

Background: Chronic inflammation is becoming a hallmark in various neurodegenerative disorders.
Results: Gemfibrozil, an FDA-approved drug for hyperlipidemia, up-regulates the expression of SOCS3, capable of suppressing inflammation, in glial cells.
Conclusion: Gemfibrozil increases SOCS3 via PI 3-kinase-mediated activation of KLF4.
Significance: Gemfibrozil may be of therapeutic benefit in different neuroinflammatory and neurodegenerative diseases.
Keywords: Kruppel-like Factor (KLF), Neurodegeneration, Neuroinflammation, Phosphatidylinositol 3-Kinase, Signal Transduction
Abstract
Glial inflammation is an important feature of several neurodegenerative disorders. Suppressor of cytokine signaling (SOCS) proteins play a crucial role in inhibiting cytokine signaling and inflammatory gene expression in various cell types, including glial cells. However, mechanisms by which SOCS genes could be up-regulated are poorly understood. This study underlines the importance of gemfibrozil, a Food and Drug Administration-approved lipid-lowering drug, in up-regulating the expression of SOCS3 in glial cells. Gemfibrozil increased the expression of Socs3 mRNA and protein in mouse astroglia and microglia in both a time- and dose-dependent manner. Interestingly, gemfibrozil induced the activation of type IA phosphatidylinositol (PI) 3-kinase and AKT. Accordingly, inhibition of PI 3-kinase and AKT by chemical inhibitors abrogated gemfibrozil-mediated up-regulation of SOCS3. Furthermore, we demonstrated that gemfibrozil induced the activation of Krüppel-like factor 4 (KLF4) via the PI 3-kinase-AKT pathway and that siRNA knockdown of KLF4 abrogated gemfibrozil-mediated up-regulation of SOCS3. Gemfibrozil also induced the recruitment of KLF4 to the distal, but not proximal, KLF4-binding site of the Socs3 promoter. This study delineates a novel property of gemfibrozil in up-regulating SOCS3 in glial cells via PI 3-kinase-AKT-mediated activation of KLF4 and suggests that gemfibrozil may find therapeutic application in neuroinflammatory and neurodegenerative disorders.
Introduction
Neurodegenerative disorders, including Alzheimer disease, Parkinson disease, Huntington disease, amyotrophic lateral sclerosis, multiple sclerosis, tauopathies, and age-related macular degeneration, can be characterized by the loss of neurons in motor, sensory, or cognitive systems (1–5). Neuropathological studies indicate that neuroinflammatory responses, including elevated levels of proinflammatory cytokines, may begin prior to significant loss of neuronal populations in the progression of these diseases (2).
The dynamic immune and inflammatory response evoked in the CNS has been attributed mostly to the activation of microglia, CNS-resident macrophage and sensor cells that function as the principal immune effector cell of the CNS responding to any pathological event like infections, trauma, stroke, toxins, and other stimuli (6). Upon activation, microglia express various proinflammatory cytokines, proinflammatory enzymes, proinflammatory adhesion molecules, etc. The excessive production of these proinflammatory molecules trigger nitrosative and oxidative stress (in case of acute inflammation) and may result in prolonged microglial activation with the sustained production of these molecules (in case of chronic inflammation), which results in perpetuated inflammatory cycle, activating additional microglia, promoting their proliferation, and resulting in further release of inflammatory factors. Thus, the cytokine-mediated inflammatory response plays an important role in initiating and/or enhancing the degenerative process in the inflamed CNS (7).
Suppressor of cytokine signaling (SOCS)2 is a group of STAT-induced STAT inhibitor family proteins and as the name suggests plays a key role in inducing a negative feedback inhibition on the cytokine-transduced signaling pathways. There are eight SOCS proteins, SOCS1, SOCS2, SOCS3, SOCS4, SOCS5, SOCS6, SOCS7, and the cytokine-induced SRC homology 2 (SH2) protein CIS. Each of the SOCS proteins contains a central SH2 domain, an amino-terminal domain with variable length and sequence, and a carboxyl-terminal 40-amino acid region termed as the SOCS box. In addition, SOCS1 and SOCS3 have a kinase inhibitory region in their amino-terminal domain. The SOCS proteins can be induced by a number of stimuli and inhibit the JAK-STAT pathway using a classic inhibitory feedback loop (8, 9). According to recent studies, SOCS3 cannot only inhibit IL-6 signaling by inhibiting the activation of STAT3 but it can also function as a positive regulator for the ERK-MAPK pathway, inhibitor of the NF-κB pathway, and may act as an antagonist to the cAMP-mediated signaling (10–13). Therefore, Socs3 gene disrupted mice show embryonic lethality at 12–16 days with severe erythrocytosis (14). SOCS3-deficient (in hemopoietic and endothelial cell) mice also exhibit IL-1-dependent arthritis, which could be suppressed by forced expression of SOCS3. Again, intracellular administration of cell-penetrating SOCS3 could inhibit the cytokine-mediated acute inflammatory response (15). SOCS3 can also inhibit chemokine-mediated chemotaxis in T-cells by binding to the chemokine receptor that attenuates the chemotaxis (16). All this evidence reinforces the fact that SOCS3 is one of the primary regulators of cytokine-induced inflammatory signaling. Hence, the up-regulation SOCS3 could be useful in suppressing the inflammation and pain associated with chronic inflammatory diseases, and identification of pharmacological drugs that could up-regulate SOCS3 is an important area of study.
Gemfibrozil, popularly known as “Lopid” in pharmacy, is long known for its ability to reduce the level of triglycerides in the blood circulation and to decrease the risk of hyperlipidemia (17–19). However, a number of recent studies reveal that apart from its lipid-lowering effects, gemfibrozil can also regulate many other signaling pathways responsible for inflammation, switching of T-helper cells, cell-to-cell contact, migration, and oxidative stress (20–22). We examined the efficacy of gemfibrozil, a Food and Drug Administration-approved lipid-lowering drug, in up-regulation of the expression of SOCS3. Here, we demonstrate for the first time that gemfibrozil is capable of increasing the expression of SOCS3 in glial cells via type IA phosphatidylinositol-3 kinase-AKT-mediated activation of KLF4.
MATERIALS AND METHODS
Reagents
DMEM/F-12 50:50 1×, Hanks' balanced salt solution, and 0.05% trypsin were purchased from Mediatech (Washington, D. C.). Fetal bovine serum (FBS) was obtained from Atlas Biologicals (Fort Collins, CO). Antibiotic-antimycotic, gemfibrozil, and Akt inhibitor (AKT-I) were obtained from Sigma. Wortmannin and LY294002 were obtained from Calbiochem.
Isolation of Mouse Primary Microglia
Microglial cells were isolated from mixed glial cultures as described earlier by us (23) according to the procedure of Giulian and Baker (24). Animal maintenance and experimental protocols were approved by the Rush University Animal Care Committee. Briefly, mixed glial cells were prepared from 7-to 9-day-old mouse pups. On day 9, the mixed glial cultures were washed three times with DMEM/F-12 and subjected to shaking at 240 rpm for 2 h at 37 °C on a rotary shaker. The floating cells were washed and seeded onto plastic tissue culture flasks and incubated at 37 °C for 1 h. The attached cells were removed by trypsinization and seeded onto new plates for further studies. To monitor purity, cells were immunostained with Abs (Pharmingen) against Mac-1 surface Ag, a marker for microglia/macrophages. 90–95% of this preparation was found to be positive for Mac-1. Mouse BV-2 microglial cells (a gift from V. Bocchini, University of Perugia, Perugia, Italy) were also maintained as indicated above. All treatments (with gemfibrozil and PI3K/AKT inhibitors) were done in serum-free DMEM/F-12.
