

Abstract
The TgCRND8 mouse model of Alzheimer’s disease exhibits progressive cortical and hippocampal β-amyloid accumulation, resulting in plaque pathology and spatial memory impairment by 3 months of age. We tested whether TgCRND8 cognitive function is disrupted prior to the appearance of macroscopic plaques in an object recognition task. We found profound deficits in 8-week-old mice. Animals this age were not impaired on the Morris water maze task. TgCRND8 and littermate controls did not differ in their duration of object exploration or optokinetic responses. Thus, visual and motor dysfunction did not confound the phenotype. Object memory deficits point to the frontal cortex and hippocampus as early targets of functional disruption. Indeed, we observed altered levels of brain-derived neurotrophic factor (BDNF) messenger ribonucleic acid (mRNA) in these brain regions of preplaque TgCRND8 mice. Our findings suggest that object recognition provides an early index of cognitive impairment associated with amyloid exposure and reduced brain-derived neurotrophic factor expression in the TgCRND8 mouse.
Keywords: Alzheimer’s disease, Object recognition memory, Entorhinal cortex, Hippocampus, Morris water maze, Optokinetic responses, TgCRND8 mice, Spatial memory, Amyloid precursor protein (APP), Brain-derived neurotrophic factor (BDNF), Real Time RT-PCR
1. Introduction
Alzheimer’s disease (AD) presents as a progressive loss in memory and general cognitive abilities. A crucial pathogenic factor is the accumulation of amyloid-β (Aβ) peptide. Processing of amyloid precursor protein (APP) into Aβ through sequential β- and γ-secretase cleavage results in the deposition of aggregated Aβ in plaques that are first observed in the temporal neocortex, hippocampus, and entorhinal cortex of the AD brain (Braak and Braak, 1991; Braak and Del Tredici, 2004; Thal et al., 2006). Transgenic mice expressing human APP are widely used to study how Aβ accumulation leads to neuronal dysfunction and cognitive impairment. The APP-transgenic TgCRND8 mouse develops a pattern of Aβ deposition similar to human AD. Plaques appear in the hippocampus and frontal cortex by 3 months and are eventually found throughout the brain, sparing the cerebellum until late in the disease (Chishti et al., 2001).
The cognitive performance of TgCRND8 mice has been widely studied (Ambree et al., 2006; Bellucci et al., 2006; Chishti et al., 2001; Janus et al., 2000). However, it remains unclear just how early relevant impairments occur. It was reported that the onset of progressive spatial memory deficits in TgCRND8 mice coincides with plaque deposition (Hyde et al., 2005). In humans, plaque burden is poorly correlated with severity of dementia (Dickson et al., 1992; Terry et al., 1991) and it has been suggested that neurotoxic oligomeric Aβ likely causes cognitive decline before plaque formation (reviewed in Hanna et al., 2009; Hardy and Selkoe, 2002; Van Dam et al., 2003; Westerman et al., 2002). To pinpoint when cognitive dysfunction first emerges in TgCRND8 mice, we sought a sensitive assay that relies on brain regions targeted by Aβ early in the pathogenic process. Among the earliest symptoms of AD is the disruption of object recognition (Done and Hajilou, 2005; Viggiano et al., 2008), a form of memory that is dependent upon reciprocal interconnections that relay multimodal sensory information between the neocortex, entorhinal cortex and the hippocampus (Charles et al., 2004; Hammond et al., 2004; Parron et al., 2006; Sipos et al., 2007; Vannucci et al., 2008; and reviewed in Dere et al., 2007).
Aβ may disrupt neural function in these regions, in part, by downregulating brain-derived neurotrophic factor (BDNF) (Christensen et al., 2008; Garzon and Fahnestock, 2007). BDNF is heavily expressed in the hippocampus and cerebral cortex (Hofer et al., 1990; Yan et al., 1997), where it plays important roles in synaptic plasticity and long term potentiation (reviewed in Nagahara et al., 2009). Decreases in BDNF are evident at preclinical stages of AD, and these reductions correlate with the rate of cognitive decline (Peng et al., 2005). We previously reported decreased levels of BDNF messenger ribonucleic acid (mRNA) in the brains of aged, plaque-bearing TgCRND8 mice (Peng et al., 2009). To determine whether BDNF downregulation is an early correlate of object recognition impairment, we measured BDNF mRNA levels in the hippocampus and frontal cortex of preplaque TgCRND8 mice.
2. Methods
2.1. Mice
TgCRND8 mice express a double mutant (Swedish: KM670/671NL plus Indiana: V717F) form of the human APP transgene under control of the Syrian hamster PrP gene promoter (Chishti et al., 2001). These mice, created by Dr. David Westaway at the Centre for Research in Neurodegerative diseases (Janus et al., 2000), exhibit progressive plaque pathology beginning at 3 months. By 7 months, they exhibit hyperphosphorylation and nitrosylation of tau (Bellucci et al., 2007) along with cholinergic cell loss (Bellucci et al., 2006). The animals were maintained on a hybrid C57BL/6/C3H background and backcrossed with C57BL/6 wild-type mice. Behavioral studies were performed on mice of the F1 generation. TgCRND8 and nontransgenic (non-Tg) littermates were housed in groups of 2 to 4 in ventilated polycarbonate clear cages under standard laboratory conditions (12/12 hour light/dark cycle with lights on at 0700 hours; room temperature of 21 °C). Food and water were available ad libitum. Tests were carried out during the light phase of the cycle in accordance with the Canadian Council on Animal Care guidelines and the Animal Care Committee at the University of Toronto.
