

Abstract
The B lymphotrophic γ-herpesvirus EBV is associated with a variety of lymphoid- and epithelial-derived malignancies, including B cell lymphomas in immunocompromised and immunosuppressed individuals. The primary oncogene of EBV, latent membrane protein 1 (LMP1), activates the PI3K/Akt pathway to induce the autocrine growth factor, IL-10, in EBV-infected B cells, but the mechanisms underlying PI3K activation remain incompletely understood. Using small molecule inhibition and siRNA strategies in human B cell lines expressing a chimeric, signaling-inducible LMP1 protein, nerve growth factor receptor (NGFR)-LMP1, we show that NGFR-LMP1 utilizes Syk to activate PI3K/Akt signaling and induce IL-10 production. NGFR-LMP1 signaling induces phosphorylation of BLNK, a marker of Syk activation. Whereas Src kinases are often required for Syk activation, we show here that PI3K/Akt activation and autocrine IL-10 production by NGFR-LMP1 involves the Src family kinase Fyn. Finally, we demonstrate that NGFR-LMP1 induces phosphorylation of c-Cbl in a Syk- and Fyn-dependent fashion. Our results indicate that the EBV protein LMP1, which lacks the canonical ITAM required for Syk activation, can nevertheless activate Syk, and the Src kinase Fyn, resulting in downstream c-Cbl and PI3K/Akt activation. Fyn, Syk, and PI3K/Akt antagonists thus may present potential new therapeutic strategies that target the oncogene LMP1 for treatment of EBV+ B cell lymphomas.
Introduction
EBV is a γ-herpesvirus that has infected >90% of the world's population. In immunocompetent hosts EBV infection is generally asymptomatic, although some adolescents develop self-limiting infectious mononucleosis [1]. Nevertheless, EBV is also linked to a variety of malignancies including Hodgkin's disease, Burkitt's lymphoma, B cell and NK cell lymphomas, and nasopharyngeal carcinoma. EBV has potent transforming ability, as infection of B lymphocytes results in the generation of immortalized lymphoblastoid cell lines (LCL) in vitro and autonomously proliferating lymphoblasts in vivo. Immunocompetent hosts readily control the expansion of EBV+ lymphoblasts through vigorous anti-viral T cell immunity. However, in immunocompromised individuals, like those infected with HIV or immunosuppressed transplant patients, EBV-infected lymphoblasts can give rise to AIDS-related lymphomas or post-transplant lymphoproliferative disorder (PTLD), respectively [2].
Latent Membrane Proteins 1 and 2a (LMP1 and LMP2a) are the two major signaling proteins of EBV, as they act as constitutively active mimics of B cell signaling molecules. LMP2a is a functional B cell receptor (BCR) mimic that propagates signals through spleen tyrosine kinase (Syk). LMP2a-derived signals function to sustain survival [3], [4] and maintain viral latency, in part by blocking BCR triggering [5], [6]. The major oncogene of EBV, LMP1, is a functional CD40 mimic. Interactions of cellular tumor necrosis factor (TNF) receptor-associated factor (TRAF) adaptor proteins, including TRAF1 [7], [8] and TRAF3 [7], [9], with the LMP1 C-terminal tail signaling domains, carboxy-terminal activating region 1 and 2 (CTAR1 or CTAR2), initiate signal transduction through a variety of pathways including the p38 [10], [11], Erk [12], [13], and JNK [14], [15] MAPK and NF-κB [16], [17] pathways.
Recent studies have also implicated LMP1 in the activation of the PI3K/Akt pathway. Given its ability to regulate cell survival and growth [18], it is unsurprising that deregulated PI3K activation is associated with a number of malignancies [19]. Activation of PI3K/Akt signaling by LMP1 results in secretion of the growth factor IL-10 in B cells [20] and is essential for the survival of LMP1-transformed Rat-1 cells [21]. Finally, inhibition of PI3K/Akt with the small molecule inhibitor LY294002 reduces proliferation and induces apoptosis of EBV+ PTLD derived B cell lines [22]. PI3K/Akt activation by LMP1 requires the CTAR1 domain [23], but the specific mechanisms by which LMP1 activates PI3K/Akt signaling remain to be determined. In the case of membrane-based receptors, Src family kinases and Syk are involved in the initial recruitment of the p85α subunit of PI3K [24]. Whether LMP1 signaling through PI3K/Akt involves the recruitment of similar helper proteins is unknown.
