

Abstract
DNA microarrays have proven extraordinarily powerful for differential expression studies across thousands of genes in a single experiment. Microarrays also have the potential for clinical applications, including the detection of infectious and immunological diseases and cancer, if they can be rendered both reliable and cost-effective. Here we report the first practical application of an active microarray based on integrated circuit technology, completely obviating the need for external measurement instrumentation while employing protocols compatible with traditional fluorescence-based surface bioassays. In a gene-expression biodosimetry study, we determine the differential activity of genes from leucocytes in irradiated human blood. Quantum dots are used as fluorescence labels to realize filterless, time-gated fluorescence detection on an active complementary metal-oxide-semiconductor (CMOS) microarray with 100-pM sensitivity. Improvements in surface chemistry should allow sensitivities that approach the microarray hardware limit of less than 10 pM. Techniques for covalent attachment of DNA capture strands to the CMOS active microarrays allow integrated sensors to be placed in immediate proximity to hybridized analyte strands, maximizing photon collection efficiencies.
Keywords: CMOS biosensor, time-resolved fluorescence detection, integrated DNA microarray
1. Introduction
Nucleic acid diagnostics are an essential ingredient of applied genomics and can impart quality and cost improvements in the fields of health care. Identifying differentially expressed genes, particularly disease-specific genes, continues to be a major focus of ongoing research, particularly for difficult and complex ailments such as cancer (Veer, Dai et al. 2002). Advances in correlating gene expression levels with clinical outcomes, combined with reduced costs of the assays, promise to place genomic analysis within the realm of routine point-of-care diagnostics. The economics of microarray technology, in particular, are limiting due to costs associated both with instrumentation and sample preparation. Traditional microarray scanners are necessarily complex and costly due to stringent optical filtering requirements and the need to image ever-decreasing feature sizes (Barbulovic-Nad, Lucente et al. 2006). Alternative signal detection modalities such as surface plasmon resonance (Homola 2003), microcantilevers (Zhang, Lang et al. 2006), and electrochemical sensing (Drummond, Hill et al. 2003), have been explored but have not gained wide acceptance for microarray applications because of comparable or higher instrumentation costs, or because of challenges in translating prototype technology to practice. At present, fluorescent labeling remains the most wide-spread and effective signal transduction technique.
Active microarray technologies allow fluorescent sensing to be incorporated into the solid support itself, tremendously reducing the cost and form factor of required instrumentation. However, optical filtering of the excitation source remains a significant challenge for fluorescence detection. Prior efforts have focused on spectrally-differentiating mechanisms based on interference filtering (Thrush, Levi et al. 2003; Fixe, Chu et al. 2004), absorption (Chediak, Luo et al. 2004), or waveguiding (Ruano, Glidle et al. 2003) principles. These optical or spectral differentiation techniques can deliver at best 60dB of background rejection (Dandin M. 2007), which is considered inadequate for practical applications. These active substrates (Thrush, Levi et al. 2003; Fixe, Chu et al. 2004; Novak, Neuzil et al. 2007; Beiderman, Tam et al. 2008; Jang, Cao et al. 2009) have consisted of simple photodiodes (Thrush, Levi et al. 2003; Fixe, Chu et al. 2004) or more complex imager arrays (Parikh, Gulak et al. 2007; Schwartz, Gong et al. 2008). In some cases, fluorescence detection is avoided through the use of bioluminescence (Eltoukhy, Salama et al. 2006) or radioactive markers (Salin, Tujasinovic et al. 2002). In this work, we report application of commodity nanoscale CMOS integrated circuit technology, which has driven tremendous improvements in cost and performance of devices for computation and communication, to fluorescence-based microarray diagnostics. The technology leverages low-cost integration of solid-state circuits for signal amplification and processing and realizes more than 70-dB of excitation rejection through time-gated fluorescence detection as shown in Fig. 1(a), eliminating the need for difficult-to-integrate optical filters (Huang, Sorgenfrei et al. 2007). These active microarrays are custom designed in a 0.18-μm CMOS technology and, through direct immobilization of DNA capture strands on the CMOS chip, allow placement of optical detectors in immediate proximity to the fluorescent labels to improve collection efficiency and to provide imaging resolution limited by pixel dimensions rather than diffraction optics. Array imaging is performed without scanning, with each pixel time-gated to collect photons only after the excitation source has been shut down. These advances have made possible the first practical application of active microarrays for a differential cDNA expression study.
