

Background: Ion transport and initiation of death in bacteria is poorly understood.
Results: Both the human milk complex HAMLET and physiological death stimuli required sodium-dependent calcium influx and kinase activation to kill pneumococci and form optimal biofilms.
Conclusion: Specific ion transport during bacterial death initiation is essential.
Significance: The study provides novel information about ion transport in bacteria and its role in bacterial physiology.
Keywords: Bacterial Protein Kinases, Calcium Transport, Cell Death, Membrane Energetics, Sodium Calcium Exchange, Streptococcus pneumoniae, α-Lactalbumin, Ion Transport, Milk, Oleic Acid
Abstract
To cause colonization or infection, most bacteria grow in biofilms where differentiation and death of subpopulations is critical for optimal survival of the whole population. However, little is known about initiation of bacterial death under physiological conditions. Membrane depolarization has been suggested, but never shown to be involved, due to the difficulty of performing such studies in bacteria and the paucity of information that exists regarding ion transport mechanisms in prokaryotes. In this study, we performed the first extensive investigation of ion transport and membrane depolarization in a bacterial system. We found that HAMLET, a human milk protein-lipid complex, kills Streptococcus pneumoniae (the pneumococcus) in a manner that shares features with activation of physiological death from starvation. Addition of HAMLET to pneumococci dissipated membrane polarity, but depolarization per se was not enough to trigger death. Rather, both HAMLET- and starvation-induced death of pneumococci specifically required a sodium-dependent calcium influx, as shown using calcium and sodium transport inhibitors. This mechanism was verified under low sodium conditions, and in the presence of ionomycin or monensin, which enhanced pneumococcal sensitivity to HAMLET- and starvation-induced death. Pneumococcal death was also inhibited by kinase inhibitors, and indicated the involvement of Ser/Thr kinases in these processes. The importance of this activation mechanism was made evident, as dysregulation and manipulation of physiological death was detrimental to biofilm formation, a hallmark of bacterial colonization. Overall, our findings provide novel information on the role of ion transport during bacterial death, with the potential to uncover future antimicrobial targets.
Introduction
There is growing evidence that bacteria live in complex communities during colonization and infection (1–3). In analogy with multicellular eukaryotes, optimal function of such communities requires tight regulation of both bacterial growth and death. Although much is known about pathways of programmed cell death (PCD)2 in eukaryotic systems, little is understood about terminal differentiation and PCD in prokaryotes and how they may be related.
Two of the best-described bacterial PCD systems are the toxin-antitoxin system and autolysis-associated death mechanisms. The toxin-antitoxin system is found throughout prokaryotic species and consists of a stable toxin and its corresponding labile antitoxin. These components exist in a delicate balance to control death of the cell for altruistic purposes during various environmental conditions (4–6). Membrane-associated holin- and antiholin-like proteins have been described in Staphylococcus aureus and other bacterial species, and have also been shown to control cell death, autolysis, and release of genomic DNA, all of which contribute to the structural integrity of biofilms and potentially to the virulence of the organism (7, 8). Autolysis is also observed in several other bacterial species, including Bacillus subtilis, Enterococcus faecalis, and S. pneumoniae. This lytic activity has recently been shown to be central during events known as “cannibalism” and “fratricide,” where a subpopulation of immature or susceptible cells is killed by sibling cells to release DNA, cellular nutrients, and intracellular toxins (such as pneumolysin) to benefit the population during events such as sporulation, genetic transformation, or biofilm formation, all of which are thought to contribute to fitness and virulence of these organisms in their natural niche (9–11).
Despite what is known about the execution of these systems, their activation mechanism(s) and signaling pathways are poorly understood. Although several ion transport processes have been shown to alter the rate of autolysis events in B. subtilis, and depolarization of the membrane potential is postulated, but not shown, to be related to holin-induced death in S. aureus, (7, 12), the specific signaling events involved are unclear. Similarly, the pathways that bacteria use to induce death during their normal life cycle in vitro and in vivo, and the role of these pathways for optimal colonization and infection are also largely unclear. Therefore, comprehensive information about death pathways in bacteria is critical for a better understanding of bacterial physiology and pathogenesis.
A major hurdle to elucidating the initiation events of bacterial death is the paucity of information on ion transport mechanisms and electrophysiology of the bacterial membrane (13, 14), combined with the difficulty of measuring these events in prokaryotic systems based on their less accessible cytoplasmic membranes. Radioisotopes have been used with some success (15, 16), whereas the loading of acetoxymethyl (AM) fluorescent indicator dyes has only been successful in a few bacterial species (17–19). Patch clamping is not realistic based on the small size of the bacteria and the presence of an inhibitory cell wall, however, some work with giant protoplasts has been performed with variable results (20, 21). Additionally, based on the lack of studies, few eukaryotic inhibitors have been evaluated for activity in bacterial systems, making interpretations with such inhibitors difficult, and fewer yet have been directly developed for bacterial transport systems.
During our studies of the anti-infectious protection conferred to infants by breastfeeding, we identified a milk fraction named HAMLET, for Human α-lactalbumin Made LEthal to Tumor cells, that induced apoptosis in tumor cells, but also killed a variety of bacterial species (22, 23). As the apoptotic effect was partly induced through a direct interaction of HAMLET with mitochondria (24), and as bacteria have long been postulated to be the evolutionary predecessors of mitochondria (25), we hypothesized that similarities in death activation pathways may exist. Indeed, similarities were identified and recently described in our study revealing apoptosis-like phenotypes in bacteria after exposure to HAMLET (26). After exposure, both tumor cells and bacteria showed hallmark morphological changes such as cell shrinkage, DNA condensation, and high-molecular weight DNA fragmentation (26). The bacterial effects were associated with a dissipation of the membrane potential, just as we observed in mitochondria, and required protease activation. These striking phenotypic parallels suggested that an apoptosis-like pathway of death exists in bacteria, which can be activated by HAMLET by targeting effectors that are potentially conserved in tumor cells and various bacterial species.
In the current study, we focused on the initiating mechanism(s) of HAMLET- and starvation-induced bacterial death. Using the pneumococcus as a model system, we found that HAMLET causes a rapid dissipation of the bacterial membrane potential and integrity. These membrane effects and bacterial death were accompanied by a sodium-dependent influx of calcium and an efflux of potassium, and were inhibited by several compounds that block ion transporters, specifically those associated with calcium transport and sodium-calcium exchange activity. Additionally, we observed that kinase activation was required for HAMLET-induced death. Importantly, we observed that all reagents that interfered with the ability of HAMLET to induce pneumococcal death and autolysis also altered the natural susceptibility of the pneumococcus to starvation-induced death and autolysis, suggesting that HAMLET is uniquely able to activate cell death machinery similar to that already in place for physiological death. Finally, dysregulation of physiological death was detrimental to biofilm formation, suggesting that tight control of pneumococcal death is required for optimal colonization and infection.
EXPERIMENTAL PROCEDURES
HAMLET Production
The HAMLET batches used for these studies were generously provided by Dr. Catharina Svanborg, Lund University, Lund, Sweden. HAMLET was produced by converting native α-lactalbumin in the presence of oleic acid (C18:1) as described (27). The preparations were stored in lyophilized form, and resuspended in 1× phosphate-buffered saline (PBS, pH 7.2; Invitrogen; 155.17 mm NaCl, 2.71 mm Na2HPO4, 1.54 mm KH2PO4) to create working stocks.
Reagents for Modulating Ion Transport
All of the following reagents are from Sigma unless noted otherwise and the majority of these reagents have never been evaluated in a bacterial system. Solvents are noted in parentheses (“aq” indicates “aqueous”).
K+ Transport Reagents
High-K+ PBS (HKPBS) containing 60 mm K+: 96.5 mm NaCl, 58.5 mm KCl, 2.7 mm Na2HPO4, 1.54 mm KH2PO4 (corresponding “regular PBS” buffer contained 58.5 mm N-methyl-d-glucamine-Cl instead of KCl); valinomycin (DMSO).
Ca2+ Transport Reagents
Ruthenium red (aq), nifedipine (DMSO), verapamil hydrochloride (aq), sodium orthovanadate (aq), and ionomycin (DMSO).
Na+ Transport Reagents
3′,4′-Dichlorobenzamil hydrochloride (DMSO); amiloride hydrochloride hydrate (DMSO); low Na+ PBS (“PBS(25%)”) was made by substituting most NaCl with N-methyl-d-glucamine chloride (aq) (35 mm NaCl, 120 mm N-methyl-d-glucamine-Cl, 2.71 mm Na2HPO4, 1.54 mm KH2PO4); monensin sodium salt (DMSO).
Mechanosensitive Channel Inhibitor
Gadolinium(III) chloride hexahydrate (DMSO).
Bacterial Strains and Growth Conditions
We used three strains of S. pneumoniae in this study: D39 (wild-type) (28), D39ΔlytA (D39 lacking the major autolysin LytA) (29), and R36A (an unencapsulated derivative of the wild-type strain D39) (28). All of these strains display equal sensitivity to HAMLET. We used D39ΔlytA throughout most of the experiments to limit the autofluorescent interference generated upon autolysin activation and to measure the lysis stimulated solely by HAMLET. The acapsular mutant R36A was used during the ion transport studies with Fura2/AM, Fluo4/AM, and PBFI/AM as the lack of capsule dramatically improved our loading protocol of these ion-sensitive dyes into the bacteria (see below). S. aureus strain I7, a clinical isolate from the University at Buffalo, State University of New York, and Escherichia coli strain JM109 were used for depolarization studies. All bacterial strains were stored as glycerol stocks at −80 °C. Stocks of pneumococci were seeded into THY broth (Todd Hewitt containing 0.5% yeast extract; Bacto, BD Diagnostics, Franklin Lakes, NJ) and allowed to grow to late logarithmic phase (A600 nm of ∼0.65) at 37 °C. Similarly, S. aureus was grown in tryptic soy broth (Bacto, BD Diagnostics), whereas E. coli was grown in Luria Bertani broth (Bacto, BD Diagnostics).
