LTQ targeted the accumulation of 3 × 104 charges for a maximum of 500 ms. The activation Q was 0.250, and the activation time was 30 ms. A normalized collisional energy of 30% for phosphopeptide analysis or 35% for experiments that targeted sumoylation adducts was used to fragment parent peptide ions.
Tandem mass spectra were extracted from raw data files by Proteome Discoverer (version 1.0, Thermo Fisher) in Mascot generic format by Xcalibur (version 2.0.5, Thermo Fisher) for database searching or manual annotation and assignment. Precursor peptide masses were calculated from the intensity-weighted ion m/z values recorded in the ICR cell by Raw2msm (version 1.10; Ref. 47). Mascot (version 2.2.04; Matrix Science, London, UK) and X! Tandem (version 2007.01.01.1; The Global Proteome Machine Organization) were used to search the International Protein Index mouse database (version 3.58; 56,619 entries). Search parameters were set to allow for two missed cleavages when trypsin was used or three missed cleavages when combined cyanogen bromide/tryptic digests were used. The search database cleavage parameter was set to cut after arginine and lysine for trypsin preparations and after arginine, lysine, and methionine when cyanogen bromide/trypsin preparations were analyzed. The mass tolerance was 8 ppm for the parent ion and 0.70 Da for the fragment ions. Oxidation on methionine, phosphorylation on serine and threonine, and homoserine lactone derivatization of carboxyl-terminal methionine residues were permitted as variable modifications, and all cysteine residues were searched for as their carbamidomethyl derivatives. Statistical validation of Mascot search results required protein confidence >99% (Protein Prophet; Ref. 48) using a minimum of two >95% confident peptides (Peptide Prophet; Ref. 49) as implemented in Scaffold (version 3.00.08; Proteome Software, Portland, OR). To identify Bcl11b sumoylation adducts, ChopNSpice (version 1.0; Ref. 50) was used to generate a custom synthetic derivative database of potential adducts on the ϵ-amine of lysine residues that was searched with Mascot. Tandem mass spectra identifying the sites of sumoylation and phosphorylation were manually validated and assigned.
DNA Constructs
FLAG-tagged Bcl11b (FLAG-Bcl11b) was described previously (35). Lysine to arginine mutants of Bcl11b including K679R, K877R, and K679R/K877R (2R) were made using the QuikChange site-directed mutagenesis kit from Stratagene (La Jolla, CA) as was the Bcl11b phosphosite mutant pMT, which harbored alanine point mutations in five phosphoacceptor sites: Thr-260, Ser-277, Ser-318, Ser-381, and Ser-664. HA-SUMO1 and HA-SUMO2 constructs were purchased from Addgene (Cambridge, MA). HA-SU-FLAG-Bcl11b was generated by PCR amplification of HA-SUMO1 with primers containing appropriate restriction sites for insertion into the amino terminus of FLAG-Bcl11b. HA-SENP1 and HA-SENP2 constructs were generously provided by Dr. Grace Gill (Tufts University). A mammalian expression vector encoding protein phosphatase 6 (PP6) was a kind gift from Dr. David Brautigan, University of Virginia (51).
To prepare the Id2-CAT reporter gene plasmid, the tk promoter was excised from pBLCAT2 vector using BamHI and XhoI restriction enzymes. This BamHI/XhoI-digested, promoterless reporter construct was used for subsequent cloning of the Id2 promoter as follows. A fragment from the mouse Id2 locus (from −2.9 kb to +152 bp relative to the transcriptional start site) was amplified using appropriate primers, mouse ES cell DNA, and the Expand Long Template PCR system (Roche Applied Science). The amplified fragment was inserted into pCR 2.1 vector using a TA cloning kit (Invitrogen) and then excised with BamHI and XhoI prior to insertion into the promoterless CAT reporter construct (also previously digested with BamHI and XhoI) described above. All constructs were verified by complete DNA sequencing.
Transfections
HEK293T cells (2 × 106 cells) were plated onto a 10-cm plate and transfected 24 h later using the calcium phosphate method. Cells were transfected with varying amounts of plasmid DNA and an appropriate empty expression vector to standardize the amount of plasmid DNA transfected. The medium was changed, and the cells were harvested 24 and 48 h after transfection, respectively. Where indicated, cells were subjected to serum starvation ∼24 h after transfection followed by P/A or vehicle treatment for varying times prior to harvest.
Immunoprecipitation and Immunoblotting
Bcl11b was immunoprecipitated from clarified nuclear extracts prepared in 20 mm HEPES, pH 7.4, 250 mm NaCl, 50 mm NaF, 2 mm EDTA, 5 mm sodium pyrophosphate, 0.05% Nonidet P-40, 100 μm PMSF, 10 μm E64, 1 μg/ml pepstatin A, 5 μg/ml leupeptin (52). For experiments where sumoylation was analyzed, the buffer was supplemented with 5 mm N-ethylmaleimide, 100 μm hemin chloride, and 1% SDS. These samples were then boiled for 5 min and sonicated. Bcl11b was immunoprecipitated from these denatured extracts after diluting 10-fold in the same buffer containing 1.1% Triton X-100 in place of SDS. Chemiluminescence-based immunoblotting procedures were as described previously (33).
Reporter Gene Assays
HEK293T cells were transfected and harvested 48 h after transfection. A β-galactosidase expression vector (pCMV-Sport-βGal, Invitrogen) was co-transfected as an internal control; β-galactosidase activity and total protein concentration were used for normalization across all samples. Relative chloramphenicol acetyltransferase activity was determined as described previously (53).
Quantitative Reverse Transcription-PCR
Total cellular RNA was extracted with the RNeasy reagent (Qiagen), and 500 ng of total RNA was reverse transcribed into cDNA using Superscript III reverse transcriptase (Invitrogen) and oligo(dT) primers. The resulting cDNA was used for the qPCR amplification reactions (see primer sets in supplemental Table S1) using an Applied Biosystems 7500 real time PCR instrument and SYBR Green methodology (Qiagen).
Chromatin Immunoprecipitation
Approximately 2 × 108 primary thymocytes were treated with P/A or vehicle for various times followed by cross-linking with formaldehyde as described previously (33). Fixed cells were lysed with a hypotonic buffer (10 mm Tris-HCl, pH 7.5, 10 mm NaCl, 3 mm MgCl2, 0.5% Nonidet P-40). Chromatin was digested briefly by treating with micrococcal nuclease in lysis buffer (10 mm Tris-HCl, pH 7.5, 10 mm NaCl, 3 mm MgCl2, 2 mm CaCl2, 2% Nonidet P-40) for 15 min at 37 °C. Approximately 2000 units of micrococcal nuclease were used to digest 1 mg of DNA/chromatin from 2 × 108 thymocytes. Micrococcal nuclease was inactivated by adding EDTA to 5 mm and NaCl to 150 mm prior to sonication. The digested chromatin was further sheared to an average fragment size of ∼500 bp using a Branson 450 sonicator. All buffers were supplemented with a complete protease inhibitor mixture (Fermentas) and 1 mm PMSF. Insoluble cell debris was removed by centrifugation. Approximately 500 μg of soluble chromatin solution was subjected to ChIP and re-ChIP analyses as described previously (52). ChIP DNA was subjected to qPCR analyses using primer sets described in supplemental Table S1.
SENP1 Interaction with Bcl11b
HEK293T cells were simultaneously transfected with vectors encoding FLAG-SENP1 and FLAG-Bcl11b. A constant amount of an expression vector encoding FLAG-SENP1 and varying amounts of expression vectors encoding wild-type Bcl11b or the Bcl11b phosphomutant pMT were used to achieve a varying expression ratio of these proteins. Bcl11b was immunoprecipitated from nuclear extracts as described above prior to immunoblotting analyses using anti-FLAG primary and IRDye 800-conjugated secondary antibodies to quantify relative amounts of FLAG-Bcl11b and FLAG-SENP1. Immunoblots were analyzed using a LI-COR Odyssey imager (LI-COR Biosystems, Lincoln, NE) with version 3.0.29 of the system software. The resulting data on the relative amount of Bcl11b in each immune complex versus the relative amount of co-immunoprecipitating SENP1 were fit to Equation 1 using a non-linear, least square procedure:
 |
where Bcl11b is the amount of Bcl11b (wild-type or mutant) in the immunoprecipitant, Bmax is the maximal amount of SENP1 co-immunoprecipitating with wild-type Bcl11b in each experiment, Bound is the amount of SENP1 in each co-IP, and K represents the amount of Bcl11b (wild-type or mutant) required to achieve 50% of Bmax in each experiment (referred to herein as an apparent association constant). An increase in the value of K is indicative of a decrease in association between the two proteins in a saturating bimolecular interaction. Statistical significance was determined by a Student's t test and analysis of three independent experiments.
RESULTS
Bcl11b Is Sumoylated in Mouse Thymocytes in a Stimulation-dependent Manner
Our previous studies implicated Bcl11b in the MAPK pathways (24). Thus, we sought to determine whether stimulation of this pathway in primary thymocytes with P/A resulted in post-translational modification of Bcl11b. Stimulation with P/A was also used as a surrogate for activation of cell signaling pathways including T-cell receptor activation (44, 45). The MAP kinases Erk1/2 and p38 were highly phosphorylated within 5 min of P/A treatment, and the level of phosphorylation of both kinases began to diminish after ∼30 min of continuous treatment (Fig. 1A). A slowly migrating species of Bcl11b became apparent at 20 min after initiation of P/A treatment (Fig. 1A, upper blot, asterisks). Although present in extracts prepared from untreated cells (Fig. 1A, upper blot, lane 1), these low mobility species of Bcl11b appeared to diminish immediately after P/A treatment (5 min; lane 3) and then began to reappear ∼20 min later (Fig. 1A, upper blot, lanes 4–6). Immunoblot analyses confirmed these low mobility forms of Bcl11b to be SUMO1 and SUMO2/3 adducts of Bcl11b (Fig. 1, B and C, respectively). P/A-induced sumoylation of Bcl11b reached a maximum level of SUMO1 (Fig. 1B, lane 3) and SUMO2/3 (Fig. 1C, lane 3) sumoylation after ∼60 min of continuous P/A treatment.
FIGURE 1.