Syk signaling was initially believed to be restricted to classical immunoreceptors of the adaptive immune response, such as the BCR and Fc receptors (FcR) [25]. These receptors associate with signaling adaptors containing a short, tyrosine-containing peptide sequence known as the immunoreceptor tyrosine-based activation motif (ITAM) [26]. Receptor ligation results in phosphorylation of the two ITAM tyrosines by the Src tyrosine kinases. Syk is recruited to, and activated by, phosphorylated ITAMs. Recent studies revealed a requirement for Syk in several innate immune functions, such as detection of fungi and tissue damage [27], and certain non-immune functions, including vascular development [28] and bone metabolism [29]. In many of these cases, classes of receptors that do not contain conventional ITAMs, including C-type lectins [30] and integrins [31], activate Syk. Some C-type lectins, like the fungal recognition receptor Dectin-1/CLEC7A [32], contain a singular YXXL motif (a ‘hemITAM’). Integrins, in contrast, do not contain either an ITAM or hemITAM, yet Syk-deficient monocytes [33], neutrophils, macrophages [34], osteoclasts [35], and platelets [36] display defective integrin signaling. These observed non-canonical Syk activation systems raise the possibility that other non-ITAM containing receptors may utilize Syk to coordinate downstream signaling pathway activation.
In the present study, we aimed to investigate how LMP1 activates PI3K. Given the recent finding that Syk can be activated in non-canonical fashion and evidence of Syk participation in PI3K activation, we asked if Syk and Src family kinases are activated in EBV+ B cell lymphomas and are required for activation of PI3K by LMP1. Using small molecule inhibition and siRNA strategies in human B cell lines expressing a signaling-inducible LMP1 protein (NGFR-LMP1), our results support the requirement for Syk and the Src family kinase Fyn in PI3K activation by LMP1.
Methods
Reagents
Streptavidin and antibodies to β-actin and NGFR (clone 20.4) were from Sigma (St. Louis, MO, USA). Biotinylated anti-NGFR was from Chromaprobe (Maryland Heights, MO, USA). Secondary antibodies, including PE-conjugated goat anti-mouse IgG, unconjugated goat-anti-mouse IgG, HRP-conjugated polyclonal goat anti-rabbit, HRP-conjugated polyclonal donkey anti-mouse, and Cy3-conjugated donkey anti-mouse antibodies were from Jackson Immunoresearch Labs, Inc (West Grove, PA, USA). Anti-LMP1 antibodies (clone CS.1-4) and isotype control antibodies were obtained from Dako (Carpinteria, CA, USA). Anti-LMP2a (clone 14B7) and rabbit anti-rat secondary antibodies were from Abcam (Cambridge, MA, USA). Anti-TRAF3 (H-20), anti-phospho-Erk (Tyr204), anti-Erk, and anti-P38 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies to Akt, phospho-Akt (Ser473), c-Cbl, phospho-c-Cbl (Tyr700), JNK, phospho-JNK (Thr183/Tyr185), BLNK, phospho-BLNK (Tyr96), Syk, and phospho-Syk (Tyr525/526) were from Cell Signaling Technology (Danvers, MA, USA), as was anti-phospho-Src family (Tyr416), a rabbit polyclonal antibody that recognizes phosphorylated Src, Lyn, Fyn, Lck, Yes, and Hck. Anti-phospho-P38 (Thr180/Tyr182), anti-Fyn (mouse mAb 25/Fyn) and anti-Lyn (mouse mAb 42/Lyn), PE-conjugated ICAM-1 (CD54), and streptavidin-PE were from BD Pharmingen (San Diego, CA, USA). F(ab)2 fragments of anti-human IgM were from Invitrogen (Carlsbad, CA, USA).
Cell Lines
The Burkitt's lymphoma line BL41 and its EBV-infected counterpart BL41.B95 were kindly provided by Dr. Elliot Kieff (Harvard Medical School) [37], [38]. BL41.NGFR-LMP1 (clone 1) expresses the chimeric NGFR-LMP1 molecule in the parental BL41 line; the generation of this line by our lab was described previously [20]. Cell lines AB5, JB7, and JC62 were generated in our lab as previously described and are spontaneously derived LCL grown from peripheral blood or lymph nodes of patients diagnosed with EBV+ PTLD [39]. All cell lines were maintained in a 5% CO2 humidified 37°C incubator in RMP1 1640 supplemented with 10% heat-inactivated FCS (Serum Source International, Charlotte, NC, USA), and 50 units/mL penicillin-streptomycin (Invitrogen). Growth media for BL41.NGFR-LMP1 was additionally supplemented with geneticin (Sigma). Prior to signaling studies, cells were grown overnight with reduced (4%) FCS.
Flow Cytometry
For cell surface staining, 1×106 cells were washed with cold FACS buffer (PBS, 1% BSA, 0.02% sodium azide) and incubated on ice for 30 minutes with 1 µg of primary antibody, washed, then incubated 30 minutes on ice with 0.4 µg of the fluorophore-conjugated secondary antibody. Stained cells were then washed and analyzed on a Becton Dickinson FACScan using Cellquest software.