Fig. 1. Implementations of integrated, active microarrays for fluorescence detection.
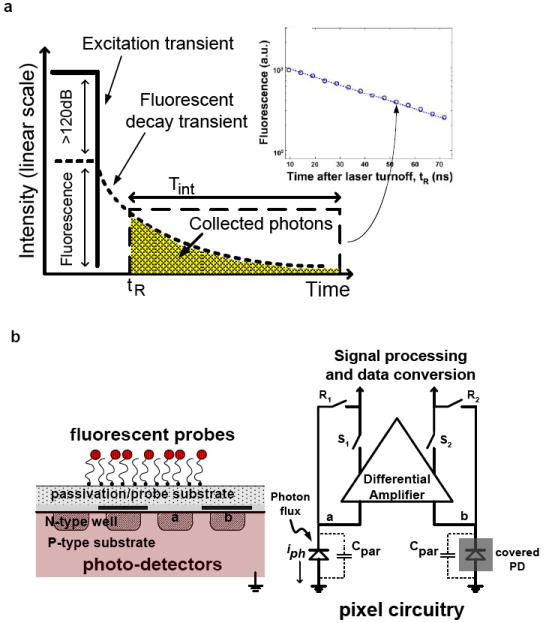
(a) The fluorescence lifetime defines the fluorescence transient after the excitation source has been removed. The photodiodes are time gated, only turning on during the “collection window,” after shutdown of the excitation source.
(b) Photograph of a packaged CMOS microarray chip. “Doughnut” epoxy encapsulation leaves the active area of the chip exposed while insulating the surrounding circuitry. Chip-micrograph with outlines of circuit architectural blocks is also shown here.
(c) Cross-sectional view of a time-gated, integrated fluorescence bioassay, as reported here. Additional optical filtering is not required because time-gating alone is used for background rejection. The photodiode geometry is “fingered” to improve its impulse response.
(d) Pixel circuit block diagram showing common-mode rejection of the differential PD, fast time-gating, and noise suppression through active reset.
2. Methods
2.1. Integrated CMOS time-resolved fluorescence detector design
The CMOS chip that was employed for the active substrate of this integrated microarray is shown packaged in Fig. 1(b). The optical sensor array consists of 64-by-64 pixels with each pixel area being 2500 μm2. It differs from conventional CMOS imagers (Kwang-Bo, Chiajen et al. 2007) in that it has both a differential photodiode (PD) design as well as support for pixel-level time-gating (Huang, Sorgenfrei et al. 2009) as shown in Fig. 1(c). In conventional CMOS PDs, the photocurrent impulse response is determined by minority carriers generated within a diffusion length of the depletion region of the PD, which create a long “tail” as they diffuse and are collected. When trying to use such PDs for fluorescence detection in the absence of optical filtering, this degraded impulse response means that deeply-generated minority carriers from the excitation source interfere with the carriers produced by the fluorescence emission, even after excitation has been turned off. To attenuate this slow diffusive component of the photocurrent response, a fingered differential PD is used in which alternating fingers are covered with metal, rendering the diffusive component of the photocurrent common-mode. This allows a faster-than-800-ps photocurrent impulse response time to be achieved. The PD is formed between the n-type well diffusion and the p-type substrate.
The differential pixel circuitry, which performs the required common-mode rejection, is shown in Fig. 1(d). Photocurrent (iph) collected within the integration period (Tint) beginning at tR appears as a differential voltage between node a (Va) and node b (Vb), where Vb-Va=iphTint/Cpar. Cpar is the total capacitance at node a and node b, and is approximately 198.7 fF for this pixel design. Active reset-noise suppression is performed by the pixel differential amplifier through the negative feedback electronic switches (R1, R2) during the reset stage. Fast time gating to define the integration window is accomplished through the action of both reset (R1, R2) and row-select switches (S1, S2). Time-gate resolution is less than 100 ps, synchronized to a laser excitation turn-off time with an accuracy exceeding 150 ps. On-chip analog-to-digital conversion allows the multiplexed data to be retrieved from the 64-by-64 array in digital form.
