

Abstract
Many clinically useful pharmaceuticals are semi-synthesized from natural products produced by actinobacteria and fungi. The synthetic protocols usually contain many complicated reaction steps and thereby result in low yields and high costs. It is therefore important to breed microorganisms that produce a compound most suitable for chemical synthesis. For a long time, desirable mutants have been obtained by random mutagenesis and mass screening. However, these mutants sometimes show unfavorable phenotypes such as low viability and low productivity of the desired compound. Fusicoccin (FC) A is a diterpene glucoside produced by the fungus Phomopsis amygdali. Both FC and the structurally-related cotylenin A (CN) have phytohormone-like activity. However, only CN exhibits anti-cancer activity. Since the CN producer lost its ability to proliferate during preservation, a study on the relationship between structure and activity was carried out, and elimination of the hydroxyl group at position 12 of FC was essential to mimic the CN-like activity. Based on detailed dissection of the biosynthetic machinery, we constructed a mutant producing a compound without a hydroxyl group at position 12 by gene-disruption. The mutant produced this compound as a sole metabolite, which can be easily and efficiently converted into an anti-cancer drug, and its productivity was equivalent to the sum of FC-related compounds produced by the parental strain. Our strategy would be applicable to development of pharmaceuticals that are semi-synthesized from fungal metabolites.
Introduction
For screening of candidate compounds useful for pharmaceutical drugs, in vitro assay methods are often employed. The recent development of robotic high throughput screening of large chemical libraries enables us to screen many compounds at once. When a compound is selected by such screening, synthesis of its derivatives would be relatively easy because the original library was chemically constructed. On the other hand, screenings from natural products still occupy an important position because many clinically useful pharmaceuticals originate from natural products. However, most natural products are chemically modified for clinical use to decrease toxicity and increase solubility. The synthetic protocols for these chemical modifications sometimes contain many complicated reactions and thereby result in low yields and high costs. To overcome this problem, a strategy of mutagenesis of the producers with mutagens such as N-Methyl-N'-nitro-N-nitrosoguanidine has been employed, expecting that a more suitable compound for chemical synthesis might be produced rather than a final product. However, this type of trial was usually unsuccessful because the mutants showed low viability and productivity. Therefore, it is essential to dissect the biosynthetic machinery of the compound and to make use of this knowledge for breeding of the producer. In this study, we applied this new strategy to a fungus producing FC A (1) (Fig. 1) [1], a diterpene glucoside.
Figure 1. Proposed biosynthetic pathways for FC A, CN A, and BC C.

Both 1 and the structurally-related cotylenin A (2) (Fig. 1) [2], [3] are diterpene glucosides produced by fungi that show phytohormone-like activity [4]. Recently, only 2 was shown to induce the differentiation of human myeloid leukemia cells and trials to apply 2-derivatives to anti-cancer drugs are in progress [5], [6], [7], [8], [9], [10]. However, the 2 producer, which was isolated more than 40 years ago and classified as Cladosporium sp. by classical methods such as morphology [5], had lost its ability to proliferate because of long preservation on a slant. We tried to isolate a producer of 2 or 2-related compounds by mass screening but could isolate only producers of 1 and brassicicene (BC) C (3) (Fig. 1) [11], a compound structurally-related to 1. This resulted in an inability to prepare a large amount of 2 for further clinical application. A study on the relationship between structure and activity was then carried out with 1 and elimination of the hydroxyl group at position 12 was found to be essential for 2-like activity. However, the sole existing synthetic protocol, which is technically applicable to industry, to remove the 12-hydroxyl group is inefficient and involves many reaction steps from FC J (13) (Fig. 1) [12], a minor biosynthetic intermediate slightly accumulated in culture broth of the 1 producer (Fig. 1). We recently developed a more efficient synthetic route from FC H (10) (Fig. 1) [13], which is another intermediate lacking a hydroxyl group at position 12 and accumulated less than 13 in the culture [10].
To estimate whether breeding of a strain that produces 10 as a main product was possible, we started to dissect its biosynthetic machinery. We previously identified a gene encoding PaFS (Orf1) from Phomopsis amygdali, which is a unique chimeric enzyme possessing both a geranylgeranyl diphosphate synthase domain and a diterpene cyclase domain [14]. In addition, three genes, α-ketoglutarate dependent dioxygenase (Orf2), cytochrome P450 (Orf3, accession number; AB68627), and short-chain dehydrogenase/reductase (Orf4) were clustered near the Orf1 gene (Fig. 2A). Orf2 was confirmed to catalyze the 16-oxydation of fusicocca-2,10(14)-diene-8β,16-diol (6) to yield 8β-hydroxyfusicocca-1,10(14)-dien-16-al (7), followed by reduction of the aldehyde to yield fusicocca-1,10(14)-diene-8β,16-diol (8) by Orf4 (Fig. 1) [15]. However, no genes related to 1 biosynthesis were identified in upstream and downstream regions by genome walking (about 50 kbp each). By draft genome sequencing, we then identified a gene (papt) at another locus that encodes a prenyltransferase (Orf11) catalyzing a reverse transfer of dimethylallyl diphosphate to the 6′-hydroxy group of glucose moiety of FC P (12) (Fig. 1) [16], [17]. Since our goal is the identification of other genes involved in 1 biosynthesis, we did not determine the complete genome sequences and were not able to estimate the distance between the two clusters. However, the remaining genes were identified in the flanking region of papt in this study. We performed detailed analysis of the enzymes catalyzing the reactions after the formation of 10. Kinetic parameters of the enzymes suggested that the enzymes could accept only intrinsic substrates and not 10. We therefore identified and disrupted a gene responsible for hydroxylation at the 12-position, which would be a biosynthetic reaction just after 10 formation. The disruptant produced 10 as a main metabolite as expected, which can be easily and efficiently converted into the anti-cancer drug, and its productivity was equivalent to the sum of FC-related compounds produced by the parental strain.