Assessing the Effect of HAMLET on Pneumococcal Viability
Upon reaching late log phase in growth media, the pneumococci were pelleted by centrifugation at 2,400 × g for 10 min and washed twice by resuspension in PBS. The bacterial pellet was resuspended in PBS to half of the original volume and energized with 50 mm glucose for 15 min at 37 °C. In each well of a 96-well microtiter plate (Falcon, BD Biosciences), a 100-μl volume of the energized bacteria was added to 100 μl of PBS containing the required HAMLET concentration and/or the specific ion transport inhibitory compound, and was subsequently incubated for 1 h at 37 °C. At the end of the incubation, serial dilutions of the bacteria were made in PBS and plated onto solid media composed of Tryptic Soy Broth (Bacto, BD Diagnostics) supplemented with 5% defibrinated sheeps blood (Bio Link Inc., Liverpool, NY) for viable counts. The concentration of HAMLET used throughout the study was that which induced between 3 and 4 log10 of death. Upon adding any inhibitory compounds, their effect on HAMLET-induced death was expressed as a percentage of the total logs of death observed in the presence of the sample treated with HAMLET alone. To determine the minimal inhibitory concentration of HAMLET, S. pneumoniae D39 was seeded at a concentration of 105 colony-forming units (CFU) per ml in THY in 96-well microtiter plates in the presence of 2-fold dilutions of HAMLET and growth was determined by measuring the optical density at 600 nm, every 5 min over 18 h in a Biotek Synergy 2 plate reader. The minimal inhibitory concentration was determined to be the lowest concentration of HAMLET where no growth was detected.
Assessing Membrane Potential and Integrity
To detect membrane potential and rupture of the pneumococcal membrane, respectively, 500 nm DiBAC4(3) (bis-(1,3-dibutylbarbituric acid) trimethine oxonol; Molecular Probes, Eugene, OR) and propidium iodide (40 μg/ml; Sigma) were added to energized pneumococci (prepared as described above). In a 96-well plate, a 100-μl volume of this bacterial suspension was then added to 100 μl of PBS containing HAMLET and/or the specific ion transport inhibitory compound in the wells of a 96-well plate yielding a final concentration of 25 mm glucose, 250 nm DiBAC4(3), 20 μg/ml of propidium iodide, and the specified concentrations of HAMLET and/or the inhibitor. The plate was then placed immediately into a pre-warmed (37 °C) Synergy 2 Multi-Mode Microplate Reader (BioTek, Winooski, VT) where fluorescence readings from DiBAC4(3) (485/20 nm excitation, 528/20 nm emission) and propidium iodide (530/25 nm excitation, 590/35 nm emission) were taken every minute for 1 h. Sodium deoxycholate (Sigma) was added as a positive control for membrane depolarization and rupture. The difference in fluorescence intensity between the untreated control and the HAMLET-treated sample was calculated for the “no inhibitor” samples and for the “inhibitor” samples using the values after 60 min. The fluorescence intensity difference for the inhibitor samples was then expressed as a percentage of the intensity of the HAMLET alone (no inhibitor) sample, describing the degree of depolarization compared with the “HAMLET alone” sample (see supplemental Fig. S1).
Fluorescent Detection of Ion Transport
Loading
The following protocol was developed. After reaching late log phase, R36A pneumococci were washed twice and resuspended in PBS containing 5 μm fluorescent indicator dye (PBFI/AM for potassium, or Fura-2/AM or Fluo-4/AM for calcium), 1× PowerLoadTM (solubilization reagent), and 1× probenecid (all from Molecular Probes). Protected from light, the pneumococci were incubated at 37 °C for 2 h to allow for loading of the dyes and cleavage of the ester, and subsequently washed twice and resuspended with PBS.
Readings
200 μl of the washed/loaded bacteria were added per well of a 96-well plate, which was then placed in the pre-warmed (37 °C) BioTek Synergy 2 plate reader where a baseline reading was taken by measuring the fluorescence every second for 1 min (Fura-2/AM, PBFI/AM, excited at 340/11 and 380/20, emission detected at 508/20; Fluo-4/AM, 485/20 excitation, 528/20 emission). If an inhibitor was present, it was added prior to this baseline reading. At the end of the baseline reading, HAMLET was added and fluorescence readings were immediately taken every second for another 4 min. Valinomycin and ionomycin (Sigma) were added as positive controls for K+ and Ca2+ transport, respectively. The degree of inhibition was expressed as a percentage of the fluorescence intensity from the sample treated with HAMLET alone (see supplemental Fig. S1 for a visual depiction of the measurements).
Radioisotope 45Ca2+ Transport Assays
D39ΔlytA was grown to log phase, washed three times, and resuspended in 1× PBS containing 0.5 mm CaCl2 (CaPBS). Alternately, for experiments with low extracellular sodium, we washed and resuspended the cells in an isoosmotic buffer containing 25% of the normal PBS Na+ concentration (PBS(25%)). No glucose was added to minimize the interference created by extrusion of Ca2+ via ATPase pumps. 45CaCl2 (PerkinElmer Life Sciences) was added to the cells at a final concentration of 2.5 μCi/ml, followed by inhibitor compound, with each addition given 2 min equilibration time. The untreated baseline sample was measured at this point. The sample was then divided, HAMLET was added to one of the tubes, and 45Ca2+ uptake was measured at various intervals. For each sample, 100 μl was dispensed onto Millipore 0.3-μm PHWP filters (EMD Millipore, Billerica, MA) presoaked in the CaPBS, and immediately washed with 9 ml of CaPBS via syringe filtration through Millipore Swinnex® filter holders. Filters were placed in scintillation vials with 5 ml of scintillation fluid, and cpm were detected on a Wallac 1409 liquid scintillation counter (Wallac Oy, Turku, Finland). The Δcpm was calculated for each sample at the indicated time points as the difference between HAMLET-treated and untreated samples, and expressed as a percentage of the Δcpm for the HAMLET alone sample without inhibitor.
Growth Curves, Autolysis
Autolysis was assessed by measuring optical density (OD) at 600 nm. Optical density has long been the established method of monitoring autolysis of bacteria; as autolysis proceeds, the cells burst and the culture will clear, producing a rapid decline in the optical density. Strains of interest were diluted from frozen stock to an A600 nm of ∼0.100 in THY broth and dispensed into a 96-well plate, with or without the addition of ionophores or inhibitor compounds of interest (monensin, ionomycin, RuR, DCB, and staurosporine; Sigma). The growth curves were plotted overnight by monitoring the A600 nm in the Synergy 2 plate reader every 5 min. To observe natural autolysis, D39 was grown to stationary phase in THY (A600 nm ≈ 1.200) prior to being added to the 96-well plate with or without the addition of ionophores of interest (ionomycin and monensin, Sigma).
Biofilm Studies
Biofilms were generated as described (30). Briefly, S. pneumoniae D39 were grown in a chemically defined medium (JRH Biosciences, Lexera, KS) to mid-logarithmic phase (A600 = 0.5), washed, and resuspended in fresh pre-warmed chemically defined medium at a density of 2 × 104 cfu in a 500-μl volume. Suspensions were used to seed sterile round glass coverslips in the bottom of polystyrene 24-well plates with a substratum of confluent NCI-H292 bronchial epithelial cells (ATCC CCL-1848; grown as previously described in Ref. 23). Biofilms were cultured at 34 °C in 5% CO2 for the indicated times, with change of culture media every 12 h, adding in fresh inhibitor with each media change to maintain their presence throughout the growth period. After 48 h, biofilms were then washed in PBS, sonicated, and detached by scraping the surface in the presence of 100 μl of PBS followed by a rinse with 100 μl of PBS. Collected cells were vortexed twice for 20 s at high speed and the dispersed biofilm cells were used to determine viable CFU/ml from diluted samples plated and grown on blood agar. Results are reported as the total number of CFU per biofilm.
RESULTS
HAMLET Kills S. pneumoniae in a Dose-dependent Manner
Although HAMLET is not universally bactericidal and fails to kill many species of bacteria including S. aureus and E. coli, it exhibits significant antibacterial activity against a handful of species, with S. pneumoniae displaying the greatest susceptibility (22). All pneumococcal wild-type strains we have tested are equally sensitive to HAMLET, regardless of capsule presence or type, or absence of genes for various virulence factors, including the major autolysin (LytA) (26). As shown in Fig. 1, D39ΔlytA (an autolysin-negative pneumococcal mutant) was killed by HAMLET in a concentration-dependent manner. The minimal inhibitory concentration for HAMLET was determined to be 5–10 μg/ml, whereas ∼110 μg/ml was required to kill 6 log10 pneumococci within 1 h (Fig. 1). These numbers are comparable with what is required to kill pneumococci with other antibiotics in the same time frame (our preliminary data and see Refs. 31 and 32) and may well be within a physiological range considering that the concentration of α-lactalbumin in milk is 2,000 μg/ml and oleic acid is the most prevalent fatty acid in human milk. For all experiments described throughout this study, we employed a concentration of HAMLET that induced between 3 and 4 log10 of death over a 1-h incubation period, unless otherwise indicated.
FIGURE 1.

HAMLET displays dose-dependent bactericidal activity against S. pneumoniae. S. pneumoniae D39ΔlytA cells were incubated in the presence of various concentrations of HAMLET. Bacterial viability was assessed and presented as log10 CFU per ml of bacterial suspension, and represents the mean of three separate experiments, with error bars representing the S.D.
Exposure to HAMLET Leads to Depolarization and Rupture of the Pneumococcal Membrane
To measure depolarization of the bacterial membrane we tested a variety of fluorescent potential-sensitive probes, including the commonly used mitochondrial probes JC-1, TMRE, and Rhodamine 123, all of which failed to produce a recordable change in membrane potential in heat-killed or detergent-treated pneumococci compared with healthy cells (not shown). However, we did find success with bis-oxonol dyes in all our bacterial cell systems and settled on using the bis-oxonol DiBAC4(3), which enters depolarized cells and fluoresces more intensely upon interacting with intracellular components.