Experiments were performed on preplaque mice at 4 – 6 weeks or 8 –9 weeks of age and on mice with advanced plaque pathology (6 – 8 months old). Groups were matched for gender and the genotype was unknown to experimenters. Genotypes were determined by dot-blot hybridization analysis of genomic DNA extracted from tail clippings using a human APP probe as described previously (Chishti et al., 2001).
2.2. Object recognition
Cohorts of TgCRND8 (n ≥ 10) and non-Tg (n ≥ 10) littermates at 4 weeks, 8 weeks, and 6 – 8 months of age were tested for object recognition. This is a test of nonspatial, episodic memory that is independent of neuromotor deficits and emotional cues. It is based on the spontaneous tendency of rodents to explore a novel object over a familiar one (Ennaceur and Delacour, 1988). Entorhinal and perirhinal regions of the cortex (the rhinal cortex) are implicated in object recognition, as is the hippocampus, although involvement of the latter is temporally delayed. The rhinal cortex is thought to support short term retention of object familiarity in concert with neocortical areas (Mumby and Pinel, 1994; Steckler et al., 1998). With delay intervals longer than 15 minutes, retention becomes dependent upon reactivation of memory traces in the hippocampus (Hammond et al., 2004). To differentiate between cortical and hippocampal components of task performance, we adapted the object memory paradigm used previously in our laboratory (Vaucher et al., 2002) by varying delay intervals. Mice were habituated to the testing arena (clear plastic mouse cages) for 15 minutes over 7 daily sessions, and were considered successfully habituated if they consumed a small piece of breakfast cereal within 2 minutes. On the test day, each animal was exposed for 10 minutes to a LEGO® construct (LEGO Group, Billund, Denmark) and a Hot Wheels® car (Mattel, Inc., El Segundo, California). The objects were predetermined to be of matched saliency to mice. Objects were fixed to the floor of the mouse cage with Velcro tape. Time spent exploring the objects was recorded. Exploration was scored when the mouse touched an object with its forepaws or snout, bit, licked, or sniffed the object from a distance of no more than 1.5 cm. Five minutes, 1 hour, or 3 hours later, mice were re-exposed for 5 minutes to 1 object from the original test pair and to a novel object. Separate cohorts (n ≥ 10 for either genotype) were tested at each retention interval. Between tests, the objects and testing cage were wiped with Virox5™ (Johnson Diversey, Inc., Sturtevant, Wisconsin) to eliminate odor cues. The possible confound of mice exhibiting preference for the right or left side of the cage was addressed by counterbalancing the placements of new objects between mice in a test group. A “memory index” (MI) was calculated as MI = (tn − tf)/(tn + tf), wherein “tn” represents time exploring a novel object and “tf” the duration of familiar object exploration.
2.3. Visual function
Visual function was assessed noninvasively in 9-week-old TgCRND8 (n = 7) and non-Tg (n = 10) littermates by testing a head tracking response to a rotating whole field stimulus. Head tracking behavior has been used to rapidly quantify spatial vision in rodents (Prusky et al., 2004) and is consistent with electrophysiology results (Thomas et al., 2004). The optokinetic apparatus was custom made and consisted of a stationary platform on which the animal was placed and where it could move freely (diameter 5 cm), surrounded by a drum (diameter 30.5 cm, height 61 cm). The inner surface of the drum was lined with alternating black and white vertical stripes covering the entire visual field. Two visual patterns with spatial frequencies of 0.13 cycle/degree and 0.26 cycle/degree were used. The weber contrast of white and black stripes (Lmax. − Lmin./Lmin.; L = luminance) was 7.55, as measured by a luminance meter (Minolta, LS-100, Aichi, Japan). A video camera was mounted on a stand overlooking the platform (Olympus digital camera, C-3000, Nagano, Japan). The drum was rotated at angular velocities ranging from 1 to 60 degrees/second either clockwise or anticlockwise using a motor (GM9413-4, Pittman, Harleysville, Pennsylvania). Angular velocity of the drum was measured with a contact tachometer (High-Accuracy Digital Contact Tachometer, McMaster-Carr, Dayton, New Jersey). Once a mouse was placed on the platform within the drum, the light was turned on and the drum was rotated clockwise or anticlockwise until head tracking was observed for a maximum of 30 seconds. A movement of the head corresponding to the direction and speed of the optokinetic stimulus was defined as head tracking. Experimenters were blind to each animal’s genotype, and all mice were tested on the same day by the same experimenters. The optokinetic stimulus was repeated 3 times in alternating directions. If a head tracking response was not observed, the stimulus was repeated 2 more times. The angular velocity of the drum was increased stepwise until a 50% response rate was achieved (Bonaventure et al., 1983; Yücel et al., 1990). Visual function was assessed by determining the optokinetic frequency at which head tracking behavior was extinguished. Frequency of extinction (FE) was calculated by multiplying spatial frequency of the visual pattern with the specific angular velocity at which a mouse responded at chance level.