Figure 2. Fusicoccin A biosynthetic gene clusters cloned in our previous (A) and this study (B), respectively.

Results
Identification and Characterization of Enzymes Responsible for Late Biosynthetic Steps of 1
To examine whether the remaining 1 biosynthetic genes were clustered with the papt gene, we searched for flanking contigs by gene walking. Consequently, we identified another gene cluster (21 kbp) containing nine genes; four cytochrome P450s (Orf5; AB686271, Orf7; AB686273, Orf10; AB686276 and Orf13; AB686278), two acetyltransferases (Orf9; AB686275 and Orf12; AB686277), a methyltransferase (Orf8; AB686274), a glycosyltransferase (Orf6; AB686272), and a prenyltransferase (Orf11) (Fig. 2B), suggesting that the 1 biosynthetic genes are scattered at two different loci; one contained four genes and the other nine.
Our goal was the breeding of a mutant accumulating 10. Based on the putative 1 biosynthetic pathway, which was estimated by analyses of intermediate compounds accumulated in the culture broth of the 1 producer [16], a 12-hydroxylation gene-disruptant was expected to accumulate 10 (Fig. 1). However, the 1 biosynthetic machinery might skip this 12-hydroxylation reaction since the “metabolic grid pathway” is well known in the biosynthesis of secondary metabolites, and accumulation of several 12-dehydroxy derivatives of 1 was also probable. Among such compounds, however, no accumulation of 12-dehydroxy FC J and 12-dehydroxy- 3′-O-deacetyl FC A (15) (Fig. 1) was suggested by the following observation: the wild type strain accumulates 15 and 13 in addition to 1 in almost the same amounts in a typical fermentation, suggesting that the catalytic activities of the two enzymes utilizing 15 (3′-O-acetylation) and 13 (19-hydroxylation) as substrates are very low even with the intrinsic substrates and that 10, the intermediate in the early biosynthetic step, would not be accepted as a substrate. Therefore, we investigated the detailed properties of a methylation enzyme and a prenylation enzyme, both of which participate in reactions after the formation of 10. To make a precise interpretation, enzymatic properties of acetylation and glycosylation enzymes were also studied and compared to those of the methylation and prenylation enzymes.
(i) Methylation
The reaction followed by the 12-hydroxylation would be a methylation of the hydroxyl group at position 16 of FC Q (11) (Fig. 1) [16] by Orf8, which has high similarity to many methyltransferases that use S-adenosyl-L-methionine (SAM) as a methyl donor. A recombinant Orf8, which was shown to be 89 kDa by SDS-PAGE (Fig. 3), was incubated with 10 and FC H aglycon (9) (Fig. 1) [13] since we did not have sufficient 11, a plausible intrinsic substrate. Although both compounds were accepted as substrates, the amount of product formed from 9 was trace (Fig. S1A) and the structure of the product from 10 was confirmed to be 16-O-methyl-10 by LC/MS analysis (Fig. S1B). When a mixture of 10 and 3-epi-10 [16] (3.7∶1) were used as substrates, only 10 was converted into methylated product (Fig. S1A), suggesting that the enzyme recognizes a stereochemistry at position 3 of the substrate. Kinetic studies were then performed with 10 as the substrate. The optimum pH and temperature were 9.0 and 35°C, respectively. The Km values were calculated as 17±1.8 µM for 10 and 56±6.0 µM for SAM with a kcat of 0.043±0.0016 S−1. The kcat/Km value was very small compared to other SAM-dependent methyltransferases, suggesting that 11 would be an intrinsic intermediate as previously reported.
Figure 3. SDS-PAGE analysis of the purified enzymes.

(A) molecular mass markers (lane 1) and purified methyltransferase (lane 2). (B) molecular mass markers (lane 1) and purified acetyltransferase (lane 2). (C) molecular mass markers (lane 1) and purified glycosyltransferase (lane 2).
(ii) Prenylation
We previously confirmed that the Orf11 product catalyzed the prenylation of 12 and 10 [17]. We also showed that the kcat/Km value for 12 was 50 times higher than for 10, suggesting that 12 is an intrinsic substrate for 13 formation in vivo.
(iii) Acetylations
Orf12 and Orf9 had significant similarity to a cytosolic acetyltransferase and another type of membrane bound acetyltransferase, respectively. Considering the structure of 1, the hydroxyl groups at the 3′-position and 19-position of dideacetyl-FC A (14) (Fig. 1) [18] should be acetylated by these enzymes. A recombinant ORF12, shown to be 103 kDa by SDS-PAGE (Fig. 3), was incubated with 14 and acetyl-CoA and the reaction product was analyzed by HPLC. A single product was detected and its structure was confirmed to be 3′-O-deacetyl-FC A (15) [19] by LC/MS analysis (Fig. S2). The optimum pH and temperature were 8.5 and 35°C, respectively. The Km values were calculated as 170±4.0 µM for 14 and 63±6.6 µM for acetyl-CoA with a very low kcat value, 3.5×10−3±0.17×10−3 S−1, which would result in significant accumulation of 14 in the culture broth of the 1 producer. We also tried to prepare a recombinant Orf9, a probable membrane bound acetyltransferase, in E. coli, Saccharomyces cerevisiae, Pichia pastoris and Aspergillus oryzae. However, no expression of recombinants was observed. Since the enzyme introducing a hydroxyl group at the 19-position probably could not accept 10 as a substrate as mentioned above and the 19-hydroxylation should precede the acetylation, we paid no more attention to this acetylation reaction from the viewpoint of 10 biosynthesis.