NGFR-LMP1 and BCR Crosslinking
NGFR-LMP1 expressing cells were resuspended at 5–10×106 cells/mL in RPMI with 4% FCS. Cells were then stimulated as previously described [20] through the addition of unconjugated mouse anti-NGFR followed goat anti-mouse F(ab)2 for the indicated amount of time at 37°C. To initiate BCR signaling, cells were resuspended at 1×106 cells/mL in RPMI with 4% FCS, and triggered to signal with 12 µg F(ab)2 fragments of anti-IgM per 106 cells for the indicated amounts of time.
Immunofluorescent Staining
Cells of interest were plated on 8-well LabTek II chamber slides (Nalge Nunc, Rochester, NY, USA) coated with poly-d-lysine (Sigma) at a concentration of 100,000–150,000 cells per well in growth medium for a minimum of 30 minutes in a 5% CO2 humidified 37°C incubator. Immunofluorescent staining was performed as previously described [40], [41] with the following antibody conditions: anti-LMP1 (Dako, 1∶250), donkey anti-mouse Cy3 (Jackson Immunoresearch, 1∶500). Slides were examined using a Zeiss 510 Meta microscope.
Inhibition of Signal Transduction Pathways
The following inhibitors were used to analyze the effect of cellular signaling pathways on phosphorylation events and IL-10 production: PP2 (Src family kinase inhibitor, 5 µM) or R406 (Syk inhibitor, 0.5 µM). Inhibition of these pathways and storage of inhibitors was done as previously described [20]. All inhibitors were obtained from Calbiochem (Gibbstown, NJ USA), except R406, the active component of fostamatinib (Rigel, South San Francisco, CA, USA and AstraZeneca, London, UK). The appropriate concentration of inhibitor for each cell line was determined in preliminary experiments based on the ability to specifically block phosphorylation of the relevant signaling molecule without inducing cell death or toxicity.
IL-10 ELISA
IL-10 levels were determined as previously described [20]. IL-10 produced per million cells was calculated based on live cell counts, as determined by trypan blue exclusion, at the time that cytokine-containing supernatants were harvested.
Immunoprecipitation and Western Blot Analysis
Western blot analysis was performed as previously described [20]. For immunoprecipitation, lysates were pre-cleared with GammaBind G sepharose (Amersham, Piscataway, NJ, USA) for 1 h at 4°C and immunoprecipitated with 1 µg/107 cells of the indicated antibody or an isotype control overnight at 4°C. Immune complexes were pulled down with GammaBind G sepharose, dissociated with 2× reducing sample buffer, and separated by SDS-PAGE prior to transfer to nitrocellulose membranes.
RNA Interference (RNAi)
Fyn validated Stealth RNAi, Stealth Select Syk RNAi, and Stealth GC-matched negative control RNAi duplexed oligoribonucleotides were obtained from Invitrogen. BL41.NGFR-LMP1 cells (2–4×106) were transfected with 100–200 nmol RNAi using the Amaxa Nucleofector 2 in 100 µl of cell line nucleofector kit V solution according to the manufacturer's instructions. A FITC-conjugated control RNA oligonucleotide confirmed transfection efficiency of at least 90% at 24 hours. Assays were initiated at 96 hours, with knockdown confirmed at this time point in cellular lysates. Western blotting of cellular lysates consistently demonstrated >80% specific knockdown of expression of the targeted gene compared to β-actin control.
Quantitative real time PCR (qRT-PCR)
Total mRNA was isolated using TRIZOL reagent (Invitrogen) and cDNA was synthesized with an oligo(dT) primer (Invitrogen), followed by quantitative real time RT-PCR on a Stratagene Mx3000P using SYBR Green PCR master mix and primers for human IL-10 and GAPDH as previously described [20]. IL-10 was normalized to GAPDH for each sample.
Results
Membrane Expression of Native LMP1 and Chimeric NGFR-LMP1 in B Cell Lymphomas
To examine LMP1 signaling we utilized the EBV-negative Burkitt's lymphoma line BL41 and its EBV-infected counterpart BL41.B95, as well as three EBV+ PTLD-derived B cell lines (AB5, JB7, and JC62). All three of the EBV+ PTLD-derived B cell lines express both EBV signaling proteins LMP1 and LMP2a, albeit to different degrees (Figure 1A). BL41.B95, however, expresses LMP1 but minimal, if any, LMP2a protein (Figure 1A). Immunofluorescent staining for LMP1 revealed that native LMP1 is found in aggregates within the membrane in the EBV+, PTLD-derived B cell lines AB5, JB7, and JC62 (Figure 1B), consistent with previous observations in latently-infected B cells [42], [43]. The EBV-negative BL41 cell line served as a negative control for LMP1 expression (Figure 1B).
Figure 1. Membrane Expression of Native LMP1 and Chimeric NGFR-LMP1 in B Cell Lymphomas.