2.4. Spatial memory
Behaviorally naive TgCRND8 (n = 5) and non-Tg (n = 5) mice were tested at 8 weeks of age in the reference memory version of the Morris water maze test. The maze apparatus and testing procedure were described previously (Janus et al., 2000). All mice underwent a day of nonspatial pretraining during which the mice learn that a submerged platform is present, how to climb onto the platform, and to perform a random swim search for the platform. The pretraining consisted of 4 trials, in each of which the mouse was released from a different quadrant and swam to a visible platform. This cued platform test was conducted to assess whether motoric or motivational factors might have confounded maze performance. One day following pretraining, mice underwent 5 days of place discrimination training with the platform hidden in the center of a single quadrant, over 4 trials per day. Following the last trial on the fifth day, the platform was removed and the mouse received a 60-second probe trial. Mean latency to platform, swim path length, and swim speed were measured with an online video tracking system (HVS Image Advanced Tracker VP200, HVS Image Ltd., Hampton, UK). An annulus-crossing index (number of passes over platform site, minus the mean of passes over sites in other quadrants) was calculated to determine the place preference during the probe trial.
2.5. Measurement of BDNF mRNA
TgCRND8 and non-Tg littermates were sacrificed at 6 weeks, 9 weeks, and 6 – 8 months of age by decapitation several days following behavioral testing. Brains were removed and the hippocampus and cortex were dissected. Tissues were flash frozen in liquid nitrogen and stored at −80 °C until analysis. Ribonucleic acid (RNA) isolation, DNase treatment, reverse transcription and absolute quantitative real time polymerase chain reaction (PCR) for measurement of BDNF mRNA in frozen cortical and hippocampal samples were completed as previously described (Peng et al., 2009). The forward and reverse primers used for total mouse BDNF mRNA were: 5′ CAG CGG CAG ATA AAA AGA and 5′ TCA GTT GGC CTT TGG ATA, product 87 bp. β-actin mRNA was used to normalize results. The forward and reverse primers used for β-actin mRNA were: 5′ CTG ACA GGA TGC AGA AGG and 5′ GAG TAC TTG CGC TCA GGA, product 85 bp. Purified PCR products derived from use of these primers were used as standards for total BDNF and β-actin. Only experiments with an R2 > 0.995 and PCR efficiency > 90% were used for analysis. All unknowns and controls were run in triplicate. A dissociation curve was created to verify that no secondary products had formed. Results were obtained as copies per ng total RNA and expressed as a ratio of BDNF/β-actin mRNA.
2.6. Statistical analysis
Object recognition and BDNF mRNA data were analyzed by unpaired Student t tests. Morris water maze data were analyzed by 2-way analysis of variance (ANOVA) with genotype as between subject factor and test day as repeated measure factor. The Mann-Whitney U test was applied to the nonparametric FE optokinetic testing scores. A significance level (α) was set to 0.05, and the 2-tailed variants of all tests were used. Data are presented as means ± standard error of the mean (SEM). All calculations were performed using GraphPad Prism version 4.0c for Macintosh (Mac OS X version by Software MacKiev™, GraphPad Software, Inc., San Diego, California).
3. Results
3.1. Progressive object recognition deficits precede amyloid plaque accumulation
TgCRND8 and non-Tg mice were tested for object recognition memory with retention intervals of 5 minutes, 1 hour, or 3 hours. We calculated a memory index, wherein a score of 0 indicates no preference for novel or familiar objects. TgCRND8 mice exhibited progressive object memory deficits that could be discerned by 8 weeks of age (Fig. 1). The performance of 4 – 6-week-old TgCRND8 mice was indistinguishable from that of non-Tg littermates in all delay interval versions of the task (p > 0.05). Performance of 8-week-old TgCRND8 mice was impaired at the 5-minute delay interval (t(18) = 2.96, p < 0.01), trended toward impairment at the 1-hour delay interval (t(18) = 1.76, p = 0.095), and was profoundly impaired when assayed with a 3-hour delay interval (t(18) = 5.92, p < 0.001). TgCRND8 mice with advanced plaque pathology (6 – 8 months of age) were impaired at delays of 5 minutes (t(38) = 4.78, p < 0.001), 1 hour (t(24) = 4.37, p = 0.001), and 3 hours (t(25) = 7.74, p < 0.001). Reductions in object recognition performance were not explained by differences between TgCRND8 and non-Tg animals in time spent exploring the initial object pairs (Fig. 2A, t(33) = 0.07, p > 0.05). The sets of object pairs were previously demonstrated to be of matched saliency (Vaucher et al., 2002), and we observed no group differences in object preference at any age during the retention phase (data not shown). Similarly, it is unlikely that our data can be explained by differences in visual function. Frequency of extinction (FE) scores of head-tracking behavior in 9-week-old TgCRND8 mice were comparable to those of age-matched non-Tg controls (Fig. 2B, U = 22.50, p > 0.05). Even at 6 – 8 months of age, TgCRND8 mice (FE = 4.55 ± 0.56) and littermate controls (FE = 5.67 ± 0.41) were indistinguishable by this task (Mann-Whitney U = 8.5, p = 0.214).
Fig. 1.

Object recognition memory assessed at preplaque and plaque ages. TgCRND8 mice exhibit no discernable memory impairment at 4 weeks (A), but a profound deficit in 3-hour object recognition memory by 8 weeks (B). By 6 – 8 months of age, TgCRND8 mice are impaired at delays of 5 minutes, 1 hour, and 3 hours following initial exposure to objects (C). Values are means ± standard error of the mean (SEM) of memory index (MI) scores, with n ≥ 10 for each genotype at each delay interval. *p < 0.01 by unpaired Student t test.
Fig. 2.