Regulated sumoylation of Bcl11b in primary thymocytes. A, time course of the appearance of slowly migrating Bcl11b species (asterisks) in stimulated thymocytes. Primary thymocytes were treated with P/A as indicated, and cells were lysed in denaturing sample buffer prior to electrophoresis and immunoblotting analyses using antibodies for Bcl11b, phospho-Erk1/2 (p-Erk), phospho-p38 kinase (p-p38), and HDAC1 indicated at right. The HDAC1 blot serves as the loading control. The filled arrowhead in the top panel depicts a P/A-induced shift in the electrophoretic mobility of Bcl11b, and the open arrowhead indicates when that electrophoretic mobility shift is reversed. B and C, P/A-induced sumoylation of Bcl11b in thymocytes. Thymocytes were treated with P/A for the indicated times prior to lysis, immunoprecipitated with the anti-SUMO antibodies indicated, and immunoblotted using an anti-Bcl11b antibody. These representative findings have been replicated multiple times using several experimental approaches. Note that mouse thymocytes express two splice variants of Bcl11b; the shorter form is composed of exons 1, 2, and 4 and migrates just under the 130-kDa marker, and a longer form is composed of exons 1, 2, 3, and 4 and is 72 amino acids longer than the short form and migrates at the 130-kDa marker.
Bcl11b Harbors Two SUMO Acceptors Sites
Tandem mass spectra identified Bcl11b Lys-679 as the adduction site for SUMO1 (Fig. 2A) and SUMO2/3 (Fig. 2B). The tryptic products of SUMO2 and SUMO3 are identical and cannot be distinguished from these spectra. However, the tryptic peptides covering the distinctive amino termini were present in sufficient number to identify both SUMO2 and SUMO3 with high confidence in the same high molecular weight bands in which the Bcl11b SUMO2/3 adduct was identified. This demonstrates that Bcl11b was adducted by SUMO1, SUMO2, and SUMO3 in native thymocytes.
FIGURE 2.

Mapping Bcl11b sumoylation sites. Identification of Bcl11b Lys-679 as a site of SUMO1 (A) and SUMO2/3 (B) adduction in primary thymocytes is shown. LTQ ion trap tandem mass spectra are shown with FT-ICR full-scan mass spectra (insets). The derived experimental mass was 2751.3200 Da (error from theoretical, 1.4 ppm) for the SUMO1 adduct and 4468.1021 Da (error from theoretical, 0.11 ppm) for the SUMO2/3 adduct. C, identification of Lys-679 and Lys-877 as Bcl11b sumoylation sites by site-directed mutagenesis. HEK293T cells were co-transfected with expression vectors encoding Bcl11b or the mutants indicated (1 μg) and HA-SUMO1 or HA-SUMO2 (2 μg). The indicated SUMO adducts were deduced from sumoylation patterns of single point mutants where “n” indicates the likely presence of SUMO chains. The results shown in C have been replicated three to five times. Note that the experiment depicted in C was conducted using the form of Bcl11b corresponding to exons 1, 2, and 4, and consequently, the SUMO adducts of this splice variant are somewhat smaller than those of native thymocytes (which express splice variants composed of exons 1, 2, and 4 and exons 1, 2, 3, and 4; see Fig. 1).
Bcl11b Lys-679 was confirmed as a site of SUMO1 and SUMO2/3 adduction by site-directed mutagenesis studies in HEK293T cells (Fig. 2C). These mutagenesis studies also revealed that Lys-877 was a site of Bcl11b sumoylation. Both SUMO1 (Fig. 2C, lanes 4 and 6) and SUMO2/3 (lanes 9 and 11) chains were evident at the Lys-679 site as observed with the wild-type protein and the Bcl11b K877R mutant, respectively. However, SUMO1 and SUMO2/3 chains were less evident at the Lys-877 site as revealed by the Bcl11b K679R mutant (Fig. 2C, lanes 5 and 10). Note that the lack of efficient sumoylation of wild-type or mutant Bcl11b in HEK293T necessitated forcing the reaction by co-transfection of expression vectors encoding HA-SUMO1 and -SUMO2.
Bcl11b Is a Dynamically Regulated Phosphoprotein in Mouse Thymocytes
An electrophoretic mobility shift of unsumoylated Bcl11b was also evident on denaturing gels after 5 min of P/A treatment (Fig. 1A, upper blot, lanes 1 and 2, filled arrowhead), temporally corresponding to induction of maximal Erk1/2 and p38 phosphorylation by P/A treatment (see Fig. 1A). The electrophoretic mobility of unsumoylated Bcl11b returned to pretreatment levels after 20 min of continuous P/A treatment (Fig. 1A, upper blot, lanes 3 and 4, open arrowhead). We hypothesized that the mobility shift of unsumoylated Bcl11b may result from an induced hyperphosphorylation state during this time frame. Indeed, Bcl11b was found to be a phosphoprotein in unstimulated, mouse thymocytes (Fig. 3A, upper blot, lane 2) and in cells stimulated with P/A (lanes 3–7). However, the kinetics of P/A-induced phosphorylation of Bcl11b were not linear.
FIGURE 3.

Phosphorylation of Bcl11b by MAPK pathways. A, time course of Bcl11b phosphorylation. Immunoprecipitation experiments were carried out using extracts from thymocytes treated with P/A as indicated. The upper blot was probed with an anti-phospho-Thr/Ser antibody (anti-pT/S). The blot was stripped and reprobed with an anti-Bcl11b antibody (lower blot). B, activated Erk and p38 kinases contributed to Bcl11b phosphorylation at 5 min. Thymocytes were preincubated with the MEK inhibitor U0126 (U0; 10 μm) or the p38 inhibitor SB202190 (SB; 10 μm) before treating with P/A for 5 min. Samples were immunoprecipitated, and immunoblots were generated as in A. C, inhibition of the Erk pathway, but not that of p38, blocks P/A-induced Bcl11b dephosphorylation at 30 min. Thymocytes were preincubated with U0126 or SB202190 as indicated before treating with P/A for 60 min. Samples were processed as in A. D, the PP1/PP2A phosphatase inhibitor calyculin A (Cal A) stimulated phosphorylation and blocked sumoylation of Bcl11b. Thymocytes were treated with 50 nm calyculin A for the indicated times, and samples were processed as in A.
Treatment of thymocytes with P/A resulted in very rapid phosphorylation of Bcl11b on serine and threonine residues that reached a maximum at around 5 min (Fig. 3A, upper blot, lane 3). This phosphorylation was transient as phospho-Bcl11b was rapidly dephosphorylated in the continued presence of P/A; the level of Bcl11b phosphorylation after 30 min of P/A treatment was less than that observed under basal conditions (Fig. 3A, upper blot, lane 4). Total levels of Bcl11b (sumoylated plus non-sumoylated) remained relatively constant over the same time course (Fig. 3A, lower blot). The phosphorylation status of Bcl11b then returned to basal levels after 1–2 h, and this level was stable in the presence of continuous P/A for up to 4 h (see Fig. 3A, upper blot, lanes 6 and 7).
Kinase inhibitors were used to identify the pathway(s) responsible for phosphorylation of Bcl11b in response to P/A stimulation in primary mouse thymocytes. U0126 and SB202190, inhibitors of the Erk1/2 and p38 pathways, respectively, decreased P/A-induced phosphorylation of Bcl11b at 5 min after initiation of treatment (Fig. 3B, upper blot, compare lanes 1–4), suggesting that both Erk1/2 and p38 pathways play a role in phosphorylating Bcl11b in response to P/A treatment. This finding is consistent with the nature of sites of Bcl11b phosphorylation identified by tandem mass spectrometry (see below), the majority of which corresponded to consensus sites for proline-directed kinases, a family of enzymes including the MAP kinases Erk1/2 and p38 (Table 1).
TABLE 1.
Bcl11b phosphorylation motifs
Motifs are aligned to the site of phosphorylation (bold underlined). Proline residues carboxyl-terminal to phosphorylation sites are shown in bold. See Fig. 4 and supplemental Fig. S1 for assigned mass spectra.
| Motif | Phosphorylation site |
|---|---|
| DKVLDKSSPPPSS | Ser-95 |
| KVLDKSSPPPSSR | Ser-96 |
| SELRRVSEPVEIG | Ser-109 |
| EDDHLLSPTKGIC | Ser-128 |
| PTSVITSPLRALG | Ser-169 |
| PASTSLTPRLTIP | Thr-260 |
| PETVAQSPLMNFL | Ser-277 |
| EGRLPGTPPLFSP | Thr-313 |
| GTPPLFSPPPRHH | Ser-318 |
| LAGNSSTPPPVSP | Thr-376 |
| STPPPVSPGRGNP | Ser-381 |
| LNPFQPSPKSPFL | Ser-398 |
| FQPSPKSPFLSTP | Ser-401 |
| KSPFLSTPPLPPP | Thr-406 |
| PPMPAGTPPPQPP | Thr-416 |
| GLSAASSPEPGTS | Ser-496 |
| KPAPLPSPGLGGP | Ser-664 |
| SPFATSSEHSSEN | Ser-731 |
| ATSSEHSSENGSL | Ser-734 |
| GSLRFSTPPGDLL | Thr-744 |
| GRSGTASGGSTPH | Ser-762 |
| TASGGSTPHLGGP | Ser-765, Thr-766a |
| GPGRPSSKEGRRS | Ser-778, Ser-779a |
a Ambiguously assigned sites.
To our great surprise, U0126, but not SB202190, also inhibited the subsequent, P/A-induced dephosphorylation of Bcl11b at 60 min after initiation of treatment (Fig. 3C, upper blot, compare lanes 1–3). Inhibition of Bcl11b dephosphorylation by U0126 also reduced the extent of Bcl11b sumoylation (Fig. 3C, lower blot, compare lanes 2–4), suggesting that the phosphorylation and sumoylation states of Bcl11b may be mutually exclusive.