Just as it induces depolarization of the mitochondrial membrane (33), HAMLET triggered a rapid dissipation of the pneumococcal membrane potential, which plateaued after about 6 min (Fig. 2A). Using the membrane-impermeable fluorescent dye propidium iodide, we simultaneously monitored membrane integrity and observed that, at about the same time that depolarization plateaued, rupture of the membrane began (Fig. 2B), with fluorescence rising steadily throughout the remainder of the incubation period. This suggests that HAMLET-induced depolarization and rupture of the pneumococcal membrane are independent events, with dissipation of polarity preceding rupture, rather than depolarization resulting from lost membrane integrity, as was seen upon treatment with the detergent sodium deoxycholate (Fig. 2).
FIGURE 2.

Disruption of pneumococcal membrane polarity and integrity. Mid-log phase D39ΔlytA pneumococci were incubated with the fluorescent indicator dyes DiBAC4(3) and propidium iodide (PI) concurrently to detect (A) membrane depolarization and (B) membrane rupture, respectively, by measuring fluorescence over time. HAMLET was added at time 0 (arrow). The detergent sodium deoxycholate (0.05%) was included as a positive control for both events. The results presented are from a representative experiment.
Depolarization Alone and K+ Efflux Are Not Sufficient for Activation of HAMLET-induced Death
Information from other bacterial systems has suggested that execution of bacterial death may be linked to initial depolarization of the bacterial membrane (7, 12). However, the specificity of depolarization as a direct trigger of death and lysis in bacteria has not been studied. We therefore evaluated the role of depolarization as a trigger for HAMLET-induced death.
While investigating pneumococcal depolarization and rupture at different concentrations of HAMLET, we observed that low-grade depolarization could be induced without membrane rupture or death, suggesting that either depolarization alone was not a trigger for bacterial death or that the depolarization at low concentrations was not sufficiently strong to trigger death. Interestingly, depolarization was also induced in the HAMLET-resistant species E. coli (Fig. 3A) and S. aureus (Fig. 3B) to a level that, when induced in S. pneumoniae (Fig. 3C), resulted in about 2 log10 of pneumococcal death (p = 0.0442) (Fig. 3D), providing further evidence that depolarization per se may not be the trigger of bacterial death.
FIGURE 3.

HAMLET-induced depolarization is not sufficient to induce death. A, E. coli JM109 (ECOLI); B, S. aureus I7 (STAAU); and C, S. pneumoniae D39ΔlytA (STRPN) were incubated with the potential-sensitive indicator dye DiBAC4(3) to monitor membrane depolarization by measuring fluorescence over time. HAMLET was added at time 0 (arrow), 250 μg/ml to the E. coli and S. aureus cultures, and 31 μg/ml to the pneumococci. After 1 h, the untreated (Untr) and HAMLET-treated (HL) bacteria were serially diluted and viability was determined (D). Viability data represent the mean of three individual experiments, with S.D. bars, and significance calculated using the paired t test with a 95% confidence interval (* = p < 0.05).
To more specifically address the role of membrane depolarization in HAMLET-induced death we investigated the role of K+ transport. In eukaryotic cells, the efflux of K+ is well described as an essential event for successful execution of apoptosis (34, 35), whereas its transport, in concert with Na+, is also central to maintaining membrane polarity (36). As no such information is available in prokaryotic systems, both of these functions were explored in relationship to HAMLET-induced bacterial death.
We first monitored intracellular K+ levels by developing a loading protocol for loading of AM ester fluorescent indicator dyes into pneumococci. AM-esters have only been successfully loaded into a handful of species but the loading efficiency varies. For pneumococci, we found that capsule expression interfered with loading of the dyes and that PowerLoadTM and probenecid (from Invitrogen) were required to obtain an optimal intracellular concentration of dye to make measurements possible.
We then successfully loaded the unencapsulated S. pneumoniae strain R36A with the AM-ester version of the K+-sensitive fluorescent indicator dye PBFI. Although the fluorescence of the bacteria from PBFI/AM remained unchanged upon treatment with PBS, the signal intensity immediately decreased following addition of HAMLET (Fig. 4A, green line), indicating a decrease in the intracellular concentration of K+ that is most likely due to an efflux of the ion, similar to what takes place during apoptosis of eukaryotic cells. The decrease in fluorescence leveled off approximately 4 min after addition of HAMLET, suggesting that this efflux occurred rapidly and that the lower intracellular concentration of K+ was sustained over time. The K+ efflux seen after HAMLET exposure was mirrored using the K+ ionophore valinomycin that allows K+ to immediately flow out of the cell down its concentration gradient (Fig. 4A, black line). This is the first time K+ transport has been directly measured in pneumococci, and contributes to the very few studies of potassium transport performed in bacterial species (37).
FIGURE 4.

HAMLET induces K+ efflux that is not central to the bactericidal activity. A, the fluorescence of PBFI-loaded (K+-indicator) S. pneumoniae strain R36A was recorded to monitor concentrations of intracellular K+. After baseline fluorescence was recorded for 1 min, either 10 μm valinomycin (positive control) or HAMLET was added (arrow) to the bacteria and the resulting fluorescence was recorded every second for the next 4 min. Tracings from a representative experiment are shown. B, D39ΔlytA pneumococci were incubated with HAMLET for 1 h in the presence of K+-related transport inhibitors, while monitoring membrane depolarization (gray bars) and death (black bars), using DiBAC fluorescence and viable counts, respectively. Membrane depolarization and death in the inhibitor-treated samples are expressed as a percentage of the HAMLET-alone treated samples (HAMLET-alone value was set to 100%, represented by dotted line). C, PBFI fluorescence was monitored in R36A to measure the HAMLET-induced decrease in intracellular K+ in the presence of ion transport inhibitors. K+ efflux after 4 min is expressed as a percentage of that observed with HAMLET alone (dotted line, 100% K+ efflux). The data in panels B and C represent the mean ± S.D. of three individual experiments with error bars, and significance was calculated using the paired t test with a 95% confidence interval (* = p < 0.05; ** = p < 0.01). Inhibitors: high-K+ PBS buffer (HKPBS; 60 mm extracellular K+), gadolinium (GAD), and RuR.
To determine the role of the detected K+ efflux in the bactericidal activity of HAMLET, we directly manipulated the transmembrane K+ gradient by creating an extracellular high-K+ PBS solution (HKPBS; 60 mm extracellular K+) that diminishes the electrochemical potential gradient of K+. This solution depolarizes the bacterial membrane and significantly limits the ability of K+ to flow out of the cells. The degree of HAMLET-induced depolarization and death in the pneumococcus (comparing the HAMLET-treated bacteria with the untreated bacteria; see supplemental Fig. S1 for a visual depiction of these calculations) in HKPBS was no different from that observed in regular PBS (Fig. 4B). On the other hand, HKPBS significantly limited the HAMLET-induced decrease in intracellular K+ levels compared with the HAMLET-induced decrease observed in bacteria bathed in PBS (p = 0.0018) (Fig. 4C). These results suggest that K+ efflux is not central to the bactericidal mechanism of HAMLET, and is likely more of a compensatory cellular response to repolarize the membrane. Furthermore, the failure of the HKPBS, which depolarizes the membrane, to either trigger death in pneumococci by itself or potentiate the effect of HAMLET treatment further demonstrates that depolarization per se is not sufficient to trigger pneumococcal death, and that more specific signaling events are required. As K+ transport was not involved in initiation of HAMLET-induced death, no further studies were performed to identify potential transport mechanisms.
Mechanosensitive Channels Play a Role in HAMLET-induced Death
During eukaryotic apoptosis, depolarization of the mitochondrial membrane causes water to fill the mitochondria, leading to swelling (33, 38). In bacteria, one of the major ion transport elements that serve to detect such swelling is the stretch-activated or mechanosensitive channel (MSC), which responds by opening and nonspecifically releasing a variety of cations (especially K+), causing water loss that relieves the cell of the increased turgor pressure. To determine whether mechanosensitive channels might be involved in HAMLET-induced death in bacteria, we treated pneumococci with HAMLET in the presence or absence of the MSC inhibitor gadolinium and observed that, although the compound had no dramatic effect on the degree of HAMLET-induced depolarization, it did rescue a significant portion of the cells from death (p = 0.0051) (Fig. 4B), suggesting that MSCs are involved in facilitating the lethal effects of HAMLET. Gadolinium also blocked more than half of the K+ efflux (p = 0.0487) (Fig. 4C), suggesting that mechanosensitive channels are involved in that event, but failed to impact Ca2+ influx (see Fig. 5D, below). However, as K+ transport was unrelated to HAMLET-induced death and gadolinium did not affect Ca2+-transport, the involvement of other ions in the trigger of bacterial death is suggested.
FIGURE 5.

HAMLET triggers influx of Ca2+ that is critical to its bactericidal mechanism. A, R36A pneumococci were loaded with the Ca2+-sensitive dye Fura-2, and B, D39ΔlytA pneumococci were incubated with the radioisotope 45Ca2+ (2.5 μCi/ml). After recording baseline readings, PBS (untreated), the Ca2+ ionophore ionomycin (1 μm in panel A and 10 μm in panel B), or HAMLET were added (arrow) to the bacteria and fluorescence or radioactivity was measured over time. Results from a representative experiment are shown for each. B, inset, HAMLET-induced 45Ca2+ uptake was recorded over 10 min, demonstrating “overshoot.” C, D39ΔlytA pneumococci were incubated with HAMLET for 1 h in the presence of RuR, nifedipine (NIF), verapamil (VPL), or sodium orthovanadate (SOV), while monitoring membrane depolarization (gray bars) and death (black bars), and were expressed as a percentage of the HAMLET-alone treated samples (HAMLET-alone value was set to 100%, represented by dotted line). The effect of ion transport inhibitors on HAMLET-induced Ca2+ uptake was assessed by monitoring (D) fluorescence (light blue bars) or 45Ca2+ uptake (dark blue bars) after 5 min of treatment. Results are expressed as the percentage of HAMLET-alone treated samples (dotted line, 100% Ca2+ influx). E, D39ΔlytA pneumococci were incubated with HAMLET for 1 h in the absence or presence of 2.5 μm ionomycin and viability was determined. F, D39ΔlytA pneumococci were incubated with HAMLET for 1 h in the presence of amiloride (Amil) or DCB, while monitoring depolarization (gray bars) and death (black bars). Results are presented as in panel C. Panels C–F, data represent the mean of three individual experiments, with S.D. error bars, and significance was calculated using the paired t test with a 95% confidence interval (* = p < 0.05; ** = p < 0.01).