Exploration of objects and visual function examined in preplaque mice at 8 –9 weeks of age. Time in seconds (s) spent exploring both left and right objects during the initial exposure period are compared (A). TgCRND8 mice (n = 16) and nontransgenic (non-Tg) mice (n = 19) did not differ in duration of object exploration (p > 0.05, unpaired Student t test). Visual function was assessed by examination of head-tracking behavior (B). Animals were placed on a stationary platform within a patterned surface drum. The speed of the drum rotation was varied stepwise. The maximal angular speed at which an optokinetic response was detected greater than 50% of the time was calculated (frequency of extinction; FE). The FE (cycle/second) was calculated by multiplying spatial frequency of the drum visual pattern with angular velocity. TgCRND8 mice (n = 7) were comparable to non-Tg mice (n = 10) in their FE scores (p > 0.05, Mann-Whitney U test). Values are means ± standard error of the mean (SEM).
3.2. Preplaque TgCRND8 mice are NOT impaired in the Morris water maze task
TgCRND8 mice were tested on the spatial reference memory version of the Morris water maze task at 8 weeks of age (Fig. 3). Consistent with previous reports (Hyde et al., 2005; Janus et al., 2000), we did not detect any differences in swim path length (F(1,9) = 0.67, p > 0.05), escape latency (F(1,9) = 0.003, p > 0.05), or swim speed (t(8) = 1.44, p = 0.187 for data from day 2, by unpaired Student t test) between preplaque TgCRND8 and control mice. Probe trial results were equivalent for TgCRND8 and non-Tg mice (data not shown).
Fig. 3.

Spatial reference memory in the Morris water maze assessed in 8-week-old mice. TgCRND8 mice (n = 5) were indistinguishable from nontransgenic (non-Tg) littermates (n = 5) in terms of their (A) swim-path length, (B) escape latency, and (C) swim speed. Data are means ± standard error of the mean (SEM) of all trials performed on each test day and were evaluated by repeated measures analysis of variance (ANOVA) (p > 0.05).
3.3. BDNF mRNA is reduced in the hippocampus and frontal cortex of young TgCRND8 mice
We measured total BDNF mRNA and β-actin mRNA by real time quantitative reverse transcriptase (RT)-PCR in the hippocampus (Fig. 4) and frontal cortex (Fig. 5) of TgCRND8 mice. These brain regions exhibit extensive plaque pathology in mature TgCRND8 mice and are crucial for object recognition memory. Hippocampal and cortical mRNA levels for the housekeeping gene β-actin did not differ between TgCRND8 and non-Tg littermates and thus were used to normalize levels of BDNF mRNA. BDNF mRNA was significantly reduced in the hippocampus (t(7) = 2.86, p < 0.05), but not in the cortex (t(10) = 0.90, p = 0.40) of 6-week-old TgCRND8 mice. By 9 weeks, BDNF mRNA levels were also reduced in the cortex of TgCRND8 mice (t(15) = 2.22, p < 0.05). Hippocampal BDNF mRNA levels did not differ between groups at 9 weeks of age (t(14) = 1.78, p > 0.05). However, in 6 – 8-month-old TgCRND8 samples, the downregulatation of hippocampal BDNF mRNA was clearly evident (t(14) = 1.78, p < 0.05).
Fig. 4.
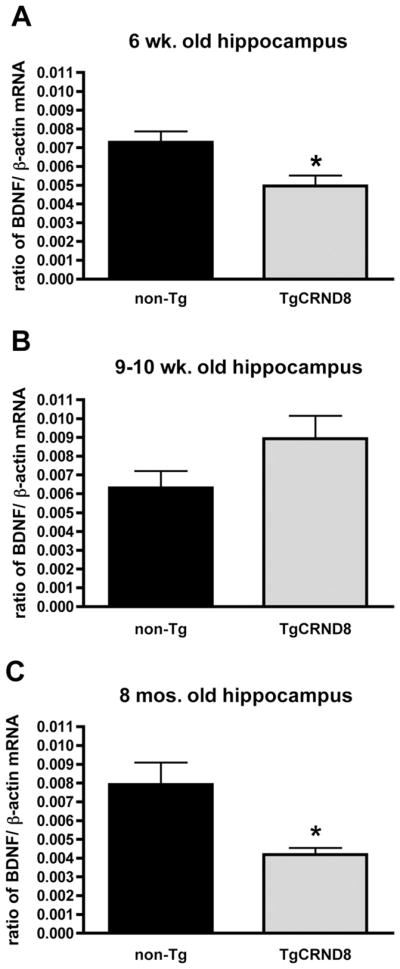
Hippocampal brain-derived neurotrophic factor (BDNF) messenger ribonucleic acid (mRNA) levels measured by absolute quantitative real-time reverse transcriptase polymerase chain reaction (RT-PCR). At 6 weeks of age (A), TgCRND8 mice (n = 5) had reduced BDNF mRNA levels in the hippocampus compared with nontransgenic (non-Tg) mice (n = 4). However at 9 weeks (B), there was no significant difference between TgCRND8 mice (n = 7) and non-Tg littermates (n = 9). By 6 – 8 months (C), hippocampal BDNF mRNA was reduced in TgCRND8 mice (n = 4) in comparison with littermate controls (n = 4). Data are expressed as a ratio of copies of BDNF mRNA/copies of β-actin mRNA. Values are means ± standard error of the mean (SEM). *p < 0.05 by unpaired Student t test.
Fig. 5.