The effectiveness of time-gating depends primarily on the lifetime of the fluorophore and the impulse response of the detector. The signal-to-background ratio (SBR) ratio (in dB) can be approximated by (see Supplementary Information):
where τfluor is the fluorescence lifetime, τdet is the detector impulse response, and tR is the time when time-gating occurs, as measured from the excitation turn-off. In this work, we use quantum dots (q-dots) (Han, Gao et al. 2001) as the fluorophore which, in addition to improved photostability and enhanced quantum yields relative to organic dyes, have lifetimes typically exceeding 10 ns. With a detector impulse response of 800 ps, this yields a SBR of more than 70 dB (see Supplementary Information), equivalent to an optical filter of more than OD 10.
2.2. Chip packaging and surface modification
Attachment of DNA capture strands requires surface modification of the CMOS microarrays compatible with their processing limitations. Modification schemes (e.g. organosilylation) used for common solid supports require harsh etches to free hydroxyl groups, deposition of thin films from solvents such as ethanol, and subsequent high-temperature anneals, all of which are damaging to CMOS chips. Instead, we deposit amino-functionalized parylene, aminomethyl-[2,2]paracyclophane (diX AM), directly on the silicon nitride passivation layer of the CMOS microarray through chemical vapor deposition at room temperature, providing a thin (~0.5 μm), stable polymer “blanket” with a functional amino group coverage exceeding 6.2×1013 cm-2 (Miwa, Suzuki et al. 2008). Before deposition of functionalized parylene, all CMOS microarrays were cleaned with 1M HCl followed by 10M NaOH for 2 min each. The chips were rinsed in ultrapure (18.2 MΩ cm) Millipore water and dried with a stream of N2. Coating of the chips was performed using a commercial parylene deposition system (PDS 2010, Specialty Coating Systems, Indianapolis, IN, USA). The dimer was vaporized at 175 °C and pyrolyzed into monomers at 690 °C. The monomers polymerize on the microarray surface at room temperature.
2.3. Immobilization of DNA strands on chip surface
DNA capture strands end-modified with carboxyl groups, catalyzed through the coupling agents EDC and NHS, are covalently attached to the amino-functionalized parylene after non-contact spotting. Capture DNA was printed on the microarrays with a Piezorray piezoelectric non-contact robotic printer (Perkin Elmer, Waltham, MA, USA). The oligo-nucleotide print concentration was 20 μM in 0.16 mM sodium phosphate buffer with 15mM NHS (N-hydroxysuccinimide) and 60mM EDC (1-(3-Dimethyl-aminopropyl)-3-ethylcarbodiimide hydrochloride). After printing, the chips were incubated in 75% humidity chamber for 12 hours, followed by vacuum storage until hybridization.
2.4. Hybridization protocols for CMOS microarrays
Silicone isolation chambers (Grace Bio-labs, Inc., Bend, OR, USA) were used in conjunction with CMOS microarrays. Total reaction volume required to fill a chamber is 60 μl. Before hybridization, microarrays were first incubated in H2O at 65° C for 15 min and then pre-hybridization solution, consisting of 6X SSPE, 0.05% Tween-20, 0.05% SDS, 20mM EDTA, 5X Denhardt’s solution, and 100ng/μl heat-treated salmon sperm DNA, for 30 min at 50° C. Hybridization solution was prepared with 6X SSPE, 0.05% Tween-20, 0.04% SDS, 20mM EDTA, 100ng/μl heat-treated salmon sperm DNA, and either synthetic oligo-nucleotides or heat-treated PCR amplified products. Hybridization was performed at room temperature for 6 hours for synthetic oligonucleotides and at 50° C overnight for PCR amplified products.
After hybridization, the active substrate was washed with 6X SSPE and 0.05% Tween-20 for 5 min at 50° C, 3X SSPE and 0.05% Tween-20 for 1 min at room temperature, 0.5X SSPE and 0.05% Tween-20 for 1 min at room temperature, and 2X PBS and 0.1% Tween-20 for 1 min at room temperature.