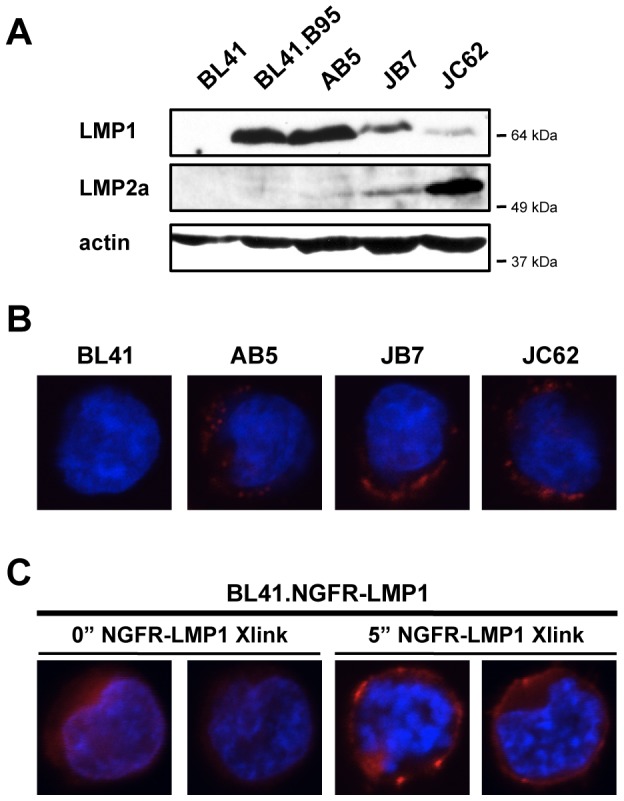
(A) Cell lysates were separated by SDS-PAGE, transferred to nitrocellulose and probed with antibodies against LMP1, LMP2a, and actin as a loading control. For all western blots, the closest molecular weight marker (e.g. 64 kDa) to the protein of interest is indicated to the right of each blot. (B) Representative fluorescent confocal images of the EBV- Burkitt's lymphoma line BL41 and the EBV+, PTLD-derived B cell lines AB5, JB7, and JC62. (C) Representative fluorescent confocal images of the stably transfected BL41.NGFR-LMP1 line, with NGFR-LMP1 crosslinked as described for the indicated amounts of time. For both (B,C), LMP1 stain is shown in red, dAPI stain is shown in blue.
To study the downstream signaling of LMP1, we utilized the EBV-negative BL41 cell line stably transfected with an NGFR-LMP1 chimeric construct [20], (Figure 1C, and Figure S1A). Chimeric NGFR-LMP1 molecules permit controlled, inducible LMP1 signaling when NGFR is crosslinked as opposed to the constitutive LMP1 signaling characteristic of the native molecule. Expression of native LMP1 increases ICAM (CD54) expression in B cells, T cells, and epithelial cells [17]. Therefore, functionality of crosslinked NGFR-LMP1 was confirmed by the upregulation of ICAM in the stably transfected, but not the parental, BL41 line (Figure S1B). Immunofluorescent staining revealed NGFR-LMP1 is expressed throughout the membrane in stably transfected cells (Figure 1C). Addition of anti-NGFR and crosslinking antibodies resulted in the aggregation of NGFR-LMP1 in the membrane of stably transfected cells (Figure 1C) in a pattern that resembled the expression of native LMP1 molecules (Figure 1B), although we also observe some NGFR-LMP1 expression outside glycosphingolipid (GSL) domains (data not shown).
NGFR-LMP1 Activates, but Does Not Physically Interact with, Syk
To assess whether LMP1 signaling activates Syk, we crosslinked NGFR-LMP1 and assayed for phosphorylation of the Syk substrate BLNK. BLNK both directly binds and is phosphorylated by Syk [44], [45]. Addition of anti-NGFR and crosslinking antibodies resulted in phosphorylation of the Syk substrate BLNK in the stably transfected lines, but not the parental BL41 line (Figure 2A, right lanes). Phosphorylation of Akt was used to confirm NGFR-LMP1 crosslinking (Figure 2A, right panel, right lane). Comparatively, NGFR-LMP1 crosslinking elicits less phosphorylation of both BLNK and Akt than BCR crosslinking, however it is able to activate each of these pathways.
Figure 2. NGFR-LMP1 Activates, but Does Not Physically Interact with, Syk.