Cortical brain-derived neurotrophic factor (BDNF) messenger ribonucleic acid (mRNA) levels in preplaque mice. At 6 weeks of age (A), TgCRND8 mice (n = 6) and nontransgenic (non-Tg) littermates (n = 6) had equivalent levels of BDNF mRNA in the cortex. By 9 weeks of age (B), TgCRND8 mice (n = 7) had reduced BDNF expression compared with non-Tg mice (n = 10). Data are expressed as a ratio of copies of BDNF mRNA/copies of β-actin mRNA. Values are means ± standard error of the mean (SEM). *p < 0.05 by unpaired Student t test.
4. Discussion
We have used an object recognition test of nonspatial, hippocampal, and cortical short-term memory to determine when cognitive deficits first occur in TgCRND8 mice. We found that object memory deficits emerge several weeks prior to the appearance of macroscopic amyloid plaques in the brain. This is the earliest cognitive deficit reported in an APP transgenic mouse. The onset of object recognition impairment coincides with reductions in BDNF mRNA in the hippocampus and cortex of pre-plaque TgCRND8 mice.
Uncovering subtle phenotypes well before widespread neurodegeneration has occurred is crucial for early diagnosis and treatment. Performance in the object recognition test is dependent on brain regions first targeted by β-amyloid and thus provides a sensitive measure of early cognitive disruption (Dodart et al., 2002; Sipos et al., 2007). Ennaceur and Delacour developed a spontaneous object recognition task for rats in 1988. Since then it has been successfully modified for use in the mouse (reviewed in Sik et al., 2003). Various object recognition paradigms may be used to investigate slightly different aspects of memory (Benice and Raber, 2008; Broadbent et al., 2004; Capsoni et al., 2000; Chopin et al., 2002; Dere et al., 2007; Dodart et al., 1997; Hammond et al., 2004; Heldt et al., 2007; Winters et al., 2004; Yuede et al., 2009).
The 1-trial, nonspatial test that we used is based on the rodent’s innate preference for novelty. Performance of the task is not associated with positive or negative reinforcement. We also undertook measures to assess the potential confounding influence of contextual, motor, or visual factors. Mice were habituated to the testing arena to reduce stress and novelty associated with the testing environment. The location of a novel object within the testing box was always identical to that of the replaced object.
We observed no difference between TgCRND8 mice and non-Tg controls in the duration of object exploration, and TgCRND8 mice did not differ from non-Tg littermates in swim speed. Thus, it is does not appear that memory deficits were confounded by differences in motor ability. Similarly, visual dysfunction was not a factor, as TgCRND8 performance was equivalent to that of non-Tg littermates in optokinetic testing. We conclude that the object recognition deficit of TgCRND8 mice can be ascribed to impaired memory.
The disruption of object recognition over short (5 minute) and long (3 hour) retention intervals suggests that both cortical and hippocampal structures are functionally disrupted early in the disease process. Steckler et al. (1998) described 2 neural networks for recognition memory in the rodent: 1 for spatial memory is encoded by the hippocampus and 1 for nonspatial memory involves the rhinal cortex and cortical association areas. Whether the hippocampus is truly irrelevant for nonspatial object memory has been questioned (Dere et al., 2007). When short delay intervals (< 15 minutes) are used, entorhinal, perirhinal, and frontal cortices are sufficient for object recognition. With longer retention intervals, the hippocampus is recruited (Hammond et al., 2004). Baker and Kim (2002) blocked long term potentiation (LTP) with an N-methyl-D-aspartate receptor antagonist in the dorsal hippocampus of rats. They reported a selective impairment of object memory when tested with a 3-hour delay but not with a 5-minute delay.
Although the profound deficit we observed in mice tested with a 3-hour delay underscores functional impairment of hippocampus in preplaque TgCRND8 mice, the water maze spatial reference memory of these mice was normal. The hippocampus is important for both spatial and nonspatial recognition memory, but the 2 tasks may differ in the degree of integrated hippocampal function required. Hippocampal lesion studies in rats have shown that more hippocampal tissue is involved in spatial memory performance than in nonspatial recognition memory (Broadbent et al., 2004). Conversely, object recognition is more sensitive to disruption of the entorhinal-hippocampal circuitry (Burwell et al., 2004; Parron et al., 2006). The entorhinal cortex relays processed information from the surrounding neo-cortex to the dentate gyrus of the hippocampus via the medial perforant pathway. Damage to this circuitry is a specific and reliable distinguishing factor between healthy aged subjects and mild/early stage AD (Chetelat and Baron, 2003; Gunten et al., 2006). As Aβ deposition is first noted in cortical and hippocampal structures of TgCRND8 mice, the object recognition task is an especially relevant and efficient method for assessing early cognitive changes in TgCRND8 mice.
Downregulation of BDNF expression may be a factor linking Aβ exposure with cognitive impairment. Site-specific deletion of the BDNF gene in dorsal hippocampus of adult mice can result in object recognition deficits (Heldt et al., 2007). Moreover, BDNF delivery to the entorhinal cortex has been shown to both mitigate degeneration in the entorhinal cortex and hippocampus, and to rescue learning and memory impairments in rodent and primate models of AD (Nagahara et al., 2009). In vitro studies have confirmed the deleterious influence of soluble Aβ on BDNF expression and signaling (Garzon and Fahnestock, 2007; Tong et al., 2004). Moreover, we previously reported that aged, plaque-bearing TgCRND8 mice exhibit very high levels of the more fibrillogenic Aβ42 relative to Aβ40 and that the relative increase in Aβ42 was inversely correlated with BDNF mRNA in the cortex (Peng et al., 2009).
