

Abstract
The p97/VCP ATPase complex facilitates the extraction and degradation of ubiquitinated proteins from larger structures. We therefore studied if p97 participates to the rapid degradation of myofibrillar proteins during muscle atrophy. Electroporation of a dominant negative p97 (DNp97), but not the WT, into mouse muscle reduced fibre atrophy caused by denervation and food deprivation. DNp97 (acting as a substrate-trap) became associated with specific myofibrillar proteins and its cofactors, Ufd1 and p47, and caused accumulation of ubiquitinated components of thin and thick filaments, which suggests a role for p97 in extracting ubiquitinated proteins from myofibrils. DNp97 expression in myotubes reduced overall proteolysis by proteasomes and lysosomes and blocked the accelerated proteolysis induced by FoxO3, which is essential for atrophy. Expression of p97, Ufd1 and p47 increases following denervation, at times when myofibrils are rapidly degraded. Surprisingly, p97 inhibition, though toxic to most cells, caused rapid growth of myotubes (without enhancing protein synthesis) and hypertrophy of adult muscles. Thus, p97 restrains post-natal muscle growth, and during atrophy, is essential for the accelerated degradation of most muscle proteins.
Keywords: atrophy, muscle growth, muscle proteolysis, myofibrillar proteins, p97/VCP
Introduction
The size and functional capacity of a muscle are determined by the balance between overall rates of protein synthesis and degradation. In response to fasting, denervation or inactivity and various systemic diseases (e.g., diabetes, cancer, sepsis, cardiac failure), skeletal muscles undergo a rapid debilitating loss of weight and contractile capacity (Mitch and Goldberg, 1996; Glass and Roubenoff, 2010). During these various types of atrophy, a common transcriptional programme is activated that leads to an increased overall rate of protein degradation that is primarily responsible for the loss of muscle mass (Mitch and Goldberg, 1996; Lecker et al, 2004). Among the atrophy-related genes (atrogenes) that are coordinately induced are a number of components of the ubiquitin-proteasome system, including two key muscle-specific ubiquitin ligases (E3), MuRF1 and atrogin-1, and also components of the autophagic/lysosomal pathway, such as Gabarapl1 and LC3 (Lecker et al, 2004; Mammucari et al, 2007; Sacheck et al, 2007; Zhao et al, 2007). This transcriptional programme is induced by activation of FoxO-transcription factors (Sandri et al, 2004). Molecules that block this activation of proteolysis could represent novel pharmacological agents to combat muscle wasting.
The major constituents of muscle are myofibrillar proteins. These highly organized components of the sarcomere are normally degraded by the ubiquitin-proteasome pathway (Solomon and Goldberg, 1996; Cohen et al, 2009), but more slowly than most cellular proteins (Zak et al, 1977). This process is markedly accelerated in atrophy, where they are disassembled and degraded in an ordered manner (Cohen et al, 2009). Moreover, since muscles continue to function even during rapid atrophy (e.g., in a fasting animal), the increased proteolysis must occur in a way that does not dramatically alter the relative amounts and functional capacities of different myofibrillar proteins. While the ubiquitin-proteasome pathway is responsible for the accelerated loss of myofibrillar proteins during atrophy (Solomon and Goldberg, 1996), autophagy is also activated and causes the degradation of some soluble proteins and organelles, especially mitochondria (Mammucari et al, 2008; Zhao et al, 2008). During disuse atrophy, thick filament components (e.g., myosin) are selectively ubiquitinated by MuRF1 while in the myofibrils and degraded (Cohen et al, 2009), but other ubiquitin ligases appear to catalyse the degradation of thin filament components (e.g., actin) (Kudryashova et al, 2005). However, the mechanisms for extraction of ubiquitinated proteins from the myofibrils and their delivery to the proteasome are still unclear, nor is it known what cellular factors catalyse this process, which must be highly selective and allows disassembly of some sarcomere components without blocking contractile function.
One attractive candidate for such a role in myofibrillar disassembly is the p97/VCP/Cdc48 ATPase complex, since it binds multiple ubiquitin ligases and ubiquitinated proteins and catalyses the ATP-driven disassembly of other protein complexes, including the extraction and degradation of endoplasmic reticulum-associated proteins (ERAD) (Rape et al, 2001; Ye et al, 2001) as well as of mitochondrial proteins (Xu et al, 2011), membrane fusion events (Ye, 2006), myofibril biogenesis (Kim et al, 2008), chromatin dynamics (Ramadan et al, 2007) and autophagy (Ju et al, 2009; Krick et al, 2010). We therefore examined whether p97 could be involved in muscle atrophy when myofibrillar proteins are rapidly degraded. p97 is a ubiquitous ATPase complex containing six identical subunits (Peters et al, 1990). A common function of p97 in these various cellular processes is to extract ubiquitinated proteins from complexes or membranes, often leading to their proteasomal degradation (Ye, 2006). The p97 complex has been proposed to disassemble also aggregates of misfolded proteins, because p97 overexpression reduces the content of polyglutamine inclusions (Nishikori et al, 2008), and p97 is found in ubiquitinated inclusions in several models of neurodegenerative diseases (Ishigaki et al, 2004). A number of proteins, including soluble cytosolic and nuclear proteins, require p97 for their degradation by the proteasomes, such as UNC45B (Janiesch et al, 2007), Aurora B kinase (Ramadan et al, 2007), the transcription factors Spt23 in yeast (Rape et al, 2001) and HIF1α in mammals (Alexandru et al, 2008), cyclin E (Dai and Li, 2001), IκB (Dai et al, 1998), misfolded secretory proteins (Ye et al, 2003), oxidatively damaged cytosolic proteins (Medicherla and Goldberg, 2008) and substrates of the N-end rule and ubiquitin-fusion degradation (UFD) pathways (Wojcik et al, 2006; Beskow et al, 2009).
The variety of p97’s actions is probably due to its ability to interact with diverse substrate-recruiting and -processing cofactors (Jentsch and Rumpf, 2007). The recruiting cofactors include p47 and Ufd1/Npl4 dimer, which associate with the N-terminus of p97 in a mutually exclusive way (Meyer et al, 2000). Substrates bound to p97 can undergo further processing by ubiquitination, deubiquitination or deglycosylation. Among these substrate-processing cofactors are many ubiquitin ligases (Zhong et al, 2004; Alexandru et al, 2008). One E3 that is of special importance in muscle is Ufd2/E4B, which binds to p97’s C-terminus and appears to add further ubiquitin molecules (termed ‘E4 activity’) to the substrate (Richly et al, 2005). In C. elegans, Ufd2 has been implicated with another E3, CHIP and p97 in the degradation of UNC45B, a myosin assembly factor (Janiesch et al, 2007).
Another strong reason for studying p97 function in skeletal muscle is that mutations in human p97 cause the multisystem disorder inclusion-body myopathy with Paget’s disease of the Bone and Frontotemporal Dementia (IBMPFD) (Watts et al, 2004), which is characterized by ubiquitin-positive inclusions in the muscle and brain (Guinto et al, 2007), abnormal vacuolation, sarcomeric disorganization, muscle weakness and functional impairment (Kimonis et al, 2008). The molecular bases for these various changes in muscle in IBMPFD are uncertain (Weihl et al, 2006; Hubbers et al, 2007).
In this study, we investigated whether p97 and its cofactors play a critical role in muscle atrophy, specifically in the rapid breakdown of the highly organized myofibrillar apparatus, during the rapid muscle wasting induced by denervation or fasting. We have studied (1) whether overproduction of WTp97 or an ATPase-deficient p97 mutant influences the atrophy process through effects on overall rates of protein synthesis and degradation; (2) whether the accelerated disassembly and degradation of myofibrillar proteins during atrophy is dependent on p97 and its cofactors; (3) how expression of p97, its cofactors and substrate, UNC45B, may change in atrophying muscles and (4) whether p97, a dominant negative p97 mutant or UNC45B also influences the sizes of normal myotubes or adult muscle fibres. These studies show that p97 plays a critical role during atrophy in the accelerated degradation of muscle proteins via both the proteasomal and autophagic pathways, and most likely functions in the disassembly and proteasomal degradation of ubiquitinated components of the myofibrillar apparatus. Moreover, in normal adult muscles and myotubes, p97 by promoting protein degradation helps limit muscle growth.
Results
Inhibition of p97 decreases muscle atrophy induced by denervation or fasting
To establish whether p97 function is essential for atrophy, we tested whether inhibition of p97 can reduce muscle wasting induced by denervation or food deprivation, by comparing the effects of expression of WTp97 and the p97K524A mutant, a dominant negative inhibitor (DNp97) (Ye et al, 2003). Mutation of lysine 524 to alanine in the D2 ATPase domain of p97 allows the binding of p97 to substrates while substantially decreasing p97’s ATPase activity, and thus preventing substrate release and degradation by proteasomes (Ye et al, 2003). In these constructs, p97 is fused to GFP, which enabled us to identify the transfected fibres. Fusion of GFP to p97’s C-terminus does not perturb its subcellular localization, propensity to form hexamers, ATPase activity or association with its cofactors p47 and Ufd1/Npl4 (Kobayashi et al, 2002). The WT and DNp97 plasmids as well as a control (GFP) plasmid were then electroporated bilaterally into Tibialis Anterior (TA) of adult mice, and at the same time, muscles of one hindlimb were denervated by sectioning the sciatic nerve. Nine days later, the muscles were collected and cryosectioned, and the areas of the transfected fibres were determined by fluorescence microscopy. At this time, denervated fibres decreased in mean area by about 40%, and muscle weight by about 35% (Figures 1A and 10C).
Figure 1.

Electroporation of DNp97 in TA blocks both denervation and starvation-induced atrophy. (A) Frequency histograms showing the distribution of cross-sectional areas of muscle fibres of TA either innervated or 9 days denervated and transfected or not with DNp97GFP. Muscles were electroporated with DNp97GFP plasmids at the same time as section of the sciatic nerve. According to the Kruskal–Wallis Test, differences (P<0.001) were found between innervated versus denervated and innervated versus denervated+DNp97. (B) Frequency histograms showing the distribution of cross-sectional areas of muscle fibres of TA either from fed or 2 days fasted mice and transfected or not with DNp97GFP. Muscles were electroporated with DNp97GFP plasmids and after 4 days mice were deprived of food for 2 days. According to the Kruskal–Wallis Test, differences (P<0.0001) were found between fed versus fasted, fed versus fasted+DNp97 and fasted versus fasted+DNp97. (C) A representative field of a transverse section of fibres expressing DNp97GFP from food-deprived mice. Scale bar represents 50 μm.