The dynamic nature of Bcl11b phosphorylation was further revealed by use of calyculin A, a potent inhibitor of class 1 and 2A phosphatases. Treatment with calyculin A in the absence of P/A dramatically increased levels of phospho-Bcl11b in a time-dependent manner (Fig. 3D, upper panel) as revealed by enhanced anti-phospho-Thr/Ser detection in Bcl11b immunoprecipitates and shifted electrophoretic mobility of unsumoylated Bcl11b (Fig. 3D, upper and lower panels). Treatment with calyculin A also resulted in a very rapid desumoylation of Bcl11b (Fig. 3D, lower blot), further supporting the mutual exclusivity of phosphorylation and sumoylation of Bcl11b. These findings demonstrate that modification of Bcl11b by both phosphorylation and sumoylation are rapid, reversible, and inter-regulated events. We interpret these findings as follows. 1) P/A stimulation of native thymocytes resulted in the activation of one or more isoforms of PKC, which then activated p38 and Erk1/2 signaling pathways, both of which contributed to the induced phosphorylation state of Bcl11b. 2) Erk1/2, but not p38, activated a type 1 or 2A phosphatase(s), which dephosphorylated Bcl11b. 3) Phosphorylation and dephosphorylation of Bcl11b were temporally out of phase, resulting in a hyperphosphorylated state of Bcl11b that persisted for ∼10 min prior to extensive dephosphorylation. 4) Dephosphorylation preceded and appeared necessary for P/A-induced sumoylation of Bcl11b. 5) Phosphorylation of Bcl11b resulted in rapid hydrolysis of basally sumoylated Bcl11b presumably by one or more SENP family proteins (SUMO hydrolases; Refs. 54 and 55), suggesting the existence of a “phospho-deSUMO” switch within Bcl11b.
Mapping Sites of Bcl11b Phosphorylation
Phosphorylation site assignments of native Bcl11b were made by tandem mass spectrometry from basal and P/A-stimulated thymocyte immunoprecipitates. We identified 23 serine and threonine phosphorylation sites with high confidence (Fig. 4, A and B, and supplemental Figs. S1 and S2); tyrosine phosphorylation of Bcl11b was not observed under these conditions. Phosphorylation of Bcl11b was predominately observed at serine and threonine residues amino-terminal of proline (Table 1), consistent with phosphorylation by Erk1/2 and p38 MAP kinases (see above). A single site (Ser-109) was consistent with direct phosphorylation by protein kinase C (Table 1 and supplemental Fig. S1B). Spectral analyses allowed all but two of these sites to be assigned at the residue level. Phosphosite assignments were manually validated and included several examples of overlapping spectra originating from co-eluting phosphosite isobaric pairs. A singly phosphorylated isobaric pair was resolved chromatographically in one example (Fig. 4A), adding confidence to its multisite assignment.
FIGURE 4.

Chromatographic resolution of isobaric phosphosite isomers of Bcl11b monophosphopeptide 310–322. A, top, FT-ICR extracted ion current (XIC) chromatogram and mass determinations of the leading and trailing peaks are presented compared with a theoretical m/z of 728.37343. Vertical lines indicate the time at which tandem mass spectra were recorded. Bottom, LTQ ion trap tandem mass spectra recorded at 26.79 min (positive ordinate) and 28.05 min (negative ordinate) elution time identify the leading peak in the top panel as Thr(P)-313 and the trailing peak as Ser(P)-318 localized to monophosphopeptide 310–322. See supplemental Fig. S1 for additional spectra identifying Bcl11b phosphorylation sites in native thymocytes. B, schematic representation of Bcl11b with 23 phosphorylation and two sumoylation sites. ZnF indicates zinc finger domains. RT, retention time. th, mass-to-charge ratio in thomsons.
The Molecular Nature of the Bcl11b Phospho-deSUMO Switch
Sumoylation is a dynamic process that is readily reversed by a family of highly active SUMO-specific proteases, the SENP family of proteins (54, 55). Phosphorylation of Bcl11b induced by either 5 min of P/A (Fig. 3, A and B) or continuous calyculin A (Fig. 3D) treatment resulted in rapid desumoylation of Bcl11b, suggesting that phosphorylated Bcl11b may be a substrate for SENP-mediated desumoylation. To test this hypothesis, we co-expressed Bcl11b and HA-SUMO1 or HA-SUMO2 together with SENP1 or SENP2. Both SENPs efficiently desumoylated SUMO1-Bcl11b and SUMO2-Bcl11b (Fig. 5A, upper blot) without affecting Bcl11b protein levels (lower blot). SENP1 also interacted stably with the Bcl11b or the Bcl11b complex (Fig. 5B, lower blot), and this interaction was not dependent on the sumoylation status of Bcl11b because the SENP1 interacted with the double SUMO site mutant 2R in a manner that was indistinguishable from that of wild-type Bcl11b (Fig. 5C, lower blot).
FIGURE 5.

SENP1 preferentially interacts with phosphorylated Bcl11b. Immunoprecipitation and immunoblotting were conducted as indicated in all panels. A, desumoylation of HA-SU1-Bcl11b and HA-SU2-Bcl11b in transfected HEK cells by both SENP1 and SENP2. The Bcl11b blot (lower image) was generated by stripping and reprobing the upper blot. B, interaction of SENP1 with Bcl11b complexes in transfected HEK293T cells. C, sumoylation is not required for the interaction between Bcl11b and SENP1. HEK293 cells were transfected with an expression vectors encoding wild-type Bcl11b or the double mutant 2R. Whole cell extracts from transfected HEK cells were immunoprecipitated with an anti-Bcl11b antibody, and the immunoprecipitates were probed with anti-Bcl11b (upper blot) or anti-FLAG (SENP1; lower blot) antibody. D, dephosphorylation of Bcl11b by HA-PP6 in transiently transfected HEK 293T cells; asterisks represent nonspecific immunoreactivity in input. E, dephosphorylation of Bcl11b inhibits interaction of SENP1 with the Bcl11b complex. The amount of expression vectors transfected in A–E were as follows: Bcl11b, 1 μg; HA-SUMO1 or -SUMO2, 2 μg; HA-SENP1 or -SENP2, 2 μg; HA-PP6, 2 μg where indicated. F, Bcl11b phosphorylation/sumoylation time course. Primary thymocytes were treated with P/A for 5 min to achieve maximum Bcl11b phosphorylation or 60 min to induce dephosphorylation and sumoylation of Bcl11b as indicated. Bcl11b was immunoprecipitated, and the immunoprecipitates were probed with anti-phospho-Thr/Ser (anti-pT/S) (upper blot) or anti-SUMO1 (middle blot) antibody, and the blot was then stripped and probed with an anti-Bcl11b antibody (lower blot). G, native SENP1 preferentially interacts with phosphorylated Bcl11b in primary thymocytes. Primary thymocytes were treated with P/A for 5 min to achieve maximum Bcl11b phosphorylation or 60 min to induce dephosphorylation of Bcl11b as indicated. H, mutation of Bcl11b phosphorylation sites decreases the affinity of SENP1 for the Bcl11b complex in transfected HEK cells. HEK293T cells were co-transfected with expression vectors encoding FLAG-tagged SENP1 (F-SENP1) and increasing amounts of FLAG-tagged WT or phosphomutant Bcl11b pMT as indicated. Protein levels of SENP1 and Bcl11b were quantified as described under “Experimental Procedures.” The graph reflects relative protein levels of immunoprecipitated Bcl11b (x axis) and co-immunoprecipitated SENP1 (y axis) at each level of Bcl11b. Asymptotic levels of WT Bcl11b and co-immunoprecipitated SENP1 were arbitrarily set as 100%. Each data point and error bar represents the mean ± S.E., respectively, of three independent determinations. The curves shown were derived from fitting data using a non-linear routine and parameters described in Equation 1. The fitted K values from the theoretical curves are 49.6 ± 6.4 and 84.2 ± 9.8 arbitrary units for wild-type Bcl11b and Bcl11b pMT, respectively, and the difference between these parameter estimates is statistically significant (p < 0.05 level; n = 3).
Next, we studied the influence of phosphorylation on the interaction between Bcl11b and SENP1. For these studies, we used a specific phosphatase, the PP2A family member PP6 (51, 56), to modulate the phosphorylation status of Bcl11b. We identified PP6 as a component of the Bcl11b complex from a proteomics screen of mouse thymocytes.7 Basal phosphorylation of Bcl11b was almost completely eliminated when cells were co-transfected with expression vectors encoding Bcl11b and PP6 (Fig. 5D, upper blot, compare lanes 3 and 4). Bcl11b, when dephosphorylated by PP6, failed to interact with SENP1 (Fig. 5E, lower blot, compare lanes 2 and 4), and SUMO1-Bcl11b could not be desumoylated by SENP1 under these conditions (Fig. 5E, upper blot, compare lanes 2 and 4). These findings are indicative of a physical lack of SENP activity in the Bcl11b complex upon Bcl11b dephosphorylation. Therefore, phosphorylation of Bcl11b appears to enhance its interaction with SENP1. To confirm this finding in a native context, we performed co-IP experiments using primary mouse thymocytes that had been treated with P/A for 5 and 60 min to achieve Bcl11b phosphorylation and dephosphorylation, respectively (Fig. 5F; see also Fig. 3A). Bcl11b co-immunoprecipitated with SENP1 weakly under basal conditions (Fig. 5G, lane 5), and this interaction was enhanced by Bcl11b phosphorylation after 5 min of P/A treatment (lane 6). The amount of Bcl11b co-immunoprecipitating with SENP1 returned to low (basal) levels upon dephosphorylation of Bcl11b after 60 min of P/A treatment (Fig. 5G, lane 7). These findings indicate that the phosphorylation status of Bcl11b influences the interaction of SENP1 with the Bcl11b complex.
Bcl11b is phosphorylated at 23 serine/threonine residues in native thymocytes after stimulation with P/A (Fig. 4B and supplemental Figs. S1 and S2). Mutation of a cluster of five phosphoacceptor sites at the core of the protein (Bcl11b pMT; mutation of Thr-260/Ser-277/Ser-318/Ser-381/Ser-664 collectively to alanine) reduced the affinity of SENP1 for the phospho-Bcl11b complex in transfected HEK293T cells by nearly 2-fold (1.7 ± 0.1-fold; see Fig. 5H), suggesting that these five phosphosites contribute to the SENP1 interaction interface of Bcl11b. We have been unsuccessful in further narrowing the specific phosphosites that confer interaction with SENP1 or SENP2.
