

Abstract
Pulmonary alveolar proteinosis (PAP) is a disease of alveolar accumulation of phospholipoproteinaceous material that results in gas exchange impairment leading to dyspnea and alveolar infiltrates. There are three forms of PAP: congenital, acquired and idiopathic; of which the latter two are predominant in the adult population. Previous case studies have found that the acquired form can be secondary to various autoimmune, infectious, malignant and environmental etiologies. Recent advances in the understanding of the pathophysiology of PAP demonstrate that the idiopathic form is due to antigranulocyte macrophage-colony stimulating factor antibodies. Therapeutic targets that replace granulocyte macrophage colony stimulating factor or remove these antibodies are being actively developed. The current standard of care is to perform whole lung lavage on these patients to clear the alveolar space to help improve respiratory physiology. A case of PAP is reported, followed by a literature review on the diagnosis and management of this rare condition with the aim of increasing awareness among physicians when treating patients who present with alveolar infiltrates.
Keywords: Alveolar infiltrates, Crazy paving, GM-CSF, Pulmonary alveolar proteinosis
Abstract
La protéinose alvéolaire pulmonaire (PAP) se caractérise par l’accumulation alvéolaire de matière phospholipoprotéinacée qui entraîne une déficience de l’échange gazeux responsable d’une dyspnée et d’infiltrats alvéolaires. Il existe trois formes de PAP : congénitale, acquise et idiopathique. Les deux dernières prédominent dans la population adulte. Des études de cas antérieures ont déterminé que la forme acquise peut être secondaire à diverses étiologies auto-immunes, infectieuses, malignes et environnementales. Les récents progrès dans la compréhension de la physiopathologie de la PAP démontrent que la forme idiopathique découle des anticorps des facteurs de stimulation des colonies de granulocytes et de macrophages. On est à mettre au point des cibles thérapeutiques qui remplacent le facteur de stimulation des colonies de granulocytes et de macrophages ou qui suppriment ces anticorps. La norme actuelle des soins consiste à exécuter un lavage pulmonaire complet afin de dégager l’espace alvéolaire pour améliorer la physiologie respiratoire. Un cas de PAP est déclaré, suivi d’une analyse bibliographique du diagnostic et de la prise en charge de cette pathologie rare, afin de mieux y sensibiliser les médecins lorsqu’ils traitent des patients qui présentent des infiltrats alvéolaires.
Learning objectives
To recognize the various presentations and appropriate management of pulmonary alveolar proteinosis (PAP).
To understand the various acquired causes of PAP.
Pre-test
What is the etiology of PAP?
What are the various differential diagnoses associated with the ‘crazy-paving’ pattern that need to be considered and ruled out before making a diagnosis of PAP?
CASE PRESENTATION
A 32-year-old Caucasian man without a significant medical history presented with a two-month history of progressively worsening dyspnea on exertion. He denied any cough, hemoptysis, chest pain, lower extremity swelling, fevers, chills, night sweats, weight change, new rashes, arthralgias or myalgias. He had no exotic travel or sick contacts. He was a 10 pack-year smoker and occasionally consumed alcohol. He denied any illicit drug use. He worked in construction, and his hobbies included woodworking, in which he was chronically exposed to cement and saw dust. Before his visit to the Mayo Clinic (Minnesota, USA), he was treated by other providers with various courses of antibiotics and prednisone without relief. A review of systems was otherwise unrevealing.
On physical examination, the patient’s vital signs were as follows: temperature 36.6°C; blood pressure 96/70 mmHg; pulse rate 80 beats/min; respiratory rate 18 breaths/min; and oxygen saturation 88% on room air. Most notably, his lung examination demonstrated scattered inspiratory crackles without any rales, rhonchi or wheezes. The cardiac, vascular, abdominal, lymphatic and integument examinations were all unremarkable.