(iv) Glycosylation
Since glycosylation of 9 forms 10, the enzymatic properties of the glycosylation enzyme (Orf6) were also investigated. A recombinant enzyme, shown to be 46 kDa by SDS-PAGE (Fig. 3), was prepared and incubated with 9 in the presence of UDP-glucose as a glucose donor. A specific peak was detected by HPLC analysis and confirmed to be 10 by LC/MS analysis (Fig. S3). The optimum pH and temperature were 5.5 and 35°C, respectively. The Km values were calculated as 44±8.1 µM for 9 and 520±46 µM for UDP-glucose with a kcat of 0.40±0.044 S−1, which is more than ten times higher than those of the methylation (Orf8), prenylation (Orf11), and acetylation (Orf12) enzymes, suggesting that the glycosylation is not a rate-limiting step.
Based on the low catalytic activities of methyltransferase and prenyltransferase calculated with 10 as the substrate and the metabolite profile of the wild strain, we expected that 12-hydroxylation gene-disruptant would accumulate 10 as a main product.
Identification of the Gene Responsible for 12-hydroxylation
Targeted gene disruption is difficult with fungi because a random integration of exogenous DNA into genomic DNA is usually more likely than a homologous recombination. We then examined the substrate specificities of five P450s (Orf3, 5, 7, 10, and 13), one of which should catalyze 12-hydroxylation, by in vitro experiments.
(i) Hydroxylation at positions 8 and 16
P450-2 (Orf5) and P450-1 (Orf3) has significant similarities to BC-Orf1 and BC-Orf7, respectively, which were identified in Alternaria brassicicola, a producer of 3 [20]. We previously confirmed that the former enzyme catalyzed the formation of fusicocca-2,10(14)-diene-8β-ol (5) by hydroxylation of fusicocca-2,10(14)-diene (4), followed by 16-hydroxylation to form fusicocca-2,10(14)-diene-8β,16-diol (6) by the latter enzyme (Fig. 1) [21]. Since 6 would be the same intermediate between 3 and 1 biosynthesis, P450-2 (Orf5) and P450-1 (Orf3) perhaps have the same catalytic activities of those of BC-Orf1 and BC-Orf7. We therefore examined the function of P450-2 (Orf5) by the same method. S. cerevisiae transformants carrying fusicocca-2,10(14)-diene synthase, cytochrome P450 reductase (AB686279), and P450-2 genes were cultivated and their product was analyzed by GC/MS analysis and confirmed to be 5 (Fig. S4), suggesting that P450-2 (Orf5) and P450-1 (Orf3) have 8- and 16-hydroxylation activities.
(ii) Hydroxylation at position 9
We previously confirmed that 6 was converted to 8 by successive reactions by the dioxygenase (Orf2) and the short chain dehydrogenase/reductase (Orf4) [15]. Therefore, the next intermediate is 9, which would be formed by Orf7, Orf10, or Orf13. To narrow this down, we expressed each of the P450 genes in microsomes of S. cerevisiae together with the cytochrome P450 reductase gene and used the isolated microsomes as enzyme sources with 8 as the substrate. We detected a specific product by HPLC analysis only when microsomes expressing P450-3 (Orf7) were used as the catalyst. The compound was identified to be 9 by LC/MS analysis (Fig. S5), suggesting that P450-3 (Orf7) is the 9-hydroxylation enzyme and that P450-4 (Orf10) and P450-5 (Orf13) should be involved in hydroxylation at 12- and 19-positions. We then examined this possibility by the same method, but no products were formed in either case.
Disruption of orf10 and orf13
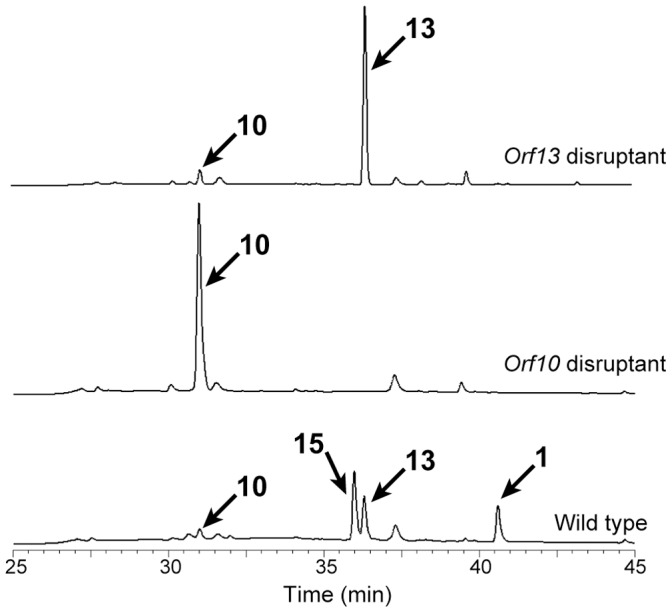
We therefore disrupted the P450-4 (orf10) and P450-5 (orf13) genes by homologous recombination, expecting that one of the disruptants would produce 10. A plasmid, in which orf10 (Fig. 4A) or orf13 (Fig. 5A) was replaced with the hygromycin resistance gene, was constructed and introduced into the 1 producer. Hygromycin-resistant transformants were selected and the homologous recombination was examined by PCR with appropriate primers and genomic DNA of transformants as a template (Fig. 4C and 5C). In both cases, the intended disruptants emerged at a frequency of less than 0.1% of transformants. Next, the product accumulated in the culture broth of the disruptants was examined. The P450-4 (orf10)-disruptant and the P450-5 (orf13)-disruptant produced 10 and 13, respectively, confirmed by LC/MS (Fig. 4B and 5B), as the main product as expected and their productivities were equivalent to the sum of 1-related compounds produced by the parental strain (Fig. 6).