We now report that BDNF is altered at earlier stages of TgCRND8 dysfunction. From 4 to 8 weeks of age tissue levels of Aβ42 double in TgCRND8 mice, and the Aβ42/Aβ40 ratio increases 1.5-fold (Chishti et al., 2001). Over the same time frame, we observed decreased BDNF mRNA in the hippocampus and cortex. Decreases were evident at 6 weeks in the hippocampus and by 9 weeks in the cortex of preplaque mice. A slight, nonsignificant increase in hippocampal BDNF mRNA was observed in TgCRND8 mice at 9 weeks. However, BDNF mRNA subsequently decreased again and low levels were observed at 6 – 8 months.
Such phasic fluctuations in BDNF may occur in response to progression through stages of amyloid pathology. Sub-lethal concentrations of soluble oligomeric Aβ functionally disrupt neurons, causing suppression of cAMP-response element-binding protein (CREB) phosphorylation and downstream BDNF expression (Garzon and Fahnestock, 2007; Tong et al., 2001). Decreases in BDNF at 6 weeks in the hippocampus and at 9 weeks in the cortex of TgCRND8 mice are consistent with the onset of Aβ-induced dysfunction in brain regions crucial for object recognition. However, this acute reduction in BDNF mRNA may be short-lived. Tissue levels of CREB and BDNF mRNA may rebound in response to a moderate load of intracellular Aβ (Arvanitis et al., 2007). In support of this notion, BDNF mRNA was found to be upregulated in glial cells surrounding plaques in APP23 transgenic mice (Burbach et al., 2004). This increase may reflect a compensatory neuroprotective response after central nervous system (CNS) injury (Lindvall et al., 1992). Human studies also indicate increased BDNF expression during early stages of Alzheimer’s disease and a decline as disease progresses (Laske et al., 2006). It has been suggested that continuing exposure to increasing levels of central nervous system Aβ leads to the neuropathology and decreased neuronal BDNF expression (Arvanitis et al., 2007; Burbach et al., 2004). In the TgCRND8 mouse, the second phasic decrease of BDNF mRNA that we observed at 6 – 8 months coincides with significant cholinergic cell loss (Bellucci et al., 2006).
In summary, the TgCRND8 mouse recapitulates early and relevant cognitive symptoms of Alzheimer’s disease. Object recognition deficits in TgCRND8 mice precede plaque accumulation and coincide with a 2-fold increase in Aβ42 levels. The early alterations in BDNF mRNA seen in preplaque TgCRND8 mice suggest that dysregulation of BDNF expression contributes to Aβ-induced cognitive impairment.
Acknowledgments
This study was supported by the Ontario Mental Health Foundation (to HM), the Paul and Adelle Deacon Ontario Graduate Studentship in Science and Technology (to BF), the K.M. Hunter Ontario Student Opportunity Trust Fund (to BF), the Alzheimer’s Association (USA) (to MF) and the Canadian Institutes of Health Research (to JM).
Footnotes
Disclosure statement
The authors of this manuscript have no conflicts of interest to disclose.
All experiments conducted with mice were carried out according to the Canadian Council on Animal Care guidelines and were approved by the Animal Care Committee at the University of Toronto.
References
- Ambree O, Touma C, Gortz N, Keyvani K, Paulus W, Palme R, Sachser N. Activity changes and marked stereotypic behaviour precede Aβ pathology in TgCRND8 Alzheimer mice. Neurobiol Aging. 2006;27:955–964. doi: 10.1016/j.neurobiolaging.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Arvanitis DN, Ducatenzeiler A, Ou JN, Grodstein E, Andrews SD, Trendulkar SR, Ribeiro-da-Silva A, Szyf M, Cuello AC. High intracellular concentrations of amyloid-beta block nuclear translocation of phosphorylated CREB. J Neurochem. 2007;103:216–228. doi: 10.1111/j.1471-4159.2007.04704.x. [DOI] [PubMed] [Google Scholar]
- Baker KB, Kim J. Effects of stress and hippocampal NMDA receptor antagonism on recognition memory in rats. Learn Mem. 2002;9:58– 65. doi: 10.1101/lm.46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci A, Luccarini I, Scali C, Prosperi C, Giovannini MG, Pepeu G, Casamenti F. Cholinergic dysfunction, neuronal damage and axonal loss in TgCRND8 mice. Neurobiol Dis. 2006;23:260–272. doi: 10.1016/j.nbd.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Bellucci A, Rosi MC, Grossi C, Fiorentini A, Luccarini I, Casamenti F. Abnormal processing of tau in the brain of aged TgCRND8 mice. Neurobiol Dis. 2007;27:328–338. doi: 10.1016/j.nbd.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Benice TS, Raber J. Object recognition analysis in mice using nose-point digital video tracking. J Neurosci Methods. 2008;168:422– 430. doi: 10.1016/j.jneumeth.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Bonaventure N, Wioland N, Bigenwald J. Involvement of GABAergic mechanisms in the optokinetic nystagmus of the frog. Exp Brain Res. 1983;50:433– 441. doi: 10.1007/BF00239210. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. Alzheimer’s disease: intraneuronal alterations precede insoluble amyloid-β-formation. Neurobiol Aging. 2004;25:713–718. doi: 10.1016/j.neurobiolaging.