A pre-labeling blocking solution was prepared consisting of 2X PBS, 0.1% Tween-20, and 1% BSA. The labeling solution was prepared through the addition of 5 nM streptavidin-conjugated QD-625 solution (Invitrogen, Inc., Carlsbad, CA, USA). The chips were treated with the blocking solution for 15 min and the labeling solution for 30 min. After labeling, the chips were washed twice with 2X PBS and 0.1% Tween-20 for 1 min, and blow-dried with a stream of N2.
2.5. Preparation of target analyte from physiological samples
Peripheral blood is drawn into 0.105 mol/l sodium citrate Vacutainer tube (Becton Dickinson and Company, Franklin Lakes, NJ) from healthy volunteers with informed consent. The blood is exposed at a rate of 0.82 Gy/min to 8 Gy γ-rays using a Gammacell-40 137Cs irradiator (AECL, Ottawa, Ontario, Canada). As control, sham-irradiated blood samples are used. Following irradiation, blood samples are diluted 1:1 with RPMI 1640 medium (Mediatech, Herndon, VA, USA) supplemented with 10% heat-inactivated fetal bovine serum (HyClone, Logan, UT, USA), and are incubated for 6 hours at 37°C in a humidified incubator with 5% CO2.
The RNA was prepared using the PerfectPure RNA Blood Kit (5 Prime Inc., Gaithersburg, MD, USA) according to the manufacturer’s recommendations. This protocol depletes globin m-RNA by differentially lysing red and white blood cells in whole blood. First the red blood cells are lysed by RBC lysis solution and the nucleic acids released are washed away followed by lysis of the white blood cells for purification of RNA. The remaining globin mRNA was further reduced using the GLOBINclear Human Kit (Ambion Inc., Austin, TX) that specifically removes both α- and β-globin mRNA. The RNA was quantified using a NanoDrop-1000 spectrophotometer, and quality was monitored with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA).
2.6. Polymerase Chain Reaction (PCR) and quantitative real-time PCR
Gene-specific primers and probe sets were designed with the aid of Primer Express software (Applied Biosystems, Foster City, CA) and TaqMan Primer Design software (GenScript Corp., Piscataway, NJ, USA). Primers-probes were synthesized with the probes containing 6-FAM (carboxyfluorescein) at 5’-end and BHQ1 (Black Hole Quencer 1) at 3’-end. 500-ng total RNA was reverse transcribed to cDNA using the High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. Regular PCR reactions were performed (MiniOpticon, CFD-3120, Bio-Rad Laboratories, Inc., Hercules, CA, USA) a reaction mixture (50 μl) composed of 1X Taq buffer, 0.5 μM of each primer, 200 μM each of dATP, dGTP and dTTP, 135 μM of dCTP, 65 μM of biotinylated dCTP (ChemCyte, Inc., San Diego, CA, USA), two units of DNA polymerase, and 100 ng of cDNA template. PCR was performed with initial heating at 95° C for 2 min, followed by 25 cycles of denaturing at 94° C for 30 sec, annealing at 55° C for 45 sec and extension at 72° C for 30 sec. A final extension at 72° C was performed for 7 min. The real-time PCR reactions were performed with the ABI 7900HT Fast Real-Time PCR System (Applied Biosystems Inc., Foster City, CA, USA) using ABI’s Universal PCR Master Mix following manufacturer’s recommendations. Standard curves were generated to optimize the amount of input cDNA for measurement of each gene (5 or 10 ng). All samples were run in duplicate and repeated 3 times on different days for each gene. The relative fold induction of target genes was calculated by the ΔΔCT method with ACTB used for normalization.
2.7. Setup for fluorescence lifetime measurement and chip readout
Quantum dots on the active substrate surface were excited at a wavelength of 408 nm with a high-speed narrow-pulsed laser (EIG1000D with PIL040, Advanced Laser Diode Systems A.L.S. GmbH, Berlin, Germany). On-chip data were acquired by the chip’s photodiode circuits, using a custom-interfaced printed-circuit board to mount the socketed chip. Digital data output from the CMOS chip are stored on a PC and processed using MATLAB (The Mathworks, Natick, MA, USA).