Semi-quantitative Reverse Transcriptase (RT)-coupled PCR
Total RNA was isolated from BV-2 and mouse primary astrocytes using RNA-Easy Qiagen (Valencia, CA) kit following the manufacturer's protocol. Semi-quantitative RT-PCR was carried out as described earlier (25) using oligo(dT)12–18 as primer and Moloney murine leukemia virus reverse transcriptase (MMLV-RT, Invitrogen) in a 20-μl reaction mixture. The resulting cDNA was appropriately amplified using Promega Master Mix (Madison, WI) and the following primers (Invitrogen) for murine genes: Socs1 sense, 5′-GGCAGCCGACAATGCGATCT-3′, and antisense, 5′-GATCTGGAAGGGGAAGGAAC-3′; Socs2 sense, 5′-GTTGCCGGAGGAACAGTCCC-3′, and antisense, 5′-ATGCTGCAGAGTGGGTGCTG-3′; Socs3 sense, 5′-CGCCTCAAGACCTTCAGCTC-3′, and antisense, 5′-CTGATCCAGGAACTCCCGAA-3′; Klf4 sense, 5′-CCCGGCGGGAAGGGAGAAGA-3′, and antisense, 5′- AACTTGCCCATCAGCCCGCC-3′; Gapdh sense, 5′-GGT GAA GGT CGG AGT CAA CG-3′, and antisense, 5′-GTG AAG ACG CCA GTG GAC TC-3′.
Amplified products were electrophoresed on 2% agarose (Invitrogen) gels and visualized by ethidium bromide (Invitrogen) staining. Response of the glyceraldehyde-3-phosphate dehydrogenase (Gapdh) gene was used as a loading control to ascertain that an equivalent amount of cDNA was synthesized from each sample.
Quantitative Real Time PCR
The mRNA quantification was performed using the ABI-Prism7700 sequence detection system (Applied Biosystems, Foster City, CA) using iTaqTM Fast Supermix With ROX (Bio-Rad) and the following 6-FAM/ZEN/IBFQ-labeled primers for murine genes as follows: Socs3, Klf4, and Gapdh (Integrated DNA Technologies Coralville, IA). The mRNA expression of the targeted genes was normalized to the level of Gapdh mRNA, and data were processed by the ABI Sequence Detection System 1.6 software.
Immunostaining
Immunocytochemistry was performed as described earlier (26). Briefly, 8-well chamber slides containing BV-2 or mouse primary microglia or astrocytes cultured to 70–80% confluence were fixed with chilled methanol (Fisher Scientific, Waltham, MA) overnight, followed by two brief rinses with filtered PBS. Samples were blocked with 2% BSA (Fisher Scientific) in PBS containing Tween 20 (Sigma) and Triton X-100 (Sigma) for 30 min and incubated at room temperature under shaking conditions for 2 h in PBS containing the following anti-mouse primary antibodies: SOCS3 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA), p-AKT (1:600; Cell Signaling, Beverly, MA), total AKT (1:600; Cell Signaling), GFAP (1:100; Santa Cruz Biotechnology), CD11b (1:500; Cedarlane Laboratories, Burlington, NC), and MAP-2 (1:50; Santa Cruz Biotechnology). After four 15-min washes in filtered PBS, slides were further incubated with Cy2-, Cy3-, or Cy5-labeled secondary antibodies (all 1:200; Jackson ImmunoResearch, West Grove, PA) for 1 h under similar shaking conditions. Following four 15-min washes with filtered PBS, cells were incubated for 4–5 min with 4′,6-diamidino-2-phenylindole (DAPI, 1:10,000; Sigma). The samples were run in an EtOH and xylene (Fisher) gradient, mounted, and observed under a Bio-Rad MRC1024ES confocal laser-scanning microscope (27).
Transfection
Cells were transfected with RNAiMax reagent (Invitrogen) according to the manufacturer's protocol. Briefly, BV-2 or astrocytes cultured in 12-well plates were transfected with 0.5–1.0 μg/well of control or KLF4 siRNA (Dharmacon RNAi Technologies) complexed with RNAiMax reagent in serum-free DMEM/F containing l-glutamine (Invitrogen) for 5 h. After 5 h, 20% FBS was added at a volume of 1:1 with the transfection media, and cells were further incubated for 36 h.
Immunoblotting
Western blotting was conducted as described earlier (28) with modifications. Briefly, cells were scraped in double-distilled H2O and SDS and electrophoresed on NuPAGE® Novex® 4–12% BisTris gels (Invitrogen), and proteins were transferred onto a nitrocellulose membrane (Bio-Rad) using the Thermo-Pierce Fast Semi-Dry Blotter. The membrane was then washed for 15 min in TBS plus Tween 20 (TBST) and blocked for 1 h in TBST containing BSA. Next, membranes were incubated overnight at 4 °C under shaking conditions with the following 1° antibodies: SOCS3 (1:250, Santa Cruz Biotechnology), AKT, p-AKT (1:600; Cell Signaling), β-actin (1:800; Abcam, Cambridge, MA), p110α, p110β, and p110γ (all 1:200; Santa Cruz Biotechnology); and KLF4 (1:500, Abcam). The next day, membranes were washed in TBST for 1 h, incubated in 2° antibodies against 1° antibody hosts (all 1:10,000; Jackson ImmunoResearch) for 1 h at room temperature, washed for 1 more h, and visualized under the Odyssey® Infrared Imaging System (Li-COR, Lincoln, NE).
Densitometric Analysis
Protein blots were analyzed using ImageJ (National Institutes of Health, Bethesda), and bands were normalized to their respective β-actin loading controls. Data are representative of the average fold change with respect to control for three independent experiments.
Cellular Membrane Extraction
Neuronal membranes were isolated to determine the recruitment of various membrane-associated proteins to the membrane. Cells were washed with PBS and scraped in phenol red-free Hanks' balanced salt solution to 5-ml ultracentrifuge tubes. The solution was then diluted with 100 mm sodium bicarbonate buffer, pH 11.5, and spun in an ultracentrifuge at 40,000 rpm for 1 h at 4 °C. The resultant supernatant liquid was aspirated, and the pellet was immersed in double-distilled H2O and SDS and stored at −80 °C overnight. The following day, the pellet was resuspended by repeated grinding and boiling.
Nuclear Extraction and Gel Shift
DNA binding activity of KLF4 was analyzed by nonradioactive electrophoretic mobility shift assay (EMSA). After treatment, cells were washed with PBS, scraped into 1.5-ml tubes, and centrifuged at 4 °C for 5 min at 500 rpm. The supernatant was aspirated, and the pellet was resuspended in a membrane lysis buffer composed of HEPES, pH 8.0, MgCl2, KCl, dithiothreitol (DTT), and protease/phosphatase inhibitors, vortexed, and centrifuged at 4 °C at 15,000 rpm for 3 min. Again, the supernatant was aspirated, and the pellet was resuspended in a high salt nuclear envelope lysis buffer composed of HEPES, pH 8.0, MgCl2, glycerol, NaCl, EDTA, DTT, and protease/phosphatase inhibitors, rotated vigorously at 4 °C for 30 min, and centrifuged at 4 °C at 15,000 rpm for 15 min. The resultant nuclear extract pellet was frozen at −80 °C overnight, resuspended in a mixture of binding buffer, 10× Orange Loading Dye (Li-Cor Biosciences), a custom-designed fluorescent KLF4-specific probe (Li-Cor Biosciences), and salmon sperm DNA (Invitrogen) and electrophoresed on custom-cast 6% polyacrylamide gels. The shift was visualized under the Odyssey® Infrared Imaging System (Li-Cor).
Immunoprecipitation
BV-2 cells were treated with 50 μm gemfibrozil for different time points, and cell extracts were first precleared with 25 μl of protein A-agarose (50%, v/v). The supernatants were immunoprecipitated with 5 μg of anti-phosphoserine (Santa Cruz Biotechnology, Inc.) overnight at 4 °C, followed by incubation with protein A-agarose for 4 h at 4 °C. Protein A-agarose-antigen-antibody complexes were collected by centrifugation at 12,000 rpm for 60 s at 4 °C. The pellets were washed five times with 1 ml of IPH buffer (20 mm Tris-HCl, pH 8.0, 137 mm NaCl, 2 mm EDTA, 1% Nonidet P-40, 10% glycerol, 0.1 mm phenylmethylsulfonyl fluoride) for 20 min each time at 4 °C. Bound proteins were resolved by SDS-PAGE, followed by Western blotting with the anti-KLF4.