Calcium Influx Is Central to the Activation of the Bactericidal Mechanism of HAMLET
The role of Ca2+ in apoptotic signaling is well-established (39); Ca2+ overload of the mitochondrial matrix through the Ca2+ uniporter leads to the opening of the permeability transition pore complex and leads to depolarization of the mitochondrial inner membrane (40). HAMLET is known to traffic to mitochondria in tumor cells (24) and induce depolarization of the mitochondrial membrane, an event that is significantly diminished in the presence of the general Ca2+ transport inhibitor compound ruthenium red (RuR) (33).
Thus, we hypothesized that HAMLET also triggers an influx of Ca2+ into the bacteria, and that this influx is important for the bactericidal mechanism and activity of HAMLET. To test this, we used our loading protocol to load S. pneumoniae strain R36A with the AM-ester version of the Ca2+-sensitive fluorescent indicator dye Fura-2 to monitor intracellular Ca2+ levels, and we also directly monitored uptake of Ca2+ into D39ΔlytA pneumococci using the radioisotope 45Ca2+. Both the 340/380 fluorescence ratio (Fig. 5A, blue line) and the 45Ca2+ isotope uptake (Fig. 5B, blue line) demonstrated that, upon HAMLET addition, intracellular Ca2+ rises immediately. A plateau of fluorescence was detected after about 4 min following addition of HAMLET, suggesting that this transport event occurred rapidly and was sustained (Fig. 5A). In the isotope experiment, we observed a brief plateau or “overshoot” of 45Ca2+ uptake that occurred around 8–10 min following HAMLET addition (Fig. 5B, inset), which is suggestive of an underlying carrier-mediated mechanism, followed by a continued rise that gradually plateaued after about 15–20 min. In both the fluorescence assay and radioisotope assay, the Ca2+-ionophore ionomycin triggered a rise in intracellular Ca2+ as expected (Fig. 5, A and B, black line), whereas Ca2+ levels in the untreated controls remained unchanged (gray line).
To investigate the importance of Ca2+ influx for the bactericidal activity of HAMLET, we introduced various inhibitors of Ca2+ transport to assess their effects on membrane depolarization, Ca2+ transport, and death. The general Ca2+ channel inhibitor RuR was the first inhibitor evaluated in the pneumococcal system based on its inhibitory role in eukaryote mitochondria (33). We observed that RuR almost completely blocked HAMLET-induced depolarization of the pneumococcal membrane (p = 0.0015) (Fig. 5C), which was directly related to an inhibition of Ca2+ influx, observed both by an almost complete inhibition of the rise in the 340/380 fluorescence ratio from cells pre-loaded with Fura-2 (p = 0.0093) and an inhibition of more than half of the 45Ca2+ uptake (p = 0.0083) (Fig. 5D). RuR was specific for calcium transport as no effect on K+ transport was seen (Fig. 4C). The inhibition of Ca2+ influx was directly related to a significant and almost complete rescue of the bacteria from death (p = 0.01) (Fig. 5C).
To further verify a role for an influx of calcium in triggering HAMLET-induced death, pneumococci were treated with ionomycin, which, by itself, increased the intracellular Ca2+ concentration (Fig. 5, A and B) without killing the bacteria, but was shown to significantly potentiate the bactericidal activity of HAMLET (p = 0.0039) (Fig. 5E), possibly by lowering the HAMLET-induced influx required to trigger death activation. Overall, the strong inhibitory effects of RuR and the potentiating effect of ionomycin suggest that Ca2+ influx plays a central role in the activation of the bactericidal activity of HAMLET.
HAMLET-induced Calcium Influx Uses a Sodium-dependent Transport Mechanism
To further elucidate the Ca2+ transport mechanism in S. pneumoniae, we first performed a BLAST search of the entire published genomes of S. pneumoniae strains TIGR4 and D39 (41, 42) in both the NCBI database and the Transporter Classification Database. The only annotations related to Ca2+ transport in the two genomes were two P-type Ca2+-ATPase pumps (SP1551 and SP1623, TIGR4), the first of which has been characterized and shown to extrude Ca2+ from the cell (43, 44). However, HAMLET-induced depolarization and death were not dramatically changed in the presence of the P-type ATPase inhibitor sodium orthovanadate; if anything, death was slightly increased (p = 0.04) (Fig. 5C). As L-type Ca2+ channels have been implicated in HAMLET-induced death of tumor cells (45), we also evaluated the effect of the L-type channel inhibitors nifedipine and verapamil, neither of which had a significant impact on HAMLET-induced depolarization or bacterial death (Fig. 5C).
In addition to P-type Ca2+-ATPase transport activity, studies by Trombe (16) have revealed that Ca2+ transport in S. pneumoniae appears to be linked to Na+ via Na+/Ca2+ antiport activity, although the transporter(s) has not yet been identified. To evaluate the potential influence of Na+ on the HAMLET-induced Ca2+ influx, we introduced the Na+ channel inhibitor amiloride and its derivate 3′,4′-dichlorobenzamil (DCB), which, compared with amiloride, has greater inhibitory specificity for a Na+/Ca2+ exchanger (NCX). As both of these compounds autofluoresce at the wavelengths used to detect Fura-2 fluorescence, we used Fluo-4/AM to measure intracellular Ca2+ levels, and also measured 45Ca2+ uptake. Both amiloride and DCB almost completely inhibited the HAMLET-induced rise in intracellular Ca2+, as indicated both by the low Fluo-4 fluorescence intensity (amiloride, p = 0.0084; DCB, p = 0.003) (Fig. 5D) and the lack of 45Ca2+ uptake (amiloride, p = 0.0059; DCB, p = 0.0024) (Fig. 5D). This provided the first evidence that Ca2+ flux is linked to Na+ following exposure of S. pneumoniae to HAMLET.
The importance of this Na+-dependent Ca2+ influx for the bactericidal activity of HAMLET was demonstrated by the fact that the presence of amiloride caused dose-dependent inhibition, with a 1 mm concentration required for complete inhibition of depolarization (p = 0.0017) and Ca2+ import, as well as more than 50% log10 rescue of HAMLET-induced death (p = 0.001), whereas the same effect was obtained using DCB at the considerably lower concentration of 100 μm (depolarization, p = 0.0201; death, p = 0.0068) (Fig. 5, D and F). This pattern of inhibition suggests that Ca2+ transport occurs through a NCX and is important for HAMLET-induced death.
To further verify the role of Na+ for Ca2+ influx, we monitored HAMLET-induced pneumococcal 45Ca2+ uptake under conditions that favor the operation of a NCX, which transports Na+ out of the pneumococcus against its electrochemical potential gradient while bringing Ca2+ into the cell in the direction of its substantial gradient (Fig. 6A) (16). First, we tested the effect of the Na+ ionophore monensin, which, under normal physiological buffer conditions (155 mm extracellular Na+), will dissipate the Na+ gradient by increasing the Na+ concentration inside the cell, decreasing the driving force against Na+ exit via the NCX. In the presence of monensin, HAMLET-induced 45Ca2+ uptake was enhanced in a concentration-dependent manner (Fig. 6B) and the facilitated import of Ca2+ was associated with an increased sensitivity to HAMLET-induced death (p = 0.0017) (Fig. 6D). Second, Ca2+ import and death were measured in pneumococci exposed to HAMLET in an isoosmotic buffer containing 25% of the normal PBS Na+ concentration (PBS(25%)), which will facilitate Na+ exit via the NCX. The 45Ca2+ uptake was greater in low-Na+ PBS compared with that observed in PBS containing a standard concentration of Na+ (Fig. 6C) and was associated with a significantly increased HAMLET-induced death (p = 0.0017) (Fig. 6D). Combined, these results confirm the role of Na+ in the bactericidal mechanism of HAMLET, and are consistent with our hypothesis that a NCX is centrally involved in this activity.
FIGURE 6.

HAMLET-induced 45Ca2+ uptake and death are sodium-dependent. A, graphical representation of the pneumococcal NCX, which transports Na+ out of the pneumococcus against its electrochemical potential gradient while bringing Ca2+ into the cell in the direction of its substantial gradient. Using the radioisotope 45Ca2+, HAMLET-induced Ca2+ uptake in D39ΔlytA pneumococci was recorded in the presence of (B) the Na+ ionophore monensin and (C) conditions of low extracellular Na+ (PBS with 25% of standard Na+ levels; PBS(25%)). cpm from representative experiments are shown for each panel. D, after 1 h of HAMLET treatment in both of these conditions, the bacteria were serially diluted for viable counts, and the log10 of HAMLET-induced death were calculated. The viability data represent three individual experiments, with S.D. indicated with error bars, and significance calculated using the paired t test with a 95% confidence interval (* = p < 0.05; ** = p < 0.01).
Inhibiting Kinase Activity Limits the Lethality of HAMLET
In eukaryotic cells, sustained elevations of intracellular Ca2+ are known to activate kinases, which, in turn, can trigger apoptosis (46, 47). Ca2+-induced kinase activation has also been recently described for conjugated linoleic acid (48), which is a fatty acid that can complex with α-lactalbumin equally as well as oleic acid and yield similar lethal effects (49). To evaluate the potential role of kinase activity in HAMLET-induced pneumococcal death, we measured the bactericidal activity of HAMLET in the presence of the pan-kinase inhibitor staurosporine, using a concentration range that has previously been reported to be effective in bacterial systems (20 μm (50)). We observed that pneumococcal viability was dramatically rescued in the presence of this inhibitor (p = 0.0017) (Fig. 7), suggesting that kinase activity is an important effector during HAMLET-induced death.
FIGURE 7.