On laboratory evaluation, the patient’s comprehensive metabolic panel and complete blood count with differential were within normal limits. Arterial blood gas analysis on room air revealed a pH of 7.47, PCO2 31.5 mmHg, PO2 43.3 mmHg and HCO3– 23 mEq/L. Pulmonary function testing demonstrated normal spirometry with decreased diffusion capacity for carbon monoxide (DLco 34% of predicted). A chest x-ray revealed new diffuse bilateral alveolar infiltrates without other abnormalities. Additional laboratory testing revealed an elevated C-reactive protein level (719 nmol/L [75.5 mg/L]); however, antinuclear antibody, rheumatoid factor, an antineutrophil cytoplasmic antibody panel and immunoglobulin levels returned normal. The patient underwent high-resolution computed tomography (HRCT) of the chest, which demonstrated centrilobular emphysema and ground-glass opacities with superimposed interlobular septal thickening.
The patient underwent bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial biopsies. There were no endobronchial abnormalities. BAL microbiology was negative, including opportunistic infections such as Pneumocystis and Nocardia. Cytology was normal. There were no significant hemosiderin-laden macrophages. Transbronchial biopsies showed chronic inflammation, thickened interlobular septae and alveolar acellular material that was negative for Grocott’s methanamine silver (GMS) stain (Figures 1 and 2). Given the clinical and pathological findings, a diagnosis of pulmonary alveolar proteinosis (PAP) was made.
Figure 1).
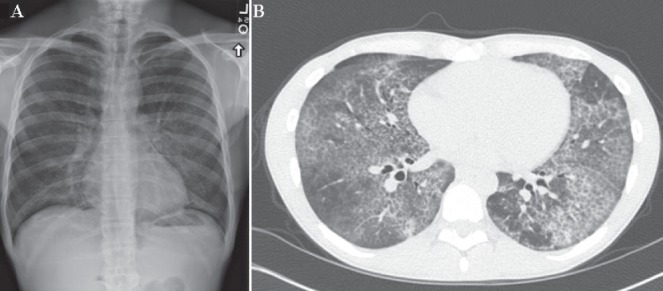
A Chest x-ray (posteroanterior view) demonstrating hazy, diffuse, bilateral alveolar infiltrates. B High-resolution computed tomography scan showing diffuse, bilateral, geographical ground-glass opacities with associated inter- and intralobular septal thickening with interspersed areas of normal lung that are indicative of ‘crazy-paving’
Figure 2).
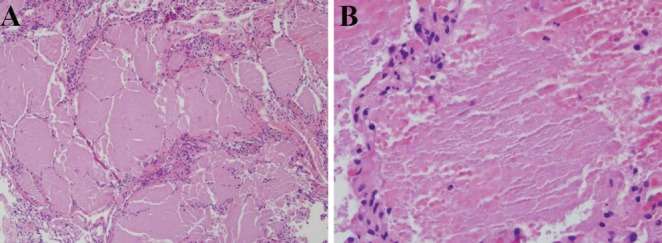
A Biopsy demonstrating dilated air spaces, thickened interlobular septae and relatively acellular, eosinophilic debris within the alveoli. Hematoxylin and eosin stain, original magnification ×100. B Higher power inspection of the alveoli demonstrating thick, amorphous material with scattered pulmonary macrophages. Hematoxylin and eosin stain, original magnification ×400. Note: Grocott’s methanamine silver staining was negative and ruled out pneumocystis pneumonia in the differential of ‘crazy-paving’
DISCUSSION
First reported in 1958, PAP is characterized by the accumulation of phospholipoproteinaceous material within the alveoli as a result of decreased protein clearance by pulmonary macrophages (1,2). Current estimates place its incidence in the United States at approximately one in 300,000 (3). Men are more commonly affected, with a median age of diagnosis of approximately 40 years and 70% of patients have a history of smoking (1).
There are three forms of PAP: congenital, acquired and idiopathic (Figure 3) (4). Congenital PAP usually presents as neonatal respiratory distress syndrome. It is believed to result from mutations/deficiencies in surfactant proteins or the granulocyte macrophage-colony stimulating factor (GM-CSF) receptor. Acquired PAP typically presents during adulthood and is associated with abnormal alveolar macrophage function due to malignancies, infections, systemic inflammatory disorders, immunosuppression or toxic exposures (Table 1) (1). Finally, idiopathic PAP is the most common form (>90%) in adults, and is believed to be due to anti-GM-CSF antibodies that inhibit alveolar macrophage function. These categories must be taken with a caveat because there is a growing body of literature to indicate that there may be an overlap between what is referred to as idiopathic and acquired PAP. For instance, various reports have demonstrated that patients with a previous exposure have also shown positive titres for GM-CSF antibodies, while those with positive antibodies have retrospectively been identified as having typical exposures that were characteristic of acquired PAP (5,6).