ChIP Assay
ChIP assays were performed using a kit (Upstate Biotechnology) according to the manufacturer's protocol. Briefly, 2 × 106 microglial cells preincubated with gemfibrozil for 2 h were stimulated with LPS. After 3 h of stimulation, cells were fixed by adding formaldehyde (1% final concentration), and cross-linked adducts were resuspended and sonicated, resulting in an average chromatin fragment size of 400 bp. ChIP was performed on the cell lysate by overnight incubation at 4 °C with 2 μg of Abs against KLF4 (and normal IgG as negative control) followed by incubation with protein G-agarose (Santa Cruz Biotechnology) for 2 h. The beads were washed and incubated with elution buffer. To reverse the cross-linking and purify the DNA, precipitates were incubated in a 65 °C incubator overnight and digested with proteinase K. DNA samples were then purified and precipitated, and precipitates were washed with 75% ethanol, air-dried, and resuspended in Tris-EDTA buffer. The following primers were used to amplify fragments flanking proximal KLF4-binding elements in the mouse Socs3 promoter: Set1 sense, 5′-GGT TGG CAC GGG ATG CTT GGA-3′, and antisense, 5′-AGT TCT AGG AGG CCA GCC GGG-3′; Set2 sense, 5′-ACG TCC CTT TTG GTT GGC ACG-3′, and antisense, 5′-GCC AGC CGG GAG AGG CT-3′. The following primers were used to amplify fragments flanking distal KLF4-binding elements in the mouse Socs3 promoter: Set1 sense, 5′-CTCCCATCGCGACGCCCC-3′, and antisense, 5′-CGCGGCGCCAGCCTT-3′; Set2 sense, 5′-CCCCGCCTCTGCCAGAAACC-3′, and antisense 5′-CAGCCTTGGCCGAGCGTTCC-3′. The PCRs were repeated by using varying cycle numbers and different amounts of templates to ensure that results were in the linear range of PCR.
Statistics
Values are expressed as means ± S.D. of at least three independent experiments. Statistical analyses for differences were performed via Student's t test. This criterion for statistical significance was p < 0.05.
RESULTS
Gemfibrozil Up-regulates SOCS3 in Mouse Glial Cells
The up-regulation of SOCS proteins could be immensely useful in inhibiting the cytokine-mediated inflammatory responses in the CNS (10). We examined if gemfibrozil could up-regulate the expression of SOCS proteins in mouse BV-2 microglial cells and primary microglia. We found that gemfibrozil indeed up-regulated the mRNA expression of Socs1, Socs2, and Socs3 within 1 h of treatment in a dose-dependent manner (Fig. 1, A and B). This induction was dose-dependent, and the maximum increase in Socs3 mRNA (almost 13-fold) was observed at 50 μm gemfibrozil (Fig. 1B). Time course study showed that induction of SOCS genes started from as early as 15 min and peaked at 60 min (∼12-fold) (Fig. 1, C and D). We further checked the SOCS3 protein levels in BV-2 cells. We observed an ∼3–5-fold increase in the protein levels of SOCS3 in BV-2 cells when treated with gemfibrozil in a dose- and time-dependent manner, which is consistent with our mRNA findings. The SOCS3 protein levels were normalized to β-actin, and densitometric analysis showed a maximum increase with 50 μm gemfibrozil (>3-fold), and the time course study revealed steady increase of SOCS3 over time (4–6-fold) (Fig. 1, E and F). Next, we examined if gemfibrozil could up-regulate the expression of SOCS3 in primary microglia. Therefore, we treated mouse primary microglia with gemfibrozil for several minutes and conducted double label immunofluorescence. Similar to that observed in BV-2 microglial cells, gemfibrozil markedly increased the expression of SOCS3 protein in primary microglia at different times of stimulation (Fig. 1G).
FIGURE 1.

Up-regulation of SOCS3 by gemfibrozil in microglia. Mouse BV-2 microglial cells were treated with different concentrations of gemfibrozil in serum-free DMEM/F-12 for 1 h followed by monitoring the mRNA expression of Socs1/2/3 by semi-quantitative RT-PCR (A), real time PCR (for Socs3) (B), and Western blot (E). Graph represents densitometric analysis of SOCS3 protein levels normalized to β-actin (loading control). BV-2 cells were treated with 50 μm gemfibrozil for different minutes under the same culture conditions followed by monitoring the mRNA expression of Socs1/2/3 by semi-quantitative RT-PCR (C), real time PCR (for Socs3) (D), and Western blot (F). Graph represents densitometric analysis of SOCS3 protein levels normalized to β-actin (loading control). G, mouse primary microglia were treated with 50 μm gemfibrozil for different minutes under the same culture conditions and were double-labeled for SOCS3 and CD11b. DAPI was used to stain nuclei. All results are mean ± S.D. of at least three independent experiments. a, p < 0.001 versus control. UN, untreated; GEM, gemfibrozil. Scale bar, 20 μm.
To further extend our findings in microglia, we took mouse primary astrocytes and conducted both mRNA and protein expression analyses for SOCS3. The quantitative RT-PCR on astrocytes treated with gemfibrozil in a dose- and time-dependent manner showed similar patterns of mRNA expression as observed in microglia. The maximum increase in Socs3 mRNA levels was with 50 μm gemfibrozil (>12-fold), and a significant increase was found as early as 15 min (∼5-fold) and a maximum was at 60 min (∼9-fold) post-treatment (Fig. 2, A and B). In the protein study, we also observed an almost 3-fold increase in the SOCS3 protein levels by gemfibrozil in a dose-dependent manner (Fig. 2C). The protein levels were normalized to β-actin, and the densitometry analysis confirmed that the maximum increase (∼3-fold) was at 50 μm gemfibrozil. We also performed a time course for SOCS3 and observed an almost 8-fold increase at 90 min of stimulation (Fig. 2D). To confirm our findings further, we performed immunofluorescence in mouse primary astrocytes. It is evident from Fig. 2E, gemfibrozil up-regulated the expression of SOCS3 protein in primary astrocytes in a time-dependent manner.
FIGURE 2.

Up-regulation of SOCS3 by gemfibrozil in astrocytes. Mouse primary astrocytes (MPA) were treated with different concentrations of gemfibrozil in serum-free DMEM/F-12 for 1 h followed by monitoring the mRNA expression of Socs3 by real time PCR (A) and Western blot (C). Graph represents densitometric analysis of SOCS3 protein levels normalized to β-actin (loading control (C)). Mouse primary astrocytes were treated with 50 μm gemfibrozil (GEM) for different minutes under the same culture conditions followed by monitoring the mRNA expression of Socs3 by real time PCR (B) and Western blot (D). Graph represents densitometric analysis of SOCS3 protein levels normalized to β-actin (loading control). E, mouse primary astrocytes were treated with 50 μm gemfibrozil for different minutes under the same culture conditions and were double-labeled for SOCS3 and GFAP. DAPI was used to stain nuclei. Results are mean ± S.D. of at least three independent experiments. a, p < 0.001 versus control. Scale bar, 50 μm. UN, untreated.
Gemfibrozil Requires the Activation of Phosphatidylinositol 3-Kinase (PI3K) to Up-regulate SOCS3
A number of cytokines and other biomolecules have been reported to induce SOCS3 (9, 29–31, 43). However, there are a few pharmacological compounds that are capable of up-regulating SOCS3, and the mechanism of drug-induced up-regulation of SOCS3 is also poorly understood. Because gemfibrozil-induced up-regulation of SOCS3 in glial cells was very rapid, and in our earlier study (32) gemfibrozil induced the activation of phosphatidylinositol-3 kinase (PI 3-kinase), a member of growth-supportive survival kinases, within minutes we decided to monitor the activation PI3K. There are two classes of PI3K. Class IA PI3K consists of a heterodimer of a regulatory 85-kDa subunit and a catalytic 110-kDa subunit (p85:p110α/p110β) (33), whereas class IB PI3K consists of a dimer of a 101-kDa regulatory subunit and a p110γ catalytic subunit (p101:p110γ) (34). The activation of PI3K causes the translocation of these subunits from the cytoplasm to the plasma membrane (35). Therefore, we monitored the recruitment of p110α, p110β, and p110γ to the membrane to ascertain the activation of class IA and class IB PI3K. Western blotting of membrane fractions for p110 subunits suggests that gemfibrozil specifically induces the recruitment of p110α (Fig. 3A), but neither p110β nor p110γ, to the plasma membrane. The densitometric analysis of the blots showed about 10-fold increase in the membrane recruitment of the p110α within 15 min of stimulation with gemfibrozil, whereas no significant recruitment of the other subunits was observed (Fig. 3B).