The Id2 Promoter: a Model for Integrating Bcl11b PTM with Transcriptional Output
Bcl11b represses expression of the Id2 gene in CD4+CD8+ (DP) thymocytes and interacts with two regions of the Id2 promoter, one site at ∼3 kb upstream of the transcriptional start site (TSS) and a second at the TSS (24). As sumoylation is often associated with transcriptional repression (11, 57, 58), we assessed the ability of SUMO site mutants of Bcl11b to repress the Id2 promoter. A fragment encompassing ∼2.9 kb of the mouse Id2 promoter (see Fig. 6A) was sufficient to recapitulate repression of this promoter mediated by wild-type Bcl11b (Fig. 6B) in the absence of co-transfection of expression vectors encoding SUMO proteins. Surprisingly, the transcriptional repressive activity of Bcl11b on the Id2 promoter was not compromised by single or compound mutation of the two sites of Bcl11b sumoylation (Fig. 6B), indicating that sumoylation does not play a role in Bcl11b-mediated repression in this context. This finding was expected because Bcl11b is not efficiently sumoylated in HEK293T cells in the absence of co-transfected SUMO proteins.
FIGURE 6.
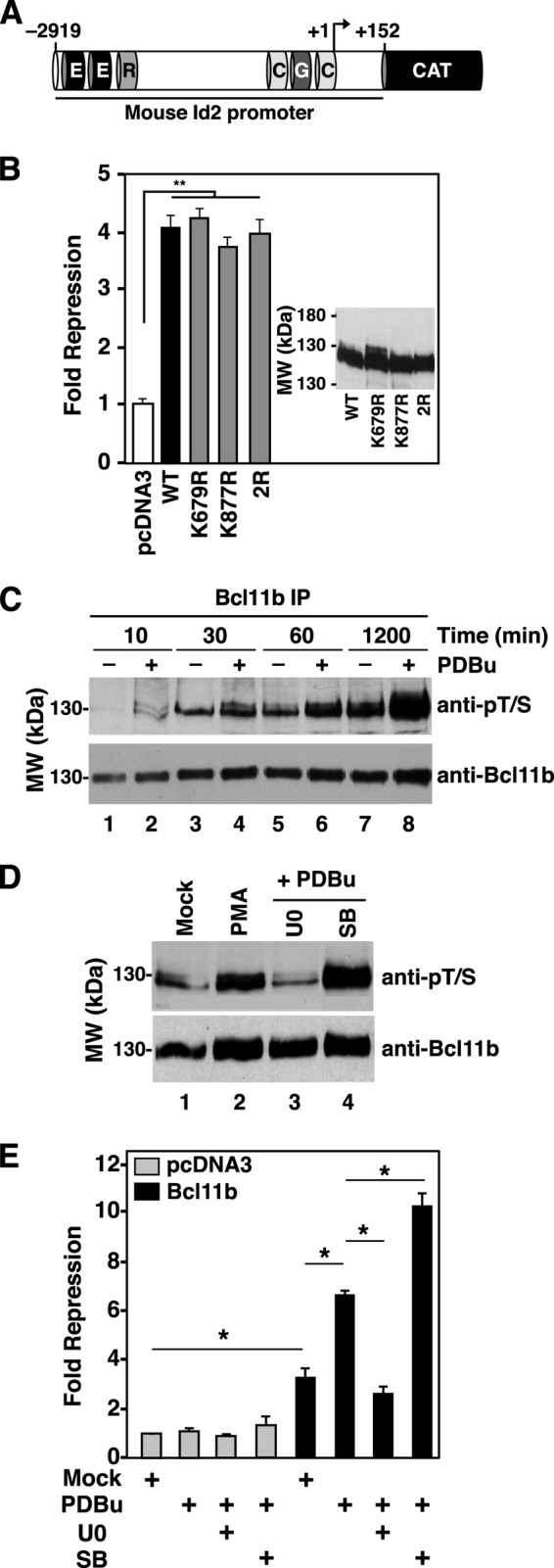
Effect of sumoylation site mutations and phosphorylation on Bcl11b-mediated transcriptional repressive activity. A, diagram of CAT reporter construct harboring the mouse Id2 promoter. An ∼3-kb fragment of the mouse Id2 promoter from −2919 bp upstream to +152 bp downstream of the TSS was cloned to a promoterless CAT reporter plasmid. Boxes represent putative binding sites for the following transcription factors: E box factors (E), RFX1 (R), GATA1 (G), and CdxA (C). B, expression vectors encoding wild-type Bcl11b, the sumoylation-deficient mutants K679R and K877R, or the double mutant K679R/K877R (400 ng of each) were co-transfected into HEK293T cells with the Id2-CAT reporter (see A) as indicated. Relative CAT expression was measured as described previously (53). Note that this experiment was conducted in the absence of a co-transfected expression vector encoding a SUMO protein. C, time course of PDBu-induced phosphorylation of Bcl11b in transfected HEK293T cells. Cultured cells were serum-starved overnight prior to addition of serum with and without PDBu (100 nm) for the indicated times. Note that addition of serum without PDBu also induced Bcl11b phosphorylation (odd-numbered lanes) that differed from PDBu-induced phosphorylation (even-numbered lanes) in the extent of Bcl11b phosphorylation. D, sensitivity of PDBu (100 nm)-induced phosphorylation of Bcl11b to inhibition by U0126 (U0; 10 μm) and SB202190 (SB; 10 μm). Cells were transfected with an expression vector encoding Bcl11b (1 μg) and treated with PDBu with or without the indicated inhibitors for 4 h. E, effect of PDBu treatment with and without inhibitors U0126 and SB202190 on Bcl11b-mediated repression of the Id2 promoter. Cells were transfected with a Bcl11b expression vector or pcDNA3 (400 ng each). All treatments were conducted for 24 h prior to harvesting and processing for reporter gene assays as described (53). Bars and error bars in panels B and E represent the average ± S.E., respectively, of at least three independent experiments. Statistical significance between the indicated pairs was determined using the Student's t test (*, p ≤ 0.05; **, p ≤ 0.01; n > 3 in all cases). anti-pT/S, anti-phospho-Thr/Ser.
The effect of phosphorylation on the transcriptional repressive activity of Bcl11b was also studied in transfected HEK293T cells using the Id2 promoter reporter. HEK293T cells differ from thymocytes in that the phospho-Bcl11b dephosphorylation reaction and therefore the resumoylation reaction are both inefficient. Thus, the phosphorylated state of Bcl11b in these cells is greatly prolonged after PDBu treatment, facilitating study of the functional consequences of this PTM.
PDBu induced rapid phosphorylation of transfected Bcl11b in HEK293T cells (Fig. 6, C and D) that was entirely reversed by co-treatment with the inhibitor of the Erk1/2 pathway, U0126 (Fig. 6D, compare lanes 1–3). However, unlike findings that utilized thymocytes, the p38 inhibitor SB202190 did not reduce but rather potentiated PDBu-induced phosphorylation of Bcl11b (Fig. 6D). These findings demonstrate that PDBu-induced phosphorylation of Bcl11b occurs downstream of the Erk1/2 pathway in HEK293T cells.
Treatment of transfected HEK293T cells with PDBu also stimulated Bcl11b-mediated repression of the Id2 promoter (Fig. 6E). Consistent with induced phosphorylation studies (Fig. 6C), stimulation of Bcl11b-mediated repression by PDBu was completely reversed by co-treatment with U0126 but stimulated by treatment with SB202190 (Fig. 6E). These findings demonstrate that Erk1/2-mediated phosphorylation of Bcl11b stimulates its repressive activity on the Id2 promoter, but sumoylation of Bcl11b at Lys-679 and/or Lys-877 is entirely dispensable for this repressive activity.
Bcl11b Interacts with the Transcriptional Co-activator p300 in a Sumoylation-dependent Manner and Is Necessary for Induction of Id2 Promoter by P/A
The lack of effect of SUMO site deletions on Bcl11b repressor activity prompted us to investigate whether P/A-induced sumoylation was instead promoting a change in Bcl11b transcriptional regulatory activity. Accordingly, treatment of primary thymocytes with P/A resulted in induction of the Id2 gene beginning at around 30 min after initiation of treatment and reaching a maximum of ∼4.5-fold at 60 min (Fig. 7A). Induction of the Id2 promoter by P/A was blocked by an inhibitor of the Erk1/2 pathway (U0126) but not by a p38 inhibitor (SB202190; Fig. 7B). Given that this time course and sensitivity to kinase inhibitors corresponded precisely with that of P/A-induced sumoylation of Bcl11b (Figs. 1, A–C, and 3, A and C) and that Id2 is repressed by Bcl11b under non-stimulated conditions (Fig. 6; see also Ref. 24), we hypothesized that sumoylation may negatively impact DNA binding by and/or the transcriptional repressive activity of Bcl11b, thus allowing for induction of the Id2 gene by P/A stimulation.
FIGURE 7.

Sumoylated Bcl11b interacts with the transcriptional co-activator p300. A, time course of Id2 induction in thymocytes by P/A treatment. Primary thymocytes were stimulated with P/A for the times indicated prior to RT-qPCR analyses of Id2 expression. Fold change in Id2 mRNA levels was calculated relative to the expression of a housekeeping gene, Gapdh. B, U0126 (U0), but not SB202190 (SB), inhibited the induction of the Id2 promoter by P/A treatment. C, effect of P/A treatment on interaction of Bcl11b and cofactors with Id2 promoter. Thymocytes were stimulated with P/A for the indicated times, and cells were subjected to ChIP analyses using the indicated antibodies or IgG. Chromatin was analyzed by qPCR using primers covering the −3-kb region of the Id2 promoter (left) that harbors a Bcl11b binding site, the TSS (middle), or 3′-intergenic region (IGR) (right). Points represent the relative ratio to input for each ChIP. D and E, P/A treatment promotes interaction of p300 with the Bcl11b complex. Primary thymocytes were treated with P/A for 60 min followed by reciprocal immunoprecipitations and immunoblotting as indicated. F, mutation of Bcl11b sumoylation sites inhibits interaction of p300 with the Bcl11b complex. HEK293T cells were transfected with 1 μg of expression vectors encoding Bcl11b (WT) or the double SUMO site deletion mutant of Bcl11b (2R) as indicated. Immunoprecipitation and immunoblotting were conducted as indicated. G, Bcl11b and p300 co-occupy Id2 promoter in thymocytes after P/A treatment. Primary thymocytes were stimulated with P/A for 60 min prior to re-ChIP analyses using anti-Bcl11b (first antibody) and anti-p300 or control IgG (second antibody). Immunoprecipitated DNA was analyzed by qPCR as indicated in C. Bars and error bars in panels A, B, and G represent the average ± S.E., respectively, of at least three independent experiments. H, induction of the Id2 promoter by P/A requires Bcl11b. P/A treatment of thymocytes was conducted, and Id2 transcripts were analyzed by RT-qPCR with relative quantification. All treatment values for each mouse are expressed relative to basal Id2 expression in thymocytes from the same mouse. Basal expression of Id2 was arbitrarily set to unity in all cases. The thymocytes used were from mice in which both alleles of the Bcl11b locus was floxed (Bcl11bL2/L2) or excised at the DP stage (Bcl11bdp−/−) (24). Bars and error bars in panel H represent the level of Id2 transcripts and S.E., respectively, in individual mice of the indicated genotype. Statistical significance throughout the figure was determined using Student's t test (*, p < 0.05; ***, p < 0.001). CalA, calyculin A.