(A) NGFR-LMP1 crosslinking or BCR crosslinking was triggered as described for 5 minutes. Lysates were separated on SDS-PAGE gels, transferred to nitrocellulose, and probed for the indicated phospho-proteins by western blotting. Blots were then stripped and reprobed for total protein levels. (B) Schematic and amino acid sequence of the C-terminal signaling tail of LMP1. TRAF binding motifs (PXQXT; black diamonds) and TRADD-interacting tyrosines (Y385, Y385; black circles) are noted in bold. C-terminal activating regions (CTAR), CTAR1 (E194 to G232) and CTAR2 (S361 to D386), are noted in underlined sections and in light grey shading. Potential JAK3 interaction sites, Box1 (P275 to P283, P302 to L316) and Box2 (P331 to K341), are noted in underlined sections and in dark grey shading. (C) Cells were treated with anti-NGFR and goat anti-mouse Ig to induce NGFR-LMP1 signaling for 5 minutes as indicated. Lysates from 10×106 cells were immunoprecipitated with anti-LMP1. Washed immunoprecipitates and flow through were separated by SDS-PAGE, transferred to nitrocellulose, and blotted for Syk, TRAF3, LMP1, and actin as a loading control (41 = BL41, B95 = BL41.B95, N-L = BL41.NGFR-LMP1). (D) Immunoprecipitation with either mIgG isotype control or anti-LMP1 Abs and western blotting of lysates was performed as described in (C). For all western blots, the closest molecular weight marker (e.g. 64 kDa) to the protein of interest is indicated to the right of each blot.
Classical Syk activation requires direct binding of the tandem SH2 domains of Syk to either an ITAM in a receptor tail or two hemITAMs on two separate receptor peptide chains. While the C-terminal, cytoplasmic tail of LMP1 contains motifs that bind adaptor molecules like the TRAFs (Figure 2B), it does not contain the canonical tandem YXXL ITAM motif. Given the absence of the canonical ITAM motifs, we asked if LMP1 physically interacts with Syk. Immunoprecipitation of native LMP1 from BL41.B95 did not co-precipitate Syk, but did co-precipitate the adaptor molecule TRAF3 (Figure 2C, B95, lane 4), known to associate with native LMP1. Similarly, immunoprecipitation of NGFR-LMP1 from BL41.NGFR-LMP1 did not co-precipitate Syk, but did co-precipitate the adaptor molecule TRAF3 after NGFR-LMP1 crosslinking (Figure 2C, N-L, lanes 5 and 6). Nevertheless, Syk was present in total cell lysates from BL41.B95 and BL41.NGFR-LMP1 (Figure 2C, lanes 1 and 2). Finally, native LMP1 did not co-precipitate Syk, but did co-precipitate TRAF3 in all three EBV+ PTLD-derived B cell lines (Figure 2D, lane 3). Taken together, these data suggest that while NGFR-LMP1 induces Syk activity, it does so without direct physical interaction with Syk.
NGFR-LMP1 Activates Syk Upstream of PI3K and IL-10
We next asked if Syk is required for the activation of PI3K by NGFR-LMP1 by utilizing R406, an ATP-competitive inhibitor of Syk. Basal phosphorylation of Syk was detected by immunoprecipitation and western blot of Syk from lysates of EBV+ PTLD-derived B cell lines; a representative blot from the JC62 cell line is shown in Figure 3A. This basal phosphorylation of Syk (Figure 3A, left lane) was reduced when EBV+ PTLD-derived B cell lines were treated with R406 (Figure 3A, right lanes).
Figure 3. NGFR-LMP1 Activates Syk Upstream of PI3K and IL-10.
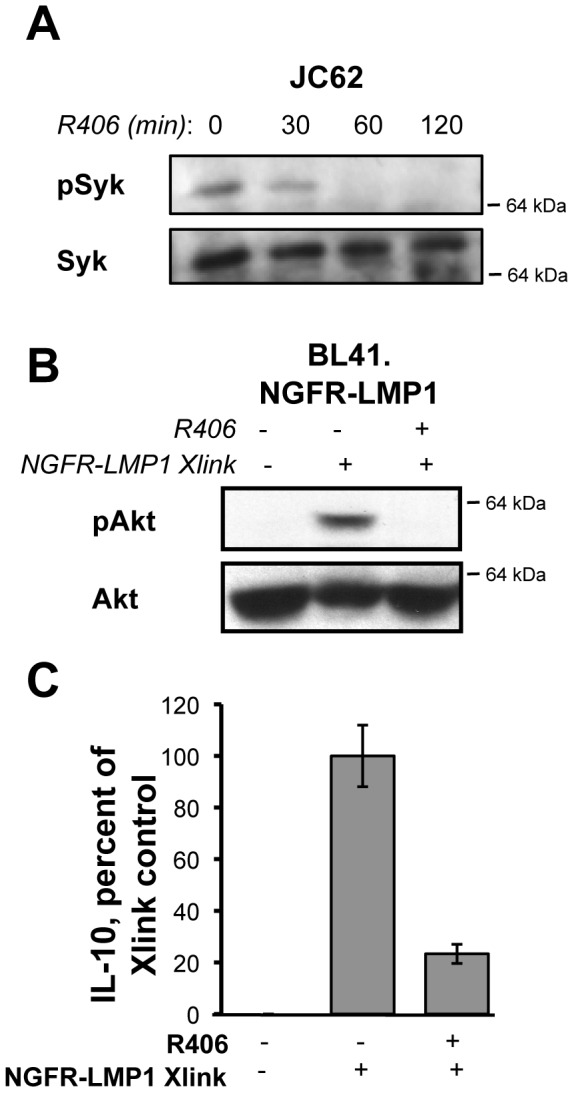
(A) Lysates from R406-treated cells (as indicated) were immunoprecipitated with anti-Syk antibodies. Washed immunoprecipitates were separated by SDS-PAGE, transferred to nitrocellulose, and blotted for pSyk and Syk. JC62 is shown here as a representative cell line. (B) NGFR-LMP1 crosslinking was triggered for 5 minutes as indicated, in the presence of DMSO control (−) or the Syk inhibitor R406 (+). Lysates were separated on SDS-PAGE gels, transferred to nitrocellulose, and probed for the indicated proteins by western blotting. The closest molecular weight marker (e.g. 64 kDa) to the protein of interest is indicated to the right of each blot. (C) Cells were pretreated for 1 hour with DMSO control (−) or the Syk inhibitor R406 (+) before induction of NGFR-LMP1 signaling. Cells were then plated at 0.5×106/ml in the presence of inhibitor. At 24 hours, supernatants were harvested and assayed for IL-10 by ELISA. IL-10 production was normalized to the number of viable cells at the time of supernatant harvest. Data is expressed as the mean percent of IL-10 produced for each group (compared to control) with standard deviations and is representative of 3–4 replicate experiments.