Although electroporation of GFP or WTp97 had little or no effect on fibre cross-sectional areas in the denervated muscles (i.e., <8% change in median area, 1946 μm2 for denervated and 2100 for denervated+WTp97, P<0.05), electroporation of the DN to inhibit p97 resulted in a marked reduction of atrophy (Figure 1A). The electroporated fibres (which comprised 60–80% of the total fibres in the different animals) had a median cross-sectional area 60% greater than that of untransfected fibres in the same denervated muscle (Figure 1A). In fact, the median cross-sectional area of denervated fibres overexpressing DNp97 did not differ from that of innervated fibres of the contralateral limb. Thus, p97 inhibition seemed to prevent completely denervation atrophy.
We therefore investigated whether p97 function is also essential for the more rapid loss of muscle mass induced by fasting. Mice were electroporated with the DNp97 plasmid, and 4 days later when no change in muscle weight was evident (data not shown), the mice were deprived of food for 48 h, and their muscles then collected. The muscles of fed mice, which had the same initial mean body weights as the fasted animals were used as controls (Figure 1B). Muscle fibres of food-deprived mice had a median cross-sectional area 33% smaller than that of fibres of fed animals (Figure 1B). Strikingly, in the starved animals, inhibition of p97 by electroporation of the DN resulted in fibres with a median cross-sectional area 70% greater than that of untransfected fibres in the same muscles (Figure 1B and C). Surprisingly, expression of DNp97 not only caused a clear inhibition of fasting-induced atrophy, but also resulted in even larger fibres than those in the TA of fed mice (Figure 1B).
Inhibition or depletion of p97 in normal muscles causes rapid fibre growth
To determine if p97 function is essential to maintain normal muscle size or if blocking it may even induce growth (as suggested in Figure 1B), we electroporated into the contralateral TA muscles of fed adult mice plasmids for either the WTp97 or DNp97 fused to GFP. Because the WTp97 was expressed more efficiently in initial control experiments, we determined by immunoblotting the amount of plasmid electroporated that gives equal expression of WT and DNp97 proteins (Figure 2E). Seven days later, the median cross-sectional area and size distribution of the TA fibres expressing WTp97 resembled that of the surrounding non-electroporated fibres (Figure 2A). By contrast, fibres expressing DNp97 had a 60% greater median area than untransfected fibres (Figure 2B). Furthermore, even though 20–40% of the fibres were not electroporated, the average weight of the TA electroporated with DNp97 in 7 days exceeded by 15% that of the muscles expressing the WTp97 (P<0.05) (Figure 2G). Thus, normal p97 function inhibits muscle growth, but increasing WTp97 content per se does not induce muscle wasting.
Figure 2.

Electroporation of DNp97 or depletion of p97 using shRNA in TA increases fibre size in 7 days. (A–D) Frequency histograms showing the distribution of cross-sectional areas of TA muscle fibres either untransfected (control) or transfected for 7 days with WTp97 (A), DNp97 (B), a non-silencing shRNA (C) or a shRNA against p97 (D). According to the Mann–Whitney Test, the differences between DNp97 versus control and p97shRNA versus control were highly significant (P<0.0001). (E) Sixty micrograms of protein lysates from TA muscles electroporated with WTp97GFP (right leg) or DNp97GFP (left leg) were loaded. The anti-GFP antibody detection shows that equal amounts of WTp97GFP or DNp97GFP were expressed per muscle. The anti-p97 antibody detection reveals two bands: the endogenous (lower band) and the exogenous (upper band) p97. Rpl26 was used as loading control. (F) Seven days after electroporation with a plasmid for a non-silencing shRNA (right leg) or a shRNA against p97 (left leg), the contralateral TA muscles were frozen in isopentane cooled-liquid nitrogen from the same animal. Scale bar represents 1 cm. (G) The average weights of TA muscles 7 days after electroporation are plotted. Error bars indicate s.e.m. Paired t-test: *P<0.05 and ***P<0.003, n=5.
Although it was quite surprising to find that reducing p97 function with the DNp97 induces fibre growth, we confirmed this conclusion by electroporating into TA muscles plasmids encoding either a shRNA for murine p97 or a non-silencing shRNA as the control (Supplementary Figure S2A, B and E). Both vectors co-expressed a GFP marker to allow the identification of the transfected fibres. Seven days later, the median cross-sectional area of fibres expressing the shRNA for p97 was ∼30% larger than untransfected fibres and those expressing the control shRNA (Figure 2C and D). Also, the average weight of the muscles electroporated with the shRNA for p97 increased by 25% in 7 days above that of controls expressing non-silencing shRNA (P<0.003) (Figure 2F and G). Because only 60–80% of fibres were transfected, these changes in muscle weight must underestimate the actual increase in the mass of the fibres when p97 is inhibited or depleted.
In contrast to the DNp97 or shRNA for p97, electroporation of a disease-associated p97 mutant gene, p97R155H (Watts et al, 2004), that is not known to reduce ATPase activity, to trap substrates or function as a dominant negative inhibitor (Halawani et al, 2009; Fernandez-Saiz and Buchberger, 2010; Manno et al, 2010), did not induce appreciable fibre growth in 7 days (i.e., <5% change in median area, 2931 μm2 for transfected fibres versus 2805 for untranfected ones, P=0.05) nor significant change in muscle weight (44.7 mg±4.1 with p97R155H versus 41.9±7.1 in controls, n=5).
p97 inhibition stimulated myotube growth by decreasing protein degradation without enhancing overall protein synthesis
In the transfected muscle fibres in mice, it is not possible to measure precisely protein synthesis and degradation as is possible with cultured cells. To test whether DNp97 also increases fibre size in myotubes as it does in adult muscles, C2C12 myotubes were infected with adenoviruses encoding GFP, WTp97 or DNp97. Increased levels of p97 protein were evident at 24 h, and subsequently p97 content continued to increase (Figure 4C). By 48 h, overexpression of DNp97 caused a 30% increase in total protein content (P<0.02) (Table I) and in mean cell diameter (P<0.0005) (Figure 3A and B) in accord with the growth-promoting effects of DNp97 in adult muscle (Figure 2B and G). By contrast, no change in cell growth occurred upon overexpression of WTp97.
Table 1. In myotubes, DNp97, but not WTp97, increases total protein content, without perturbing protein synthesis.
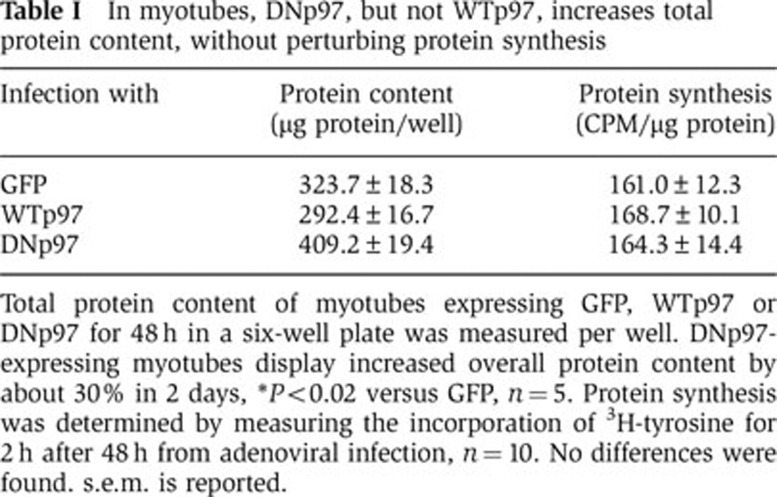
Total protein content of myotubes expressing GFP, WTp97 or DNp97 for 48 h in a six-well plate was measured per well. DNp97-expressing myotubes display increased overall protein content by about 30% in 2 days, *P<0.02 versus GFP, n=5. Protein synthesis was determined by measuring the incorporation of 3H-tyrosine for 2 h after 48 h from adenoviral infection, n=10. No differences were found. s.e.m. is reported.
Figure 3.

DNp97 increases cell diameter of myotubes. (A) Brightfield images of myotubes expressing GFP or DNp97 for 72 h are shown. Scale bar represents 50 μm. (B) The average diameter of myotubes expressing GFP or DNp97 for 72 h is plotted. ***P<0.0005, n=100. Error bars indicate s.e.m.
We then examined whether the increase in myotube size caused by the DNp97 was due to an overall enhancement of protein synthesis as is characteristic of growing cells, and/or a reduction in overall protein degradation. Although we anticipated finding an increase in protein synthetic rate, when we assayed the rates of incorporation of 3H-tyrosine into total cell proteins, no clear difference was observed upon overexpression of GFP, WTp97 or DNp97 (Table I). Furthermore, no increase in the content or phosphorylation of AKT or mTOR’s downstream effectors was found in the myotubes overexpressing the DNp97 (Supplementary Figure S5A and B).