The influence of P/A treatment on Bcl11b interaction with the Id2 locus was interrogated by time-dependent ChIP. A Bcl11b ChIP signal was detected at both the −3 kb site and TSS but not at the 3′-intergenic region (Fig. 7C) of Id2 as reported previously (24). P/A treatment dramatically stimulated Bcl11b interaction with the Id2 promoter, and this was most evident 60 min after initiation of treatment and at the TSS (Fig. 7C, middle panel). This finding is inconsistent with the hypothesis that P/A treatment and subsequent sumoylation impair the DNA binding activity of Bcl11b. Thus, we considered the possibility that sumoylated Bcl11b may play an active role in the induction of the Id2 promoter by P/A, and this idea gained additional credence with our finding that Bcl11b, particularly sumoylated Bcl11b, reciprocally co-immunoprecipitated with the transcriptional coactivator protein p300 in a manner that was stimulated by treatment of primary thymocytes with P/A (Fig. 7, D and E). P/A treatment did not influence the interaction of Bcl11b with the core NuRD component proteins MTA2 and HDAC1 (Fig. 7, D and E, lower panels; see also proteomics data in supplemental Table S2). P/A-stimulated interaction of p300 with the Bcl11b complex required sumoylation of Bcl11b as mutation of both SUMO acceptor sites of Bcl11b destroyed interaction of p300 with the Bcl11b complex (Fig. 7F, upper panel, compare lanes 1 and 2 with lanes 3 and 4). Moreover, hyperphosphorylation of wild-type (WT) Bcl11b, which was induced by treatment with the phosphatase inhibitor calyculin A (see Fig. 3D), blocked interaction of p300 with the Bcl11b complex (Fig. 7F, upper panel, compare lanes 1 and 2) presumably by facilitated recruitment of a SENP protein(s) to the Bcl11b complex followed by rapid desumoylation of Bcl11b.
The histone acetyltransferase p300 appeared to be recruited to both the −3 kb region and the TSS of the Id2 promoter by P/A treatment in a manner that mirrored that of Bcl11b and MTA2 (Fig. 7C). This was confirmed by re-ChIP studies in which p300 and Bcl11b were found to co-occupy both the −3 kb site and the TSS of the Id2 promoter in a manner that was strongly stimulated by P/A treatment (Fig. 7G). ChIP signals were also observed for acetylated histones H3 and H4 at the TSS, consistent with the recruitment of a histone acetyltransferase, such as p300, and the activated state of this promoter (Fig. 7C, middle panel).
The above findings suggest that Bcl11b may function as a bimodal regulator of Id2 expression in native thymocytes and that sumoylation of Bcl11b could play a key role in switching Bcl11b from a repressor to an activator of Id2 expression. If true, then loss of Bcl11b should compromise the induction of the Id2 promoter by P/A, and we assessed this by using thymocytes specifically lacking Bcl11b at the DP stage (Bcl11bdp−/− mice; Ref. 24). Induction of the Id2 promoter by P/A was nearly entirely abrogated in the absence of Bcl11b (Fig. 7H), supporting a central role for Bcl11b in the activation of Id2 expression, which occurs downstream of Erk1/2 activation in thymocytes (Fig. 7B).
Fusion to SUMO1 Abrogates Bcl11b-mediated Repression
The above data indicated that sumoylated Bcl11b interacted with p300 on the Id2 promoter and played a key role in the induction of the promoter by cell signaling pathways. To determine whether sumoylated Bcl11b may function as a transcriptional activator of the Id2 promoter, we fused SUMO1 to the amino terminus of Bcl11b and tested this on a reporter construct harboring a fragment of the Id2 promoter (see Fig. 6A). Although SUMO1-Bcl11b did not overtly activate this Id2 promoter construct in HEK293T cells, fusion to SUMO1 did reduce the transcriptional repressive activity of Bcl11b by ∼2-fold (Fig. 8A, upper panel) without influencing expression levels (Fig. 8A, lower panel) or the nuclear/subnuclear distribution of the Bcl11b (Fig. 8B). Moreover, co-expression of SUMO1-Bcl11b attenuated repression of the Id2 promoter by wild-type Bcl11b (Fig. 8C) possibly by heterocomplex formation as revealed by co-IP analyses (Fig. 8D, upper panel). Similar data were obtained for a SUMO2-Bcl11b fusion protein (Fig. 8E).
FIGURE 8.

Fusion to SUMO1 and SUMO2 abrogates Bcl11b-mediated repression. A, fusion to SUMO1 abrogates Bcl11b-mediated repression. HEK293T cells were co-transfected with the Id2-CAT reporter construct and 200, 400, or 800 ng of expression vectors encoding wild-type Bcl11b or the HA-SUMO1-Bcl11b (HA-SU1-Bcl11b) fusion construct as indicated. Similar levels of Bcl11b and SUMO1-Bcl11b protein were expressed as shown in the immunoblot below the figure. B, immunocytochemical nuclear/subnuclear distribution of Bcl11b and SU1-Bcl11b. Fusion to SUMO1 did not influence expression levels or the nuclear/subnuclear distribution of Bcl11b. HEK293T cells were transfected with either Bcl11b or SU1-Bcl11b as indicated, and then cells were fixed and subjected to immunocytochemistry analysis using anti-Bcl11b antibody (green). The nuclei were counterstained with DAPI. The white size bar in the lower left panel corresponds to 5 μm. C, co-transfection of HA-SU1-Bcl11b impairs the transcriptional repression activity of wild-type Bcl11b on the Id2 promoter. The amounts of transfected expression vectors encoding Bcl11b and HA-SU1-Bcl11b (200 or 400 ng) are indicated below. D, SUMO1-Bcl11b interacts with wild-type Bcl11b. Transfections, immunoprecipitations, and immunoblotting were conducted as indicated. The amounts of transfected expression vector encoding Bcl11b and HA-SU1-Bcl11b (200 or 400 ng) are indicated above the immunoblot. E, fusion to SUMO2 abrogates Bcl11b-mediated repression. The amounts of transfected expression vectors encoding Bcl11b and HA-SU2-Bcl11b (200 or 400 ng) are indicated below the figure. Experiments were conducted as described in A. Similar levels of Bcl11b and the HA-SU2-Bcl11b fusion protein were expressed as shown in the blot below the figure. F, SENP1-mediated desumoylation enhances Bcl11b-mediated repression. Expression vectors encoding wild-type Bcl11b or the SUMO-deficient mutant 2R were co-transfected (400 ng of each) with those encoding HA-SUMO1 (800 ng) and the Id2-CAT reporter plasmid (2 μg) with or without a co-transfected expression vector encoding HA-SENP1 (800 ng) as indicated. Note that the expression vector for HA-SUMO1 was co-transfected in all lanes to promote maximum sumoylation of Bcl11b. Bars and error bars in panels A, C, E, and F represent mean relative -fold repression values ±S.E., respectively, (n = 3). Statistical significance throughout the figure was determined using Student's t test (*, p < 0.05; **, p < 0.01).
We have shown that SUMO1- and SUMO2-Bcl11b can be desumoylated by SENP1. Therefore, we next determined whether desumoylation of Bcl11b influenced Bcl11b-mediated repression. An expression vector encoding HA-SUMO1 was co-transfected with the Bcl11b expression vector in these studies to promote maximum sumoylation of Bcl11b. Desumoylation of HA-SUMO1-Bcl11b by SENP1 enhanced Bcl11b-mediated repression of the Id2 promoter (Fig. 8F). In contrast, the transcriptional repressive activity of the double SUMO acceptor site mutant (Bcl11b 2R) was not significantly affected by co-transfection with the SENP1 expression vector (Fig. 8F) presumably because the 2R mutant is not sumoylated (see Fig. 2B).
Collectively, these findings indicate that sumoylation compromised the transcriptional repressive activity of Bcl11b on the Id2 promoter, which may allow for induction of the Id2 promoter downstream of cell signaling pathways including T-cell receptor activation. Sumoylation of Bcl11b resulted in recruitment of the transcriptional coactivator p300 to the Bcl11b complex present on the Id2 promoter, further supporting a role for sumoylated Bcl11b in transcriptional induction of the Id2 gene in response to P/A treatment.
Targeting Sumoylated Bcl11b for Degradation
Sumoylated Bcl11b appeared to be relatively stable and functionally involved in promoting Id2 mRNA induction after P/A treatment. However, prolonged treatment of mouse thymocytes with P/A resulted in degradation of Bcl11b as demonstrated by the appearance of proteolytic cleavage products that were evident after 4 h of P/A treatment (Fig. 9A, lanes 4 and 6). Degradation of Bcl11b was accompanied by Bcl11b ubiquitination, which was undetectable after 3 h of P/A treatment but was extensive by 6 h after initiation of P/A treatment (Fig. 9B).
FIGURE 9.

Prolonged activation of thymocytes results in ubiquitination and degradation of Bcl11b. A, degradation of Bcl11b. Thymocytes were stimulated with P/A as indicated prior to immunoblotting using the anti-Bcl11b and anti-HDAC1 (loading control) antibodies. B, time course of Bcl11b ubiquitination in stimulated thymocytes. Time of P/A treatment, immunoprecipitation, and immunoblotting were as indicated. The immunoprecipitates were probed with anti-ubiquitin (Ub) (upper blot), and the blot was then stripped and probed with an anti-Bcl11b antibody (lower blot). Similar results were obtained in two to four additional independent experiments.