Figure 3).
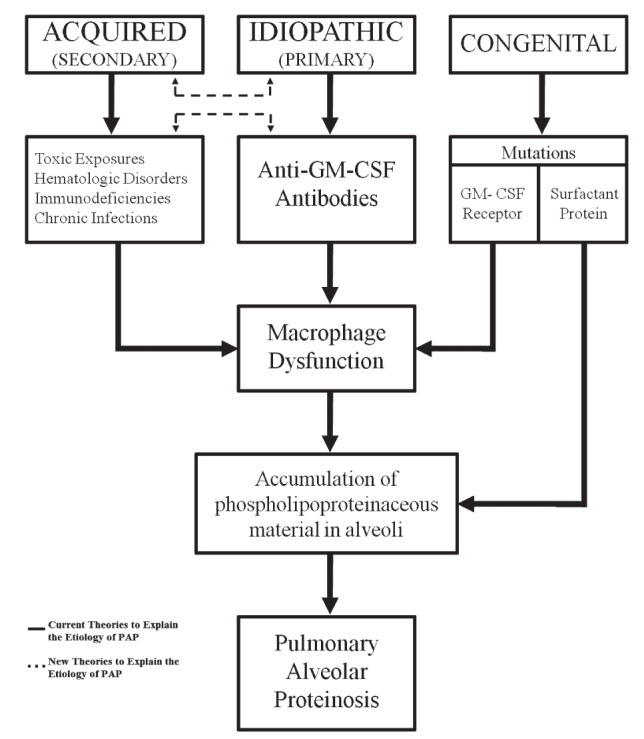
Categories, etiologies and pathophysiology of the various forms of pulmonary alveolar proteinosis (PAP). GM-CSF Granulocyte macrophage-colony stimulating factor
TABLE 1.
Acquired causes of pulmonary alveolar proteinosis
| Toxic exposures | Hematological disorders | Immunodeficiencies | Chronic infections |
|---|---|---|---|
| Aluminum dust Cement dust Chlorine Fertilizer Nitrogen dioxide Paint Petroleum Sawdust Silica dust Varnish |
Lymphoid leukemias Myeloid leukemias Non-Hodgkin’s lymphoma Myelodysplastic syndromes Multiple myeloma Waldenstrom’s macroglobulinemia Amyloidosis Idiopathic thrombocytopenic purpura Aplastic anemia |
AIDS Severe combined immunodeficiency Immunoglobulin A deficiency Thymic lymphoplasia |
Norcardia Pneumocystis jirovecii Histoplasmosis Cryptococcus Mycobacterial species Cytomegalovirus |
The main symptoms include progressive exertional dyspnea and nonproductive cough. Inspiratory crackles may be heard on physical examination. Pulmonary function testing usually demonstrates a non-specific spirometry or a mild restrictive pattern with a decrease in diffusion capacity. Similar to our case, a relatively normal or only mildly abnormal spirometry compared with the significant reduction in diffusing capacity is likely due to the nature of the disease, in which the accumulation of phospholipoproteinaceous material within the alveoli causes the gas exchange dysfunction more than the dynamic airflow abnormality. Chest radiography will demonstrate bilateral alveolar infiltrates out of proportion to the clinical presentation. Typical HRCT findings include ground-glass opacifications with superimposed interlobular septal thickening adjacent to normal lung parenchyma, colloquially referred to as ‘crazy-paving’ because of its resemblance to irregularly laid cobblestone paths (1). Despite the well-known association between ‘crazy-paving’ patterns and PAP, this radiographic finding is neither sensitive nor specific for the diagnosis of PAP. It can be observed in alveolar sarcoidosis, lipoid pneumonia, bronchioalveolar carcinoma, pneumocystis pneumonia, Nocardia infection, chronic eosinophilic pneumonia, acute interstitial pneumonia, acute respiratory distress syndrome or even hemorrhage (7–9). It must also be noted that PAP can present as interstitial, focal, nodular or ground-glass opacities, and even fibrosis; thus, it should not be ruled out if distinct alveolar infiltrates are not seen (1,10).