Figure 4. Construction of the orf10 disruptant and LC/MS analyses of the product accumulated in a culture broth of the orf10 disruptants.

(A) A strategy for disruption of the orf10 by a double crossover is schematically shown. Arrows indicate the primers (Table S1) used in PCR analysis, which correspond to the upstream and downstream of the orf10. (B) Disruption was confirmed by agarose gel electrophoresis of the PCR-amplified fragment. (C) The product accumulated in a culture broth of the orf10 disruptant (peak A) was confirmed to be 10 by LC/MS analysis.
Figure 5. Construction of the orf13 disruptant and LC/MS analyses of the product accumulated in a culture broth of the orf13 disruptants.

(A) A strategy for disruption of the orf13 by a double crossover is schematically shown. Arrows indicate the primers (Table S1) used in PCR analysis, which correspond to the upstream and downstream of the orf13. (B) Disruption was confirmed by agarose gel electrophoresis of the PCR-amplified fragment. (C) The product accumulated in a culture broth of the orf10 disruptant (peak B) was confirmed to be 13 by LC/MS analysis.
Figure 6. HPLC traces of the broths of the orf13 disruptant (upper), the orf10 disruptant (middle), and the parental strain (lower).
Discussion
To date, production of many useful compounds by microorganisms has been achieved. High-titer production of amino acids, nucleic acids, and antibiotics, producers of which were mostly bred by classical mutagenesis and mass screenings, are representative. Recently, synthetic biology and metabolic engineering has enabled successful production of specific, valuable compounds, including those of eukaryotic origin, with Escherichia coli and yeast as hosts [22], [23], although expression optimization for each of the genes/enzymes introduced into the hosts is indispensable and greater titer is essential for industrial application. Besides these processes, in this study, we demonstrated an additional process: production of an intermediate compound suitable for semi-synthesis by a mutant constructed by disruption of a specific gene by homologous recombination. It is worth mentioning that the disruptants produced the intermediates as sole products and that their productivities were the identical to the end product produced by the parental strain (0.6 g/L broth). From the viewpoint of industrial application, 10 accumulated in the culture broth of the P450-4 (orf10)-disruptant easily crystallized after filtration and successive ethyl acetate extraction, which enabled its direct use for semi-synthesis at low cost.
There are few reports on the productivities of intermediate compounds produced by engineered strains bred by classical mutagenesis and/or metabolic engineering. Moreover, to the best of our knowledge, there are no reports on detailed enzymatic properties of a series of biosynthetic enzymes responsible for a secondary metabolite. In this study, we demonstrated the strict substrate specificities of 1 biosynthetic enzymes. This would be the reason why the disruptant accumulated the specific intermediate as the sole product without utilizing the “metabolic grid pathway”, which is sometimes observed in the biosynthesis of secondary metabolites. In gibberellin biosynthesis, high accumulation of ent-kaurene, ent-kaurenoic acid, and gibberellin 14 by Fusarium fujikuroi mutants, in which the P450-4, P450-1, and P450-2 genes responsible for gibberellin biosynthesis were disrupted by homologous recombination, were also reported [24]. Therefore, fungi would generally have the potential to accumulate an intermediate compound, at least for isoprenoid secondary metabolites.
We have been studying biosynthetic machinery of actinobacteria such as Streptomyces strains [25], [26], [27], [28], counterparts of natural product producers, and have tried to breed isoprenoid producers that accumulate intermediate compounds by the same methods employed in this study. In these cases, however, the engineered strains usually produced very small amounts of or no metabolites [29]. We do not know why engineered fungi and actinobacteria show these different phenotypes, but they might be caused by differences in transcription machinery between prokaryotes and eukaryotes. In prokaryotes, including actinobacteria, several genes are usually transcribed polycistronically; thus, disruption of a gene might affect transcription/translation of the other genes in an operon even though the disruption is designed to be an in-frame deletion. In contrast, all genes in fungi are transcribed monocistronically, and thereby unaffected by expression of neighboring genes. If these are the reasons, the strategy employed in this study should be generally applicable to all types of compounds produced by fungi.
Materials and Methods
General
Sequence analysis of PCR fragments was performed by the dideoxy chain termination method with an automatic DNA sequencer (Li-Cor, model 4000L). Cell disruption was performed with an Ultrasonic Disruptor (TOMY, UD-200). Analysis of the samples during protein purification was performed using SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and the proteins were visualized by Coomassie brilliant blue staining. Protein concentration was determined by the Bradford method with bovine serum albumin as a standard.
Strain and Plasmids
P. amygdali Niigata-2 was used for production of 1 and preparation of genomic DNA and cDNA. Draft genome sequences were previously determined [17] and used to search for 1 biosynthetic genes. Total RNA of the strain was isolated using the TRIzol® reagent (Invitrogen, USA) according to the manufacturer’s protocol. The fragments containing 5′- or 3′-termini of cDNA were obtained using the SMART™ RACE cDNA Amplification Kit (Clontech, USA) and GeneRacer™ Kit (Invitrogen).
Cloning and in vitro Assay of Methyltransferase (Orf8)
After determination of the coding region by the RACE method, the full length of cDNA of orf8 was amplified with P. amygdali 5′-Ready cDNA as the template and a primer set listed in Table S1 by PCR. The EcoRI-SalI-digested fragment was inserted into the same site of pMAL-p2X (N-terminal MBP-fused, New England Biolabs). Expression and purification conditions for the recombinant enzyme were essentially the same as those described in the manufacturer’s protocols. After a purity of the recombinant enzyme was checked by SDS-PAGE, the enzyme was used for in vitro assay.