DISCUSSION
Our results provide the first evidence of post-translational modification of Bcl11b by signal transduction pathways leading to an alteration in the transcriptional regulatory activity of the protein on a natural target gene in primary cells. As Bcl11b is absolutely required for T-cell development at critical stages of commitment and positive selection (at a minimum) and is implicated in development of T-cell acute lymphoblastic leukemia, elucidation of the pathways and consequences of regulating Bcl11b has important implications for understanding both immune system development and leukemogenesis. This work defines a central regulatory pathway linking stimulation of thymocytes through MAP kinase activation to Bcl11b-dependent regulation of gene expression in T-cell development.
Stimulation of thymocytes with P/A to mimic TCR-dependent thymocyte selection resulted in sequential and linked PTMs of Bcl11b, namely phosphorylation, desumoylation, dephosphorylation, resumoylation, and ubiquitination. Within the context of a model target gene, Id2, the net effect of these PTMs appears to be the conversion of Bcl11b from a potent repressor of transcription (in the unmodified or phosphorylated state) to a dramatically weakened repressor (sumoylated state) that participates in the P/A-induced activation of Id2 expression and ultimately to a degraded protein (ubiquitinated state).
We directly identified 23 sites of phosphorylation and one site of sumoylation (modified by SUMO1 and SUMO2/3) on Bcl11b as natively expressed in mouse thymocytes. We identified a second sumoylation site by site-directed mutagenesis. Composite phosphorylation and sumoylation were dynamically regulated by stimulation of native thymocytes with P/A. Moreover, sumoylation at both SUMO acceptor sites was inversely related to the global phosphorylation status of Bcl11b, forming the basis of a phospho-deSUMO switch within the protein.
Direct Identification of a Sumoylation Site in a Native System
Our identification of Bcl11b sumoylation represents a unique instance in which the site of SUMO adduction has been directly identified in a native mammalian tissue. Such direct identification of PTMs in natively sourced tissue is desirable to avoid artifacts that may occur in overexpressed and non-native, exogenous cell systems.
Although mass spectrometry (MS) has been used extensively to identify sites of small mass PTMs, such as phosphorylation, direct analysis of sumoylation sites is more difficult. Sumoylation results in the adduction of a large polypeptide to the ϵ-amino group of acceptor lysines, creating branched chain proteins. The resulting tandem mass spectra contain fragment ions originating from both the parent and SUMO adduct peptides, complicating its interpretation and precluding the use of standard, proteomics database-searching techniques. Further complicating direct analysis of sumoylation is the low stoichiometry of these highly dynamic modifications and the lability of the SUMO moiety (53). Because of these difficulties, mammalian sumoylation site analyses have relied on inferential techniques (e.g. modified SUMO adducts and overexpression of the target substrate, SUMO adduct proteins, and/or conjugating enzyme) in exogenous expression systems.
Although direct analysis is preferred, our lack of observation of SUMO adduction at Lys-877 of Bcl11b in native thymocytes is perhaps an example of the limitation of SUMO site identification by MS. Unlike Lys-679, the predicted trypsin-cleaved SUMO adduct at Lys-877 would be too large to identify by methods used in this study. Nevertheless, Lys-877 is clearly sumoylated in HEK293T cells (Fig. 2C) and appears to be sumoylated in thymocytes in response to P/A treatment (see Fig. 1).
The Bcl11b Phosphorylation-deSUMO Switch
Phosphorylation and sumoylation of Bcl11b appeared to be mutually exclusive processes, implying the existence of a phospho-deSUMO switch within the protein. At the heart of this switch is the phosphorylation-dependent recruitment of SENP proteins to the Bcl11b complex, which precedes rapid hydrolysis of SUMO-Bcl11b. Both SENP1 and SENP2 interacted with the Bcl11b complex and catalyzed the desumoylation of Bcl11b. The interaction of SENP1 with the Bcl11b complex was studied most extensively, and we found that this interaction was independent of the sumoylation status of Bcl11b. This finding was unexpected as interactions between SENPs and most substrates have been reported to be transient because of the SUMO-dependent nature of the interaction of these enzymes with substrates and the high catalytic activity of SENP proteins, which autoterminates SENP-substrate interactions (59). In contrast, interaction of SENP1 with Bcl11b does not appear to be autoterminated by the enzymatic activity of the SENP protein, which likely prolongs the lifetime of the SENP-Bcl11b interaction and the unsumoylated state of Bcl11b following hyperphosphorylation.
PP6, a Bcl11b Phosphatase
The induced, hyperphosphorylated state of Bcl11b was transient, persisting for around 10–15 min after P/A stimulation and prior to profound dephosphorylation. The latter was catalyzed by PP6, a newly identified Bcl11b phosphatase, and perhaps by other unidentified phosphatases. The Bcl11b phosphatase(s) appeared to be activated by Erk1/2, but the time course of activation and/or onset of catalytic activity toward phospho-Bcl11b was slightly delayed relative to phosphorylation of Bcl11b. This delay served to define the lifetime of hyperphosphorylated Bcl11b.
Why does the MAPK pathway phosphorylate Bcl11b so robustly only to have it extensively dephosphorylated minutes later? We speculate that the answer lies in creating a bigger “delta” between the basal and stimulated states of expression of the Id2 gene and possibly other genes that are repressed by Bcl11b in the basal state. Erk1/2-dependent phosphorylation stimulates the transcriptional repressive activity of Bcl11b on the Id2 promoter in HEK293T cells (Fig. 6E) and we presume in thymocytes. Within the latter, this phosphorylation is followed by extensive dephosphorylation of the phospho-Bcl11b pool, which becomes evident 20–30 min after stimulation. Dephosphorylation of Bcl11b may have at least two effects: 1) attenuation of enhanced Bcl11b-mediated repression and 2) dismissal of SENP proteins from the Bcl11b complex. The latter would facilitate stable resumoylation of Bcl11b, which is required for recruitment of p300 to the Bcl11b complex and derepression of the Id2 promoter in response to P/A treatment and presumably T-cell receptor activation. We do not presently know whether our results using the Id2 promoter can be generalized to the other genes that are repressed by Bcl11b in DP cells under basal conditions (24). However, the scenario described above may allow Bcl11b to “sense” the intensity and duration of stimulation of TCR by MHC peptides, thus contributing to the threshold that must be surmounted for DP cells to initiate the CD4 and CD8 SP differentiation programs during positive selection.
Sumoylation Attenuates the Transcriptional Repressive Activity of Bcl11b
Protein sumoylation most commonly leads to loss of transcriptional activation potential or enhancement of the repressive activity of transcription factors. The latter is generally mediated through recruitment of repressors by direct protein-protein interaction with the sumoylated protein on target promoters (60). For example, sumoylation of Elk1 (11, 61), androgen receptor (62), MafG (63), and KAP1 (64) promotes association of these factors with HDACs, leading to gene silencing. However, the situation with Bcl11b is rather different. Sumoylation compromised the transcriptional repressive activity of Bcl11b most likely by promoting recruitment of histone acetyltransferase p300 to the Bcl11b-NuRD complex and to the Id2 promoter, contributing to acetylation of histones associated with the transcriptional start site and activation of transcription. These data indicate that the effect of sumoylation on the activity of transcriptional regulatory proteins is substrate- and/or context-dependent. In this regard, Bcl11b is most similar to Ikaros (65), NFAT1 (66), Oct4 (67), and JunB (68) for which sumoylation is associated with dampened transcriptional repressive activity and enhanced activation potential.
We identified two sites of sumoylation within Bcl11b (Lys-679 and Lys-877); however, SUMO chains were observed at only one of these sites (Lys-679), and these chains were composed of both SUMO1 and SUMO2/3. The functional significance of poly-SUMO chains on Bcl11b Lys-679 is presently unknown. However, poly-SUMO chains have higher affinity for proteins harboring multiplexed SUMO-interacting motifs (69, 70). Thus, formation of poly-SUMO chains on Bcl11b Lys-679 may serve to restrict the pool of cellular proteins capable of interaction with this form of the protein.
Integration of Bcl11b into Cell Signaling Pathways in Thymocytes
A summary of all data presented herein describing the role of sequential, signaling-dependent PTMs in regulating the transcriptional activity of Bcl11b in developing thymocytes is shown in Fig. 10. The principal components of this summary are as follows. 1) Bcl11b exists in an ensemble of PTM states in the basal state, phosphorylated, sumoylated, and neither phosphorylated nor sumoylated, and in all of these contexts Bcl11b associates with the NuRD complex. However, we have no evidence that NuRD component proteins become modified as a consequence of P/A treatment or play a role in induced Bcl11b PTMs. The net transcriptional outcome of interaction of the Bcl11b-NuRD complex with the Id2 promoter under basal conditions is repression. 2) Upon cell stimulation, either by TCR or growth factor receptor activation, Bcl11b becomes rapidly phosphorylated by the MAPK pathways, and this drives collapse of all three states into a single, highly phosphorylated state. 3) Phospho-Bcl11b recruits SENP proteins to the Bcl11b complex, resulting in rapid hydrolysis of SUMO-Bcl11b within this complex. 4) In parallel with step 2, PP6 and/or other Bcl11b phosphatases become phosphorylated and activated by Erk1/2. 5) Activated PP6 dephosphorylates phospho-Bcl11b, causing dismissal of SENP1 from the Bcl11b complex. 6) Bcl11b is then conjugated to SUMO1 or SUMO2/3 by the E2 SUMO ligase Ubc9. 7) SUMO-Bcl11b recruits the histone acetyltransferase/transcriptional coactivator p300 to the Bcl11b complex on target promoters. 8) p300 acetylates promoter-associated histones, leading to activation of the Id2 promoter. 9) Prolonged cellular stimulation results in ubiquitination and degradation of Bcl11b.
FIGURE 10.

Coordinated regulation of Bcl11b transcriptional activity by reversible phosphorylation and sumoylation in thymocytes. This figure summarizes the relevant findings presented herein. Note that SENPx refers to either SENP1 or SENP2 and perhaps other SENP family members. The term “kinase” refers to Erk1/2 and p38, but other kinases may be implicated in phosphorylation of Bcl11b. Similarly, PPTase refers to the phosphatase PP6, but other phosphatases may be involved in the dephosphorylation of phospho-Bcl11b. Time domains are represented in italicized text, and signaling components between TCR and the MAP kinases Erk1/2 and p38 were omitted for clarity. See discussion for further details of this summary. p-p38, phospho-p38; pErk1/2, phospho-Erk1/2; pPPTase, phosphorylated phosphoprotein phosphatase.