FIGURE 3.

Activation of PI3K and AKT by gemfibrozil in microglia. A, mouse BV-2 microglial cells were treated with 50 μm gemfibrozil (Gem) under serum-free conditions for different minutes followed by analysis of the recruitment of p110α (panel i), p110β (panel ii), and p110γ (panel iii) to the cellular membrane via Western blot. TLR2 was used as a loading control for membrane fragments. B, densitometric analysis of dose-dependent change (relative to TLR2) of PI3K subunits by gemfibrozil treatment. C, BV-2 cells were treated with 50 μm gemfibrozil for different minutes followed by monitoring the activation of AKT by Western blot with antibodies against phospho-AKT and total AKT. D, densitometric analysis of dose-dependent change (relative to TLR2) of PI3K subunits by gemfibrozil treatment. Mouse primary microglia were treated with 50 μm gemfibrozil for different minutes followed by double-labeling for CD11b and either phospho-AKT (E) or total AKT (F). DAPI was used to stain nuclei. Results are means ± S.D. of at least three independent experiments. a, p < 0.001 versus control. Scale bar, 20 μm. UN, untreated.
Next, we examined if gemfibrozil required PI3K for the up-regulation of SOCS3 in mouse glial cells. We used pharmacological inhibitors of PI3K and monitored any change in the level of induction of Socs3 by gemfibrozil. The quantitative RT-PCR analysis showed a marked decline in the level of induction of SOCS3 by gemfibrozil when wortmannin (Fig. 4A) and LY294002 (Fig. 4B) were used. To confirm our mRNA findings we performed Western blot analysis to check the protein levels of SOCS3 in both mouse primary astrocytes and BV-2 in presence of the inhibitor (Fig. 4, D and E). We observed an almost 5-fold increase with gemfibrozil treatment and a sharp decline of the SOCS3 protein levels in presence of the inhibitor in both cell types.
FIGURE 4.

Up-regulation of SOCS3 by gemfibrozil in glial cells via PI3K-AKT pathway. BV-2 microglial cells preincubated with different concentrations of wortmannin (Wo) and LY294002 (LY) (PI3K inhibitors) and AKT-I for 1 h were treated with 50 μm gemfibrozil (Gem) in serum-free conditions. After 1 h of treatment, the mRNA expression of Socs3 was monitored by quantitative real time PCR (A, wortmannin; B, LY294002; C, AKT-I). D, BV-2 cells pretreated with different concentrations of LY294002 and AKT-I for 1 h were treated with 50 μm gemfibrozil for 90 min followed by Western blot for SOCS3. Graph represents densitometric analysis of change in SOCS3 levels relative to β-actin. E, mouse primary astrocytes pretreated with different concentrations of LY294002 and AKT-I for 1 h were treated with 50 μm gemfibrozil for 90 min followed by Western blot for SOCS3. Graph represents densitometric analysis of change in SOCS3 levels relative to β-actin. Results are mean ± S.D. of three independent experiments. a, p < 0.001 versus control; b, p < 0.01 versus gemfibrozil.
AKT Is Involved in Gem-mediated Up-regulation of SOCS3 in Mouse Glial Cells
AKT, which is activated by phosphorylation, is known to be the downstream kinase for PI3K in the signaling cascade (36). Therefore, it was logical to look for the involvement of AKT in gemfibrozil-induced up-regulation of SOCS3. We examined whether gemfibrozil could induce the activation of AKT in microglial cells by monitoring levels of phosphorylated AKT (p-AKT). We found an ∼6-fold increase in phospho-AKT levels as early as 15 min, while the level of the total AKT remained constant over the course of treatment (Fig. 3, C and D). We double-labeled primary microglia for both pAKT and CD11b and total AKT and CD11b separately for different time points, and we observed a gradual increase of pAKT over time with maximum levels at 30 min post-treatment with gemfibrozil, although total AKT remained constant throughout (Fig. 3, E and F).
Now, we examined whether AKT was involved in this gemfibrozil-induced SOCS3 up-regulation. The increase in expression of Socs3 mRNA by gemfibrozil was reduced by the addition of AKT-I, a specific inhibitor of AKT (Fig. 4C). The protein levels of SOCS3 were also checked in both BV-2 and mouse astrocytes upon treatment with AKT-I. Western blots for both BV-2 (Fig. 4D) and astrocytes (Fig. 4E) showed drastic decrease (from 5- to 1.5-fold) in gemfibrozil-induced expression of SOCS3 in the presence of AKT-I. Together, these results suggest that gemfibrozil-induced up-regulation of SOCS3 requires the activation of the type IA p110α-dependent PI3K/AKT pathway.
Gemfibrozil Induces the Activation of Krüppel-like Factor-4 (KLF4) in Glial Cells
Apart from its role in development, KLF4 has also been found to be important in tumorigenesis and inflammation (37–42). When we analyzed the promoter region of the Socs3 gene, we found two distinct binding sites for KLF4. Therefore, we investigated whether gemfibrozil has any effect on the nuclear translocation and activation of KLF4. As evident from Western blot analysis of nuclear extracts, gemfibrozil rapidly induced the translocation of KLF4 into the nucleus of BV-2 (Fig. 5A) cells as well as mouse primary astrocytes (Fig. 5C) within 30 min of treatment. It was also observed that gemfibrozil marginally increased the levels of total KLF4 in those cells as well (Fig. 5, B and D). Furthermore, mouse primary astrocytes were double-labeled for KLF4 and GFAP. We found that translocation of KLF4 to the nucleus was induced by gemfibrozil after 30 min of treatment with gradual accumulation up to 60 min (Fig. 5E). These results show an increased accumulation of KLF4 in the nucleus following gemfibrozil treatment over time.
FIGURE 5.

Gemfibrozil induces the nuclear translocation of KLF4 in glial cells. BV-2 microglial cells were treated with 50 μm gemfibrozil (Gem) under serum-free conditions and at different minutes, and the level of KLF4 protein was monitored in nuclear extracts (A) and whole cell extract by Western blot (B). Histone 3 (H3) and β-actin were used as a loading control for nuclear extract and whole cell extract. respectively. Graphs represent densitometric analysis of the nuclear and total KLF4 normalized to their respective controls. Mouse primary astrocytes were treated with 50 μm gemfibrozil under serum-free conditions and at different minutes, and the level of KLF4 protein was monitored in nuclear extracts (C) and whole cell extract by Western blot (D). Histone 3 and β-actin were used as a loading control for nuclear extract and whole cell extract, respectively. Graphs represent densitometric analysis of the nuclear and total KLF4 normalized to their respective controls. E, mouse primary astrocytes were treated with 50 μm gemfibrozil, and at different time points the level of KLF4 was monitored by double label immunofluorescence for GFAP and KLF4. Arrows indicate the presence of KLF4 inside the nucleus. Results are mean ± S.D. of three independent experiments.a, p < 0.001 versus control; Scale bar, 20 μm. UN, untreated.
It has been reported that during the activation process KLF4 is phosphorylated at its serine residue (40, 41). Therefore, we examined if gemfibrozil induced the phosphorylation of KLF4. Because no antibody was available against phospho-KLF4, we performed immunoprecipitation with antibodies against phosphoserine followed by immunoblot analysis with anti-KLF4 antibody, and we observed a time-dependent increase in serine phosphorylation of KLF4 in gemfibrozil-treated microglial cells (Fig. 6A). These results were specific as we could not detect any KLF4 in control IgG immunoprecipitates (Fig. 6A).
FIGURE 6.