This lack of a clear increase in protein synthesis raised the possibility that the DNp97 induced growth primarily by inhibiting overall proteolysis. In fact, by 36 h after adenoviral infection of DNp97, but not of WTp97, polyubiquitinated proteins began to accumulate in the myotubes (as shown using the FK1 antibody, which does not recognize monoubiquitinated or multiple monoubiquitinated proteins) (Figure 4C). Such an accumulation of ubiquitin chains would be expected if p97 plays a general role in the degradation of ubiquitinated proteins by proteasomes, and because the DNp97 complex binds ubiquitin conjugates but, unlike the WT, fails to release them (Ye et al, 2003; Wojcik et al, 2004). We therefore measured the rates of degradation of long-lived proteins (the bulk of cell proteins) as well as short-lived ones by pulse-chase method using 3H-tyrosine (Zhao et al, 2007). Myotubes were infected for 36 h with adenoviruses expressing GFP, WTp97 or DNp97. To follow the degradation of short-lived cell components, myotube proteins were labelled with a 20-min-pulse of 3H-tyrosine, and to selectively label the long-lived ones, cells were exposed to the labelled precursor for 20 h. The cells were then washed and resuspended in a large excess of not labelled tyrosine. Overexpression of WTp97 did not increase the degradation of either short- or long-lived proteins (Figure 4A and B). Therefore, the level of p97 is not rate-limiting for overall protein degradation. By contrast, expression of the DNp97 for 36 h, but not 24 h (not shown), decreased the degradation of short- (Figure 4A) and also long-lived proteins (Figure 4B). These 20–35% reductions in rates of proteolysis were highly reproducible, and if sustained for several days, should have major effects on muscle size. Thus, inhibition of p97 function seems to increase the protein content of myotubes primarily by decreasing overall proteolysis without significantly altering rates of synthesis.
Figure 4.

In myotubes, DNp97, but not WTp97, decreases degradation of both short- and long-lived proteins. (A, B) Myotubes were infected with adenoviruses encoding GFP (dotted line), WTp97 (dashed line) or DNp97 (full line) for 36 h and then incubated with 3H-tyrosine for the last 20 min (A) or 20 h (B) to differentially label short- and long-lived proteins, respectively. Error bars indicate s.d. n=6. (C) Equal amounts of protein lysates from myotubes expressing WTp97 or DNp97 were loaded and blotted with antibodies against: RGSH4, polyubiquitinated proteins (FK1) and GAPDH as loading control. Thirty-six hours after infection with DNp97-expressing virus, myotubes start to accumulate ubiquitinated proteins.
p97 inhibition decreases FoxO3-stimulated proteasomal and lysosomal proteolysis
A critical factor in the induction of muscle atrophy is the activation of the transcription factor FoxO3 (Sandri et al, 2004). Overexpression of a constitutively active FoxO3 mutant (caFoxO3) by itself causes profound atrophy by enhancing protein degradation and the expression of genes involved in the ubiquitin-proteasome pathway and autophagy (Mammucari et al, 2007; Zhao et al, 2007). To test whether inhibition of p97 can block the increased proteolysis induced by FoxO3, we compared myotubes expressing GFP, WTp97 or DNp97 for 24 h and then labelled these same cells for 24 h with 3H-tyrosine and concomitantly infected the cells with adenoviruses encoding GFP or caFoxO3. In myotubes, DNp97 overexpression blocked the increase in degradation of long-lived proteins by caFoxO3 (Figure 5A). Thus, p97 function appears essential for the stimulation of overall proteolysis by FoxO3 that is critical in multiple types of atrophy.
Figure 5.

In myotubes, DNp97, unlike WTp97, completely blocks the stimulation of proteolysis by caFoxO3, without altering the content of some atrogenes. (A) Myotubes expressing GFP, WTp97 or DNp97 for 24 h were given fresh media containing a second adenovirus expressing GFP or caFoxO3 supplemented with 3H-tyrosine to label long-lived proteins. Error bars indicate s.d. ANOVA was performed followed by Tukey’s HSD procedure. *P<0.05 DN–GFP versus GFP–GFP, **P<0.05 GFP–caFoxO3 versus GFP–GFP, ***P<0.05 DN–caFoxO3 versus GFP–caFoxO3, n=4. The DNp97-induced decrease in proteolysis was calculated by subtracting the protein degradation rate in the DNp97 overproducing cells from that in the controls (GFP–GFP or GFP–caFoxO3 expressing cells). Error bars indicate s.d. *P<0.001. (B) Overexpression of caFoxO3 in myotubes does not change the endogenous level of p97 protein. Equal amounts of protein from myotubes expressing for 48 h GFP (first three lanes) or caFoxO3 (last three lanes) were loaded. Endogenous p97, FoxO3 and GAPDH were immunoblotted. (C) The content of some atrogenes (atrogin-1, MuRF1, LC3 and Gabarapl1) increases upon overexpression of caFoxO3 but does not change in myotubes expressing WTp97 or DNp97 for 48 h. A lysate of cells treated with Bortezomib for 2 h has been also loaded as additional control for accumulation of polyubiquitin conjugates. Bortz: Bortezomib.
Because caFoxO3 overexpression nearly doubled p97-dependent proteolysis (Figure 5A), but did not change the amount of p97 in the cell (Figure 5B), the capacity of the p97 complex to support proteolysis in the myotubes is not saturated. It is also noteworthy that this inhibitory effect of DNp97 on proteolysis did not involve any alteration in the content of FoxO3-induced atrogenes, including the key muscle-specific ubiquitin ligases, atrogin-1 and MuRF1, and the proteins involved in autophagy, Gabarapl1 and LC3 (Figure 5C). Thus, inhibiting p97 did not reduce FoxO-mediated gene expression but blocks the ability of these atrogenes to catalyse proteolysis.
Although in atrophying adult muscles, the ubiquitin-proteasome pathway is important in myofibrillar degradation (Solomon and Goldberg, 1996; Cohen et al, 2009), in myotubes, where such proteins are not abundant, the autophagic pathway makes a greater contribution to overall proteolysis (Zhao et al, 2007). To learn how p97 influences these two processes, we measured the FoxO3-induced degradation of long-lived proteins in myotubes and dissected the relative importance of lysosomal and proteasomal pathways using inhibitors of the proteasome (1 μM Bortezomib/Velcade) or lysosomal acidification (10 mM NH4Cl) (Zhao et al, 2007). DNp97 expression decreased both proteasomal and lysosomal pathways in control myotubes as well as ones atrophying due to the overexpression of caFoxO3 (Figure 6A–C).
Figure 6.

In myotubes, DNp97 blocks the FoxO3-induced proteolysis by inhibiting both proteasomal and lysosomal degradation. (A) Myotubes treated as described in Figure 5A were given fresh DMEM (control) containing 2% HS with or without the proteasomal (Bortezomib=Bortz) or lysosomal (ammonium chloride=NH4Cl) inhibitors. Rates of proteolysis were determined starting 2 h later. Error bars indicate s.d. n=4. (B) The amount of proteolysis sensitive to each inhibitor represented the amount of proteasome or lysosome-mediated degradation and was calculated from data in Figure 6A by subtracting the rates of proteolysis in cells treated with the inhibitors from those of untreated cells. Error bars indicate s.d. (C) The DNp97-induced decrease in proteolysis was calculated by subtracting the amount of lysosomal or proteasomal protein degradation rate (i.e., the inhibitor-sensitive component) in the DNp97 overproducing cells from that in the controls (GFP-expressing cells). ***P<0.005 and *P<0.05 versus GFP–GFP controls. Error bars indicate s.d.
p97 inhibition causes muscle growth independently of the myosin-chaperone UNC45B
One possible mechanism by which inhibiting proteolysis might promote muscle growth is by causing a buildup of UNC45B, the chaperone necessary for myosin assembly. In muscles of C. elegans, UNC45B is a substrate of p97, and mutations in p97 impair UNC45B degradation (Janiesch et al, 2007). However, during atrophy induced by either denervation or food deprivation (Figure 7A), we did not observe any change in the content of UNC45B as shown by quantitation of bands in SDS–PAGE. Nevertheless, we tested whether the increased fibre size upon inhibition of p97 was in part due to stabilization of UNC45B and thus, to a possible increase in myosin incorporation into myofibrils. When GFP or UNC45B were overexpressed for 7 days, the fibres overexpressing UNC45B were reproducibly only about 10% larger in median area than control fibres (Figure 7B), and no difference was found in the weights of these muscles (Figure 7C). Thus, at most UNC45B can cause a small increase in fibre size, and an UNC45B buildup clearly cannot account for the 30–60% increase in fibre size observed upon p97 inhibition for 7 days (Figure 2B and D). Nevertheless, to further examine if UNC45B might mediate the growth induced by DNp97, we electroporated a plasmid carrying a shRNA against UNC45B (Supplementary Figure S2C and D) with or without a plasmid expressing DNp97 into adult muscles. Although depletion of endogenous UNC45B caused a small (<8%) increase in median fibre cross-sectional area for unknown reasons (Figure 7D), DNp97 expression still resulted in about a 60% increase in fibre area in 7 days despite the depletion of endogenous UNC45B (Figure 7E). Thus, p97 inhibition must trigger muscle growth independently of UNC45B.
Figure 7.

The myosin-chaperone UNC45B, a substrate of p97, does not alter muscle fibre size as DNp97. (A) Equal amounts of protein from normal and denervated TA muscles or from TA muscles of fed or food-deprived mice were analysed at the indicated times. Band quantitation analysis showed that the content of UNC45B does not change during two types of atrophy. (B) Frequency histograms showing the distribution of cross-sectional areas of TA muscle fibres either untransfected (control) or expressing UNC45B for 7 days. According to the Mann–Whitney Test, the differences between UNC45B versus control were significant (P<0.0001). (C) The average weights of TA muscles expressing for 7 days GFP or UNC45B are plotted. Error bars indicate s.e.m. No differences were found. n=5. (D, E) Frequency histograms showing the distribution of cross-sectional areas of TA muscle fibres either untransfected (control) or expressing for 7 days a shRNA for UNC45B (D) or shRNA for UNC45B and a plasmid for DNp97 (E). According to the Mann–Whitney Test, the differences between shRNA for UNC45B versus control (P<0.005) and between shRNA for UNC45B+DNp97 versus control (P<0.0001) were significant.