Phosphorylation of Akt after NGFR-LMP1 crosslinking (Figure 3B, lanes 1,2) in the stably transfected line is reduced in the presence of the Syk inhibitor R406 (Figure 3B, lane 3). IL-10 is a critical autocrine growth factor for EBV-infected cells, and is required for the autonomous proliferation of EBV+, PTLD derived B cell lines [39]. We have previously shown that IL-10 production by LMP1 is PI3K-dependent [20]. To further establish the role for Syk in PI3K/Akt activation by NGFR-LMP1, we asked if Syk inhibition affects IL-10 production by NGFR-LMP1. Indeed, Syk inhibition reduced NGFR-LMP1-dependent IL-10 production by approximately 80% (Figure 3C). These results suggest that Syk is activated by NGFR-LMP1 upstream of PI3K.
NGFR-LMP1 Activates Src Family Kinases
Classical Syk activation requires the action of Src tyrosine kinases. To determine if Src family kinases also participate in PI3K/Akt activation following NGFR-LMP1 signaling, we first investigated whether Src family kinases are activated in latently infected B cells. Western blot analysis with a pan-specific antibody that recognizes phosphorylated Src family members Src, Lyn, Fyn, Lck, Yes, and Hck, revealed constitutive Src family kinase activation in latently infected cells (Figure 4A). In a time course analysis following NGFR-LMP1 crosslinking, we observed rapid phosphorylation of Src family kinases that coincided with Akt phosphorylation (Figure 4B, right panels). C-Cbl, an E3 ubiquitin ligase often concurrently activated with Src family kinases [46], contains multiple phosphorylation sites required for association with the p85α subunit of PI3K [47]. We observed a rapid, time-dependent phosphorylation of c-Cbl at Tyr700 that peaked 5–10 minutes after LMP1 signaling was initiated (Figure 4B). Inhibition of Src family kinases by the small molecule inhibitor PP2 ablated phosphorylation of Akt by NGFR-LMP1 crosslinking (Figure 4C) and diminished the induction of IL-10 by NGFR-LMP1 crosslinking (Figure 4D). Taken together, these data indicate that NGFR-LMP1 activates Src family kinases upstream of PI3K activation.
Figure 4. NGFR-LMP1 Activates Src Family Kinases.