2.8. DNA oligonucleotide sequences
The sequences of the 5’-end carboxylated 25-mer DNA oligonucleotide probes (Trilink Biotechnologies, San Diego, CA, USA) used in our CMOS microarray experiments are as follows: P1 5’-GGA GCT GGA AGC AGC CGT GGC CAT C -3’, P2 5’-TGT CTG GCC CAC ACC TTC TTT AGT C and P3 5’-GTA TTT TAA GTG TCC CAT ATC CGC A-3’. The target sequence for microarray platform characterization is: T1 5’-GAT GGC CAC GGC TGC TTC CAG CTC C-3’. Sequences of forward and reverse primers (Operon Biotech, Inc., Huntsville, AL, USA) for PCR and qRT-PCR are: F1 5’- CAC TCT TCC AGC CTT CCT TC-3’, R1 5’- GGA TGT CCA CGT CAC ACT TC-3’, F2 5’- CAG GAC ACG GAA GTG AGA GA-3’, R2 5’- CAG GAC ACG GAA GTG AGA GA-3’, F3 5’-ATT GCG GAT ATG GGA CAC TT-3’, and R3 5’-GCTGGCATCTTAGAAGCAGTTC-3’. Sequences of Taqman probes (Operon Biotech, Inc., Huntsville, AL, USA) are: P1: 5’- TGCCACAGGACTCCATGCCC-3’, P2: 5’- TCCAAGGCCTTGTCTGGCCC-3’, and P3: 5’-TCATCCTCGATCTTGGGAGCCA-3’.
3. Results and discussion
3.1. DNA hybridization detection through time-resolved fluorescence lifetime measurements
Characterizations of this integrated microarray with synthesized oligo-nucleotides are performed to determine platform dynamic range and sensitivity, and verified with independent, external fluorescence measurements. The 25-mer probe P1 is 5’ modified with carboxyl group while target T1 is 5’ modified with biotin. After hybridization, streptavidin-conjugated q-dots were applied to quantify hybridization.
CMOS microarray chips, hybridized with known analyte target concentrations between 100 pM and 300 nM, were observed under an epifluorescence microscope fitted with a cooled CCD camera as shown in Fig. 2(a). Thus, both external epifluorescence and on-chip measurements could be performed simultaneously. The CMOS microarray measures integrated photons from the decaying fluorescence emission with an integration window beginning at tR after gain-switched shut off of the excitation laser at t=0. These results, when numerically differentiated with respect to time (see Supplementary Information), yield the fluorescence decay transient associated with the q-dot fluorophore lifetime (48 ns). The total integrated fluorescence intensity for tR= 10 ns is plotted in Fig. 2(b) against analyte target concentrations. A close correlation is observed between CMOS and epi-fluorescence measurements. The sensitivity limit (100 pM) is determined by the non-specific adsorption of streptavidin-labelled q-dots, while measurable target concentrations are limited at 300 nM because of steric issues related to the size of the q-dot fluorophores. With an average q-dot diameter of 20 nm, a maximum single-layer surface coverage of 2.5 × 1011 cm-2 is permitted. The CMOS microarray’s true optical sensitivity, at 12 photons per pixel, and dynamic range, at more than three orders of magnitude, extends well beyond the range allowed by the current surface chemistry and labeling protocol. The hardware sensitivity limit is achieved by averaging 4096-point data sets to reduce photon shot noise (both from the excitation source and the fluorescence) and thermal noise from the electronics. Fig. 2(b) overlays results from such an independent optical characterization (Huang, Sorgenfrei et al. 2009), which extrapolate to a molecular sensitivity of 7.2 pM if the detection limit is determined only by the detector sensitivity (see Supplementary Information).
Fig. 2. Sensitivity and dynamic range analysis of the active CMOS microarray.