The DNp97 associates tightly with myofibrils, especially during denervation atrophy
Since DNp97 binds ubiquitinated proteins but fails to release them, it can be used to ‘trap’ p97 substrates (Ye et al, 2003). To test if the degradation of myofibrillar proteins involves p97, we electroporated either WTp97 or DNp97 into the contralateral TA muscles and 7 days later isolated the myofibrillar fractions. The active and inactive p97s were overexpressed to similar extents, as shown by GFP fluorescence and immunoblotting (Figure 2E). These myofibrillar preparations appeared quite pure since they contained all the characteristic major components of the myofibrils, as shown by Coomassie staining (Cohen et al, 2009) and immunoblotting (Figure 8A), but no markers of the nucleus (HDAC1) or mitochondrial membranes (VDAC1) (Figure 8A). When the subcellular distributions of the GFP-labelled WT and DNp97 were determined, striking differences were observed. While nearly all the WTp97 was found in the soluble fraction, most of the DNp97 co-sedimented with the myofibrillar fractions and remained tightly associated with them even after extraction with 300 mM KCl (Figure 8B). Thus, the WTp97 probably associates transiently with myofibrillar proteins, but the non-dissociating ATPase-deficient mutant remains bound presumably to ubiquitinated myofibrillar components (see below). These results were also confirmed by confocal microscopic analysis with another dominant negative p97 mutant (p97E578Q) that, unlike the WT, showed a striation-like distribution upon expression for 7 days in adult muscles (Figure 8D).
Figure 8.

DNp97 becomes tightly associated to myofibrils in mouse TA (even more after denervation) and causes accumulation of ubiquitinated actin and MyLC2 in myotubes. (A) The purity of the myofibrils extracted by adult muscle is assayed by immunoblotting for proteins localized to nucleus (HDAC1) or to the mitochondrial membrane (VDAC1). Sol: soluble. Myo: myofibrils. (B) Myofibril isolation from paired mouse TA expressing WTp97GFP (left leg) or DNp97GFP (right leg) for 7 days. Fluorescence measurements of the soluble, the myofibrillar fraction and the washes from myofibrils with increasing salt concentration were plotted as a percentage of total fluorescence. The changes shown are typical of results obtained three times, although the absolute amounts of p97 in these fractions varied among experiments. (C) Gentle myofibril isolation from paired mouse muscles expressing DNp97GFP and denervated or not for 9 days (*P<0.005, n=3). Error bars indicate s.e.m. (D) Representative images by confocal microscopy of longitudinal sections of TA muscle expressing WTp97–MycHis or DNp97–MycHis (p97E578Q) for 7 days were stained with anti-Myc antibodies (green). While WTp97 shows a diffuse and nuclear distribution, the DN mutant clearly displays a striation-like pattern. Scale bar represents 5 μm. (E) Soluble and myofibrillar fractions (before and after 0.6 M KI extraction) from whole-muscle homogenate were stained for MyHC, MyLC2, actin, p97 and HDAC1. (F) In extracts of myotubes expressing GFP or DNp97 for the indicated times, the contractile proteins were assayed by immunoblotting. Both actin and MyLC2 (unlike MyHC and MyBP-C) were found in part as ubiquitinated species upon 72 h expression of DNp97. No appreciable difference of the MyHC/actin ratio of Coomassie Blue-stained myofibrils from muscles expressing WTp97 or DNp97 was found, suggesting that both thin and thick filaments are degraded by p97-dependent mechanisms (WTp97 muscles: 1.74±0.31; DNp97 muscles: 1.99±0.19, n=3, P=0.52). Figure source data can be found with the Supplementary data.
To test if this association with myofibrillar fractions is related to the degradation of these proteins during atrophy, we prepared myofibrils with gentler washing than used above (see Materials and methods) from control muscles and ones denervated for 9 days (i.e., when myofibrillar proteins are degraded rapidly). In these atrophying muscles, even more of the DNp97 was recovered in the myofibrillar fractions than from contralateral controls and less in the soluble fraction (Figure 8C) (P<0.005). Using this gentle method to isolate the myofibrils, we were also able to detect appreciable amounts of endogenous p97 in the myofibrillar fractions in the denervated as well as in control muscles (Figure 8E).
To examine whether p97 was associated with contractile proteins, these myofibrils were extracted with 0.6 M potassium iodide (KI), which depolymerizes and solubilizes both thin and thick filaments (Szent-Gyorgyi, 1951). After extraction and re-centrifugation, the endogenous p97 was found in the wash together with the solubilized myofibrillar components, including myosin heavy chain (MyHC), myosin light chain 2 (MyLC2) and actin (Figure 8E). No such release of p97 or contractile proteins was observed if KI was not present in the extraction buffer (Figure 8E). Therefore, p97 was associated with the contractile proteins and not with the desmin cytoskeleton or some minor undetected insoluble contaminant.
The DNp97 causes accumulation of ubiquitinated myofibrillar components and forms distinct complexes with different contractile proteins and cofactors
Because the decrease in protein content during atrophy involves mainly a loss of myofibrillar proteins, it is most likely that DNp97 in decreasing atrophy blocks a key p97-dependent step in myofibrillar breakdown. Furthermore, p97 seems to be directly involved in myofibrillar turnover because the non-dissociating DNp97 mutant, while blocking atrophy and inhibiting proteolysis, became tightly associated with the myofibrillar proteins. Based upon p97’s roles in the disassembly and degradation of other protein complexes, it seems very likely that this non-dissociating mutant blocked myofibrillar degradation by binding to ubiquitinated components and preventing their release from the sarcomere and delivery to the proteasome. Accordingly, we found that the expression of the DNp97 (but not GFP or WTp97) in myotubes, resulted in a small but reproducible accumulation of ubiquitinated MyLC2 and actin (Figure 8F and Supplementary Figure S6). When we immunoprecipitated actin from these infected myotubes, we found that the substrate-trapping DNp97 was associated with actin, which was heavily ubiquitinated (Figure 9A; Supplementary Figure S4A). Furthermore, when we immunoprecipitated the MyLC2 from the myotubes where p97 was inhibited with the DN for 48 h (Supplementary Figure S4B) or 72 h (Figure 9B), the MyLC2 was found associated with DNp97 and was ubiquitinated.
Figure 9.

In myotubes, DNp97 binds actin (which also binds Ufd1) or MyLC2 or MyLC1 (which also binds p47) and results in accumulation of actin or MyLC2 as ubiquitinated species. (A–D) Immunoprecipitation of actin (A) or MyLC2 (B) or Ufd1 (C) or p47 (D) was performed from myotubes overexpressing GFP, WTp97–RGSH4 (in the presence of DMSO or 1 μM Bortezomib for 3 h) or DNp97–RGSH4 for 48–72 h. As control, immunoprecipitation of the same samples with appropriate IgG was carried out. Actin, RGSH4, polyubiquitinated proteins (FK1), MyLC2 and 1,Ufd1, p47 and MyHC were immunoblotted. Figure source data can be found with the Supplementary data.
Thus, ubiquitinated MyLC2 and actin behave as typical p97 substrates. Accordingly, the DNp97 could not be immunoprecipitated with the MyLC2 or actin, if ATP was omitted from the lysis buffer (Supplementary Figure S4C). As was reported for p97 binding to other substrates (Ye et al, 2003), ATP binding to the intact D1 domain in DNp97 is required for the interaction with these contractile proteins, probably because ATP is essential for stabilization of p97 hexamers (Wang et al, 2003).
By contrast, when we immunoprecipitated actin or MyLC2 from myotubes overexpressing the WTp97 or myotubes treated with Bortezomib/Velcade for 3 h to block proteasomal degradation, we found no accumulation of these ubiquitinated proteins (Supplementary Figure S4A and B). Although treatment with the proteasome inhibitor for 3 h, like p97 inhibition, caused an accumulation of ubiquitin conjugates (Supplementary Figure S4A and B), no ubiquitinated actin or MyLC2 was detected, as would be expected since actin and MyLC2 are long-lived proteins and should not undergo significant ubiquitination in 3 h. Thus, their buildup in ubiquitinated forms appears to be a selective response to the expression of the DNp97. It is interesting that under these same conditions, other thick filament components (e.g., MyHC and myosin binding protein C) (Figure 8F) did not show any clear change indicative of ubiquitination, even though in adult muscle, these proteins are ubiquitinated by the same E3, and myosin binding protein C is degraded at about the same time after denervation as MyLC2 (Cohen et al, 2009).
In order to identify the cofactors that may also be present with p97 in association with actin and MyLC2, we immunoprecipitated Ufd1 or also p47 from the infected myotubes. Interestingly, the DNp97 and actin could be co-immunoprecipitated with Ufd1, but p47 or MyLC2 could not (Figure 9C). However, when p47 was immunoprecipitated, some DNp97 and MyLC1 were also pulled down, but Ufd1, actin, MyHC or MyLC2 were not (Figure 9D). Thus, actin and MyLCs appear to be novel p97 substrates that form distinct p97 complexes with different cofactors, p97–Ufd1—actin and p97–p47–MyLC1. These results together suggest that these different p97–cofactor complexes associate with and extract these ubiquitinated proteins from the myofibrils and thus facilitate their degradation by proteasomes, but that the non-dissociating p97 blocks this process and thereby prevents their loss during atrophy.
The muscle content of p97 and two of its cofactors increase after denervation
Together, these findings all indicate an important role of p97 during atrophy. We therefore measured the muscle’s content of p97 and these cofactors in mouse Gastrocnemius at 3 days after sectioning the sciatic nerve, when the atrophy-specific ubiquitin ligases, MuRF1 and atrogin-1, are maximally induced (Bodine et al, 2001; Sacheck et al, 2007), and at 10 days when muscle weight decreased by 40% (Figure 10C) and when myofibrillar proteins, especially thick filaments, are rapidly degraded (Cohen et al, 2009). Although p97 levels did not change at 3 days, they increased by three-fold at 10 days (Figure 10A; P<0.05).
Figure 10.

The content of p97 and two of its cofactors are induced together with proteasomal subunits and the shuttling factor mR23A in Gastrocnemius 10 days after denervation. (A, B) Levels of p97 and its cofactors Ufd1, p47 and Ufd2 (A) and of the proteasomal subunit of the 19S regulatory particle Rpt1, the 20S catalytic core and the shuttling factor mR23A (B) were measured by immunoblotting on extracts from mouse Gastrocnemius at different times after cutting the sciatic nerve. Equal amounts of protein were loaded. Densitometry data and statistical analysis of protein levels are plotted. Error bars indicate s.e.m. (A) *P<0.05, n=6. (B) **P<0.01 and ***P<0.005, n=7. (C) Gastrocnemius weight was decreased 10 days after denervation. *P<0.001, n=4.