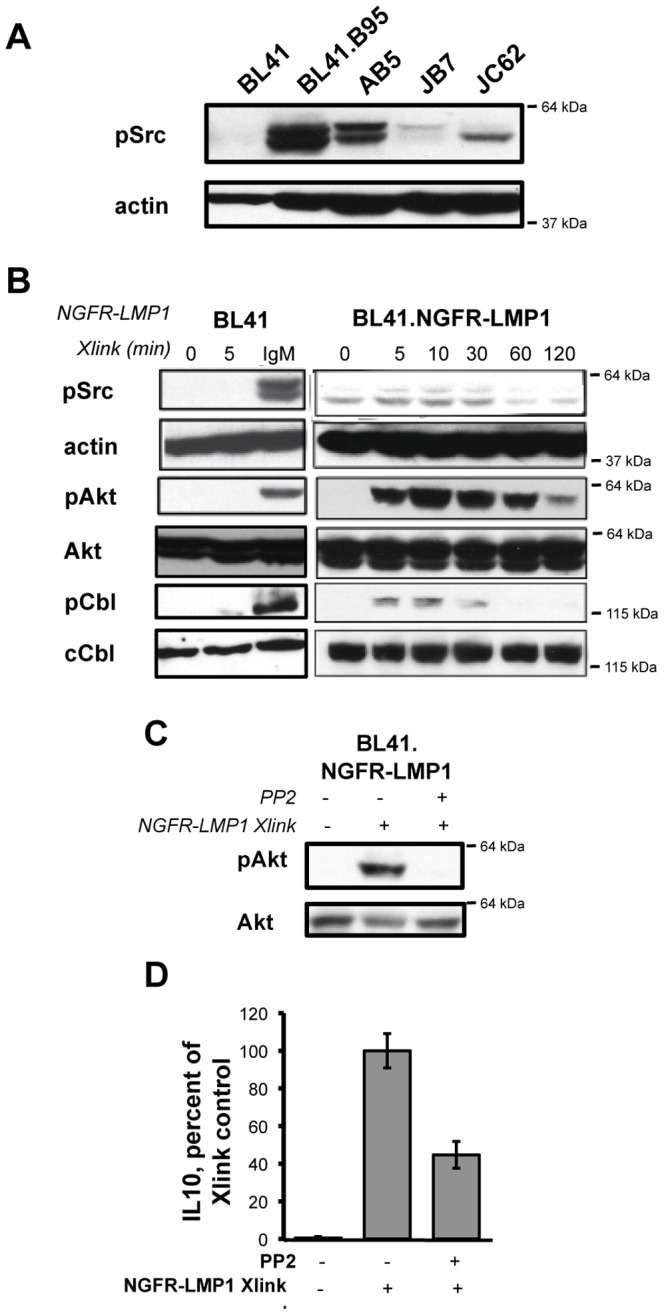
(A) Cell lysates were separated by SDS-PAGE, transferred to nitrocellulose, and probed with indicated antibodies. (B) NGFR-LMP1 signaling or BCR signaling (IgM) was triggered for the indicated amount of time. Lysates were separated on SDS-PAGE gels, transferred to nitrocellulose, and probed for the indicated proteins by western blotting. (C) Cells were pretreated for 1 hour with DMSO control (−) or the Src inhibitor PP2 (+) before triggering NGFR-LMP1 signaling or BCR signaling for 5 minutes in BL41.NGFR-LMP1 cells as described. Lysates were separated on SDS-PAGE gels, transferred to nitrocellulose, and probed for the indicated proteins by western blotting. For all western blots, the closest molecular weight marker (e.g. 64 kDa) to the protein of interest is indicated to the right of each blot. (D) Cells were pretreated for 1 hour with DMSO control (−) or the Src inhibitor PP2 (+) before induction of NGFR-LMP1 signaling. Cells were then plated at 0.5×106/ml in the presence of inhibitor. At 24 hours, supernatants were harvested and assayed for IL-10 by ELISA. IL-10 production was normalized to the number of viable cells at the time of supernatant harvest. Data is expressed as the mean percent of IL-10 produced for each group (compared to control) with standard deviations and is representative of 3–4 replicate experiments.
NGFR-LMP1 Activates the Src Kinase Fyn
The Src family tyrosine kinase inhibitor PP2 can inhibit all of the Src family kinases - Src (p60), Fgr (p58), Fyn (p59), Hck (p59/p61), Lck (p56), Lyn (p53/p56), Yes (p62), and Yrk (p60). Additionally, the phosphorylated forms of Src, Fyn, Hck, Lyn, and Yes are each recognized by the anti-phospho-Src family antibody. Thus, it was important to determine the specific Src family kinase phosphorylated by NGFR-LMP1 crosslinking. We first determined whether any phosphorylated Src family kinase member could directly associate with native LMP1 or chimeric NGFR-LMP1. We found that a phosphorylated Src family kinase of approximately 55–60 kDa co-precipitated with LMP1 (Figure 5A, top panel). This Src family kinase co-precipitated with native LMP1 in EBV-infected BL41.B95 cells (Figure 5A top panel, lane 2) and its association with NGFR-LMP1 was increased upon NGFR-LMP1 crosslinking (Figure 5A, top panel, lanes 3 and 4).
Figure 5. NGFR-LMP1 Activates the Src Kinase Fyn.