Potential Role of the Bcl11b PTMs in Positive Selection
Bcl11b is essential for T-cell development, playing an essential role in early progenitors at the TCR-independent commitment stage (25, 26) and in DP cells at the TCR-dependent stage of positive selection (24, 32). Interestingly, many of the genes that are repressed by Bcl11b in DP cells are highly expressed in differentiated SP cells (e.g. Zbtb7b and Runx3 in CD4+ and CD8+ cells, respectively) even though Bcl11b protein levels are comparable in DP and SP cells (24). The results presented herein provide a potential mechanistic explanation for the reversal of Bcl11b-mediated repression of gene expression during thymocyte maturation. This pathway appears to facilitate derepression of repressed Bcl11b target genes as DP cells receive a TCR signal and initiate SP differentiation programs during positive selection. Thus, regulation of Bcl11b activity through the pathway described herein represents a novel strategy linking MAPK signaling with the latter stages of T-cell differentiation.
Acknowledgments
We thank David Brautigan for generously providing expression vectors encoding HA-PP6 subunits and anti-PP6 antibodies, Grace Gill for sharing mammalian expression vector encoding FLAG-SENP protein and bacterial expression vectors for GST-SUMO1 and -SUMO2, and Mark Zabriskie for encouragement and continuous support of this project. The Mass Spectrometry Facility at Oregon State University is supported by NIEHS, National Institutes of Health Center Grant ES000210 to the Environmental Health Sciences Center at Oregon State University.
This work was supported, in whole or in part, by National Institutes of Health Grant GM096243 (to T. M. F.). This work was also supported by an award from the Medical Research Foundation of Oregon (to M. L.) and funds provided by the Oregon State University College of Pharmacy.

This article contains supplemental Figs. S1 and S2 and Tables S1 and S2.
W. K. Vogel and M. Leid, manuscript in preparation.
- PTM
- post-translational modification
- SUMO
- small ubiquitin-like modifier
- PDBu
- phorbol 12,13-dibutyrate
- NuRD
- nucleosome remodeling and deacetylation
- DN
- double negative
- DP
- double positive
- SP
- single positive
- HDAC
- histone deacetylase
- TCR
- T-cell receptor
- MAP
- mitogen-activated protein
- IP
- immunoprecipitation
- P/A
- PDBu and A23187
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- FT-ICR
- Fourier transform-ion cyclotron mass analyzer
- LTQ
- linear ion trap mass analyzer
- 2R
- K679R/K877R
- PP6
- protein phosphatase 6
- CAT
- chloramphenicol acetyltransferase
- qPCR
- quantitative PCR
- TSS
- transcriptional start site
- SU
- SUMO.
REFERENCES
- 1. Deribe Y. L., Pawson T., Dikic I. (2010) Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666–672 [DOI] [PubMed] [Google Scholar]
- 2. Johnson L. N., Lewis R. J. (2001) Structural basis for control by phosphorylation. Chem. Rev. 101, 2209–2242 [DOI] [PubMed] [Google Scholar]
- 3. Narayanan A., Jacobson M. P. (2009) Computational studies of protein regulation by post-translational phosphorylation. Curr. Opin. Struct. Biol. 19, 156–163 [DOI] [PubMed] [Google Scholar]
- 4. Meulmeester E., Melchior F. (2008) Cell biology: SUMO. Nature 452, 709–711 [DOI] [PubMed] [Google Scholar]
- 5. Tempé D., Piechaczyk M., Bossis G. (2008) SUMO under stress. Biochem. Soc. Trans. 36, 874–878 [DOI] [PubMed] [Google Scholar]
- 6. Wilkinson K. A., Henley J. M. (2010) Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 428, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perry J. J., Tainer J. A., Boddy M. N. (2008) A SIM-ultaneous role for SUMO and ubiquitin. Trends Biochem. Sci. 33, 201–208 [DOI] [PubMed] [Google Scholar]
- 8. Hietakangas V., Anckar J., Blomster H. A., Fujimoto M., Palvimo J. J., Nakai A., Sistonen L. (2006) PDSM, a motif for phosphorylation-dependent SUMO modification. Proc. Natl. Acad. Sci. U.S.A. 103, 45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kang J., Gocke C. B., Yu H. (2006) Phosphorylation-facilitated sumoylation of MEF2C negatively regulates its transcriptional activity. BMC Biochem. 7, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tremblay A. M., Wilson B. J., Yang X. J., Giguère V. (2008) Phosphorylation-dependent sumoylation regulates estrogen-related receptor-α and -γ transcriptional activity through a synergy control motif. Mol. Endocrinol. 22, 570–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang S. H., Jaffray E., Hay R. T., Sharrocks A. D. (2003) Dynamic interplay of the SUMO and ERK pathways in regulating Elk-1 transcriptional activity. Mol. Cell 12, 63–74 [DOI] [PubMed] [Google Scholar]
- 12. Wu H., Sun L., Zhang Y., Chen Y., Shi B., Li R., Wang Y., Liang J., Fan D., Wu G., Wang D., Li S., Shang Y. (2006) Coordinated regulation of AIB1 transcriptional activity by sumoylation and phosphorylation. J. Biol. Chem. 281, 21848–21856 [DOI] [PubMed] [Google Scholar]
- 13. Li X., Lee Y. K., Jeng J. C., Yen Y., Schultz D. C., Shih H. M., Ann D. K. (2007) Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J. Biol. Chem. 282, 36177–36189 [DOI] [PubMed] [Google Scholar]
- 14. Gill G. (2004) SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 18, 2046–2059 [DOI] [PubMed] [Google Scholar]
- 15. Huang T. T., D'Andrea A. D. (2006) Regulation of DNA repair by ubiquitylation. Nat. Rev. Mol. Cell Biol. 7, 323–334 [DOI] [PubMed] [Google Scholar]
- 16. Prudden J., Pebernard S., Raffa G., Slavin D. A., Perry J. J., Tainer J. A., McGowan C. H., Boddy M. N. (2007) SUMO-targeted ubiquitin ligases in genome stability. EMBO J. 26, 4089–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lallemand-Breitenbach V., Jeanne M., Benhenda S., Nasr R., Lei M., Peres L., Zhou J., Zhu J., Raught B., de Thé H. (2008) Arsenic degrades PML or PML-RARα through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 10, 547–555 [DOI] [PubMed] [Google Scholar]
- 18. Leid M., Ishmael J. E., Avram D., Shepherd D., Fraulob V., Dollé P. (2004) CTIP1 and CTIP2 are differentially expressed during mouse embryogenesis. Gene Expr. Patterns 4, 733–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Golonzhka O., Leid M., Indra G., Indra A. K. (2007) Expression of COUP-TF-interacting protein 2 (CTIP2) in mouse skin during development and in adulthood. Gene Expr. Patterns 7, 754–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arlotta P., Molyneaux B. J., Jabaudon D., Yoshida Y., Macklis J. D. (2008) Ctip2 controls the differentiation of medium spiny neurons and the establishment of the cellular architecture of the striatum. J. Neurosci. 28, 622–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Enomoto T., Ohmoto M., Iwata T., Uno A., Saitou M., Yamaguchi T., Kominami R., Matsumoto I., Hirota J. (2011) Bcl11b/Ctip2 controls the differentiation of vomeronasal sensory neurons in mice. J. Neurosci. 31, 10159–10173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Golonzhka O., Liang X., Messaddeq N., Bornert J. M., Campbell A. L., Metzger D., Chambon P., Ganguli-Indra G., Leid M., Indra A. K. (2009) Dual role of COUP-TF-interacting protein 2 in epidermal homeostasis and permeability barrier formation. J. Invest. Dermatol. 129, 1459–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Golonzhka O., Metzger D., Bornert J. M., Bay B. K., Gross M. K., Kioussi C., Leid M. (2009) Ctip2/Bcl11b controls ameloblast formation during mammalian odontogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 4278–4283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kastner P., Chan S., Vogel W. K., Zhang L. J., Topark-Ngarm A., Golonzhka O., Jost B., Le Gras S., Gross M. K., Leid M. (2010) Bcl11b represses a mature T-cell gene expression program in immature CD4+ CD8+ thymocytes. Eur. J. Immunol. 40, 2143–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li L., Leid M., Rothenberg E. V. (2010) An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science 329, 89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li P., Burke S., Wang J., Chen X., Ortiz M., Lee S. C., Lu D., Campos L., Goulding D., Ng B. L., Dougan G., Huntly B., Gottgens B., Jenkins N. A., Copeland N. G., Colucci F., Liu P. (2010) Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science 329, 85–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Przybylski G. K., Dik W. A., Wanzeck J., Grabarczyk P., Majunke S., Martin-Subero J. I., Siebert R., Dölken G., Ludwig W. D., Verhaaf B., van Dongen J. J., Schmidt C. A., Langerak A. W. (2005) Disruption of the BCL11B gene through inv(14)(q11.2q32.31) results in the expression of BCL11B-TRDC fusion transcripts and is associated with the absence of wild-type BCL11B transcripts in T-ALL. Leukemia 19, 201–208 [DOI] [PubMed] [Google Scholar]
- 28. Kamimura K., Mishima Y., Obata M., Endo T., Aoyagi Y., Kominami R. (2007) Lack of Bcl11b tumor suppressor results in vulnerability to DNA replication stress and damages. Oncogene 26, 5840–5850 [DOI] [PubMed] [Google Scholar]