Gemfibrozil induces the phosphorylation and activation of KLF4 in glial cells. A, BV-2 microglial cells were treated with 50 μm gemfibrozil (Gem) under serum-free conditions, and at different minutes, total cellular extracts were immunoprecipitated (IP) by antibodies against phospho-Ser (P-Ser) followed by Western blot (WB) of immunoprecipitates with antibodies against KLF4. Normal IgG was used as control. Graph represents densitometric analysis of the bands normalized to untreated (UN) sample. B, BV-2 microglial glial cells preincubated with different concentrations of LY294002 (LY) and AKT-I for 1 h were treated with 50 μm gemfibrozil for 60 min under serum-free conditions followed by monitoring the serine phosphorylation via immunoprecipitation. Graph represents densitometric analysis of the bands normalized to the untreated sample. C, BV-2 cells were treated with 50 μm gemfibrozil for different time points, and total cell extracts were immunoprecipitated separately using antibody against AKT and KLF4, followed by Western blot for AKT and KLF4. Appropriate IgG antibodies were used as control. Results are mean ± S.D. of three independent experiments. a, p < 0.001 versus control; b, p < 0.001 versus gemfibrozil.
Gemfibrozil Induces the Activation of KLF4 in Microglial Cells via PI3K-AKT Pathway
AKT is a serine/threonine kinase. Because gemfibrozil activated the PI3K-AKT pathway and induced the serine phosphorylation of KLF4, we examined if gemfibrozil induced the phosphorylation of KLF4 via the PI3K-AKT pathway. We found that the phosphorylation status of KLF4 was drastically reduced in the presence of both PI3K inhibitor (LY294002) and AKT-I (Fig. 6B). Next we further checked whether AKT was physically associated with KLF4 in gemfibrozil-stimulated cells. Cell extracts were subjected to immunoprecipitation by both AKT and KLF4 antibodies separately, followed by Western blot. The Western blots showed increased presence of KLF4 in the precipitate when pulled down with AKT antibody (Fig. 6C). Similarly increased abundance of AKT was also observed in the precipitates when extracts were pulled down with KLF4 antibody (Fig. 6C). These results clearly suggest a physical interaction between AKT and KLF4. As mentioned earlier, activation of KLF4 includes both phosphorylation and subsequent translocation to nucleus. Therefore, we wanted to confirm whether nuclear translocation of KLF4 was hampered upon inhibiting the PI3K-AKT pathway. Nuclear extracts from BV-2 cells treated with gemfibrozil in the presence of PI3K and AKT inhibitors (LY294002 and AKT-I) were subjected to immunoblot for KLF4. The data when normalized to the respective controls showed a decline of levels of nuclear KLF4 from 3-fold (only gemfibrozil treatment) to 1–1.5-fold (in the presence of inhibitors) compared with untreated cells (Fig. 7A). Similar results were obtained in whole cell extract of BV-2 cells upon inhibition of PI 3-kinase and AKT (Fig. 7B).
FIGURE 7.

Gemfibrozil induces the activation of KLF4 in glial cells via PI3K-AKT pathway. BV-2 microglial glial cells preincubated with different concentrations of LY294002 (LY) and AKT-I for 1 h were treated with 50 μm gemfibrozil (Gem) for 60 min under serum-free conditions followed by monitoring the nuclear translocation of KLF4 (A) and the levels of total KLF4 as described above (B). C, BV-2 cells were treated with different concentrations of gemfibrozil under serum-free conditions for 60 min followed by monitoring the DNA binding activity of KLF4 by EMSA. D, at different minutes of gemfibrozil treatment, the DNA binding activity of KLF4 was monitored by EMSA. Graphs represent densitometric analysis of the bands compared with untreated (UN) sample. Results are mean ± S.D. of three independent experiments. a, p < 0.001 versus control. E, BV-2 cells preincubated with different concentrations of LY for 1 h were treated with 50 μm gemfibrozil for 60 min followed by monitoring the DNA binding activity of KLF4 by EMSA. F, graph represents densitometric analysis of the bands compared with untreated sample. Results are means ± S.D. of three independent experiments. a, p < 0.001 versus control; b, p < 0.01 versus gemfibrozil.
Next, we examined whether gemfibrozil induced the DNA binding activity of KLF4 in microglial cells. It is clearly evident from EMSA that gemfibrozil alone induced the DNA binding activity of KLF4 in both a dose-dependent (Fig. 7C, 1st panel) and time-dependent (Fig. 7D, 1st panel) manner. The densitometric analysis of the gels showed a 3-fold increase in binding with 50 μm gemfibrozil treatment (Fig. 7C, 2nd panel) and a gradual increase (from 2-fold to about 10-fold) in DNA binding of KLF4 over time (30–120 min) with 50 μm gemfibrozil treatment (Fig. 7D, 2nd panel). We also observed reduced DNA binding activity of KLF4 when PI3K was inhibited by LY294002 (Fig. 7E). Densitometry analysis showed a sharp decline from 5-fold to 2-fold in DNA binding in the presence of LY294002 (Fig. 7F). Taken together, these results suggest that gemfibrozil induces the activation of KLF4 via PI3K-AKT pathway.
Gemfibrozil Up-regulates SOCS3 in Glial Cells via KLF4
Next, we investigated if gemfibrozil required KLF4 for the up-regulation of SOCS3. We used 0.5 and 1.0 μg of KLF4 siRNA for this purpose, and 1.0 μg of scrambled siRNA was used as control. As expected, KLF4 siRNA, but not control siRNA, suppressed the expression of Klf4 as well as Socs3 in gemfibrozil-treated and -untreated BV-2 microglial cells (Fig. 8A, 1st panel) and mouse astrocytes (Fig. 8C, 1st panel) as observed from the RT-PCR data. The quantitative RT-PCR for Socs3 in BV-2 cells showed a comparable increase of Socs3 in cells treated with gemfibrozil with or without the presence of control siRNA (about 12–13-fold) but only about 2-fold induction in the presence of even 0.5 μg of KLF4 siRNA (Fig. 8A, 2nd panel). Similar results were obtained with mouse primary astrocytes as well where the induction of Socs3 by gemfibrozil, both in presence and absence of control siRNA (16–18-fold), was abrogated in presence of 0.5 μg of KLF4 siRNA (2–4-fold) (Fig. 8C, 2nd panel). Collectively, these data suggest that KLF4 is in fact required for gemfibrozil-mediated up-regulation of SOCS3 in glial cells.
FIGURE 8.
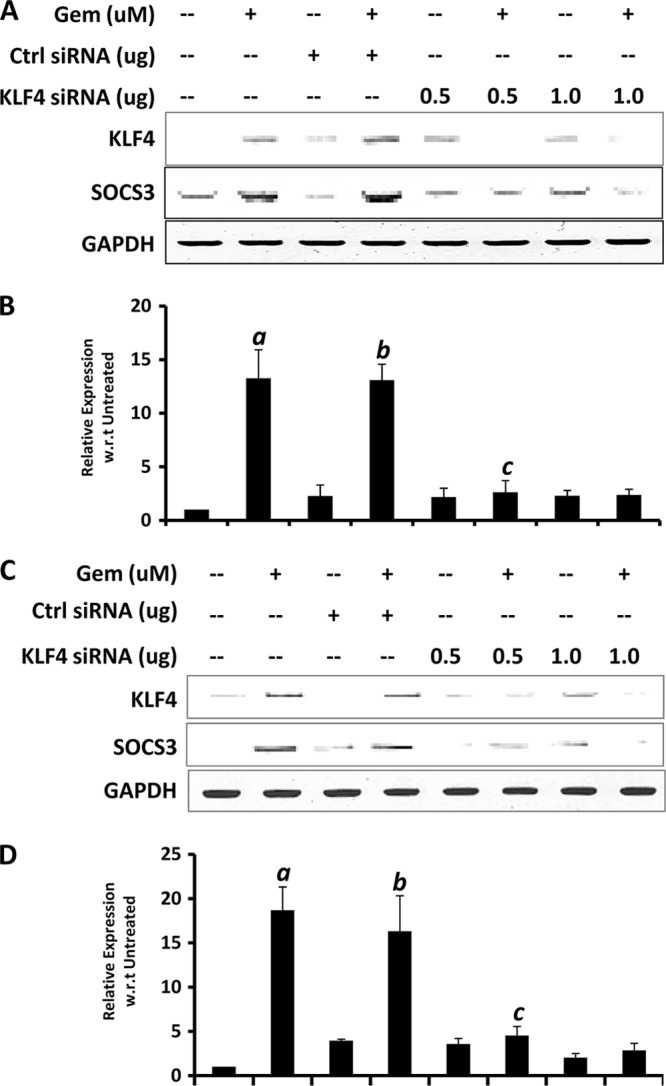
siRNA knockdown of KLF4 abrogates gemfibrozil-induced up-regulation of SOCS3 in glial cells. Mouse BV-2 microglial cells (A and B) and primary astrocytes (C and D) were transfected with either control (Ctrl) or KLF4 siRNA. Forty eight hours after transfection, cells were treated with 50 μm gemfibrozil (Gem) for 60 min under serum-free conditions followed by monitoring the mRNA expression of Socs3 by semi-quantitative RT-PCR (A and C) and quantitative real time PCR (B and D). Results are mean ± S.D. of at least three independent experiments. a, p < 0.001 versus untreated; b, p < 0.001 versus control siRNA treatment; c, p < 0.001 versus control siRNA + gemfibrozil treatment.