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Broadbent NJ, Squire LR, Clark RE. Spatial memory, recognition memory, and the hippocampus. Proc Natl Acad Sci U S A. 2004;101:14515–14520. doi: 10.1073/pnas.0406344101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbach G, Hellweg R, Haas C, Del Turco D, Deicke U, Abramowski D, Jucker M, Staufenbiel M, Deller T. Induction of Brain-derived neurotrophic factor in plaque-associated glial cells of aged APP23 transgenic mice. J Neurosci. 2004;24:2421–2430. doi: 10.1523/JNEUROSCI.5599-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burwell RD, Saddoris MP, Bucci DJ, Wiig KA. Corticohippocampal contributions to spatial and contextual learning. J Neurosci. 2004;24:3826–3836. doi: 10.1523/JNEUROSCI.0410-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capsoni S, Ugolini G, Comparini A, Ruberti F, Berardi N, Cattaneo A. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc Natl Acad Sci U S A. 2000;97:6826– 6831. doi: 10.1073/pnas.97.12.6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles DP, Browning PG, Gaffan D. Entorhinal cortex contributes to object-in-place scene memory. Eur J Neurosci. 2004;20:3157–3164. doi: 10.1111/j.1460-9568.2004.03777.x. [DOI] [PubMed] [Google Scholar]
- Chetelat G, Baron JC. Early diagnosis of Alzheimer’s disease: Contribution of structural neuroimaging. Neuroimage. 2003;18:525–541. doi: 10.1016/s1053-8119(02)00026-5. [DOI] [PubMed] [Google Scholar]
- Chishti MA, Yang D, Janus C, Phinney A, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser P, Carlson G, St George-Hyslop P, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Chopin P, Colpaert F, Marien M. Effects of acute and subchronic administration of dexefaroxan, an alpha2-adrenoceptor antagonist, on memory performance in young adult and aged rodents. J Pharmacol Exp Ther. 2002;301:187–196. doi: 10.1124/jpet.301.1.187. [DOI] [PubMed] [Google Scholar]
- Christensen R, Marcussen AB, Wortwein G, Knudsen GM, Aznar S. Aβ (1– 42) injection causes memory impairment, lowered cortical and serum BDNF levels, and decreased hippocampal five-HT2A levels. Exp Neurol. 2008;210:164–171. doi: 10.1016/j.expneurol.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Dere E, Huston JP, De Souza Silva MA. The pharmacology, neuroanatomy and neurogenetics of one-trial object recognition in rodents. Neurosci Biobehav Rev. 2007;31:673–704. doi: 10.1016/j.neubiorev.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neuobiol Aging. 1992;13:179–189. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Mathis C, Bales KR, Paul SM. Does my mouse have Alzheimer’s disease? Genes Brain Behav. 2002;1:142–155. doi: 10.1034/j.1601-183x.2002.10302.x. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Mathis C, Ungerer A. Scopolamine-induced deficits in a two-trial object recognition task in mice. Neuroreport. 1997;8:1173–1178. doi: 10.1097/00001756-199703240-00023. [DOI] [PubMed] [Google Scholar]
- Done D, Hajilou B. Loss of high-level perceptual knowledge of object structure in DAT. Neuropsychologia. 2005;43:60– 68. doi: 10.1016/j.neuropsychologia.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioural data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- Garzon DJ, Fahnestock M. Oligomeric amyloid decreases basal levels of brain-derived neurotrophic factor (BDNF) mRNA via specific downregulation of BDNF transcripts IV and V in differentiated human neuroblastoma cells. J Neurosci. 2007;27:2628–2635. doi: 10.1523/JNEUROSCI.5053-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond RS, Tull LE, Stackman RW. On the delay-dependent involvement of the hippocampus in object recognition memory. Neurobiol Learn Mem. 2004;82:26–34. doi: 10.1016/j.nlm.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Hanna A, Horne P, Yager D, Eckman C, Eckman E, Janus C. Amyloid-β and impairment in multiple memory systems in older transgenic APP TgCRND8 mice. Genes Brain Behav. 2009;8:676– 684. doi: 10.1111/j.1601-183X.2009.00510.x. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;279:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Heldt SA, Stanek L, Chhatwal JP, Ressler KJ. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry. 2007;12:656– 670. doi: 10.1038/sj.mp.4001957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde YA. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J. 1990;9:2459–2464. doi: 10.1002/j.1460-2075.1990.tb07423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde LA, Kazdoba TM, Grilli M, Lozza G, Brussa R, Zhang Q, Wong GT, McCool MF, Zhang L, Parker EM, Higgins GA. Age-progressing cognitive impairments and neuropathology in transgenic CRND8 mice. Behav Brain Res. 2005;160:344–355. doi: 10.1016/j.bbr.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Gunten A, Kovari E, Bussiere T, Rivara C, Gold G, Bouras C, Hof P, Giannakopoulos P. Cognitive impact of neuronal pathology in the entorhinal cortex and CA1 field in Alzheimer’s disease. Neurobiol Aging. 2006;27:270–277. doi: 10.1016/j.neurobiolaging.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HTJ, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D. Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Laske C, Stransky E, Leyhe T, Eschweiler G, Wittorf A, Richartz E, Bartels M, Buchkremer G, Schott K. Stage-dependent BDNF serum concentrations in Alzheimer’s disease. J Neural Transm. 2006;113:1217–1224. doi: 10.1007/s00702-005-0397-y. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Ernfors P, Bengzon J, Kokaia Z, Smith ML, Siesjo BK, Persson H. Differential regulation of mRNAs for nerve growth factor, brain-derived neurotrophic factor, and neurotrophin 3 in the adult rat brain following cerebral ischemia and hypoglycemic coma. Proc Natl Acad Sci U S A. 1992;89:648– 652. doi: 10.1073/pnas.89.2.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumby DG, Pinel JP. Rhinal cortex lesions and object recognition in rats. Behav Neurosci. 1994;108:11–18. doi: 10.1037//0735-7044.108.1.11. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynksi MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parron C, Poucet B, Save E. Cooperation between the hippocampus and the entorhinal cortex in spatial memory: A disconnection study. Behav Brain Res. 2006;170:99–109. doi: 10.1016/j.bbr.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Peng S, Garzon DJ, Marchese M, Klein W, Ginsberg SD, Francis BM, Mount HTJ, Mufson EJ, Salehi A, Fahnestock M. Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer’s disease. J Neurosci. 2009;29:9321–9329. doi: 10.1523/JNEUROSCI.4736-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J Neurochem. 2005;93:1412–1421. doi: 10.1111/j.1471-4159.2005.03135.x. [DOI] [PubMed] [Google Scholar]
- Prusky GT, Alam NM, Beekman S, Douglas RM. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci. 2004;45:4611– 4616. doi: 10.1167/iovs.04-0541. [DOI] [PubMed] [Google Scholar]
- Sik A, Nieuwehuyzen P, Prickaerts J, Blockland A. Performance of different mouse strains in an object recognition task. Behav Brain Res. 2003;147:49–54. doi: 10.1016/s0166-4328(03)00117-7. [DOI] [PubMed] [Google Scholar]
- Sipos E, Kurunczi A, Kasza A, Horvath J, Felszeghy K, Laroche S, Toldi J, Parducz A, Penke B, Penke Z. β-amyloid pathology in the entorhinal cortex of rats induces memory deficits: Implications for Alzheimer’s disease. Neuroscience. 2007;147:28–36. doi: 10.1016/j.neuroscience.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Steckler T, Drinkenburg WHIM, Sahgal A, Aggleton JP. Recognition memory in rats – II. Neuroanatomical substrates Prog Neurobiol. 1998;54:313–332. doi: 10.1016/s0301-0082(97)00061-0. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alteration in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Tredici K, Braak H. The development of amyloid β-protein deposits in the aged brain. Sci Aging Knowl Environ. 2006;2006:1–9. doi: 10.1126/sageke.2006.6.re1. [DOI] [PubMed] [Google Scholar]
- Thomas BB, Seiler MJ, Sadda SR, Coffey PJ, Aramant RB. Optokinetic test to evaluate visual acuity of each eye independently. J Neurosci Methods. 2004;138:7–13. doi: 10.1016/j.jneumeth.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Tong L, Balazs R, Thornton PL, Cotman CW. β-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci. 2004;24:6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L, Thornton PL, Balazs R, Cotman CW. β-amyloid-(1-42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at concentrations in which cell survival is not compromised. J Biol Chem. 2001;276:17301–17306. doi: 10.1074/jbc.M010450200. [DOI] [PubMed] [Google Scholar]
- Van Dam D, D’Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, De Deyn PP. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci. 2003;17:388–396. doi: 10.1046/j.1460-9568.2003.02444.x. [DOI] [PubMed] [Google Scholar]
- Vannucci M, Pezer N, Helmstaedter C, Schaller K, Viggiano M, Elger C, Grunwald T. Hippocampal response to visual objects is related to visual memory functioning. Neuroreport. 2008;19:965–968. doi: 10.1097/WNR.0b013e328302c89c. [DOI] [PubMed] [Google Scholar]
- Vaucher E, Fluit P, Chishti MA, Westaway D, Mount HTJ, Kar S. Object recognition memory and cholinergic parameters in mice expressing human presenilin 1 transgenes. Exp Neurol. 2002;175:398– 406. doi: 10.1006/exnr.2002.7915. [DOI] [PubMed] [Google Scholar]
- Viggiano M, Galli G, Righi S, Brancati C, Gori G, Cincotta M. Visual recognition memory in Alzheimer’s disease: Repetition-lag effects. Exp Aging Res. 2008;34:267–281. doi: 10.1080/03610730802070241. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. The relationship between Aβ and Memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters BD, Forwood SE, Cowell RA, Saksida LM, Bussey TJ. Double dissociation between the effects of peri-postrhinal cortex and hippocampal lesions on tests of object recognition and spatial memory: heterogeneity of function within the temporal lobe. J Neurosci. 2004;24:5901–5908. doi: 10.1523/JNEUROSCI.1346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Rosenfeld RD, Matheson CR, Hawkins NA, Lopez OT, Bennett L, Welcher AA. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience. 1997;78:431– 448. doi: 10.1016/s0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]
- Yücel YH, Jardon B, Kim MS, Bonaventure N. Directional asymmetry of the horizontal monocular head and eye optokinetic nystagmus: Effects of picrotoxin. Vis Res. 1990;30:549–555. doi: 10.1016/0042-6989(90)90067-u. [DOI] [PubMed] [Google Scholar]
- Yuede CM, Zimmerman SD, Dong H, Kling MJ, Bero AW, Holtzman DM, Timson BF, Csernansky JG. Effects of voluntary and forced exercise on plaque deposition, hippocampal volume, and behavior in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol Dis. 2009;35:426– 432. doi: 10.1016/j.nbd.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]