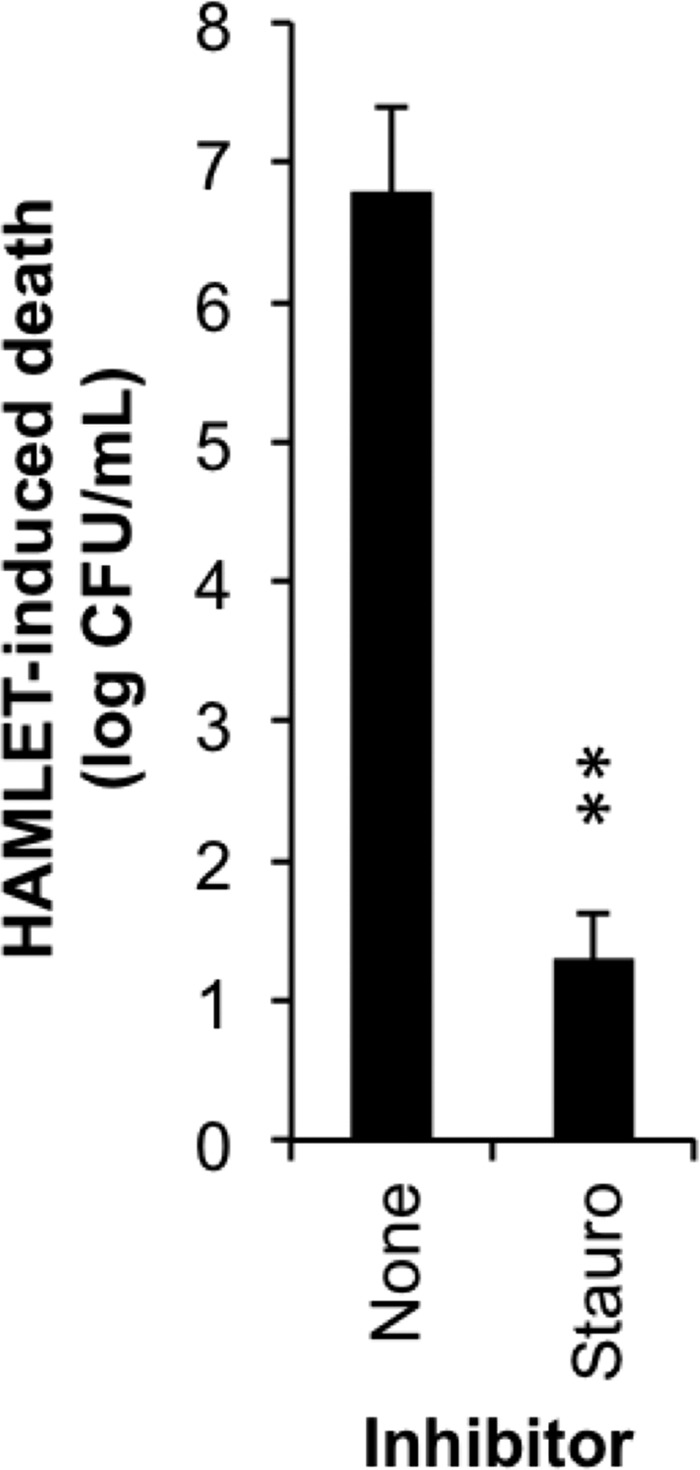
Inhibition of kinase activity protects pneumococci from HAMLET-induced death. S. pneumoniae D39 were incubated with HAMLET in the absence or presence of the protein kinase inhibitor staurosporine (20 μm). After 1 h, cells were serially diluted and plated for viable counts, which are presented as log10 of HAMLET-induced death. The data represent the mean ± S.D. of three individual experiments with error bars, and significance was calculated using the paired t test with a 95% confidence interval (** = p < 0.01).
Pneumococcal Physiological Death and Autolysis Use the Same Activation Mechanism as HAMLET-induced Death
Previous work by Marie-Claude Trombe (51) has shown that Ca2+ is important for effective induction of competence and autolysis. As the results of our studies point to a central importance of Ca2+ and Na+ as well as kinase activity for the bactericidal mechanism of HAMLET, we explored the possibility that this same activation mechanism is also responsible for physiological death that results in autolysis. To do this, we took the same conditions that enhanced or inhibited HAMLET-induced death and evaluated their effect on stationary phase starvation-induced death and subsequent autolysis.
Enhancement
Above, we showed that the Na+ ionophore monensin enhanced HAMLET-induced 45Ca2+ influx (Fig. 6B) and both monensin and the Ca2+ ionophore ionomycin enhanced death (Figs. 5E and 6D) in the pneumococcus. To determine whether monensin and ionomycin also enhance stationary-phase autolysis of the pneumococcus, we evaluated the effect of their presence during growth of wild-type D39 pneumococci, by monitoring the optical density over time. The concentrations of ionophore used did not affect growth in the logarithmic phase. The optical density tracings (Fig. 8A) demonstrate that the bacteria died and autolysed sooner and more rapidly in the presence of monensin, as well as ionomycin, suggesting that conditions that favor the influx of Ca2+ through a Na+-dependent transport mechanism to reach a triggering concentration, enhance autolysis in the pneumococcus. These results suggest that conditions that enhance the bactericidal activity of HAMLET also enhance starvation-induced death and autolysis in stationary phase growth of pneumococci.
FIGURE 8.

Augmenting sensitivity to the natural pneumococcal death pathway autolysis. A, pneumococci were grown to stationary phase, at which point 1× PBS (Untreated), 10 μm ionomycin, or 100 μm monensin was added to samples of the culture, with the A600 nm subsequently plotted over time. B, pneumococci were grown from lag phase in the presence of broth alone (Untreated), 30 μm RuR or 15 μm DCB, with the A600 nm after late stationary phase displayed in the graph. C, pneumococci were grown from lag phase in the absence (Untreated) or presence of 20 μm staurosporine, with the A600 nm after late stationary phase displayed in the graph.
Inhibition
As addition of the Ca2+-transport inhibitor RuR and the Na+-inhibitor DCB each inhibited the bactericidal activity of HAMLET, including Ca2+ influx, depolarization, and death (Fig. 5, C, D, and F), we predicted that these inhibitors would have a similar inhibitory effect on stationary phase death and autolysis. As shown in Fig. 8B, the activation of death and autolysis was significantly delayed, causing the continual extension of the stationary phase. Similarly, pneumococci that were grown in the presence of the kinase inhibitor staurosporine, which blocked HAMLET-induced death (Fig. 7), also displayed a prolonged stationary phase, delaying the activation of autolysis (Fig. 8C). Thus, conditions that inhibit the bactericidal pathway of HAMLET-induced death also appear to inhibit pneumococcal autolysis.
Inhibition of Biofilm Formation
We have recently shown that pneumococci form complex biofilms during colonization of the murine nasopharynx and that the ability of strains to form biofilms on epithelial cells, but not on abiotic surfaces, is directly associated with their ability to cause nasopharyngeal colonization in vivo (30). As colonization and infection through biofilm formation in vivo is thought to require a tight regulation of death and survival of the bacteria (1), we sought to determine how modulation of cell death affects biofilm formation on epithelial cells. D39 bacteria were seeded on top of NCI-H292 bronchial carcinoma cells in the presence or absence of RuR and DCB, and biofilm formation over 48 h was recorded as the total biomass. Even though these inhibitors caused prolonged survival of planktonic pneumococci in stationary phase (Fig. 7B), the pneumococci formed less structured biofilms of significantly lower biomass in the presence of these inhibitors (RuR, p = 0.0171; DCB, p = 0.0078) (Fig. 9). The significance of these findings are, first, that we have identified components of a novel bactericidal activation mechanism induced by HAMLET that is also used by pneumococci during physiological death, and second, that a tight regulation of the execution of this death pathway by physiological stimuli is required for optimal biofilm formation and, by extension, the potential ability of bacteria to cause colonization and infection in vivo.
FIGURE 9.
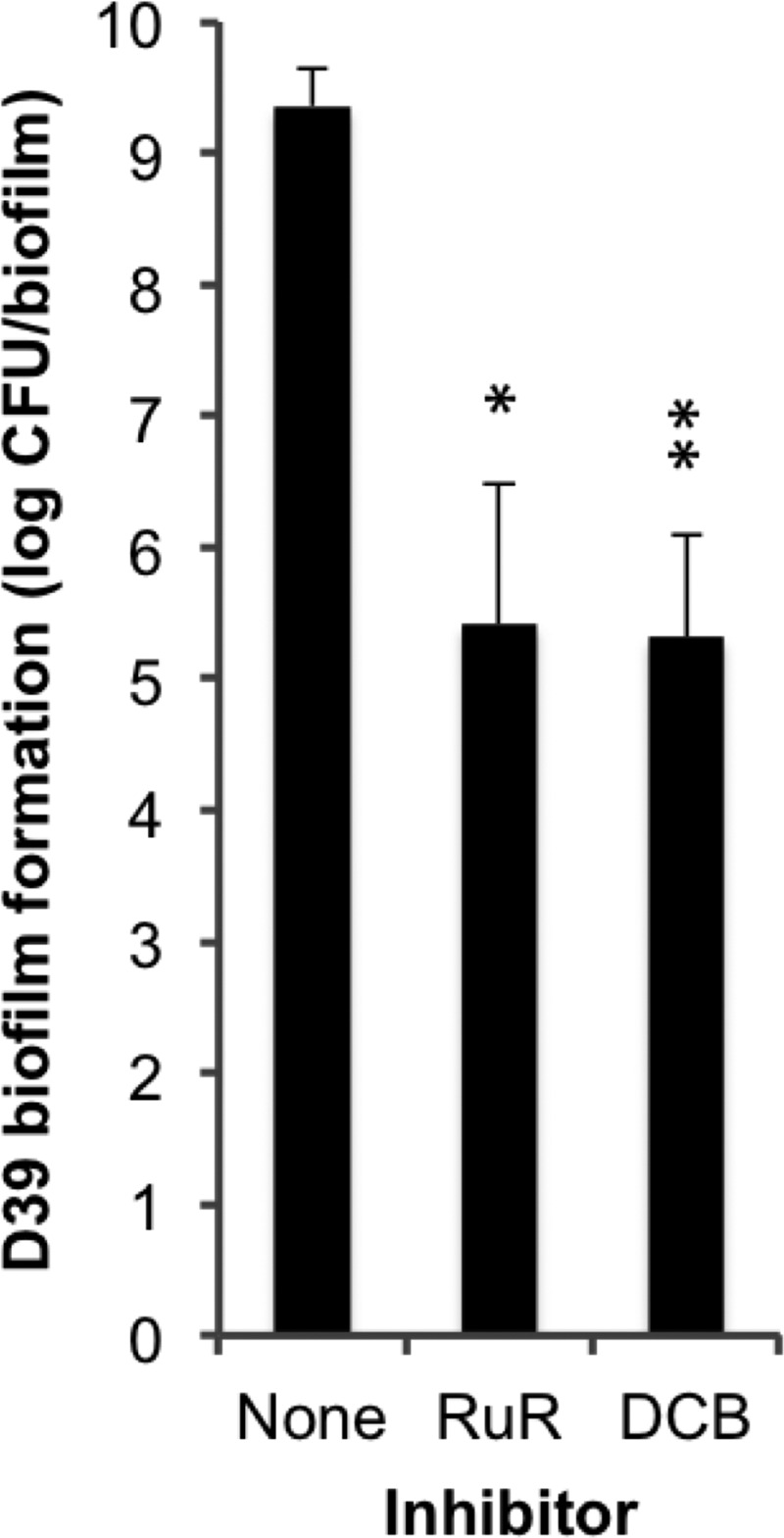
Inhibitors of autolysis limit pneumococcal biofilm formation. D39 was seeded into biofilms over a prefixed substratum of NCI-H292 cells in chemically defined medium. At each media change, PBS (negative control), 30 μm RuR, or 100 μm DCB was added so that it was present throughout the 48-h growth period. The data represent the total biomass of three individual biofilms per sample after 48 h of incubation, with S.D. indicated with error bars and significance calculated using the paired t test with a 95% confidence interval (* = p < 0.05; ** = p < 0.01).