Adjunctive routine laboratory studies are rarely helpful in honing the diagnosis. However, if idiopathic PAP is suspected, serum anti-GM-CSF antibody titres or BAL fluid can be a helpful diagnostic tool (1); although its clinical utility needs to be validated in a large cohort study. Currently, the testing is not widely available and can be accomplished at specialized centres via sending a specifically packaged blood sample. Most commonly, bronchoscopy with BAL and transbronchial biopsy can be performed to confirm the diagnosis of PAP and rule out other conditions (1). Due to the the patchy nature of the disease, sampling error may result; therefore, if suspicion is high, video-assisted thoracoscopy or open lung biopsy should be performed. Typically, BAL reveals cloudy, milk-like fluid that when allowed to stand, separates into a translucent supernatant and thick sediment (1). Similar alveolar proteinaceous material may be observed in various inhalational exposures including silica, titanium or aluminum, and can also be seen in pneumocystis pneumonia, pulmonary nocardiosis and leukemia. Pathological differentiation of PAP from these other entities requires cytospin analysis, which usually reveals periodic acid-Schiff-positive, GMS stain-negative, eosinophilic, granular, acellular material with a few enlarged foamy macrophages (1).
Once diagnosed, the underlying etiology of the PAP should be determined to guide management. Currently, the standard of care for the immediate treatment of any form of PAP is whole-lung lavage (WLL) under anesthesia (1). Unfortunately, congenital PAP responds poorly to WLL and, at this time, the only other option is lung transplantation (11). In cases of acquired PAP, definitive treatment rests on addressing the underlying disorder or avoiding the suspected environmental agent (1). Recently, studies have demonstrated that GM-CSF (inhalation or subcutaneous) can result in improvement in some patients with idiopathic PAP (12,13). Other options focus on reducing the amount of autoantibodies via B-cell depletion therapy (rituximab) or removal of autoantibodies (plasmapheresis); however, the exact role of these modalities remains to be defined (14,15). Furthermore, the use of lung transplantation for noncongenital PAP has not been clearly studied and recurrence after transplantation has been reported (16,17).
Our patient underwent WLL and his dyspnea and oxygen saturation improved. It is unknown whether his chronic exposure to cement and saw dust was directly associated with the development of PAP; however, he was advised to change his occupational environment and refrain from woodworking. On follow-up, the patient’s dyspnea on exertion was no longer present and he continues to do well. The present case highlights the need to consider all differential diagnoses when alveolar infiltrates are seen and add PAP to the list of possibilities because the diagnosis can lead to the search for potential secondary etiologies and the treatment can result in significant clinical improvement.
Post-test
- What is the etiology of PAP?
- ○ The current state of research suggests that PAP results from a decrease in the clearance of phospholipoproteins from the alveolar space. Mechanistically, three various forms of PAP exist, but all of them uniformly result in macrophage dysfunction, whether it be from direct macrophage effect or indirectly via mutations or antibodies to the GM-CSF receptor resulting in accumulation of the proteinaceous material.
- What are the various differential diagnoses associated with the ‘crazy-paving’ pattern that need to be considered and ruled out before making a diagnosis of PAP?
- ○ ‘Crazy-paving’ is the colloquial term for interlobular septal thickening with superimposed ground-glass opacities. The most common differential diagnosis that should be considered outside of PAP included bronchioalveolar carcinoma, Pneumocystis jirovecii infection, hemorrhage and, rarely, alveolar sarcoid. Appropriate diagnostic testing should ensue to rule out these etiologies, and definitive diagnosis of PAP should be made by demonstrating acellular, amorphous, eosinophilic, periodic acid-Schiff-positive material.
Footnotes
DISCLOSURE: The authors have no financial disclosures or conflicts of interest to declare.
AUTHORSHIP CRITERIA STATEMENT: All authors have met criteria for authorship and had a role in preparing the manuscript.