The muscle’s content of three main p97 cofactors, Ufd1, Ufd2 and p47, also did not change by 3 days after denervation, but by 10 days, Ufd1 content had also increased about three-fold and p47 about two-fold (P<0.05), while the amount of Ufd2 did not change (Figure 10A). It is noteworthy that this accumulation of p97, Ufd1 and p47 coincided with the time (10–14 days) after denervation, when breakdown of myofibrillar proteins is rapid (Cohen et al, 2009) and further suggests an involvement of the p97 complex in this process. Interestingly, at this time, the amounts of the 26S proteasome also seemed to increase about two-fold since the content of Rpt1, a 19S ATPase subunit and the 20S particle’s α-subunits were both two- to three-fold higher than in contralateral control muscle, and nearly all of these subunits are found in intact proteasomes (Altun et al, 2010). In addition, these denervated muscles had an increased content of the Uba–Ubl protein, murine Rad23A (mR23A) (Figure 10B; P<0.005); in yeast, Rad23 appears to facilitate the delivery of ubiquitinated proteins from p97 to the proteasome (Chen and Madura, 2002). This induction of p97 and its cofactors result from changes in gene expression, since mRNA for p97, Ufd1, Ufd2 and especially p47, increased before the proteins accumulated (Supplementary Figure S1C; P<0.05).
However, the increased content of p97, Ufd1 or p47 is not a general feature of all atrophying muscles, but seems specific to denervation atrophy. During the rapid weight loss induced by 1–2 days of fasting, there was no increase in the amount of these proteins in the Gastrocnemius muscle (Supplementary Figure S1A). Similarly, in fasting, there was no increase in the content of the proteasome subunit Rpt1 (Supplementary Figure S1A). Interestingly, p97 mRNA content (but not that of its cofactors) rose by 48 h after food deprivation even though the protein did not rise by this time (Supplementary Figure S1B; P<0.005). Since the mRNA of p97 increases both upon denervation and food deprivation, as well as in the muscle wasting seen in aged rats (Altun et al, 2010), the p97 gene may be classified as an atrogene (Lecker et al, 2004; Sacheck et al, 2007), whose expression rises or falls similarly in many types of atrophy. However, p97 mRNA and protein increased more slowly than the well-studied atrogenes (e.g., atrogin-1 or MuRF1) (Jagoe et al, 2002; Sacheck et al, 2007), perhaps because the transcription of p97 is not regulated by the transcription factor, FoxO3 (Figure 5B). Although the importance of these changes in mRNA will require further study, these findings on fasting clearly indicate that marked loss of muscle proteins does not require an increased content of p97 or its major cofactors.
Discussion
p97 complex plays a major role in muscle atrophy
The present findings clearly demonstrate that p97 is essential for the loss of muscle mass upon denervation as well as food deprivation (Figure 1). These two types of atrophy are quite different physiological responses, resulting from either a lack of contractile function in a specific muscle or systematically from decreased levels of insulin and IGF1 and increased levels of glucocorticoids. However, the present findings are very likely to apply to multiple types of atrophy since similar cellular mechanisms contribute to muscle wasting in multiple systemic diseases (e.g., cancer cachexia, renal failure, sepsis), which are all characterized by insulin resistance and high glucocorticoids levels. Also, rodent models of many different catabolic states reveal a similar activation of protein degradation (Mitch and Goldberg, 1996), of the atrogene transcriptional programme (Lecker et al, 2004; Sacheck et al, 2007) and of FoxO-induced transcription (Sandri et al, 2004). The attenuation of muscle wasting upon p97 inhibition is due largely to the resulting fall in overall protein degradation and the blocking of FoxO-induced proteolysis (Figure 5A), which is essential for atrophy upon fasting and denervation (Sandri et al, 2004). Notably, this blockage of FoxO-dependent proteolysis occurred even though FoxO-induced expression of atrogin-1 and MuRF1 occurred normally (Figure 5C). Thus, p97 is not essential for signalling atrogene transcription, but for the resulting activation of proteolysis.
Despite this essential role in atrophy, the increase in p97 content per se is not sufficient to induce muscle wasting (Figure 2A) or to accelerate protein degradation (Figure 4A and B). Therefore, its level is not rate-limiting for proteolysis by the ubiquitin-proteasome pathway or autophagy, even though p97 activity is absolutely necessary for the progression of atrophy and the associated acceleration of proteolysis by these systems (Figure 6).
It seems important that the levels and expression of p97, Ufd1 and p47 increase during denervation atrophy at the time (i.e., 10 days after nerve section) when myofibrillar components begin to be rapidly degraded (Cohen et al, 2009). By this time, muscle mass has already decreased significantly, but mainly through the loss of soluble proteins (Figure 10). Interestingly, this increase in the levels of the p97 complexes coincided with an increase in the content of 26S proteasomes and the shuttling factor, mR23A (Figure 10B), which in yeast functions in the ERAD pathway to enhance the delivery of p97-processed ubiquitin conjugates to the proteasome (Chen and Madura, 2002). Together, these findings strongly suggest a role for p97, Ufd1 and p47 in the enhanced degradation of myofibrillar components by the ubiquitin-proteasome pathway. However, upon the more rapid atrophy with fasting (which could only be studied for 2 days, for ethical reasons), the content of p97, its cofactors and proteasomes does not increase (Supplementary Figure S1), despite a general activation of proteolysis by the ubiquitin-proteasome pathway (Tawa et al, 1997) and increased mRNA for some proteasomal subunits and p97 (Supplementary Figure S1B) (Medina et al, 1995). Normally, p97 and proteasomes are highly abundant cell constituents (about 1–2% of cell proteins), and apparently are present in sufficient amounts to support the increased proteolysis in fasting, although not for the rapid breakdown of the myofibrillar apparatus late after denervation. Perhaps, in fasting, their enhanced gene expression serves to maintain these complexes at high levels in the face of their accelerated destruction due to the enhanced proteolysis.
Interestingly, in rat muscles from aged rats that have undergone the marked systemic wasting (sarcopenia) associated with advanced age, we also found an increased content of p97 and p47 and greater expression of these genes (Altun et al, 2010), as well as an increased proteasome content and other changes indicative of an enhanced activity of the ubiquitin-proteasome system (Altun et al, 2010). Perhaps, the slow muscle wasting in the aged, where there is a general reduction in contractile activity, involves similar p97-dependent mechanisms as denervation/disuse-atrophy. Interestingly, p97 mutations have also been implicated in 1–2% of cases of familial Amyotrophic Lateral Sclerosis, where motor neurons degenerate, producing a type of denervation/disuse atrophy (Johnson et al, 2010).
Previously, we have coined the term ‘atrogenes’ to refer to the set of atrophy-related genes that are coordinately regulated in all types of rapid muscle atrophy. The increases in p97 mRNA upon denervation and fasting, though modest, raise the possibility that p97 is such a gene (Supplementary Figure S1B and C). Nonetheless, further analysis of mRNA levels of p97 in other catabolic states will be needed to determine if p97 is an atrogene. However, p97, unlike the dramatically induced atrogenes studied previously, is not induced upon overexpression of caFoxO3 in myotubes (Figure 5B). So, its accumulation may occur through action of other transcription factors or reduced proteolysis.
p97 and the degradation of ubiquitinated myofibrillar proteins
Nearly all p97-dependent substrates identified previously are short-lived misfolded or regulatory proteins (Yamanaka et al, 2012). However, we showed here that the destruction of long-lived structural components (e.g., myofibrillar proteins) and short-lived proteins are reduced similarly upon p97 inhibition. Myofibrillar proteins are normally among the most stable intracellular proteins, and even when their breakdown is accelerated during atrophy, they have half-lives of days. Following p97 inhibition, ubiquitinated forms of two myofibrillar components, actin and MyLCs became evident (Figures 8F and 9; Supplementary Figures S4 and S6). After denervation, thick filament components are ubiquitinated by MuRF1, leading first to degradation of the regulatory proteins, myosin binding protein C and myosin light chains, followed by MyHCs (Cohen et al, 2009). Thin filament components (e.g., actin) are then degraded via a distinct ubiquitin ligase, probably Trim32 (Kudryashova et al, 2005; Cohen et al, 2009). The present findings suggest that p97 plays a role in the degradation of at least one component of both types of filaments.
Because the DNp97 mutant studied here is defective in dissociating from substrates, it can be used to trap substrates in cells (Ye et al, 2003). Its strong and specific binding to (presumably ubiquitinated) myofibrils in muscles (Figure 8) therefore implies a role in turnover of these components in normal muscle and especially after denervation. Although the p97 cofactors, Ufd1 and p47 (Figure 10), both bind ubiquitin conjugates (Madsen et al, 2009), they were found to form complexes with p97 and different myofibrillar components, Ufd1 with actin, and p47 with the thick filament protein MyLC1 (Figure 9). The p97–Ufd1 and/or p97–p47 complexes thus probably act on myofibrils after ubiquitination during atrophy and to a lesser extent in normal muscle. In other cellular processes, p97 and ubiquitin-binding cofactors catalyse the ATP-dependent extraction of ubiquitinated components from chromatin, the ER membrane (in the ERAD pathway) or mitochondria prior to delivery to the 26S proteasome (Rape et al, 2001; Ye et al, 2001). By analogy, therefore, it is very likely that these different p97–cofactor complexes catalyse the extraction of these contractile proteins from the sarcomere prior to delivery to the 26S proteasome. However, conclusive evidence for this likely role in extracting ubiquitinated myofibrillar components and delivery to the proteasome is presently lacking, and such studies (e.g., by cell-free reconstitution of this process) will be quite difficult or impossible for multiple technical reasons.