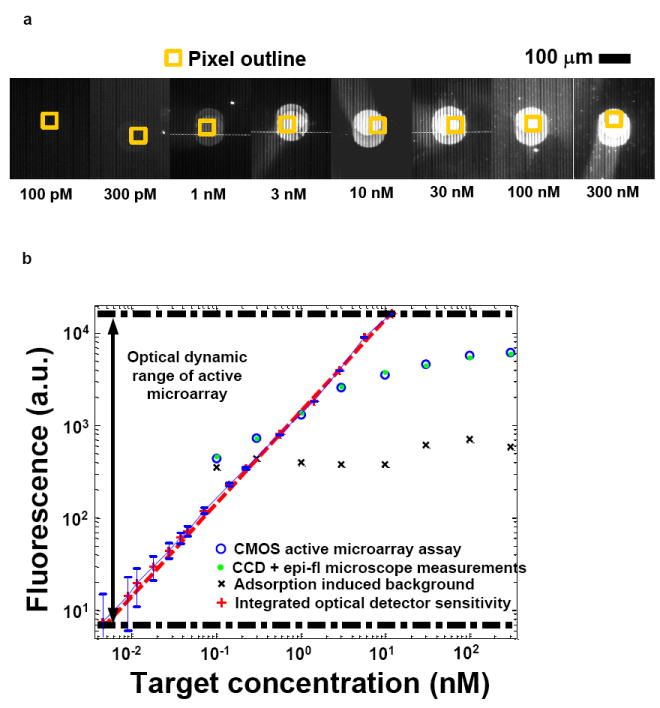
(a) Images of hybridized spots on the CMOS array with varying target concentrations, as seen under an epi-fluorescence microscope. The photodiode area is outlined by the yellow square. Also evident are the metal lines associated with the differential photodiode design.
(b) Fluorescence intensity as measured by the CMOS microarray at varying target concentrations. On-chip measurements are plotted in blue x and exhibit a compressed dynamic range due to non-specific adsorption and steric issues of q-dots conjugates. The blue curve shows the dynamic range and sensitivity as determined from independent measurement of the optical detector alone.
3.2. Gene expression profiling of physiological samples
The CMOS microarrays were used to perform differential gene expression profiling of physiological samples from a radiation biodosimetry study (Paul and Amundson 2008). We focused on semi-quantitative detection of two sentinel genes: human damage-specific DNA binding protein (DDB2) and proliferating cell nuclear antigen (PCNA), using β-actin (ACTB) as a reference target. In this demonstrative experiment, as outlined in Fig. 3(a), half of a whole-blood sample from one healthy consenting donor is irradiated ex vivo with a high dose (8 Gy) of γ-rays. The irradiated and untreated control blood is then incubated for 6 hours at 37 °C to allow temporal gene expression responses to cellular radiation damage to occur. Total RNA was extracted from each (untreated and irradiated) sample and reverse transcribed into total cDNA. Gene expression profiling, through determination of quantitative fold-changes in selected genes, was performed using both the active CMOS microarray and established quantitative reverse transcription-polymerase chain reaction (qRT-PCR) protocols (Schmittgen, Zakrajsek et al. 2000). In the CMOS microarray assay, 50 ng of total cDNA was used as a starting template for multiplex PCR. Selected PCR product sequences of 85-140 bp in length from sentinel and reference genes were amplified, with biotin-labeled nucleotides randomly incorporated during the amplification. 25 cycles of PCR proved sufficient to amplify target sequences above the non-specific adsorption limited sensitivity of 100 pM, without saturating the sensor. As we are able to improve the surface chemistry of the active array and approach sensor-limited detection limits, we expect that PCR will no longer be required in the sample preparation protocols. Microarray experiments adhered to standard nucleic acid based hybridization protocols, with q-dot conjugates added following overnight hybridization.
Fig. 3. Differential expression profiling with the integrated CMOS microarray.