The assay mixture for methyltransferase contained, in a final volume of 100 µL, 0.5 mM of 9, or 0.5 mM 10, 5 mM of S-adenosyl-L-methionine (SAM), 50 mM Tris–HCl (pH 7.5), and a suitable amount of methyltransferase. The reaction mixture was incubated at 30°C for 2 h and the products were analyzed by HPLC. Analytical conditions were as follows: Merck Mightisil RP-18GP Aqua column (250 mm × 4.6 mm) (Kanto Chemicals, Japan); mobile phase of acetonitrile in water (0 to 20 min, 30% AcCN; 20 to 30 min, 30 to 40%; 30 to 50 min, 40 to 70%; 50 to 60 min, 70 to 100%); flow rate, 1.0 mL/min; detection, 205 nm. The steady-state kinetic parameters were determined by fitting to the Michaelis-Menten equation. An assay for determination of the kinetic parameters of 10 contained, in a final volume of 100 µL, 50 mM HEPES (pH 7.5), 5 mM SAM, 0.145 mM methyltransferase, and 7.5 µM to 0.25 mM 10. The mixture was incubated at 30°C for 15 min. When the concentration of 10 was fixed at 0.5 mM, the concentration of SAM was varied from 10 µM to 2.5 mM. Triplicate sets of enzyme assays were performed at each substrate concentration, and the Hanes-Woolf plot was used for estimation of kinetic constants.
Cloning and in vitro Assay of Acetyltransferase-2 (Orf12)
After detemination of the coding region by the RACE method, the full length of cDNA for orf8 was amplified with P. amygdali 5′-Ready cDNA as the template and a primer set listed in Table S1 by PCR. The KpnI-PstI-digested fragment was inserted into the same site of pCold TF vector (TaKaRa, Japan) to make pCold-AT-2. Expression and purification conditions for the recombinant enzyme were essentially the same as those described in the manufacturer’s protocols. After a purity of the recombinant enzyme was checked by SDS-PAGE, the enzyme was used for in vitro assay.
The assay mixture for acetyltransferase-2 contained, in a final volume of 100 µL, 50 µM 14, 1 mM acetyl-coenzyme A, 137 mM NaCl, 2.7 mM KCl, 12.4 mM phosphate buffer (pH 7.4), and a suitable amount of purified enzyme. The mixture was incubated at 30°C for overnight and the reaction product was analyzed by HPLC. Analytical conditions were same as those of methyltransferase.
The steady-state kinetic parameters were determined by fitting to the Michaelis-Menten equation. An assay for determination of the kinetic parameters of 14 contained, in a final volume of 100 µL, 50 mM Tris–HCl (pH 8.5), 5.06 mM acetyl-coenzyme A, 7.68 µM acetyltransferase, and 50.9 µM to 1.02 mM 14. The mixture was incubated at 35°C for 30 min. When the concentration of 14 was fixed at 203 µM, the concentration of acetyl-coenzyme A was varied from 23.1 µM to 1.16 mM. Triplicate sets of enzyme assays were performed at each substrate concentration, and the Hanes-Woolf plot was used for estimation of kinetic constants.
Cloning and in vitro Assay of Glycosyltransferase (Orf6)
After determination of the coding region by the RACE method, the full length of cDNA for orf6 was amplified with P. amygdali 5′-Ready cDNA as the template and a primer set listed in Table S1 by PCR. The NdeI-BamHI-digested fragment was inserted into the same site of pET15b (Merck, Germany) to make pET15-GLY. The E. coli cells having pET15-GLY was grown in LB medium supplemented 100 µg/ml ampicillin. The culture was grown at 37°C until OD600 reached 0.5. After addition of 0.5 mM of isopropyl β-D-thiogalactopyranoside, the cultivation was continued for additional 18 hours at 25°C. His-tagged enzyme was purified by using the manufacturer’s protocol. After a purity of the recombinant enzyme was checked by SDS-PAGE, the enzyme was used for in vitro assay.
The assay mixture for glycosyltransferase contained, in a final volume of 100 µL, 0.5 mM of 9, 2.5 mM of UDP-glucose, 50 mM Tris–HCl (pH 7.5), and a suitable amount of glycosyltransferase. The mixture was incubated at 30°C for 2 h and the products were analyzed by HPLC. Analytical conditions were same as those of methyltransferase. The steady-state kinetic parameters were determined by fitting to the Michaelis-Menten equation. An assay for determination of the kinetic parameters of 9 contained, in a final volume of 100 µL, 50 mM MES (pH 6.0), 5 mM UDP-glucose, 0.58 mM glycosyltransferase, and 5 µM to 100 µM 9. The mixture was incubated at 30°C for 15 min. When the concentration of 9 was fixed at 0.1 mM, the concentration of UDP-glucose was varied from 25 µM to 2.5 mM. Triplicate sets of enzyme assays were performed at each substrate concentration, and the Hanes-Woolf plot was used for estimation of kinetic constants.