The ATPases in the proteasome and the p97 complex are homologous members of the AAA family of hexameric ATPases. By themselves, the proteasomal ATPases can unfold and translocate globular substrates into the 20S proteolytic chamber (Smith et al, 2011). Presumably, p97, by similar mechanisms, extracts and unfolds ubiquitinated proteins from large macromolecular complexes, such as the thick and thin filaments, that for some unknown reasons cannot be processed directly by the 26S proteasomes. Recent studies suggest that p97 is necessary for the degradation of proteins that lack a loose C-terminus (Beskow et al, 2009). This p97-dependent disassembly process seems crucial in muscle, where many proteins are tightly associated in the very rigid, highly organized sarcomere.
p97 inhibition causes hypertrophy of normal muscles
Rapid, muscle growth upon p97 inhibition was a completely unexpected finding, especially since loss of p97 function has deleterious effects on most cells and is lethal during murine embryogenesis (Muller et al, 2007). In yeast, Cdc48 ablation prevents growth and causes cell-cycle defects (Moir et al, 1982), whereas in various mammalian cells, p97 knockdown or treatment with p97 inhibitors leads to multiple defects and apoptosis (Kobayashi et al, 2002; Wojcik et al, 2004; Bursavich et al, 2010; Chou et al, 2011). In several cell lines, we also observed that overexpression of DNp97 with the same adenovirus used here caused toxicity. Myotubes and hepatocytes (Acharya et al, 2011) are notable exceptions. In myotubes, treatment with DNp97 or shRNA caused no evident cell death, did not reduce overall protein synthesis (Table I; Supplementary Figure S5), and did not cause an accumulation of oxidatively damaged proteins (Supplementary Figure S3). Most surprisingly, the loss of p97 function in myotubes increased protein content and fibre diameter (Table I; Figure 3) and in adult muscle caused rapid increases in fibre size (Figure 2). Thus, p97 function appears important for most protein degradation in muscles and in this way limits the growth of myotubes and muscle fibres in the adult.
In our initial studies where we electroporated equal amounts of the plasmids for the WT and DNp97 plasmids into muscle, we found that at least 15 times more WT protein was expressed. Thus, the mutant form must be much more short-lived than the WT protein. Consequently, in the present studies, the amounts of these plasmids electroporated were adjusted to insure that equal amounts of WT and DNp97 were compared (Figure 2E). Interestingly, when we electroporated the WTp97 in great excess (∼15 ×), the muscle fibres hypertrophied (not shown), as was seen with low levels of DNp97. Thus, at very high concentration the WT behaves like the DN forms, presumably because p97 only functions in protein degradation together with the BS1 (binding site 1) family of cofactors (e.g., p47 and Ufd1), and the excess p97 must inhibit the formation of such functional complexes.
Normal growth of muscle requires the IGF1/insulin-signalling pathway to enhance protein synthesis and reduce FoxO3-activated proteolysis (Sandri et al, 2004; Stitt et al, 2004; Glass, 2010). It is quite surprising that myotubes and presumably muscle can grow simply through an inhibition of overall proteolysis, in other words by reducing both proteasomal and autophagic pathways, without any clear increase in overall protein synthetic rates (Table I), activation of the PI3K/AKT pathway (Supplementary Figure S5) or increased content of the 60S ribosomal marker protein Rpl26 (Figure 2E).
One possible explanation for this surprising growth of muscles and myotubes upon p97 inhibition was more efficient incorporation of myosin into myofibrils by UNC45B, the chaperone catalysing myosin assembly. However, its stabilization caused little or no increase in mass, and its depletion did not reduce the muscle growth upon p97 inhibition (Figure 7). Therefore, the stabilization of the bulk of cell proteins and of key regulatory factors probably accounts for this hypertrophy. Interestingly, the content of UNC45B (Figure 7) as well as of Ufd2 (Figure 10) and CHIP (Altun et al, 2010), which function in UNC45B degradation (Janiesch et al, 2007), do not change during denervation atrophy when p97 and certain of its cofactors increase. Thus, during atrophy certain p97 complexes accumulate, presumably those apparently involved in sarcomere degradation, but not those catalysing myofibrillar formation.
p97 and disease
Because several p97 substrates are crucial for cell-cycle progression and tumorigenesis (i.e., Aurora B kinase, cyclinE and IkBα) (Dai et al, 1998; Dai and Li, 2001; Ramadan et al, 2007), and p97 upregulation correlates with a poor prognosis in many cancers (Tsujimoto et al, 2004; Yamamoto et al, 2004), inhibitors of p97 are being investigated as anti-cancer therapeutics (Bursavich et al, 2010; Chou et al, 2011). Unlike many anti-cancer agents that inhibit the growth processes (e.g., PI3K or mTOR pathways), p97 inhibitors should not have undesirable catabolic effects on muscle, and might even reduce the muscle wasting characteristic of cancer cachexia, which seems to contribute to the downhill course of patients (Houten and Reilley, 1980; Anker et al, 1997; Zhou et al, 2010). Therefore, p97 inhibitors may not only be anti-neoplastic but may also be beneficial by decreasing muscle proteolysis and myofibrillar loss.
p97 mutations that cause IBMPFD lead to sarcomeric disorganization, inclusion formation and muscle wasting (Kimonis et al, 2008). These p97 mutants show an increased propensity to bind the cofactors, ataxin-3, p47 and Npl4, but decreased binding of ubiquitin ligases (Fernandez-Saiz and Buchberger, 2010). It is controversial whether these p97 mutants have increased (Halawani et al, 2009; Manno et al, 2010) or normal ATPase activity (Weihl et al, 2006; Fernandez-Saiz and Buchberger, 2010), and their effects on the rates or selectivity of protein degradation are unknown. Interestingly, overexpression of certain p97 cofactors can recapitulate some of the features of p97-induced disease (Nagahama et al, 2003; Song et al, 2005). The induction of muscle growth seen here upon p97 inhibition clearly differs from the changes characteristic of inclusion-body myopathy. For example, electroporation of the disease-associated p97 mutant gene, p97R155H, unlike the DNp97 mutation, did not induce muscle growth. Also, in our studies, no inclusions were found upon p97 inhibition (although they lasted only 10 days) (Figure 1C). Thus, it seems unlikely that the IBMPFD-associated myopathy results simply from loss of p97 ATPase activity and a resulting failure to degrade proteins that eventually accumulate as toxic aggregates.
Future work hopefully will illuminate the precise role of p97 and its cofactors in recognition of different ubiquitinated myofibrillar proteins and processing them for proteasomal degradation. Our finding that lysosomal proteolysis also decreased with DNp97 is in agreement with the recent report (Krick et al, 2010) that in other cells p97 functions in the autophagic/lysosomal pathway, although its precise role in lysosomal function is unclear. In IBMPFD, there is abnormal vacuolization with inclusions containing proteins that are normally degraded by lysosomes (Ju et al, 2009) and increased number of autophagosomes (Tresse et al, 2010). However, since p97 plays multiple roles in proteolysis, defects in the breakdown of many proteins, including myofibrillar components, can contribute to the functional and structural abnormalities in these patients.
Materials and methods
Cell culture and adenoviral infection
C2C12 myoblasts (ATCC) were cultured in DMEM+10% FBS (Invitrogen) and penicillin/streptomycin, differentiated in DMEM with 2% Horse Serum (HS) (Invitrogen) for 3 or 4 days before infection. The infection efficiency was typically >90%. Adenoviruses expressing WTp97 and DNp97 fused to arginine, glycine, serine and four histidines (RGSH4) were kindly provided by Professor W Lencer (Kothe et al, 2005). For those cultures infected with a second virus (GFP or caFoxO3) (Sandri et al, 2004), the first virus was removed from the myotubes at 24 h, the cells washed once, and the second virus in fresh differentiation medium added.
Plasmids
Plasmids for GFP, WTp97GFP and p97K524AGFP were a generous gift of Professor A Kakizuka and described in Kobayashi et al (2002). Plasmids for WTp97–MycHis and p97E578Q–MycHis were kindly provided by Professor C Weihl. The non-silencing shRNA (a) or the shRNA for murine p97 (b) or for murine UNC45B (c) in GIPZ vector were purchased from Open Biosystem. The plasmid encoding for UNC45B was kindly provided by Professor D Winkelmann and described in Srikakulam et al (2008). Hairpin sequences:
ATCTCGCTTGGGCGAGAGTAAGTGCTGTTGACAGTGAGCGATCTCGCTTGGGCGAGAG TAAGTAGTGAAGCCACAGATGTACTTACTCTCGCCCAAGCGAGAGTGCCTACTGCCTCGGA
TGCTGTTGACAGTGAGCGCGGTCCTGAAATCATGAGCAAATAGTGAAGCCACAGATGT ATTTGCTCATGATTTCAGGACCATGCCTACTGCCTCGGA
TGCTGTTGACAGTGAGCGAGCACCATCTATGTTGTAGATATAGTGAAGCCACAGATGT ATATCTACAACATAGATGGTGCGTGCCTACTGCCTCGGA
Adult mouse skeletal muscle electroporation
TA muscles of adult male CD1 mice (28–30 g) (Charles River) were in vivo transfected (Sandri et al, 2004). The muscle was isolated through a small hindlimb incision, and 20 μg of plasmid DNA was injected along the muscle length. Electric pulses were then applied by two stainless steel spatula electrodes placed on each side of the isolated muscle belly (50 Volts/cm, 5 pulses, 200 ms intervals). Muscles were analysed 7 or 9 days later. For denervation studies, at the time of electroporation, the sciatic nerve was cut and the downstream muscles were allowed to atrophy for 3–14 days. For starvation studies, animals of the fed and fasted group were matched in order to have same initial body weights. The Animal Research Committee at Harvard Medical School approved all the experimental protocols.
Immunohistochemistry and fibre size measurements
Ten-micrometre-thick cryosections of electroporated muscles were fixed by 4% paraformaldehyde and stained for nuclei with Hoechst (Sigma). Fluorescent images were collected in the Nikon Imaging Center at Harvard Medical School. Using ImageJ software (National Institutes of Health), cross-sectional areas of transfected and untransfected fibres from the same muscle were measured for a total of 250–500 fibres each condition. Myotube diameter was measured using ImageJ software on brightfield images. Ten-micrometre-thick longitudinal muscle sections were decorated with anti-Myc antibody (GeneTex) and Alexa 488-conjugated secondary antibody (Invitrogen).