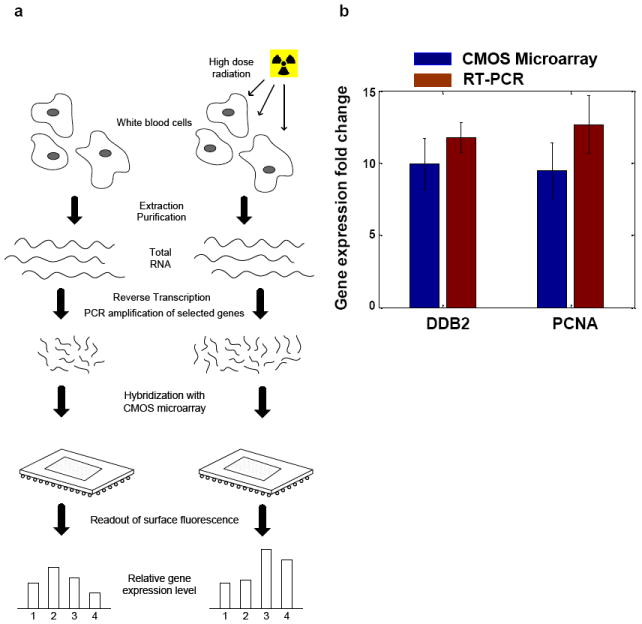
(a) Preparation of physiological samples. Total RNA was extracted and reverse-transcribed into cDNA. Selected genetic sequences were amplified and quantified through hybridization with a CMOS microarray.
(b) Measured gene expression fold changes for DDB2 and PCNA, using ACTB as a reference gene, were obtained from both CMOS microarray and RT-PCR assays in ex vivo (8Gy) irradiated and control blood samples.
Fluorescent intensities at each assay site were measured through the active CMOS microarray under equal excitation condition, using the same optical and electronic settings for each measurement. The gene expression fold change (GEFC) of the sentinel genes is defined as the fluorescence intensity ratio between irradiated and untreated samples, normalized by the reference intensity (Schmittgen, Zakrajsek et al. 2000):
Gene expression profiling of identical samples was also realized through a standard real time PCR (qRT-PCR) assay using dual-labeled fluorogenic probes and the comparative CT (ΔΔCT) method (Schmittgen and Livak 2008). Experimental results of gene expression fold changes from both the CMOS microarray and RT-PCR are plotted and compared in Fig. 3(b). The plot shows that the experimental results from CMOS microarray match those from RT-PCR assay within experimental uncertainties. Experimental uncertainties for the CMOS microarray include both measurement and spatial variations of hybridized intensity within each spot, while experimental uncertainties from qRT-PCR assay are derived from standard deviations in the comparative CT values. This demonstrates that relative gene expression measurements obtained by CMOS microarray are equivalent to those of real-time PCR, the current gold-standard in gene expression quantification.
4. Conclusions
Here we have demonstrated how low-cost commodity CMOS integrated circuit technology can be leveraged for biotechnology applications, in the form of affinity-based assays in which traditional passive solid supports are replaced by active integrated circuit chips. Integrated electronics allows an imager to be designed that utilizes fast time-gating for fluorescence imaging without the need for optical filters. Direct use of the chip as a solid-support allows further simplification of the platform put presents additional challenges to the surface chemistry, which we have begun to address in this work.
Supplementary Material
Acknowledgments
This work was supported in part by Grant Number HG003089 from the National Human Genome Research Institute, Grant Number BES-428544 from the National Science Foundation, Center for Circuit and System Solutions (C2S2) and by NYSTAR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barbulovic-Nad I, Lucente M, et al. Bio-Microarray Fabrication Techniques - A Review. Critical Reviews In Biotechnology. 2006;26(4):237–259. doi: 10.1080/07388550600978358. [DOI] [PubMed] [Google Scholar]
- Beiderman M, Tam T, et al. A Low noise CMOS image sensor with an emission filter for fluorescence applications. IEEE International Symposium on Circuits and Systems. 2008:1100–1103. [Google Scholar]
- Chediak JA, Luo Z, et al. Heterogeneous integration of CdS filters with GaN LEDs for fluorescence detection microsystems. Sensors and Actuators A: Physical. 2004;111(1):1–7. [Google Scholar]
- Dandin MAP, Smela E. Optical filtering technologies for integrated fluorescence sensors. Lab on a Chip. 2007;7:955–977. doi: 10.1039/b704008c. [DOI] [PubMed] [Google Scholar]
- Drummond TG, Hill MG, et al. Electrochemical DNA sensors. Nat Biotechnol. 2003;21:1192–1199. doi: 10.1038/nbt873. [DOI] [PubMed] [Google Scholar]
- Eltoukhy H, Salama K, et al. A 0.18 μm CMOS bioluminescence detection lab-on-chip. Solid-State Circuits, IEEE Journal of. 2006;41:651–662. [Google Scholar]