Cloning and Characterization of P450-2 (Orf5)
We examined the function of cytochrome P450-2 (orf5) by a heterologous expression in yeast, the detailed procedure of which was described previously [21]. The coding region of the P450-2 was determined using the RACE (Rapid Amplification of cDNA End) method. Then, approximately 1.6 kb of full length DNA fragment was amplified with P. amygdali 5′-Ready cDNA as the template and a primer set listed in Table S1. The BamHI-XhoI-digested fragment was inserted into pESC-URA to yield pESC-URA-P450-2. For functional analyses of cytochrome P450 enzymes, a cytochrome P450 reductase is essential. We identified a cytochrome P450 reductase gene in draft genome database of the FC producer. The 2.1 kb of full length DNA fragment was amplified with P. amygdali 5′-Ready cDNA as the template and a primer set listed in Table S1. The EcoRI-BglII-digested fragment was inserted into the same sites of the pESC-URA-P450-2. The constructed plasimd was introduced into S. cerevisiae YPH500 (his−, leu−, trp−, ura−, ade− and lys−) together with pESC-TRP-orf8-ADH, which was previously constructed and carried an fusicocca-2,10(14)-diene synthase gene [21]. After cultivation, pentane extract of the culture broth was analyzed by GC/MS, which was conducted with QP2010 apparatus (Shimazu), using a DB-1MS capillary column (0.32 mm × 30 m, 0.25 µm film thickness; J&W Scientific). Each sample was injected into the column at 100°C in a split less mode. After a three-min isothermal hold at 100°C, the column temperature was increased to 250°C at a ratio of 16°C/min, followed by a 3-min isothermal hold at 250°C. The flow rate of the helium carrier was 1 mL/min.
Cloning and in vitro Assay of P450-3 (Orf7)
After determination of the coding region by the RACE method, the full length of cDNA for orf7 was amplified with P. amygdali 5′-Ready cDNA as the template and a primer set listed in Table S1 by PCR. The 1.5 kb PCR product was cloned into pGEM-T Easy vector (Promega Corporation, USA) to yield pGEM450-3. The SpeI-EcoRI-digested fragment from pGEM450-3 was inserted into the same site of pESC-HIS to construct pESC-P450-3. The constructed pESC-P450-3 was introduced into S. cerevisiae YPH500 (his−, leu−, trp−, ura−, ade− and lys−). After cultivation of the transformants in the presence of galactose, microsomal fractions were prepared and incubated with fusicocca-1,10(14)-diene-8β,16-diol. After 48 h incubation at 30°C, the products were analyzed by HPLC. Analytical conditions of HPLC were previously reported [1].
Transformation of P. amygdali
Protoplast preparation: P. amygdali Niigata-2 was cultivated by the same method as described previously [15]. Mycelia from 50 ml culture were collected by centrifugation (5,000 × g, 10 min), washed with 0.8 M NaCl twice and then incubated in 10 ml of the same medium with yatalase (5 mg/ml, Takara Bio) and lysing enzyme (10 mg/ml, Novozyme) at 30°C for 2 h. Protoplast formation was monitored microscopically. Protoplasts were recovered by filtration with Miracloth (Calbiochem), centrifuged at 1,800 × g and 4°C for 10 min, and washed twice with ice-cold 0.8 M NaCl. They were gently resuspended in 300 µl of STC buffer (1.2 M of Sorbitol, 10 mM of Tris-HCl (pH 7.5), and 10 mM of CaCl2) and immediately used for transformation. Transformation was done with 10 µg of plasmid DNA in 10 µl of 10 mM of TE buffer to which a 100-µl suspension of 108 protoplasts was added. After the mixture was left for 10 min on ice, 1 ml of 60% (vol/vol) polyethylene glycol 4000 in a buffer containing Tris-HCl (10 mM, pH 7.5) and CaCl2 (10 mM) was added and mixed gently by pipetting. The mixture was appropriately diluted with STC buffer and 100-µl samples were plated on YPSA plate containing 0.1% yeast extract, 0.1% bacto tryptone, 27.7% sucrose, 0.005% hygromycin, and 2% agar. These plates were incubated at 30°C for 20 h and then overlaid with 5 ml of nutrient soft agar (0.8%).
Gene Disruption
The orf10 and the orf13 genes were disrupted by a double-crossover event. To construct the orf10 and orf13 gene disruption plasmids, two 3-kb and 2-kb DNA fragments, carrying upstream and downstream regions of the target genes, respectively, were amplified with the primer set listed in Table S1 by PCR. After sequence confirmation, these fragments were inserted into appropriate restriction sites of pGEM-T Easy in the same direction as in the genomic region. A 3.0-kb DNA fragment containing a Hygromycin resistance gene controlled by a TrpC promoter and terminator was amplified by PCR with pSH75 [30] as the template and the primer set listed in Table S1. After sequence confirmation, this 3.0-kb fragment was then inserted between the above-mentioned two fragments. The constructed plasmids were used to transform the 1 producer (Fig. 4A and 5A). Gene disruption of the transformant was confirmed by PCR analysis (Fig. 4C and 5C) and the PCR product was confirmed by DNA sequencing.
Structural Analysis of the Reaction Product
The reaction products formed by in vitro assays were subjected to LC-MS analysis. The analytical conditions were as follows: Develosil RPAQUEOUS-AR-5 column (150×2.0 mm); column temperature, 30°C; detection, 250 nm and positive mode; mobile phase, 0.1% formic acid:acetonitrile = 10:90 at 0 min, and a linear gradient to 50:50 for an additional 30 min; flow rate, 0.3 ml/min.
Supporting Information
(A) HPLC analyses of the product formed by in vitro methyltransferase assay using FC H aglycon (9) (upper), FC H (10) (middle), and a mixture of 10 and 3-epi-10 (lower) as substrates. (B) The reaction products formed from 9 (peak A) and 10 (peak B) were confirmed to be 16-O-methyl-9 and 16-O-methyl-10 by LC/MS analysis, respectively.
(DOC)
HPLC (A) and LC/MS (B) analyses of the product formed by in vitro acetyltransferase assay. The reaction product formed from dideacetyl-FC A (14) was confirmed to be 15 by LC/MS analysis.