Protein degradation measurement in C2C12 myotubes
As we have done previously (Sacheck et al, 2004; Zhao et al, 2007), C2C12 myotubes expressing for 36 h GFP, WTp97 or DNp97 were incubated with 3H-tyrosine (4 μCi/ml; Perkin-Elmer) for 20 min (pulse medium) to label short-lived proteins or 20 h to label long-lived proteins and then washed three times with chase medium (containing 2 mM unlabelled tyrosine), before collecting aliquots at specific times. Alternatively, pulse medium containing GFP or caFoxO3 adenoviruses was added to myotubes expressing GFP, WTp97 or DNp97 for 24 h, and media samples were collected over 32 h and combined with TCA (10% final concentration) to pellet overall proteins. Proteasomal and lysosomal proteolysis rates were determined precisely by treating with 1 μM Bortezomib (Millennium Pharmaceuticals) and 10 mM NH4Cl (Sigma), respectively (Zhao et al, 2007). The inhibitors were added 2 h prior to measuring protein degradation. Results are expressed as mean±s.d. of six replicates. Experiments were repeated 2–4 times with highly reproducible results so only a representative one is shown.
Protein synthesis measurement in C2C12 myotubes
Total protein synthesis was measured upon incorporation of 3H-tyrosine into cell proteins (White et al, 1988). C2C12 myotubes expressing GFP, WTp97 or DNp97 for 48 h were then exposed to 4 μCi/ml 3H-tyrosine for 2 h and then the media was discarded. Cells were washed twice with PBS, collected in 1 ml of 10% TCA per well and incubated for 30 min at 4°C to precipitate proteins. Cells were then spun for 5 min at 12 000 g at 4°C. The resulting pellet was washed with 1 ml of 95% ethanol, solubilized with 0.1 N NaOH and the amount of radioactivity measured by scintillation counting. Protein content was measured using the Bradford method (Pierce).
Protein extraction and immunoblotting
Muscles were homogenized with Polytron in ice-cold lysis Buffer A (1% Triton X-100, 30 mM sodium pyrophosphate, 50 mM NaF, 10 mM Tris (pH 7.6), 50 mM NaCl, 2 mM ATP, 10 mM MgCl2, 0.1 mM Na3VO4 and a protease inhibitor cocktail (Roche)). Myofibrillar proteins were isolated from TA, as described in Cohen et al (2009), but all the washes were not performed with Polytron but vortexed for 1 min and the pellet then spun at 1500 g for 12 min at 4°C. In the more gentle washing method, the samples were not vortexed but tapped and then spun at 1500 g for 12 min at 4°C. Fluorescence measurements were performed using FLUOstar Galaxy plate reader (BMG). The extraction with KI (0.6 M) was performed by incubating the myofibrils with Buffer A with 0.6 M KI for 15 min in ice and then spun at 1500 g for 12 min at 4°C. Lysates from myotubes were prepared in Buffer A except the ones in Figure 8F and Supplementary Figure S6 that are total extracts obtained with Buffer A with 4% SDS. Total protein was measured using BCA Protein or Coomassie Plus Assay Kit (Pierce). Equal amounts of total protein per lane were immunoblotted with the following primary antibodies: anti-Ufd2 and anti-FoxO3 (Cell Signaling); anti-MyHC, anti-mR23A and anti-Rpl26 (Abcam); anti-GFP (Santa Cruz Biotechnology); anti-p97 (BD Biosciences); anti-RGSH4 (Qiagen); anti-MyLC2, anti-UNC45B and anti-LC3 (Novus Biologicals); anti-GAPDH and anti-actin (Sigma); anti-VDAC1 (Millipore); anti-atrogin-1 (ECM Biosciences); anti-Gabarapl1 (ProteinTech Group); anti-MyLC1 (Hybridoma Bank); anti-HDAC1 (BioVision); anti-Rpt1, anti α 1–7 subunits of the 20S proteasome and anti-polyubiquitinated proteins (FK1) (Biomol). MyBP-C antibodies were kindly provided by Dr Greaser. Anti-Ufd1 and anti-p47 antibodies were kindly provided by Professor Meyer. The MuRF1 antibody was raised against rat MuRF1 and was described previously in Bodine et al (2001). Secondary antibodies were conjugated to alkaline phosphatase (Promega). Band intensities were analysed using ImageJ software (National Institutes of Health).
Immunoprecipitation
Myotube lysate was prepared in Buffer A with 2 mM PMSF (Sigma), 2 mM ATP and 10 mM MgCl2 (Sigma) (Buffer B) and precleared with appropriate IgG (Santa Cruz Biotechnology) and Protein A-Agarose (Roche) or Protein G PLUS-Agarose (Santa Cruz Biotechnology) for 1 h at 4°C. Actin, MyLC2 or p47 were in turn immunoprecipitated with the corresponding antibody (from Epitomics, Novus Biologicals and Santa Cruz Biotechnology clone E-16, respectively) coupled to Protein A-Agarose overnight at 4°C. To immunoprecipitate Ufd1, the antibody (Santa Cruz Biotechnology, clone C-20) was coupled to Protein G PLUS-Agarose. The immunoprecipitated material was washed five times with Buffer B, before heating at 95°C for 5 min in sample buffer (Invitrogen), 50 mM DTT and 20% 2-Mercaptoethanol (Sigma).
Statistical analysis and image acquisition
Differences among samples were assessed by the Student’s two-tailed t-test for independent samples. Statistical significance was assessed using paired Student’s t-test (for comparisons of TA electroporated with different plasmids in the same mouse) or ANOVA or the Kruskal–Wallis Test (non-parametric ANOVA) or the Mann–Whitney Test followed by Tukey’s HSD procedure where appropriate. As typically done, we show the medians for cross-sectional areas for fibre size distributions but performed statistics assuming that these distributions were normal, which is justified because median and means were quite similar. Images were processed using Adobe Photoshop CS3, version 10.0.1.
Supplementary Material
Acknowledgments
This work was supported by grants from the Muscular Dystrophy Association, the Packard Foundation and National Institutes of Health (AR055255). We are very grateful to JJ Brault, S Cohen, R Hourez, J Zhao and LM Selfors for advice and assistance. We thank Professor A Kakizuka, Professor W Lencer, Professor D Winkelmann, Professor H Meyer, Professor C Weihl and Millennium Pharmaceuticals for kindly providing reagents. We thank the Nikon Imaging Center at Harvard Medical School for help with light microscopy.
Author contributions: RP designed, performed and interpreted the experiments. ALG designed and interpreted the experiments. RP and ALG wrote the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Acharya P, Liao M, Engel JC, Correia MA (2011) Liver cytochrome P450 3A endoplasmic reticulum-associated degradation: a major role for the p97 AAA ATPase in cytochrome P450 3A extraction into the cytosol. J Biol Chem 286: 3815–3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, Deshaies RJ (2008) UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell 134: 804–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun M, Besche HC, Overkleeft HS, Piccirillo R, Edelmann MJ, Kessler BM, Goldberg AL, Ulfhake B (2010) Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin-proteasome pathway. J Biol Chem 285: 39597–39608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, Harrington D, Kox WJ, Poole-Wilson PA, Coats AJ (1997) Wasting as independent risk factor for mortality in chronic heart failure. Lancet 349: 1050–1053 [DOI] [PubMed] [Google Scholar]
- Beskow A, Grimberg KB, Bott LC, Salomons FA, Dantuma NP, Young P (2009) A conserved unfoldase activity for the p97 AAA-ATPase in proteasomal degradation. J Mol Biol 394: 732–746 [DOI] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708 [DOI] [PubMed] [Google Scholar]
- Bursavich MG, Parker DP, Willardsen JA, Gao ZH, Davis T, Ostanin K, Robinson R, Peterson A, Cimbora DM, Zhu JF, Richards B (2010) 2-Anilino-4-aryl-1,3-thiazole inhibitors of valosin-containing protein (VCP or p97). Bioorg Med Chem Lett 20: 1677–1679 [DOI] [PubMed] [Google Scholar]
- Chen L, Madura K (2002) Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol Cell Biol 22: 4902–4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TF, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, Patricelli MP, Hodder P, Rosen H, Deshaies RJ (2011) Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci USA 108: 4834–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E, Goldberg AL (2009) During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol 185: 1083–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai RM, Chen E, Longo DL, Gorbea CM, Li CC (1998) Involvement of valosin-containing protein, an ATPase Co-purified with IkappaBalpha and 26 S proteasome, in ubiquitin-proteasome-mediated degradation of IkappaBalpha. J Biol Chem 273: 3562–3573 [DOI] [PubMed] [Google Scholar]
- Dai RM, Li CC (2001) Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol 3: 740–744 [DOI] [PubMed] [Google Scholar]
- Fernandez-Saiz V, Buchberger A (2010) Imbalances in p97 co-factor interactions in human proteinopathy. EMBO Rep 11: 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass D, Roubenoff R (2010) Recent advances in the biology and therapy of muscle wasting. Ann NY Acad Sci 1211: 25–36 [DOI] [PubMed] [Google Scholar]
- Glass DJ (2010) Signaling pathways perturbing muscle mass. Curr Opin Clin Nutr Metab Care 13: 225–229 [DOI] [PubMed] [Google Scholar]
- Guinto JB, Ritson GP, Taylor JP, Forman MS (2007) Valosin-containing protein and the pathogenesis of frontotemporal dementia associated with inclusion body myopathy. Acta Neuropathol 114: 55–61 [DOI] [PubMed] [Google Scholar]
- Halawani D, LeBlanc AC, Rouiller I, Michnick SW, Servant MJ, Latterich M (2009) Hereditary inclusion body myopathy-linked p97/VCP mutations in the NH2 domain and the D1 ring modulate p97/VCP ATPase activity and D2 ring conformation. Mol Cell Biol 29: 4484–4494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten L, Reilley AA (1980) An investigation of the cause of death from cancer. J Surg Oncol 13: 111–116 [DOI] [PubMed] [Google Scholar]
- Hubbers CU, Clemen CS, Kesper K, Boddrich A, Hofmann A, Kamarainen O, Tolksdorf K, Stumpf M, Reichelt J, Roth U, Krause S, Watts G, Kimonis V, Wattjes MP, Reimann J, Thal DR, Biermann K, Evert BO, Lochmuller H, Wanker EE et al. (2007) Pathological consequences of VCP mutations on human striated muscle. Brain 130(Part 2): 381–393 [DOI] [PubMed] [Google Scholar]
- Ishigaki S, Hishikawa N, Niwa J, Iemura S, Natsume T, Hori S, Kakizuka A, Tanaka K, Sobue G (2004) Physical and functional interaction between Dorfin and Valosin-containing protein that are colocalized in ubiquitylated inclusions in neurodegenerative disorders. J Biol Chem 279: 51376–51385 [DOI] [PubMed] [Google Scholar]