Gemfibrozil Induces the Recruitment of KLF4 to the Socs3 Promoter
Because upon analysis of the mouse Socs3 promoter using MatInspector we have observed two consensus KLF4-binding sites (Fig. 9), we next examined if gemfibrozil treatment could induce the recruitment of KLF4 to the Socs3 promoter. Using ChIP analysis, we first tested whether gemfibrozil induced the recruitment of KLF4 to the proximal one (Fig. 9B) and/or the distal KLF4-binding site (Fig. 9C). Interestingly, after repeated ChIP analysis using various primers spanning the proximal KLF4-binding site, we did not find any recruitment of KLF4 in response to gemfibrozil treatment (Fig. 9B), indicating that gemfibrozil does not involve the proximal KLF4-binding site of the Socs3 promoter to up-regulate the transcription of Socs3. However, gemfibrozil markedly induced the recruitment of KLF4 to the distal site (Fig. 9C). This effect was specific as it was not observed with control IgG (Fig. 9C). We next examined if gemfibrozil induced the recruitment of KLF4 to the distal KLF4-binding site of Socs3 via the PI3K-AKT pathway. As evident from Fig. 9C, both LY294002 and AKT-I markedly abrogated the ability of gemfibrozil to recruit KLF4 to the Socs3 promoter. These results suggest that gemfibrozil induces the recruitment of KLF4 to the distal KLF4-binding site of the Socs3 promoter via the PI3K-AKT pathway.
FIGURE 9.

Gemfibrozil induces the recruitment of KLF4 to the distal KLF4-binding site on the Socs3 promoter. A, DNA sequence of the Socs3 promoter region containing the KLF4-binding sites with positions of the primers used for the ChIP analysis. Mouse BV-2 microglial cells pretreated with different concentrations of LY294002 (LY) and AKT-I for 1 h were stimulated with 50 μm gemfibrozil for 30 min under serum-free conditions. The recruitment of KLF4 to proximal (B) and distal (C) KLF4-binding sites of the Socs3 promoter was monitored by ChIP analysis as described under “Materials and Methods.” Normal IgG was used as control.
DISCUSSION
Immunological activation of glial cells in the CNS results in the production of cytokines and chemokines that leads to the initiation of the cytokine-mediated signaling pathway. Cytokine signaling in immune cells may lead to immune clearance of the toxins, but prolonged or unregulated signaling may negatively impact the glial and neuronal function. Recent studies suggest that SOCS3 protein has an important role in mitigating pathogenic effects of cytokine-induced immune and inflammatory responses (8–10, 43). Hence up-regulation of SOCS3 is considered beneficial for various neuroinflammatory and neurodegenerative diseases. Accordingly, identifying pharmacological compounds capable of up-regulating SOCS3 protein and understanding the cellular signaling mechanisms involved in the drug-induced up-regulation of SOCS3 in brain cells are important steps for the therapeutic intervention in neuroinflammatory and neurodegenerative diseases. Here, we delineate for the first time that gemfibrozil, a Food and Drug Administration-approved lipid-lowering drug, is capable of up-regulating SOCS3 in mouse glial cells. The observation has been confirmed in both mRNA studies and protein studies in microglia and astrocytes. Because a precise regulation of both the magnitude and duration of cytokine signaling is essential for orchestration of numerous biological processes, including innate and adaptive immune responses, our study provides a potentially important mechanism to ameliorate the pathogenic effects of cytokine signaling in the CNS by gemfibrozil.
Intracellular pathways that regulate the expression of pro- or anti-inflammatory molecules in glial cells are critical for studying the underlying mechanisms for inflammation. PI3K is a key signaling molecule implicated in the regulation of a broad array of biological responses, including receptor-stimulated mitogenesis, oxidative burst, and cell survival. For class IA PI3K, the p85 regulatory subunit acts as an interface by interacting with the insulin receptor substrate-1 through its SH2 domain and thus recruits the p110 catalytic subunit to the cell membrane through its SH2 domain. In contrast, for class IB PI3K, the p110γ is activated by the engagement of G protein-coupled receptors (34). The p110 then catalyzes the reaction to release phosphatidylinositol (3,4,5)-trisphosphate as the second messenger using phosphatidylinositol (4,5)-bisphosphate as the substrate and activates downstream signaling molecules like AKT/protein kinase B and p70 ribosomal S6 kinase (44). Because we have already reported that gemfibrozil induces the activation of PI3K and that PI3K is responsible for its anti-inflammatory activity (32), we were prompted to investigate the role of PI3K in gemfibrozil-mediated up-regulation of SOCS3. In this study, we have demonstrated the significant activation of type IA p110α PI3K by gemfibrozil and its subsequent downstream activation of AKT, resulting in SOCS3 up-regulation. This conclusion is based on the following observations. First, gemfibrozil induced the recruitment of type IA p110α subunit of PI3K to the plasma membrane. Second, gemfibrozil induced the activation of AKT via PI3K. Third, chemical inhibitors of PI3K and AKT abrogated the effect of gemfibrozil on SOCS3 up-regulation. Collectively, these data suggest an essential role of the PI3K/AKT pathway in the induction of SOCS3 by gemfibrozil. However, we are not aware of the mechanism for gemfibrozil-mediated activation of p85α-associated p110α PI3K in microglia. In general, tyrosine phosphorylation of growth factor receptors creates docking sites for binding of p85α through its SH2 domains, resulting in the activation of p85α-associated PI3K (33). Because gemfibrozil is inducing the activation of PI3K at an early time point, it may not be surprising if gemfibrozil uses any of these growth factor receptors to activate PI3K.
Now that we have established the PI3K/AKT pathway to be critical for the up-regulation of SOCS3, we tried to find out the transcription factor involved in this pathway. The Socs-3 promoter contains numerous potential regulatory elements (45, 46). A careful analysis of its promoter sites revealed two potential binding sites for KLF4 and multiple binding sites for AP-1 and NF-κB. However, in our earlier studies (32, 47), we have observed that gemfibrozil suppresses the activation of NF-κB and AP-1, ruling out the possible involvement of NF-κB and AP-1 in gemfibrozil-induced up-regulation of SOCS3 in glial cells. Therefore, we focused on KLF4. Krüppel is a zinc finger-containing transcription factor in Drosophila melanogaster that is crucial for controlling embryogenesis as well as diverse biological processes, including proliferation, apoptosis, differentiation, and also tumorigenesis and inflammation (39, 40). The KLF family consists of at least 16 different members, which are in turn separated into a few structurally related subgroups. The largest of them includes at least seven proteins as follows: erythroid KLF (EKLF or KLF1); lung KLF (LKLF or KLF2); basic KLF (BKLF or KLF3); gut KLF (GKLF/EZF or KLF4); intestinal KLF (IKLF/BTEB2 or KLF5); core promoter element-binding protein (COPEB/Zf9 or KLF6), and ubiquitous KLF (UKLF or KLF7) (48). Here, we have delineated that gemfibrozil up-regulates SOCS3 via KLF4. Gemfibrozil alone induced the activation of KLF4 in glial cells as evident from an increase in serine phosphorylation of KLF4, translocation of KLF4 to the nucleus, and DNA binding activity of KLF4. This KLF4 activation by gemfibrozil was abrogated by inhibitors of PI3K and AKT, suggesting that KLF4 is indeed activated via the PI3K-AKT pathway following gemfibrozil treatment. The actual physical interaction between AKT and KLF4 as shown in the co-immunoprecipitation study also proves that AKT directly phosphorylates KLF4 upon stimulation by gemfibrozil. Abrogation of gemfibrozil-mediated up-regulation of Socs3 by siRNA knockdown of Klf4 and recruitment of KLF4 to the promoter of Socs3 by gemfibrozil via PI3K- and AKT-sensitive pathways demonstrate for the first time that gemfibrozil induces the up-regulation of SOCS3 in glial cells via a novel PI3K-AKT-KLF4 pathway (summarized in Fig. 10).