DISCUSSION
This study characterized the activation mechanism of pneumococcal death induced by HAMLET, a human milk protein-lipid complex, and comprehensively demonstrated the importance of specific ion transport mechanisms and kinase activation in triggering bacterial death (see Fig. 10 for a summary of our findings). These mechanistic features also appeared to be the same or overlapping with the mechanism used to trigger physiological death from starvation of the bacteria in stationary phase cultures, and constitutes the first study to address the activation events responsible for initiating death in bacteria from natural stimuli. The importance of this death mechanism was further verified when its modulation resulted in significantly decreased biofilm formation, suggesting an important role for a tight regulation of bacterial growth and death during colonization and infection.
FIGURE 10.

Summary model of membrane changes, ion transport events, and kinase activation during HAMLET-induced pneumococcal death. HAMLET encounters S. pneumoniae and triggers membrane perturbations including depolarization (detected by monitoring DiBAC fluorescence) and loss of integrity (propidium iodide fluorescence), with depolarization not sufficient for inducing death. Specific ion fluxes that are induced include an efflux of K+ (PBFI fluorescence) and an influx of Ca2+ (Fura-2 and Fluo-4 fluorescence; 45Ca2+ uptake). Although the efflux of K+ does not appear to be required for the bactericidal activity of HAMLET, the influx of Ca2+ does, and is tightly linked to sodium gradients, suggesting a sodium-calcium exchange mechanism. These HAMLET-induced transport events and death can be modulated by introducing related channel inhibitors or altering ion gradients. Additionally, staurosporine, the calcium-related kinase inhibitor, blocks bactericidal activity of HAMLET.
It is well-documented that breastfeeding protects infants against infections, including those of the upper respiratory tract, where S. pneumoniae is a major pathogen (52, 53). During our studies of this protective activity we identified HAMLET, a complex of human α-lactalbumin in a partially unfolded state that is stabilized with the human milk-specific fatty acids oleic and linoleic acid that constitutes ∼75% of the fatty acid content in human milk (27, 54). Active complex can be isolated from human milk, that in itself has antibacterial activity (23, 54), but the specific concentration of HAMLET present in human milk is difficult to quantify as milk contains other antibacterial components besides HAMLET, as the purification process has the potential to induce HAMLET complex formation, and as we lack specific tools to differentiate the bactericidal conformation of α-lactalbumin from the natively folded version. Still, the concentrations required to kill pneumococci (5–100 μg/ml) lie well within the physiological range, as the concentration of α-lactalbumin is especially high in human milk (2,000 μg/ml).
Our earlier studies have shown that HAMLET binds to the pneumococcal surface and induces death with morphological changes that are similar to those observed during eukaryote apoptosis (26). In this study, we characterized the initiation events leading to HAMLET-induced bacterial death, with a focus on membrane-associated events leading to depolarization and ion transport events that we already know are involved in HAMLET-induced tumor cell apoptosis involving mitochondria (33).
Membrane polarity and ion transport have been comprehensively studied in eukaryotic systems and are centrally responsible for various cellular processes and events, including apoptosis (55). Furthermore, methodology to study these events is well established. However, this is not true for prokaryotic systems. There is a significant paucity in the literature of studies concerning ion transport in bacteria and its role in cellular signaling (13), especially as it concerns bacterial death. With the exception of the studies by Trombe and co-workers (16, 51) of the role of calcium in pneumococcal autolysis, and previous work demonstrating that, in a few bacterial systems, various ionophores can enhance autolysis (12, 56), the role of ion transport in bacterial death has not been studied and no studies are available describing the specific ion transport events and signals required for autolysis activation under natural circumstances. Similarly, virtually nothing is known about the specific ion fluxes involved in the activation of bacterial death. Much of this can be attributed to logistical challenges, as the smaller size of bacteria is incompatible with standard methods of measurement (i.e. patch clamping) and makes ion transport studies in prokaryotes very difficult, which, we speculate, accounts for the lack of literature describing bacterial ion transport. The central novelty of this study, besides its findings, lies in the utilization and establishment of methodology to comprehensively study these events in Gram-positive organisms, providing the most comprehensive evaluation and characterization of membrane polarity and ion transport in a prokaryotic system to date.
Our results demonstrated that HAMLET associates with the bacterial surface and triggers dissipation of membrane polarity, with a subsequent loss of membrane integrity and eventual rupture. Some groups have speculated that alterations in membrane energy or polarity are involved in activating bacterial PCD, including pneumococcal autolysis (12, 57), but no specific data supporting this has been provided. However, several of our results suggested that depolarization in and of itself is not sufficient to activate HAMLET-induced death. First, high potassium in the extracellular environment (HKPBS), which caused membrane depolarization by itself, did not induce pneumococcal death, nor did it potentiate the bactericidal activity of HAMLET. Second, HAMLET effectively depolarized the membrane of S. aureus and E. coli, two species that are not killed by HAMLET; when the same degree of depolarization was induced in S. pneumoniae, it caused about 2 log10 of death. This suggested that membrane depolarization during HAMLET-induced death of pneumococci is an event that is parallel or secondary to specific ion transport events involved in triggering bacterial death.
Besides the role of potassium in maintaining membrane polarity, its efflux is an essential aspect of apoptosis activation in higher eukaryotes and some fungi, and is required for caspase and nuclease activation during the execution phase of cell death (35, 58, 59). In this study, we showed for the first time that potassium is transported out of a bacterial cell when induced to die, but also showed that this efflux was not required for HAMLET to induce pneumococcal death. This suggests that cell death regulation performed through the cytoplasmic concentration of potassium ions is potentially confined to eukaryotes and appeared at a later stage in evolution. Although the mechanism of potassium efflux in HAMLET-treated pneumococci is not fully clear, MSCs played a significant role due to the ability of gadolinium to inhibit a large portion of this efflux. Using gadolinium as an inhibitor, MSCs also appeared to play some role in HAMLET-induced death, which was independent of potassium. It can be speculated that MSCs could potentially transport other ions over the membrane that facilitate HAMLET-induced death or that gadolinium inhibits other transport mechanisms of importance for HAMLET-induced death. This will be investigated in more detail in the future.
As HAMLET causes a calcium-dependent depolarization of the mitochondrial membrane (33), and given the well-established role of calcium as an important signaling molecule involved in a wide variety of cellular processes (60, 61), we investigated the role of calcium in HAMLET-induced pneumococcal death. HAMLET caused a rapid and sustained increase in the intracellular concentration of calcium. Inhibition of this calcium influx by the calcium channel inhibitor RuR rescued the pneumococci from HAMLET-induced death. The potent inhibition of calcium transport and death seen also by the sodium transport inhibitors amiloride and DCB, combined with increased calcium influx and death associated with a decreased sodium gradient, strongly indicated a role for sodium in facilitating calcium influx, potentially through a sodium-calcium exchanger. An interesting parallel is that mitochondria express a NCX that is important for mitochondrial Ca2+ regulation during apoptosis (40). Sodium-calcium exchange activity has been suggested in earlier studies of the pneumococcus as the primary transport mechanism responsible for calcium uptake in the pneumococcus, with a direct link to optimal activation of competence and autolysis (16, 51, 62). Intriguingly, sodium-dependent calcium influx was also required for activation of pneumococcal death in stationary phase, as RuR and DCB both prolonged the duration of pneumococcal survival and integrity during this phase, whereas monensin and ionomycin (which facilitated HAMLET-induced death) induced earlier death in stationary phase growth without affecting growth in the logarithmic phase.
The specific transporter involved is still unidentified. No sodium/calcium motif transporters are annotated in any pneumococcal genome and attempts to isolate the transporter by co-immunoprecipitations with HAMLET have failed. We have also produced mutants resistant to high concentrations of DCB and sequenced them (data not shown). However, all mutants had point mutations in the gene annotated as cardiolipin synthetase, which likely results in pleiotropic membrane effects that indirectly affect transporter activity and HAMLET accessibility; no mutations were found in open reading frames that contain transmembrane domains.