REFERENCES
- 1.Juvet SC, Hwang D, Waddell TK, Downey GP. Rare lung disease II: Pulmonary alveolar proteinosis. Can Respir J. 2008;15:203–10. doi: 10.1155/2008/528948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958;258:1123–42. doi: 10.1056/NEJM195806052582301. [DOI] [PubMed] [Google Scholar]
- 3.Hunt S, Miller AL, Schissel S, Ross JJ. A crazy cause of dyspnea. N Engl J Med. 2010;363:e38. doi: 10.1056/NEJMimc1008281. [DOI] [PubMed] [Google Scholar]
- 4.Inoue Y, Trapnell BC, Tazawa R, et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med. 2008;177:752–62. doi: 10.1164/rccm.200708-1271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costabel U, Nakata K. Pulmonary alveolar proteinosis associated with dust inhalation: Not secondary but autoimmune? Am J Respir Crit Care Med. 2010;181:427–8. doi: 10.1164/rccm.200912-1800ED. [DOI] [PubMed] [Google Scholar]
- 6.Cummings KJ, Donat WE, Ettensohn DB, Roggli VL, Ingram P, Kreiss K. Pulmonary alveolar proteinosis in workers at an indium processing facility. Am J Respir Crit Care Med. 2010;181:458–64. doi: 10.1164/rccm.200907-1022CR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossi SE, Erasmus JJ, Volpacchio M, Franquet T, Castiglioni T, McAdams HP. “Crazy-paving” pattern at thin-section CT of the lungs: Radiologic-pathologic overview. Radiographics. 2003;23:1509–19. doi: 10.1148/rg.236035101. [DOI] [PubMed] [Google Scholar]
- 8.Ebara H, Ikezoe J, Johkoh T, et al. Chronic eosinophilic pneumonia: Evolution of chest radiograms and CT features. J Comput Assist Tomogr. 1994;18:737–44. [PubMed] [Google Scholar]
- 9.Johkoh T, Muller NL, Taniguchi H, et al. Acute interstitial pneumonia: Thin-section CT findings in 36 patients. Radiology. 1999;211:859–63. doi: 10.1148/radiology.211.3.r99jn04859. [DOI] [PubMed] [Google Scholar]
- 10.Holbert JM, Costello P, Li W, Hoffman RM, Rogers RM. CT features of pulmonary alveolar proteinosis. AJR Am J Roentgenol. 2001;176:1287–94. doi: 10.2214/ajr.176.5.1761287. [DOI] [PubMed] [Google Scholar]
- 11.Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med. 2002;166:215–35. doi: 10.1164/rccm.2109105. [DOI] [PubMed] [Google Scholar]
- 12.Ohashi K, Sato A, Takada T, et al. Direct evidence that GM-CSF inhalation improves lung clearance in pulmonary alveolar proteinosis. Respiratory medicine. 2012;106:284–93. doi: 10.1016/j.rmed.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 13.Venkateshiah SB, Yan TD, Bonfield TL, et al. An open-label trial of granulocyte macrophage colony stimulating factor therapy for moderate symptomatic pulmonary alveolar proteinosis. Chest. 2006;130:227–37. doi: 10.1378/chest.130.1.227. [DOI] [PubMed] [Google Scholar]
- 14.Kavuru MS, Malur A, Marshall I, et al. An open-label trial of rituximab therapy in pulmonary alveolar proteinosis. Eur Respir J. 2011;38:1361–7. doi: 10.1183/09031936.00197710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luisetti M, Rodi G, Perotti C, et al. Plasmapheresis for treatment of pulmonary alveolar proteinosis. Eur Respir J. 2009;33:1220–2. doi: 10.1183/09031936.00097508. [DOI] [PubMed] [Google Scholar]
- 16.Parker LA, Novotny DB. Recurrent alveolar proteinosis following double lung transplantation. Chest. 1997;111:1457–8. doi: 10.1378/chest.111.5.1457. [DOI] [PubMed] [Google Scholar]
- 17.Gal AA, Bryan JA, Kanter KR, Lawrence EC. Cytopathology of pulmonary alveolar proteinosis complicating lung transplantation. J Heart Lung Transplant. 2004;23:135–8. doi: 10.1016/s1053-2498(03)00032-9. [DOI] [PubMed] [Google Scholar]