- Fixe F, Chu V, et al. An on-chip thin film photodetector for the quantification of DNA probes and targets in microarrays. Nucl Acids Res. 2004;32(9):e70. doi: 10.1093/nar/gnh066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M, Gao X, et al. Quantum-dot-tagged microbeads for multiplexed optical coding of biomolecules. Nature Biotechnology. 2001;19:631–635. doi: 10.1038/90228. [DOI] [PubMed] [Google Scholar]
- Homola J. Present and future of surface plasmon resonance biosensors. Analytical and Bioanalytical Chemistry. 2003;377(3):528–539. doi: 10.1007/s00216-003-2101-0. [DOI] [PubMed] [Google Scholar]
- Huang T, Sorgenfrei S, et al. A CMOS array sensor for sub-800-ps time-resolved fluorescence detection. Custom Integrated Circuits Conference; San Jose, CA. 2007. pp. 829–832. [Google Scholar]
- Huang TcD, Sorgenfrei S, et al. A 0.18-μm CMOS Array Sensor for Integrated Time-Resolved Fluorescence Detection. Solid-State Circuits, IEEE Journal of. 2009;44(5):1644–1654. doi: 10.1109/JSSC.2009.2016994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang B, Cao P, et al. A CMOS fluorescent-based biosensor microarray. Solid-State Circuits Conference - Digest of Technical Papers, 2009. ISSCC 2009; IEEE International; 2009. [Google Scholar]
- Kwang-Bo C, Chiajen L, et al. A 1/2.5 inch 8.1Mpixel CMOS Image Sensor for Digital Cameras. Solid-State Circuits Conference, 2007. ISSCC 2007. Digest of Technical Papers; IEEE International; 2007. [Google Scholar]
- Miwa J, Suzuki Y, et al. Adhesion-Based Cell Sorter With Antibody-Coated Amino-Functionalized-Parylene Surface. Microelectromechanical Systems, Journal of. 2008;17(3):611–622. [Google Scholar]
- Novak L, Neuzil P, et al. An integrated fluorescence detection system for lab-on-a-chip applications. Lab on a Chip. 2007;7:27–29. doi: 10.1039/b611745g. [DOI] [PubMed] [Google Scholar]
- Parikh S, Gulak G, et al. A CMOS Image Sensor for DNA Microarrays. Custom Integrated Circuits Conference, 2007. CICC ’07; IEEE; 2007. [Google Scholar]
- Paul S, Amundson S. Development of gene expression signatures for practical radiation biodosimetry. Int J Radiat Oncol Biol Phys. 2008;71(4):1236–1244. doi: 10.1016/j.ijrobp.2008.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruano JM, Glidle A, et al. Design and fabrication of a silica on silicon integrated optical biochip as a fluorescence microarray platform. Biosensors and Bioelectronics. 2003;18(2-3):175–184. doi: 10.1016/s0956-5663(02)00170-7. [DOI] [PubMed] [Google Scholar]
- Salin H, Tujasinovic T, et al. A novel sensitive microarray approach for differential screening using probes labelled with two different radioelements. Nucleic Acids Res. 2002;30(4):e17. doi: 10.1093/nar/30.4.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen T, Zakrajsek B, et al. Quantitative Reverse Transcription-Polymerase Chain Reaction to Study mRNA Decay: Comparison of Endpoint and Real-Time Methods. Analytical Biochemistry. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protocols. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Schwartz DE, Gong P, et al. Time-resolved Forster-resonance-energy-transfer DNA assay on an active CMOS microarray. Biosensors and Bioelectronics. 2008;24(3):383–390. doi: 10.1016/j.bios.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrush E, Levi O, et al. Integrated bio-fluorescence sensor. Journal of Chromatography A. 2003;1013(1-2):103–110. doi: 10.1016/s0021-9673(03)01361-x. [DOI] [PubMed] [Google Scholar]
- Veer LJ, Dai H, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature Biotechnology. 2002;415:530–535. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lang HP, et al. Rapid and label-free nanomechanical detection of biomarker transcripts in human RNA. Nat Nano. 2006;1(3):214–220. doi: 10.1038/nnano.2006.134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.