Calcium is a major signaling ion in many eukaryotic pathways that often targets downstream kinases (46, 47). As a recent example, sodium-calcium exchange activity was shown to activate mitogen-activated protein kinases (MAPK), including p38 kinases, in a calcium-dependent manner, leading to elevation of reactive oxygen species and subsequent apoptotic death (63). Calcium-induced kinase activation has also recently been described for conjugated linoleic acid (48), a fatty acid that can associate with α-lactalbumin equally well as oleic acid with similar physiological effects (49). The fact that staurosporine, a kinase inhibitor, blocked HAMLET-induced death and also displayed the ability to extend stationary phase during normal growth of D39 bacteria, provides an additional link between physiological death and HAMLET-induced death. We have identified a potential staurosporine-sensitive kinase and are currently investigating its role in the bactericidal mechanism of HAMLET.3
As death is tightly regulated in bacterial communities during colonization or infection, our recent findings that pneumococci colonizing the murine nasopharyngeal tissue in vivo or epithelial cells in vitro grow in organized biofilms (30), combined with the potential overlap in molecules involved in HAMLET-induced death and physiological death, led us to predict that modulating components involved in the activation pathway of HAMLET would affect biofilm formation in vitro. As expected, we found that increasing the survival of the bacteria by inhibiting calcium influx to delay autolysis activation caused a significant decrease in biofilm biomass in vitro. This supports the postulated idea that bacteria that live in complex communities (e.g. biofilms) during colonization and infection require tight regulation of both bacterial survival and death for optimal function. It also supports the importance of the PCD systems that have been identified in bacteria and have been tied to a variety of important functions (5, 64, 65), including biofilm integrity. However, our studies are the first to mechanistically address the initiation of bacterial death.
HAMLET was recently shown to cause phenotypic changes in both S. pneumoniae and Haemophilus influenzae similar to those observed during eukaryotic apoptosis. Both bacterial species displayed cell shrinkage, DNA condensation, and high molecular weight fragmentation in association with HAMLET-induced death, and, at least for pneumococci, this was partially dependent on serine protease activity (26). We also have evidence that these organisms, as well as the HAMLET-insensitive species S. aureus and E. coli, all respond to starvation-induced death with apoptosis-like DNA fragmentation. Thus, even though HAMLET per se cannot trigger death in all bacteria, it is possible that the elements that make up the core pathway of death are conserved throughout a wider pool of bacterial species, but that the components responsible for activation of this pathway are presented with varying degrees of accessibility to HAMLET, resulting in varying degrees of bactericidal activation and corresponding susceptibility.
This concept is supported by the identification of apoptosis-like features of death seen in all organisms tested, and supports the widely accepted endosymbiotic theory that links mitochondria with bacteria. Furthermore, as HAMLET also specifically triggers apoptosis in tumor cells, we may speculate on the potential overlap in death pathways that exist between these two systems, with a more primitive, evolutionarily conserved PCD pathway that can be activated in bacteria.
Overall, the results of our present study provide a comprehensive framework for future investigation of the ion transport targets that are central to the activity of HAMLET and/or to activation of natural physiological bacterial death. Further elucidation of these pathways is a highly attractive endeavor, as the bacterial components that are responsible for executing these programs, possibly in many different bacterial species, hold enormous potential as novel drug targets and could be exploited by new antimicrobial and antitumor therapies to activate death in both systems.
Acknowledgments
We thank Dr. Catharina Svanborg for fruitful discussions of parallels that exist between HAMLET-induced death of eukaryotic and prokaryotic cells, and Drs. Svanborg and Ann-Kristin Mossberg for providing HAMLET preparations.
This work was supported, in whole or in part, by National Institutes of Health (NIDCD) Fellowship F31DC011218 (to E. A. C.), Bill and Melinda Gates Foundation Grant 53085, the J. R. Oishei Foundation, and American Lung Association Grant RG-123721-N (to A. P. H.).

This article contains supplemental Fig. S1.
L. R. Marks, E. A. Clementi, and A. P. Hakansson, unpublished observations.
- PCD
- programmed cell death
- AM
- acetoxymethyl
- MSC
- mechanosensitive channel
- DCB
- dichlorobenzamil
- HAMLET
- Human α-lactalbumin Made LEthal to Tumor cells
- HKPBS
- high-potassium PBS preparation
- RuR
- ruthenium red
- NCX
- Na+/Ca2+ exchanger
- DMSO
- dimethyl sulfoxide
- CFU
- colony-forming units.
REFERENCES
- 1. Costerton J. W., Stewart P. S., Greenberg E. P. (1999) Bacterial biofilms. A common cause of persistent infections. Science 284, 1318–1322 [DOI] [PubMed] [Google Scholar]
- 2. Stoodley P., Sauer K., Davies D. G., Costerton J. W. (2002) Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 56, 187–209 [DOI] [PubMed] [Google Scholar]
- 3. Wolcott R. D., Ehrlich G. D. (2008) Biofilms and chronic infections. JAMA 299, 2682–2684 [DOI] [PubMed] [Google Scholar]
- 4. Gerdes K., Christensen S. K., Løbner-Olesen A. (2005) Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 3, 371–382 [DOI] [PubMed] [Google Scholar]
- 5. Lewis K. (2008) Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 322, 107–131 [DOI] [PubMed] [Google Scholar]
- 6. Van Melderen L., Saavedra De Bast M. (2009) Bacterial toxin-antitoxin systems. More than selfish entities? PLoS Genet. 5, e1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bayles K. W. (2007) The biological role of death and lysis in biofilm development. Nat. Rev. Microbiol. 5, 721–726 [DOI] [PubMed] [Google Scholar]
- 8. Ranjit D. K., Endres J. L., Bayles K. W. (2011) Staphylococcus aureus CidA and LrgA proteins exhibit holin-like properties. J. Bacteriol. 193, 2468–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Claverys J. P., Håvarstein L. S. (2007) Cannibalism and fratricide. Mechanisms and raisons d'être. Nat. Rev. Microbiol. 5, 219–229 [DOI] [PubMed] [Google Scholar]
- 10. López D., Vlamakis H., Losick R., Kolter R. (2009) Cannibalism enhances biofilm development in Bacillus subtilis. Mol. Microbiol. 74, 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thomas V. C., Hiromasa Y., Harms N., Thurlow L., Tomich J., Hancock L. E. (2009) A fratricidal mechanism is responsible for eDNA release and contributes to biofilm development of Enterococcus faecalis. Mol. Microbiol. 72, 1022–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jolliffe L. K., Doyle R. J., Streips U. N. (1981) The energized membrane and cellular autolysis in Bacillus subtilis. Cell 25, 753–763 [DOI] [PubMed] [Google Scholar]
- 13. Dominguez D. C. (2004) Calcium signaling in bacteria. Mol. Microbiol. 54, 291–297 [DOI] [PubMed] [Google Scholar]
- 14. Corratgé-Faillie C., Jabnoune M., Zimmermann S., Véry A. A., Fizames C., Sentenac H. (2010) Potassium and sodium transport in non-animal cells. The Trk/Ktr/HKT transporter family. Cell. Mol. Life Sci. 67, 2511–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trombe M. C., Lanéelle G., Sicard A. M. (1984) Characterization of a Streptococcus pneumoniae mutant with altered electric transmembrane potential. J. Bacteriol. 158, 1109–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trombe M. C. (1993) Characterization of a calcium porter of Streptococcus pneumoniae involved in calcium regulation of growth and competence. J. Gen. Microbiol. 139, 433–439 [DOI] [PubMed] [Google Scholar]
- 17. Futsaether C. M., Johnsson A. (1994) Using fura-2 to measure intracellular free calcium in Propionibacterium acnes. Can. J. Microbiol. 40, 439–445 [DOI] [PubMed] [Google Scholar]
- 18. Tisa L. S., Adler J. (1995) Chemotactic properties of Escherichia coli mutants having abnormal Ca2+ content. J. Bacteriol. 177, 7112–7118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Werthén M., Lundgren T. (2001) Intracellular Ca2+ mobilization and kinase activity during acylated homoserine lactone-dependent quorum sensing in Serratia liquefaciens. J. Biol. Chem. 276, 6468–6472 [DOI] [PubMed] [Google Scholar]
- 20. Szabó I., Petronilli V., Zoratti M. (1992) A patch-clamp study of Bacillus subtilis. Biochim. Biophys. Acta 1112, 29–38 [DOI] [PubMed] [Google Scholar]
- 21. Zoratti M., Petronilli V., Szabo I. (1990) Stretch-activated composite ion channels in Bacillus subtilis. Biochem. Biophys. Res. Commun. 168, 443–450 [DOI] [PubMed] [Google Scholar]
- 22. Håkansson A., Svensson M., Mossberg A. K., Sabharwal H., Linse S., Lazou I., Lönnerdal B., Svanborg C. (2000) A folding variant of α-lactalbumin with bactericidal activity against Streptococcus pneumoniae. Mol Microbiol 35, 589–600 [DOI] [PubMed] [Google Scholar]
- 23. Håkansson A., Zhivotovsky B., Orrenius S., Sabharwal H., Svanborg C. (1995) Apoptosis induced by a human milk protein. Proc. Natl. Acad. Sci. U.S.A. 92, 8064–8068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Håkansson A., Andréasson J., Zhivotovsky B., Karpman D., Orrenius S., Svanborg C. (1999) Multimeric α-lactalbumin from human milk induces apoptosis through a direct effect on cell nuclei. Exp. Cell Res. 246, 451–460 [DOI] [PubMed] [Google Scholar]