FIGURE 10.

Graphical representation of pathways by which gemfibrozil up-regulates the level of SOCS3 in glial cells.
SOCS3 mainly regulates IL-6 family of cytokine-mediated signaling by inhibiting STAT3 as well as signal transduction induced by LPS, IL-12, IL-4, and IFN-γ (43, 49, 50). SOCS3 not only acts as a mere inhibitor of cytokine signaling, it also has various other roles. For example, SOCS3 can regulate chemokine expression as well as inhibit chemokine-mediated chemotaxis in cells like T-cells. Hence, the up-regulation of SOCS3 in the glial cells may also attenuate the chemotactic migration of the other inflammatory cells into the CNS. Furthermore, SOCS3 has a broad range of actions as it inhibits the activation of NF-κB, antagonizes cAMP-mediated signaling, and enhances signaling through the ERK/MAPK pathways (11). SOCS3 levels were found to be lower in B6 mice with chronic experimental autoimmune encephalomyelitis without complete remission in comparison with SJL mice with relapsing-remitting experimental autoimmune encephalomyelitis and failure of B6 mice to completely recover may be due to the lower levels of SOCS3 (51). Simvastatin was also found to increase SOCS3 in monocytes of relapsing-remitting multiple sclerosis patients, causing reduced STAT-3 phosphorylation and decreased IL-6 and IL-23 expression (52). A number of studies have reported that SOCS3 can be up-regulated in response to proinflammatory stimuli like LPS or IFN-γ probably via the JAK-STAT pathway, resulting in feedback inhibition of LPS- and IFN-γ-induced cytokine signaling (43, 53). But in inflammatory diseases, the balance between proinflammatory and anti-inflammatory molecules is lost, thereby causing a rapid progression of the disease. In such cases, a pharmacological drug could be used to up-regulate SOCS proteins to inhibit the cytokine signaling and counteract detrimental effects of inflammatory cytokines. This study provides such an option with gemfibrozil via the induction of type IA p110α PI3K-AKT-KLF4 pathway (Fig. 10).
This work was supported, in whole or in part, by National Institutes of Health Grants AT6681, NS64564, and NS71479.
- SOCS
- suppressor of cytokine signaling
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- SH2
- SRC homology 2
- GFAP
- glial fibrillary acidic protein
- KLF
- Krüppel-like factor
- AKT-I
- AKT inhibitor.
REFERENCES
- 1. Tansey M. G., McCoy M. K., Frank-Cannon T. C. (2007) Neuroinflammatory mechanisms in Parkinson disease. Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 208, 1–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Frank-Cannon T. C., Alto L. T., McAlpine F. E., Tansey M. G. (2009) Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener 4, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Martin J. B. (1999) Molecular basis of the neurodegenerative disorders. N. Engl. J. Med. 340, 1970–1980 [DOI] [PubMed] [Google Scholar]
- 4. Cummings J. L., Vinters H. V., Cole G. M., Khachaturian Z. S. (1998) Alzheimer disease. Etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology 51, S2–S17 [DOI] [PubMed] [Google Scholar]
- 5. Jana A., Pahan K. (2010) Fibrillar amyloid-β-activated human astroglia kill primary human neurons via neutral sphingomyelinase. Implications for Alzheimer disease. J. Neurosci. 30, 12676–12689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hanisch U. K., Kettenmann H. (2007) Microglia. Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394 [DOI] [PubMed] [Google Scholar]
- 7. Jana M., Palencia C. A., Pahan K. (2008) Fibrillar amyloid-β peptides activate microglia via TLR2: implications for Alzheimer disease. J. Immunol. 181, 7254–7262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alexander W. S. (2002) Suppressors of cytokine signaling (SOCS) in the immune system. Nat. Rev. Immunol. 2, 410–416 [DOI] [PubMed] [Google Scholar]
- 9. Chen X. P., Losman J. A., Rothman P. (2000) SOCS proteins, regulators of intracellular signaling. Immunity 13, 287–290 [DOI] [PubMed] [Google Scholar]
- 10. Baker B. J., Akhtar L. N., Benveniste E. N. (2009) SOCS1 and SOCS3 in the control of CNS immunity. Trends Immunol. 30, 392–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou H., Miki R., Eeva M., Fike F. M., Seligson D., Yang L., Yoshimura A., Teitell M. A., Jamieson C. A., Cacalano N. A. (2007) Reciprocal regulation of SOCS1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin. Cancer Res. 13, 2344–2353 [DOI] [PubMed] [Google Scholar]
- 12. Dominguez E., Mauborgne A., Mallet J., Desclaux M., Pohl M. (2010) SOCS3-mediated blockade of JAK/STAT3 signaling pathway reveals its major contribution to spinal cord neuroinflammation and mechanical allodynia after peripheral nerve injury. J. Neurosci. 30, 5754–5766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoshimura A., Naka T., Kubo M. (2007) SOCS proteins, cytokine signaling, and immune regulation. Nat. Rev. Immunol. 7, 454–465 [DOI] [PubMed] [Google Scholar]
- 14. Takahashi Y., Carpino N., Cross J. C., Torres M., Parganas E., Ihle J. N. (2003) SOCS3. An essential regulator of LIF receptor signaling in trophoblast giant cell differentiation. EMBO J. 22, 372–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wong P. K., Egan P. J., Croker B. A., O'Donnell K., Sims N. A., Drake S., Kiu H., McManus E. J., Alexander W. S., Roberts A. W., Wicks I. P. (2006) SOCS3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J. Clin. Invest. 116, 1571–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qin H., Niyongere S. A., Lee S. J., Baker B. J., Benveniste E. N. (2008) Expression and functional significance of SOCS1 and SOCS3 in astrocytes. J. Immunol. 181, 3167–3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Robins S. J., Collins D., Wittes J. T., Papademetriou V., Deedwania P. C., Schaefer E. J., McNamara J. R., Kashyap M. L., Hershman J. M., Wexler L. F., Rubins H. B. (2001) Relation of gemfibrozil treatment and lipid levels with major coronary events. VA-HIT, a randomized controlled trial. JAMA 285, 1585–1591 [DOI] [PubMed] [Google Scholar]
- 18. Rubins H. B., Robins S. J. (1992) Effect of reduction of plasma triglycerides with gemfibrozil on high density lipoprotein-cholesterol concentrations. J. Intern. Med. 231, 421–426 [DOI] [PubMed] [Google Scholar]
- 19. Rubins H. B., Robins S. J., Collins D., Fye C. L., Anderson J. W., Elam M. B., Faas F. H., Linares E., Schaefer E. J., Schectman G., Wilt T. J., Wittes J. (1999) Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high density lipoprotein cholesterol. Veterans Affairs High Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 341, 410–418 [DOI] [PubMed] [Google Scholar]
- 20. Roy A., Pahan K. (2009) Gemfibrozil, stretching arms beyond lipid lowering. Immunopharmacol. Immunotoxicol. 31, 339–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dasgupta S., Roy A., Jana M., Hartley D. M., Pahan K. (2007) Gemfibrozil ameliorates relapsing-remitting experimental autoimmune encephalomyelitis independent of peroxisome proliferator-activated receptor-α. Mol. Pharmacol. 72, 934–946 [DOI] [PubMed] [Google Scholar]