- Jagoe RT, Lecker SH, Gomes M, Goldberg AL (2002) Patterns of gene expression in atrophying skeletal muscles: response to food deprivation. FASEB J 16: 1697–1712 [DOI] [PubMed] [Google Scholar]
- Janiesch PC, Kim J, Mouysset J, Barikbin R, Lochmuller H, Cassata G, Krause S, Hoppe T (2007) The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy. Nat Cell Biol 9: 379–390 [DOI] [PubMed] [Google Scholar]
- Jentsch S, Rumpf S (2007) Cdc48 (p97): a ‘molecular gearbox’ in the ubiquitin pathway? Trends Biochem Sci 32: 6–11 [DOI] [PubMed] [Google Scholar]
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez-Lage M, Falcone D, Hernandez DG, Arepalli S, Chong S, Schymick JC, Rothstein J, Landi F et al. (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68: 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, Weihl CC (2009) Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 187: 875–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lowe T, Hoppe T (2008) Protein quality control gets muscle into shape. Trends Cell Biol 18: 264–272 [DOI] [PubMed] [Google Scholar]
- Kimonis VE, Fulchiero E, Vesa J, Watts G (2008) VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys Acta 1782: 744–748 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Tanaka K, Inoue K, Kakizuka A (2002) Functional ATPase activity of p97/valosin-containing protein (VCP) is required for the quality control of endoplasmic reticulum in neuronally differentiated mammalian PC12 cells. J Biol Chem 277: 47358–47365 [DOI] [PubMed] [Google Scholar]
- Kothe M, Ye Y, Wagner JS, De Luca HE, Kern E, Rapoport TA, Lencer WI (2005) Role of p97 AAA-ATPase in the retrotranslocation of the cholera toxin A1 chain, a non-ubiquitinated substrate. J Biol Chem 280: 28127–28132 [DOI] [PubMed] [Google Scholar]
- Krick R, Bremer S, Welter E, Schlotterhose P, Muehe Y, Eskelinen EL, Thumm M (2010) Cdc48/p97 and Shp1/p47 regulate autophagosome biogenesis in concert with ubiquitin-like Atg8. J Cell Biol 190: 965–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudryashova E, Kudryashov D, Kramerova I, Spencer MJ (2005) Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J Mol Biol 354: 413–424 [DOI] [PubMed] [Google Scholar]
- Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL (2004) Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18: 39–51 [DOI] [PubMed] [Google Scholar]
- Madsen L, Seeger M, Semple CA, Hartmann-Petersen R (2009) New ATPase regulators—p97 goes to the PUB. Int J Biochem Cell Biol 41: 2380–2388 [DOI] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6: 458–471 [DOI] [PubMed] [Google Scholar]
- Mammucari C, Schiaffino S, Sandri M (2008) Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy 4: 524–526 [DOI] [PubMed] [Google Scholar]
- Manno A, Noguchi M, Fukushi J, Motohashi Y, Kakizuka A (2010) Enhanced ATPase activities as a primary defect of mutant valosin-containing proteins that cause inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Genes Cells 15: 911–922 [DOI] [PubMed] [Google Scholar]
- Medicherla B, Goldberg AL (2008) Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. J Cell Biol 182: 663–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina R, Wing SS, Goldberg AL (1995) Increase in levels of polyubiquitin and proteasome mRNA in skeletal muscle during starvation and denervation atrophy. Biochem J 307(Part 3): 631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G (2000) A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J 19: 2181–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitch WE, Goldberg AL (1996) Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway. N Engl J Med 335: 1897–1905 [DOI] [PubMed] [Google Scholar]
- Moir D, Stewart SE, Osmond BC, Botstein D (1982) Cold-sensitive cell-division-cycle mutants of yeast: isolation, properties, and pseudoreversion studies. Genetics 100: 547–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller JM, Deinhardt K, Rosewell I, Warren G, Shima DT (2007) Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem Biophys Res Commun 354: 459–465 [DOI] [PubMed] [Google Scholar]
- Nagahama M, Suzuki M, Hamada Y, Hatsuzawa K, Tani K, Yamamoto A, Tagaya M (2003) SVIP is a novel VCP/p97-interacting protein whose expression causes cell vacuolation. Mol Biol Cell 14: 262–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikori S, Yamanaka K, Sakurai T, Esaki M, Ogura T (2008) p97 Homologs from Caenorhabditis elegans, CDC-48.1 and CDC-48.2, suppress the aggregate formation of huntingtin exon1 containing expanded polyQ repeat. Genes Cells 13: 827–838 [DOI] [PubMed] [Google Scholar]
- Peters JM, Walsh MJ, Franke WW (1990) An abundant and ubiquitous homo-oligomeric ring-shaped ATPase particle related to the putative vesicle fusion proteins Sec18p and NSF. EMBO J 9: 1757–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan K, Bruderer R, Spiga FM, Popp O, Baur T, Gotta M, Meyer HH (2007) Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature 450: 1258–1262 [DOI] [PubMed] [Google Scholar]
- Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, Jentsch S (2001) Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell 107: 667–677 [DOI] [PubMed] [Google Scholar]
- Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S (2005) A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120: 73–84 [DOI] [PubMed] [Google Scholar]
- Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, Lecker SH, Goldberg AL (2007) Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J 21: 140–155 [DOI] [PubMed] [Google Scholar]
- Sacheck JM, Ohtsuka A, McLary SC, Goldberg AL (2004) IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am J Physiol Endocrinol Metab 287: E591–E601 [DOI] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117: 399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DM, Fraga H, Reis C, Kafri G, Goldberg AL (2011) ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell 144: 526–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon V, Goldberg AL (1996) Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J Biol Chem 271: 26690–26697 [DOI] [PubMed] [Google Scholar]
- Song EJ, Yim SH, Kim E, Kim NS, Lee KJ (2005) Human Fas-associated factor 1, interacting with ubiquitinated proteins and valosin-containing protein, is involved in the ubiquitin-proteasome pathway. Mol Cell Biol 25: 2511–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikakulam R, Liu L, Winkelmann DA (2008) Unc45b forms a cytosolic complex with Hsp90 and targets the unfolded myosin motor domain. PLoS One 3: e2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ (2004) The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14: 395–403 [DOI] [PubMed] [Google Scholar]
- Szent-Gyorgyi AG (1951) The reversible depolymerization of actin by potassium iodide. Arch Biochem Biophys 31: 97–103 [DOI] [PubMed] [Google Scholar]
- Tawa NE Jr., Odessey R, Goldberg AL (1997) Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest 100: 197–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, Dantuma NP, Taylor JP (2010) VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6: 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, Tomita Y, Hoshida Y, Kono T, Oka T, Yamamoto S, Nonomura N, Okuyama A, Aozasa K (2004) Elevated expression of valosin-containing protein (p97) is associated with poor prognosis of prostate cancer. Clin Cancer Res 10: 3007–3012 [DOI] [PubMed] [Google Scholar]
- Wang Q, Song C, Li CC (2003) Hexamerization of p97-VCP is promoted by ATP binding to the D1 domain and required for ATPase and biological activities. Biochem Biophys Res Commun 300: 253–260 [DOI] [PubMed] [Google Scholar]
- Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36: 377–381 [DOI] [PubMed] [Google Scholar]
- Weihl CC, Dalal S, Pestronk A, Hanson PI (2006) Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum Mol Genet 15: 189–199 [DOI] [PubMed] [Google Scholar]
- White ME, Allen CE, Dayton WR (1988) Effect of sera from fed and fasted pigs on proliferation and protein turnover in cultured myogenic cells. J Anim Sci 66: 34–40 [DOI] [PubMed] [Google Scholar]
- Wojcik C, Rowicka M, Kudlicki A, Nowis D, McConnell E, Kujawa M, DeMartino GN (2006) Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol Biol Cell 17: 4606–4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik C, Yano M, DeMartino GN (2004) RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J Cell Sci 117(Part 2): 281–292 [DOI] [PubMed] [Google Scholar]
- Xu S, Peng G, Wang Y, Fang S, Karbowski M (2011) The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol Biol Cell 22: 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Tomita Y, Hoshida Y, Iizuka N, Kidogami S, Miyata H, Takiguchi S, Fujiwara Y, Yasuda T, Yano M, Nakamori S, Sakon M, Monden M, Aozasa K (2004) Expression level of valosin-containing protein (p97) is associated with prognosis of esophageal carcinoma. Clin Cancer Res 10: 5558–5565 [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Sasagawa Y, Ogura T (2012) Recent advances in p97/VCP/Cdc48 cellular functions. Biochim Biophys Acta 1823: 130–137 [DOI] [PubMed] [Google Scholar]
- Ye Y (2006) Diverse functions with a common regulator: ubiquitin takes command of an AAA ATPase. J Struct Biol 156: 29–40 [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414: 652–656 [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2003) Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol 162: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak R, Martin AF, Prior G, Rabinowitz M (1977) Comparison of turnover of several myofibrillar proteins and critical evaluation of double isotope method. J Biol Chem 252: 3430–3435 [PubMed] [Google Scholar]
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL (2007) FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6: 472–483 [DOI] [PubMed] [Google Scholar]
- Zhao J, Brault JJ, Schild A, Goldberg AL (2008) Coordinate activation of autophagy and the proteasome pathway by FoxO transcription factor. Autophagy 4: 378–380 [DOI] [PubMed] [Google Scholar]
- Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang S (2004) AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem 279: 45676–45684 [DOI] [PubMed] [Google Scholar]
- Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, Lacey DL, Goldberg AL, Han HQ (2010) Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142: 531–543 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.