- 25. Margulis L. (1975) Symbiotic theory of the origin of eukaryotic organelles. Criteria for proof. Symp. Soc. Exp. Biol. 29, 21–38 [PubMed] [Google Scholar]
- 26. Hakansson A. P., Roche-Hakansson H., Mossberg A. K., Svanborg C. (2011) Apoptosis-like death in bacteria induced by HAMLET, a human milk lipid-protein complex. PLoS One 6, e17717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Svensson M., Håkansson A., Mossberg A. K., Linse S., Svanborg C. (2000) Conversion of α-lactalbumin to a protein inducing apoptosis. Proc. Natl. Acad. Sci. U.S.A. 97, 4221–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Avery O. T., Macleod C. M., McCarty M. (1944) Studies on the chemical nature of the substance inducing transformation of pneumococcal types. Induction of transformation by a desoxyribonucleic acid fraction isolated from Pneumococcus type III. J. Exp. Med. 79, 137–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berry A. M., Lock R. A., Hansman D., Paton J. C. (1989) Contribution of autolysin to virulence of Streptococcus pneumoniae. Infect. Immun. 57, 2324–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marks L. R., Parameswaran G. I., Hakansson A. P. (2012) Pneumococcal interactions with epithelial cells are crucial for optimal biofilm formation and colonization in vitro and in vivo. Infect. Immun., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spangler S. K., Jacobs M. R., Appelbaum P. C. (1997) Time-kill studies on susceptibility of nine penicillin-susceptible and -resistant pneumococci to cefditoren compared with nine other β-lactams. J. Antimicrob. Chemother. 39, 141–148 [DOI] [PubMed] [Google Scholar]
- 32. Johansen H. K., Jensen T. G., Dessau R. B., Lundgren B., Frimodt-Moller N. (2000) Antagonism between penicillin and erythromycin against Streptococcus pneumoniae in vitro and in vivo. J. Antimicrob. Chemother. 46, 973–980 [DOI] [PubMed] [Google Scholar]
- 33. Köhler C., Gogvadze V., Håkansson A., Svanborg C., Orrenius S., Zhivotovsky B. (2001) A folding variant of human α-lactalbumin induces mitochondrial permeability transition in isolated mitochondria. Eur. J. Biochem. 268, 186–191 [DOI] [PubMed] [Google Scholar]
- 34. Bortner C. D., Hughes F. M., Jr., Cidlowski J. A. (1997) A primary role for K+ and Na+ efflux in the activation of apoptosis. J. Biol. Chem. 272, 32436–32442 [DOI] [PubMed] [Google Scholar]
- 35. Hughes F. M., Jr., Bortner C. D., Purdy G. D., Cidlowski J. A. (1997) Intracellular K+ suppresses the activation of apoptosis in lymphocytes. J. Biol. Chem. 272, 30567–30576 [DOI] [PubMed] [Google Scholar]
- 36. Franks C. J., Pemberton D., Vinogradova I., Cook A., Walker R. J., Holden-Dye L. (2002) Ionic basis of the resting membrane potential and action potential in the pharyngeal muscle of Caenorhabditis elegans. J. Neurophysiol. 87, 954–961 [DOI] [PubMed] [Google Scholar]
- 37. Bakker E. P., Harold F. M. (1980) Energy coupling to potassium transport in Streptococcus faecalis. Interplay of ATP and the protonmotive force. J. Biol. Chem. 255, 433–440 [PubMed] [Google Scholar]
- 38. Petit P. X., Goubern M., Diolez P., Susin S. A., Zamzami N., Kroemer G. (1998) Disruption of the outer mitochondrial membrane as a result of large amplitude swelling. The impact of irreversible permeability transition. FEBS Lett. 426, 111–116 [DOI] [PubMed] [Google Scholar]
- 39. Giorgi C., Romagnoli A., Pinton P., Rizzuto R. (2008) Ca2+ signaling, mitochondria and cell death. Curr. Mol. Med. 8, 119–130 [DOI] [PubMed] [Google Scholar]
- 40. Miyamoto S., Howes A. L., Adams J. W., Dorn G. W., 2nd., Brown J. H. (2005) Ca2+ dysregulation induces mitochondrial depolarization and apoptosis. Role of Na+/Ca2+ exchanger and AKT. J. Biol. Chem. 280, 38505–38512 [DOI] [PubMed] [Google Scholar]
- 41. Tettelin H., Nelson K. E., Paulsen I. T., Eisen J. A., Read T. D., Peterson S., Heidelberg J., DeBoy R. T., Haft D. H., Dodson R. J., Durkin A. S., Gwinn M., Kolonay J. F., Nelson W. C., Peterson J. D., Umayam L. A., White O., Salzberg S. L., Lewis M. R., Radune D., Holtzapple E., Khouri H., Wolf A. M., Utterback T. R., Hansen C. L., McDonald L. A., Feldblyum T. V., Angiuoli S., Dickinson T., Hickey E. K., Holt I. E., Loftus B. J., Yang F., Smith H. O., Venter J. C., Dougherty B. A., Morrison D. A., Hollingshead S. K., Fraser C. M. (2001) Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293, 498–506 [DOI] [PubMed] [Google Scholar]
- 42. Lanie J. A., Ng W. L., Kazmierczak K. M., Andrzejewski T. M., Davidsen T. M., Wayne K. J., Tettelin H., Glass J. I., Winkler M. E. (2007) Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J. Bacteriol. 189, 38–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rosch J. W., Sublett J., Gao G., Wang Y. D., Tuomanen E. I. (2008) Calcium efflux is essential for bacterial survival in the eukaryotic host. Mol. Microbiol. 70, 435–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neef J., Andisi V. F., Kim K. S., Kuipers O. P., Bijlsma J. J. (2011) Deletion of a cation transporter promotes lysis in Streptococcus pneumoniae. Infect. Immun. 79, 2314–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hakansson A. (1999) Apoptosis induced by a human milk protein complex. Cellular and structural studies in tumour cells and bacteria. Ph.D. thesis, Department of Laboratory Medicine, Lund University Medical School, Lund, Sweden [Google Scholar]
- 46. Spat A., Fulop L., Koncz P., Szanda G. (2009) When is high-Ca+ microdomain required for mitochondrial Ca+ uptake? Acta Physiol. 195, 139–147 [DOI] [PubMed] [Google Scholar]
- 47. Brnjic S., Olofsson M. H., Havelka A. M., Linder S. (2010) Chemical biology suggests a role for calcium signaling in mediating sustained JNK activation during apoptosis. Mol. Biosyst. 6, 767–774 [DOI] [PubMed] [Google Scholar]
- 48. Hsu Y. C., Ip M. M. (2011) Conjugated linoleic acid-induced apoptosis in mouse mammary tumor cells is mediated by both G protein-coupled receptor-dependent activation of the AMP-activated protein kinase pathway and by oxidative stress. Cell. Signal. 23, 2013–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Svensson M., Mossberg A. K., Pettersson J., Linse S., Svanborg C. (2003) Lipids as cofactors in protein folding. Stereo-specific lipid-protein interactions are required to form HAMLET (human α-lactalbumin made lethal to tumor cells). Protein Sci. 12, 2805–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nováková L., Sasková L., Pallová P., Janecek J., Novotná J., Ulrych A., Echenique J., Trombe M. C., Branny P. (2005) Characterization of a eukaryotic type serine/threonine protein kinase and protein phosphatase of Streptococcus pneumoniae and identification of kinase substrates. FEBS J. 272, 1243–1254 [DOI] [PubMed] [Google Scholar]
- 51. Trombe M. C., Clavé C., Manias J. M. (1992) Calcium regulation of growth and differentiation in Streptococcus pneumoniae. J. Gen. Microbiol. 138, 77–84 [DOI] [PubMed] [Google Scholar]
- 52. Aniansson G., Alm B., Andersson B., Håkansson A., Larsson P., Nylén O., Peterson H., Rignér P., Svanborg M., Sabharwal H. (1994) A prospective cohort study on breastfeeding and otitis media in Swedish infants. Pediatr. Infect. Dis. J. 13, 183–188 [DOI] [PubMed] [Google Scholar]
- 53. Hanson L. A. (1998) Breastfeeding provides passive and likely long-lasting active immunity. Ann. Allergy Asthma Immunol. 81, 523–533 [DOI] [PubMed] [Google Scholar]
- 54. Svensson M., Sabharwal H., Håkansson A., Mossberg A. K., Lipniunas P., Leffler H., Svanborg C., Linse S. (1999) Molecular characterization of α-lactalbumin folding variants that induce apoptosis in tumor cells. J. Biol. Chem. 274, 6388–6396 [DOI] [PubMed] [Google Scholar]
- 55. Lehen'kyi V., Shapovalov G., Skryma R., Prevarskaya N. (2011) Ion channnels and transporters in cancer. 5. Ion channels in control of cancer and cell apoptosis. Am. J. Physiol. Cell Physiol. 301, C1281–1289 [DOI] [PubMed] [Google Scholar]
- 56. Harold F. M. (1972) Conservation and transformation of energy by bacterial membranes. Bacteriol. Rev. 36, 172–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Frias M. J., Melo-Cristino J., Ramirez M. (2009) The autolysin LytA contributes to efficient bacteriophage progeny release in Streptococcus pneumoniae. J. Bacteriol. 191, 5428–5440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bortner C. D., Cidlowski J. A. (2007) Cell shrinkage and monovalent cation fluxes. Role in apoptosis. Arch. Biochem. Biophys. 462, 176–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Andrés M. T., Viejo-Díaz M., Fierro J. F. (2008) Human lactoferrin induces apoptosis-like cell death in Candida albicans. Critical role of K+-channel-mediated K+ efflux. Antimicrob. Agents Chemother. 52, 4081–4088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Carafoli E. (1987) Intracellular calcium homeostasis. Annu. Rev. Biochem. 56, 395–433 [DOI] [PubMed] [Google Scholar]
- 61. Smith R. J. (1995) Calcium and bacteria. Adv. Microb. Physiol. 37, 83–133 [DOI] [PubMed] [Google Scholar]
- 62. Trombe M. C., Rieux V., Baille F. (1994) Mutations which alter the kinetics of calcium transport alter the regulation of competence in Streptococcus pneumoniae. J. Bacteriol. 176, 1992–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nashida T., Takuma K., Fukuda S., Kawasaki T., Takahashi T., Baba A., Ago Y., Matsuda T. (2011) The specific Na+/Ca2+ exchange inhibitor SEA0400 prevents nitric oxide-induced cytotoxicity in SH-SY5Y cells. Neurochem. Int. 59, 51–58 [DOI] [PubMed] [Google Scholar]
- 64. Lewis K. (2000) Programmed death in bacteria. Microbiol. Mol. Biol. Rev. 64, 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Engelberg-Kulka H., Amitai S., Kolodkin-Gal I., Hazan R. (2006) Bacterial programmed cell death and multicellular behavior in bacteria. PLoS Genet. 2, e135. [DOI] [PMC free article] [PubMed] [Google Scholar]