(DOC)
HPLC (A) and LC/MS (B) analyses of the product formed by in vitro glycosyltransferase assay. The reaction product formed from FC H aglycon (9) was confirmed to be 10 by LC/MS analysis.
(DOC)
GC-MS analyses of pentane extract of the broth of S. cerevisiae transformant carrying fusicocca-2,10(14)-diene synthase gene, cytochrome P450 reductase gene, and P450-2 gene. Chromatograms were recorded in the TIC mode (A and B). (A) Authentic fusicocca-2,10(14)-diene-8β-ol (5). (B) Extract of the transformant. (C) Mass spectrum of authentic 5. (D) Mass spectrum of the transformant.
(DOC)
HPLC (A) and LC/MS (B) analyses of the product formed by in vitro P450-3 assay. The reaction product formed from fusicocca-1,10(14)-diene-8β,16-diol (8) was confirmed to be 9 by LC/MS analysis.
(DOC)
Primers used for PCR.
(DOC)
Funding Statement
This work was supported in part by the Program for the Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation, Japan, a Grant-in-Aid for Scientific Research on Innovative Areas, the Global COE Program (Project no. B01: Catalysis as the Basis for Innovation in Materials Science), MEXT, Japan, the Naito Foundation, and the Foundation NAGASE Science Technology Development. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ballio A, Brufani M, Casinovi CG, Cerrini S, Fedeli W, et al. (1968) The structure of fusicoccin A. Experientia. 24: 631–635. [DOI] [PubMed] [Google Scholar]
- 2. Sassa T (1971) Cotylenins, leaf growth substances produced by a fungus. I. Isolation and characterization of cotylenins A and B. Agric Biol Chem 35: 1415–1418. [Google Scholar]
- 3. Sassa T, Ooi T, Nukina M, Kato N (1998) Structural Confirmation of Cotylenin A, a Novel Fusicoccane-diterpene Glycoside with Potent Plant Growth-regulating Activity from Cladosporium Fungus sp. 501–7W. Biosci Biotechnol Biochem 62: 1815–1818. [DOI] [PubMed] [Google Scholar]
- 4. Marre E (1979) Fusicoccin: A Tool in Plant Physiology. Ann Rev Plant Physiol 30: 273–288. [Google Scholar]
- 5. Asahi K, Honma Y, Hazeki K, Sassa T, Kubohara Y, et al. (1997) Cotylenin A, a plant-growth regulator, induces the differentiation in murine and human myeloid leukemia cells. Biochem Biophys Res Commun 238: 758–763. [DOI] [PubMed] [Google Scholar]
- 6. Honma Y (2002) Cotylenin A–a plant growth regulator as a differentiation-inducing agent against myeloid leukemia. Leuk Lymphoma 43: 1169–1178. [DOI] [PubMed] [Google Scholar]
- 7. Honma Y, Ishii Y, Yamamoto-Yamaguchi Y, Sassa T, Asahi K (2003) Cotylenin A, a differentiation-inducing agent, and IFN-alpha cooperatively induce apoptosis and have an antitumor effect on human non-small cell lung carcinoma cells in nude mice. Cancer Res 63: 3659–3666. [PubMed] [Google Scholar]
- 8. Honma Y, Kasukabe T, Yamori T, Kato N, Sassa T (2005) Antitumor effect of cotylenin A plus interferon-alpha: possible therapeutic agents against ovary carcinoma. Gynecol Oncol 99: 680–688. [DOI] [PubMed] [Google Scholar]
- 9. Matsunawa M, Ishii Y, Kasukabe T, Tomoyasu S, Ota H, et al. (2006) Cotylenin A-induced differentiation is independent of the transforming growth factor-beta signaling system in human myeloid leukemia HL-60 cells. Leuk Lymphoma 47: 733–740. [DOI] [PubMed] [Google Scholar]
- 10. Kawakami K, Hattori M, Inoue T, Maruyama Y, Ohkanda J, et al. (2012) A novel fusicoccin derivative preferentially targets hypoxic tumor cells and inhibits tumor growth in xenografts. Anticancer Agents Med Chem in press. (PMID:22263802).. [DOI] [PubMed] [Google Scholar]
- 11. Mackinnon SL, Keifer P, Ayer WA (1999) Components from the phytotoxic extract of Alternaria brassicicola, a black spot pathogen of canola. Phytochemistry 51: 215–221. [Google Scholar]
- 12. Barrow KD, Barton DHR, Chain EB, Bageenda-Kasujja D, Mellows G (1975) Fusicoccin. Part IV. The structure of fusicoccin J. J Chem Soc Perkin Trans 1 877–883. [Google Scholar]
- 13. Barrow KD, Barton DHR, Chain EB, Ohnsorge UF, Sharma RP (1973) Fusicoccin. Part III. The structure of fusicoccin H. J Chem Soc Perkin Trans 1 1590–1599. [PubMed] [Google Scholar]
- 14. Toyomasu T, Tsukahara M, Kaneko A, Niida R, Mitsuhashi W, et al. (2007) Fusicoccins are biosynthesized by an unusual chimera diterpene synthase in fungi. Proc Natl Acad Sci U S A 104: 3084–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ono Y, Minami A, Noike M, Higuchi Y, Toyomasu T, et al. (2011) Dioxygenases, key enzymes to determine the aglycon structures of fusicoccin and brassicicene, diterpene compounds produced by fungi. J Am Chem Soc 133: 2548–2555. [DOI] [PubMed] [Google Scholar]