- 29. Huang X., Chen S., Shen Q., Yang L., Li B., Zhong L., Geng S., Du X., Li Y. (2010) Analysis of the expression pattern of the BCL11B gene and its relatives in patients with T-cell acute lymphoblastic leukemia. J. Hematol. Oncol. 3, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. De Keersmaecker K., Real P. J., Gatta G. D., Palomero T., Sulis M. L., Tosello V., Van Vlierberghe P., Barnes K., Castillo M., Sole X., Hadler M., Lenz J., Aplan P. D., Kelliher M., Kee B. L., Pandolfi P. P., Kappes D., Gounari F., Petrie H., Van der Meulen J., Speleman F., Paietta E., Racevskis J., Wiernik P. H., Rowe J. M., Soulier J., Avran D., Cavé H., Dastugue N., Raimondi S., Meijerink J. P., Cordon-Cardo C., Califano A., Ferrando A. A. (2010) The TLX1 oncogene drives aneuploidy in T cell transformation. Nat. Med. 16, 1321–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wakabayashi Y., Watanabe H., Inoue J., Takeda N., Sakata J., Mishima Y., Hitomi J., Yamamoto T., Utsuyama M., Niwa O., Aizawa S., Kominami R. (2003) Bcl11b is required for differentiation and survival of αβ T lymphocytes. Nat. Immunol. 4, 533–539 [DOI] [PubMed] [Google Scholar]
- 32. Albu D. I., Feng D., Bhattacharya D., Jenkins N. A., Copeland N. G., Liu P., Avram D. (2007) BCL11B is required for positive selection and survival of double-positive thymocytes. J. Exp. Med. 204, 3003–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Topark-Ngarm A., Golonzhka O., Peterson V. J., Barrett B., Jr., Martinez B., Crofoot K., Filtz T. M., Leid M. (2006) CTIP2 associates with the NuRD complex on the promoter of p57KIP2, a newly identified CTIP2 target gene. J. Biol. Chem. 281, 32272–32283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cismasiu V. B., Adamo K., Gecewicz J., Duque J., Lin Q., Avram D. (2005) BCL11B functionally associates with the NuRD complex in T lymphocytes to repress targeted promoter. Oncogene 24, 6753–6764 [DOI] [PubMed] [Google Scholar]
- 35. Senawong T., Peterson V. J., Avram D., Shepherd D. M., Frye R. A., Minucci S., Leid M. (2003) Involvement of the histone deacetylase SIRT1 in chicken ovalbumin upstream promoter transcription factor (COUP-TF)-interacting protein 2-mediated transcriptional repression. J. Biol. Chem. 278, 43041–43050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Avram D., Fields A., Pretty On Top K., Nevrivy D. J., Ishmael J. E., Leid M. (2000) Isolation of a novel family of C2H2 zinc finger proteins implicated in transcriptional repression mediated by chicken ovalbumin upstream promoter transcription factor (COUP-TF) orphan nuclear receptors. J. Biol. Chem. 275, 10315–10322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rothenberg E. V., Taghon T. (2005) Molecular genetics of T cell development. Annu. Rev. Immunol. 23, 601–649 [DOI] [PubMed] [Google Scholar]
- 38. Bommhardt U., Basson M. A., Krummrei U., Zamoyska R. (1999) Activation of the extracellular signal-related kinase/mitogen-activated protein kinase pathway discriminates CD4 versus CD8 lineage commitment in the thymus. J. Immunol. 163, 715–722 [PubMed] [Google Scholar]
- 39. Crompton T., Gilmour K. C., Owen M. J. (1996) The MAP kinase pathway controls differentiation from double-negative to double-positive thymocyte. Cell 86, 243–251 [DOI] [PubMed] [Google Scholar]
- 40. Sharp L. L., Schwarz D. A., Bott C. M., Marshall C. J., Hedrick S. M. (1997) The influence of the MAPK pathway on T cell lineage commitment. Immunity 7, 609–618 [DOI] [PubMed] [Google Scholar]
- 41. Alberola-Ila J., Takaki S., Kerner J. D., Perlmutter R. M. (1997) Differential signaling by lymphocyte antigen receptors. Annu. Rev. Immunol. 15, 125–154 [DOI] [PubMed] [Google Scholar]
- 42. Priatel J. J., Teh S. J., Dower N. A., Stone J. C., Teh H. S. (2002) RasGRP1 transduces low-grade TCR signals which are critical for T cell development, homeostasis, and differentiation. Immunity 17, 617–627 [DOI] [PubMed] [Google Scholar]
- 43. Fischer A. M., Katayama C. D., Pagès G., Pouysségur J., Hedrick S. M. (2005) The role of erk1 and erk2 in multiple stages of T cell development. Immunity 23, 431–443 [DOI] [PubMed] [Google Scholar]
- 44. Ohoka Y., Kuwata T., Tozawa Y., Zhao Y., Mukai M., Motegi Y., Suzuki R., Yokoyama M., Iwata M. (1996) In vitro differentiation and commitment of CD4+ CD8+ thymocytes to the CD4 lineage, without TCR engagement. Int. Immunol. 8, 297–306 [DOI] [PubMed] [Google Scholar]
- 45. Takahama Y., Nakauchi H. (1996) Phorbol ester and calcium ionophore can replace TCR signals that induce positive selection of CD4 T cells. J. Immunol. 157, 1508–1513 [PubMed] [Google Scholar]
- 46. Nielsen M. L., Vermeulen M., Bonaldi T., Cox J., Moroder L., Mann M. (2008) Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat. Methods 5, 459–460 [DOI] [PubMed] [Google Scholar]
- 47. Olsen J. V., de Godoy L. M., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M. (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4, 2010–2021 [DOI] [PubMed] [Google Scholar]
- 48. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 49. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 50. Hsiao H. H., Meulmeester E., Frank B. T., Melchior F., Urlaub H. (2009) “ChopNSpice,” a mass spectrometric approach that allows identification of endogenous small ubiquitin-like modifier-conjugated peptides. Mol. Cell. Proteomics 8, 2664–2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kajino T., Ren H., Iemura S., Natsume T., Stefansson B., Brautigan D. L., Matsumoto K., Ninomiya-Tsuji J. (2006) Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J. Biol. Chem. 281, 39891–39896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang L. J., Liu X., Gafken P. R., Kioussi C., Leid M. (2009) A chicken ovalbumin upstream promoter transcription factor I (COUP-TFI) complex represses expression of the gene encoding tumor necrosis factor α-induced protein 8 (TNFAIP8). J. Biol. Chem. 284, 6156–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dowell P., Ishmael J. E., Avram D., Peterson V. J., Nevrivy D. J., Leid M. (1997) p300 functions as a coactivator for the peroxisome proliferator-activated receptor α. J. Biol. Chem. 272, 33435–33443 [DOI] [PubMed] [Google Scholar]
- 54. Bailey D., O'Hare P. (2004) Characterization of the localization and proteolytic activity of the SUMO-specific protease, SENP1. J. Biol. Chem. 279, 692–703 [DOI] [PubMed] [Google Scholar]
- 55. Drag M., Salvesen G. S. (2008) DeSUMOylating enzymes—SENPs. IUBMB Life 60, 734–742 [DOI] [PubMed] [Google Scholar]
- 56. Cohen P. T. (1997) Novel protein serine/threonine phosphatases: variety is the spice of life. Trends Biochem. Sci. 22, 245–251 [DOI] [PubMed] [Google Scholar]
- 57. Ross S., Best J. L., Zon L. I., Gill G. (2002) SUMO-1 modification represses Sp3 transcriptional activation and modulates its subnuclear localization. Mol. Cell 10, 831–842 [DOI] [PubMed] [Google Scholar]
- 58. Kuo H. Y., Chang C. C., Jeng J. C., Hu H. M., Lin D. Y., Maul G. G., Kwok R. P., Shih H. M. (2005) SUMO modification negatively modulates the transcriptional activity of CREB-binding protein via the recruitment of Daxx. Proc. Natl. Acad. Sci. U.S.A. 102, 16973–16978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Witty J., Aguilar-Martinez E., Sharrocks A. D. (2010) SENP1 participates in the dynamic regulation of Elk-1 SUMOylation. Biochem. J. 428, 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Valin A., Gill G. (2007) Regulation of the dual-function transcription factor Sp3 by SUMO. Biochem. Soc. Trans. 35, 1393–1396 [DOI] [PubMed] [Google Scholar]
- 61. Yang S. H., Sharrocks A. D. (2004) SUMO promotes HDAC-mediated transcriptional repression. Mol. Cell 13, 611–617 [DOI] [PubMed] [Google Scholar]
- 62. Yang Y., Tse A. K., Li P., Ma Q., Xiang S., Nicosia S. V., Seto E., Zhang X., Bai W. (2011) Inhibition of androgen receptor activity by histone deacetylase 4 through receptor SUMOylation. Oncogene 30, 2207–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Motohashi H., Katsuoka F., Miyoshi C., Uchimura Y., Saitoh H., Francastel C., Engel J. D., Yamamoto M. (2006) MafG sumoylation is required for active transcriptional repression. Mol. Cell. Biol. 26, 4652–4663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ivanov A. V., Peng H., Yurchenko V., Yap K. L., Negorev D. G., Schultz D. C., Psulkowski E., Fredericks W. J., White D. E., Maul G. G., Sadofsky M. J., Zhou M. M., Rauscher F. J., 3rd (2007) PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 28, 823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gómez-del Arco P., Koipally J., Georgopoulos K. (2005) Ikaros SUMOylation: switching out of repression. Mol. Cell. Biol. 25, 2688–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Terui Y., Saad N., Jia S., McKeon F., Yuan J. (2004) Dual role of sumoylation in the nuclear localization and transcriptional activation of NFAT1. J. Biol. Chem. 279, 28257–28265 [DOI] [PubMed] [Google Scholar]
- 67. Wei F., Schöler H. R., Atchison M. L. (2007) Sumoylation of Oct4 enhances its stability, DNA binding, and transactivation. J. Biol. Chem. 282, 21551–21560 [DOI] [PubMed] [Google Scholar]
- 68. Garaude J., Farrás R., Bossis G., Charni S., Piechaczyk M., Hipskind R. A., Villalba M. (2008) SUMOylation regulates the transcriptional activity of JunB in T lymphocytes. J. Immunol. 180, 5983–5990 [DOI] [PubMed] [Google Scholar]
- 69. Tatham M. H., Geoffroy M. C., Shen L., Plechanovova A., Hattersley N., Jaffray E. G., Palvimo J. J., Hay R. T. (2008) RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 10, 538–546 [DOI] [PubMed] [Google Scholar]
- 70. Li Y. J., Stark J. M., Chen D. J., Ann D. K., Chen Y. (2010) Role of SUMO:SIM-mediated protein-protein interaction in non-homologous end joining. Oncogene 29, 3509–3518 [DOI] [PMC free article] [PubMed] [Google Scholar]