- 22. Pahan K., Jana M., Liu X., Taylor B. S., Wood C., Fischer S. M. (2002) Gemfibrozil, a lipid-lowering drug, inhibits the induction of nitric-oxide synthase in human astrocytes. J. Biol. Chem. 277, 45984–45991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jana M., Jana A., Pal U., Pahan K. (2007) A simplified method for isolating highly purified neurons, oligodendrocytes, astrocytes, and microglia from the same human fetal brain tissue. Neurochem. Res. 32, 2015–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Giulian D., Baker T. J. (1986) Characterization of amoeboid microglia isolated from developing mammalian brain. J. Neurosci. 6, 2163–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jana M., Pahan K. (2005) Redox regulation of cytokine-mediated inhibition of myelin gene expression in human primary oligodendrocytes. Free Radic. Biol. Med. 39, 823–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saha R. N., Jana M., Pahan K. (2007) MAPK p38 regulates transcriptional activity of NF-κB in primary human astrocytes via acetylation of p65. J. Immunol. 179, 7101–7109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dasgupta S., Jana M., Zhou Y., Fung Y. K., Ghosh S., Pahan K. (2004) Antineuroinflammatory effect of NF-κB essential modifier-binding domain peptides in the adoptive transfer model of experimental allergic encephalomyelitis. J. Immunol. 173, 1344–1354 [DOI] [PubMed] [Google Scholar]
- 28. Saha R. N., Liu X., Pahan K. (2006) Up-regulation of BDNF in astrocytes by TNF-α. A case for the neuroprotective role of cytokine. J. Neuroimmune Pharmacol. 1, 212–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fukushima A., Kajiya H., Izumi T., Shigeyama C., Okabe K., Anan H. (2010) Pro-inflammatory cytokines induce suppressor of cytokine signaling-3 in human periodontal ligament cells. J. Endod. 36, 1004–1008 [DOI] [PubMed] [Google Scholar]
- 30. Peraldi P., Filloux C., Emanuelli B., Hilton D. J., Van Obberghen E. (2001) Insulin induces suppressor of cytokine signaling-3 tyrosine phosphorylation through Janus-activated kinase. J. Biol. Chem. 276, 24614–24620 [DOI] [PubMed] [Google Scholar]
- 31. Steppan C. M., Wang J., Whiteman E. L., Birnbaum M. J., Lazar M. A. (2005) Activation of SOCS3 by resistin. Mol. Cell. Biol. 25, 1569–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jana M., Jana A., Liu X., Ghosh S., Pahan K. (2007) Involvement of phosphatidylinositol 3-kinase-mediated up-regulation of IκBα in anti-inflammatory effect of gemfibrozil in microglia. J. Immunol. 179, 4142–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Geering B., Cutillas P. R., Nock G., Gharbi S. I., Vanhaesebroeck B. (2007) Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc. Natl. Acad. Sci. U.S.A. 104, 7809–7814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andrews S., Stephens L. R., Hawkins P. T. (2007) PI3K class IB pathway. Sci. STKE 2007, cm2. [DOI] [PubMed] [Google Scholar]
- 35. Sasaki A. T., Chun C., Takeda K., Firtel R. A. (2004) Localized Ras signaling at the leading edge regulates PI3K, cell polarity, and directional cell movement. J. Cell Biol. 167, 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo J., Manning B. D., Cantley L. C. (2003) Targeting the PI3K-AKT pathway in human cancer. Rationale and promise. Cancer Cell 4, 257–262 [DOI] [PubMed] [Google Scholar]
- 37. Liu J., Liu Y., Zhang H., Chen G., Wang K., Xiao X. (2008) KLF4 promotes the expression, translocation, and release of HMGB1 in RAW264.7 macrophages in response to LPS. Shock 30, 260–266 [DOI] [PubMed] [Google Scholar]
- 38. Liu J., Zhang H., Liu Y., Wang K., Feng Y., Liu M., Xiao X. (2007) KLF4 regulates the expression of interleukin-10 in RAW264.7 macrophages. Biochem. Biophys. Res. Commun. 362, 575–581 [DOI] [PubMed] [Google Scholar]
- 39. Liu Y., Liu M., Liu J., Zhang H., Tu Z., Xiao X. (2010) KLF4 is a novel regulator of the constitutively expressed HSP90. Cell Stress Chaperones 15, 211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yan F. F., Liu Y. F., Liu Y., Zhao Y. X. (2008) KLF4. A novel target for the treatment of atherosclerosis. Med. Hypotheses 70, 845–847 [DOI] [PubMed] [Google Scholar]
- 41. Ai W., Liu Y., Langlois M., Wang T. C. (2004) Kruppel-like factor 4 (KLF4) represses histidine decarboxylase gene expression through an upstream Sp1 site and downstream gastrin-responsive elements. J. Biol. Chem. 279, 8684–8693 [DOI] [PubMed] [Google Scholar]
- 42. Hagos E. G., Ghaleb A. M., Kumar A., Neish A. S., Yang V. W. (2011) Expression profiling and pathway analysis of Krüppel-like factor 4 in mouse embryonic fibroblasts. Am. J. Cancer Res. 1, 85–97 [PMC free article] [PubMed] [Google Scholar]
- 43. Qin H., Roberts K. L., Niyongere S. A., Cong Y., Elson C. O., Benveniste E. N. (2007) Molecular mechanism of lipopolysaccharide-induced SOCS3 gene expression in macrophages and microglia. J. Immunol. 179, 5966–5976 [DOI] [PubMed] [Google Scholar]
- 44. Hannan K. M., Thomas G., Pearson R. B. (2003) Activation of S6K1 (p70 ribosomal protein S6 kinase 1) requires an initial calcium-dependent priming event involving formation of a high molecular mass signaling complex. Biochem. J. 370, 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barclay J. L., Anderson S. T., Waters M. J., Curlewis J. D. (2007) Characterization of the SOCS3 promoter response to prostaglandin E2 in T47D cells. Mol. Endocrinol. 21, 2516–2528 [DOI] [PubMed] [Google Scholar]
- 46. Gatto L., Berlato C., Poli V., Tininini S., Kinjyo I., Yoshimura A., Cassatella M. A., Bazzoni F. (2004) Analysis of SOCS3 promoter responses to interferon γ. J. Biol. Chem. 279, 13746–13754 [DOI] [PubMed] [Google Scholar]
- 47. Pahan K. (2006) Lipid lowering drugs. Cell. Mol. Life Sci. 63, 1165–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wei D., Kanai M., Huang S., Xie K. (2006) Emerging role of KLF4 in human gastrointestinal cancer. Carcinogenesis 27, 23–31 [DOI] [PubMed] [Google Scholar]
- 49. Yasukawa H., Ohishi M., Mori H., Murakami M., Chinen T., Aki D., Hanada T., Takeda K., Akira S., Hoshijima M., Hirano T., Chien K. R., Yoshimura A. (2003) IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat. Immunol. 4, 551–556 [DOI] [PubMed] [Google Scholar]
- 50. Yan C., Cao J., Wu M., Zhang W., Jiang T., Yoshimura A., Gao H. (2010) Suppressor of cytokine signaling 3 inhibits LPS-induced IL-6 expression in osteoblasts by suppressing CCAAT/enhancer-binding protein β activity. J. Biol. Chem. 285, 37227–37239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stark J. L., Cross A. H. (2006) Differential expression of suppressors of cytokine signaling-1 and -3 and related cytokines in central nervous system during remitting versus nonremitting forms of experimental autoimmune encephalomyelitis. Int. Immunol. 18, 347–353 [DOI] [PubMed] [Google Scholar]
- 52. Ramgolam V. S., Markovic-Plese S. (2011) Regulation of suppressors of cytokine signaling as a therapeutic approach in autoimmune diseases, with an emphasis on multiple sclerosis. J. Signal. Transduct. 635721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bluyssen H. A., Rastmanesh M. M., Tilburgs C., Jie K., Wesseling S., Goumans M. J., Boer P., Joles J. A., Braam B. (2010) IFNγ-dependent SOCS3 expression inhibits IL-6-induced STAT3 phosphorylation and differentially affects IL-6 mediated transcriptional responses in endothelial cells. Am. J. Physiol. Cell Physiol. 299, C354–C362 [DOI] [PubMed] [Google Scholar]