- 16. Sassa T, Tajima N, Sato M, Takahashi A, Kato N (2002) Fusicoccins P and Q, and 3-epifusicoccins H and Q, new polar fusicoccins from isolate Niigata 2-A of a peach Fusicoccum canker fungus. Biosci Biotechnol Biochem 66: 2356–2361. [DOI] [PubMed] [Google Scholar]
- 17. Noike M, Liu C, Ono Y, Hamano Y, Toyomasu T, et al. (2012) An enzyme catalyzing O-prenylation of the glucose moiety of fusicoccin A, a diterpene glucoside produced by the fungus Phomopsis amygdali . Chembiochem 13: 66–573. [DOI] [PubMed] [Google Scholar]
- 18. Barrow KD, Barton DHR, Chain E, Conlay C, Smale TC, et al. (1971) Fusicoccin. Part I. The nature of the substituent groups. J Chem Soc C 1259–1264. [Google Scholar]
- 19. Tajima N, Kume H, Kanematsu S, Kato N, Sassa T (2001) Chemical identification of fusicoccins from a Japanese isolate Niigata 2 of peach Fusicoccum canker fungus (Phomopsis amygdali) and production of 3'-deacetylfusicoccin A by the fungus. J Pestcide Sci 27: 64–67. [Google Scholar]
- 20. Minami A, Tajima N, Higuchi Y, Toyomasu T, Sassa T, et al. (2009) Identification and functional analysis of brassicicene C biosynthetic gene cluster in Alternaria brassicicola . Bioorg Med Chem Lett 19: 870–874. [DOI] [PubMed] [Google Scholar]
- 21. Hashimoto M, Higuchi Y, Takahashi S, Osada H, Sakaki T, et al. (2009) Functional analyses of cytochrome P450 genes responsible for the early steps of brassicicene C biosynthesis. Bioorg Med Chem Lett 19: 5640–5643. [DOI] [PubMed] [Google Scholar]
- 22. Tyo KE, Alper HS, Stephanopoulos GN (2007) Expanding the metabolic engineering toolbox: more options to engineer cells. Trends Biotechnol 25: 132–137. [DOI] [PubMed] [Google Scholar]
- 23. Ajikumar PK, Xiao WH, Tyo KE, Wang Y, Simeon F, et al. (2010) Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli . Science 330: 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tudzynski B (2005) Gibberellin biosynthesis in fungi: genes, enzymes, evolution, and impact on biotechnology. Appl Microbiol Biotechnol 66: 597–611. [DOI] [PubMed] [Google Scholar]
- 25. Hamano Y, Kuzuyama T, Itoh N, Furihata K, Seto H, et al. (2002) Functional analysis of eubacterial diterpene cyclases responsible for biosynthesis of a diterpene antibiotic, terpentecin. J Biol Chem 277: 37098–37104. [DOI] [PubMed] [Google Scholar]
- 26. Kawasaki T, Hayashi Y, Kuzuyama T, Furihata K, Itoh N, et al. (2006) Biosynthesis of a natural polyketide-isoprenoid hybrid compound, furaquinocin A: identification and heterologous expression of the gene cluster. J Bacteriol 188: 1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hayashi Y, Onaka H, Itoh N, Seto H, Dairi T (2007) Cloning of the gene cluster responsible for biosynthesis of KS-505a (longestin), a unique tetraterpenoid. Biosci Biotechnol Biochem 71: 3072–3081. [DOI] [PubMed] [Google Scholar]
- 28. Hayashi Y, Matsuura N, Toshima H, Itoh N, Ishikawa J, et al. (2008) Cloning of the gene cluster responsible for the biosynthesis of brasilicardin A, a unique diterpenoid. J Antibiot (Tokyo) 61: 164–174. [DOI] [PubMed] [Google Scholar]
- 29. Dairi T (2005) Studies on biosynthetic genes and enzymes of isoprenoids produced by actinomycetes. J Antibiot (Tokyo) 58: 227–243. [DOI] [PubMed] [Google Scholar]
- 30. Kimura N, Tsuge T (1993) Gene cluster involved in melanin biosynthesis of the filamentous fungus Alternaria alternata . J Bacteriol 175: 4427–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) HPLC analyses of the product formed by in vitro methyltransferase assay using FC H aglycon (9) (upper), FC H (10) (middle), and a mixture of 10 and 3-epi-10 (lower) as substrates. (B) The reaction products formed from 9 (peak A) and 10 (peak B) were confirmed to be 16-O-methyl-9 and 16-O-methyl-10 by LC/MS analysis, respectively.
(DOC)
HPLC (A) and LC/MS (B) analyses of the product formed by in vitro acetyltransferase assay. The reaction product formed from dideacetyl-FC A (14) was confirmed to be 15 by LC/MS analysis.
(DOC)
HPLC (A) and LC/MS (B) analyses of the product formed by in vitro glycosyltransferase assay. The reaction product formed from FC H aglycon (9) was confirmed to be 10 by LC/MS analysis.
(DOC)
GC-MS analyses of pentane extract of the broth of S. cerevisiae transformant carrying fusicocca-2,10(14)-diene synthase gene, cytochrome P450 reductase gene, and P450-2 gene. Chromatograms were recorded in the TIC mode (A and B). (A) Authentic fusicocca-2,10(14)-diene-8β-ol (5). (B) Extract of the transformant. (C) Mass spectrum of authentic 5. (D) Mass spectrum of the transformant.
(DOC)
HPLC (A) and LC/MS (B) analyses of the product formed by in vitro P450-3 assay. The reaction product formed from fusicocca-1,10(14)-diene-8β,16-diol (8) was confirmed to be 9 by LC/MS analysis.
(DOC)
Primers used for PCR.
(DOC)