(A) Cell lysates were prepared 15 minutes after the induction of NGFR-LMP1 signaling. Lysates were immunoprecipitated overnight as indicated. Washed immunoprecipitates were separated on SDS-PAGE gels, transferred to nitrocellulose, and probed for phospho-Src by western blotting. (B–D) Three million cells were transfected with 200 nM RNAi specific for either Fyn or Syk 96 hours prior to analysis. (B, C) Knockdown confirmation and downstream signaling analysis. NGFR-LMP1 signaling was triggered in RNAi transfected cells for 30 minutes. Lysates were separated on SDS-PAGE gels, transferred to nitrocellulose, and probed for the indicated proteins by western blotting. For all western blots, the closest molecular weight marker (e.g. 64 kDa) to the protein of interest is indicated to the right of each blot. (D) IL-10 production. RNAi transfected cells were treated with anti-NGFR and goat anti-mouse Ig. At 24 hours, supernatants were harvested and assayed for IL-10 by ELISA. IL-10 cytokine production was normalized as previously described, with data expressed as the mean percent of IL-10 produced for each group (compared to Xlink control group) with standard deviations and represents the combined results of 3–4 replicate experiments. IL-10 mRNA expression was evaluated in parallel cultures by quantitative RT-PCR following 18 hours of NGFR-LMP1 signaling. Data is expressed as the mean percent of IL-10/GAPDH for each group (compared to the control group) with standard deviations and represents the combined results of 2 experiments.
We then analyzed the activation of specific Src family kinases by immunoprecipitation and western blot. Based on the molecular weight of the LMP1-associated protein and their known expression in B cells, Lyn and Fyn were the mostly likely Src family members. Immunoprecipitated Lyn was hyper-phosphorylated in EBV-infected BL41.B95 cells (Figure 5A, middle panel, lane 2), but NGFR-LMP1 crosslinking did not increase Lyn phosphorylation in BL41.NGFR-LMP1 cells (Figure 5A, middle panel, lanes 3 and 4). These data indicate that Lyn is likely not the Src family kinase involved in LMP1 signaling. In contrast, immunoprecipitated Fyn kinase was found to be hyper-phosphorylated in BL41.NGFR-LMP1 cells only after induction of NGFR-LMP1 signaling (Figure 5A, bottom panel, lanes 3 and 4). Taken together, these results strongly suggest that Fyn is the Src family kinase involved in activation of PI3K by NGFR-LMP1.
NGFR-LMP1 Activates Fyn Upstream of PI3K and IL-10
To confirm the role of Syk and Fyn in NGFR-LMP1 activation of PI3K, we utilized siRNA to knockdown Syk and Fyn in NGFR-LMP1 expressing cells. We routinely achieved knockdown of ∼90% for Fyn and ∼80% for Syk compared to the siRNA control in BL41.NGFR-LMP1 cells (Figure 5B). Fyn knockdown also decreased the levels of phospho-Src in NGFR-LMP1-crosslinked cells (Figure 5C), consistent with the interpretation that Fyn is the upstream Src kinase involved in LMP1 signaling. Syk knockdown did not affect the levels of phospho-Src in NGFR-LMP1-crosslinked cells (Figure 5C), suggesting that Syk is downstream of the Src family kinases.
In concordance with our inhibitor data, at least a 50% decrease in NGFR-LMP1-dependent Akt phosphorylation was observed in cells in which either Syk or Fyn was knocked down (Figure 5C). As the downstream molecules of interest (Akt, c-Cbl, Jnk, p38, and Erk) are not activated without NGFR-LMP1 crosslinking (Figure 5C, left lanes), we used crosslinked cells treated with control siRNA as our negative control for these experiments. Knockdown of either Syk or Fyn decreased NGFR-LMP1-dependent IL-10 cytokine secretion by ∼40% and ∼60%, respectively (Figure 5D, left panel). Fyn or Syk knockdown also inhibited NGFR-LMP1-dependent IL-10 mRNA expression (Figure 5D, right panel) in agreement with our earlier published results that LMP1 regulates IL-10 transcription [20]. Taken together with the previous data, these findings demonstrate that both Fyn and Syk play essential roles in NGFR-LMP1 signaling upstream of PI3K.
As discussed previously, we observed rapid c-Cbl phosphorylation following NGFR-LMP1 crosslinking (Figure 4B). Since both Fyn and Syk are known to be c-Cbl kinases, and since phosphorylated c-Cbl can interact with the p85α subunit of PI3K [48], we asked whether NGFR-LMP1-dependent c-Cbl phosphorylation levels correlated with NGFR-LMP1-dependent Akt phosphorylation levels. Indeed, in both the Fyn knockdown and the Syk knockdown cells, decreased Akt phosphorylation was accompanied by significantly decreased c-Cbl phosphorylation (down 70–80%) (Figure 5C). These data indicate that both Fyn and Syk activation are required for maximal phosphorylation of c-Cbl following NGFR-LMP1 signaling.
LMP1 is also a potent inducer of the MAPK pathways in EBV-infected B cells but the specific signaling intermediates that participate in MAPK activation have not been determined. Interestingly, our results reveal a difference in the requirements for Fyn and Syk in the NGFR-LMP1-dependent activation of MAPK. Whereas maximal JNK activation required Fyn but not Syk (Figure 5C), p38 activation by NGFR-LMP1crosslinking was not dependent on either Fyn or Syk (Figure 5C). In contrast, Fyn and Syk were each required for optimal Erk activation following NGFR-LMP1 crosslinking (Figure 5C). It has been previously reported that, during TCR signaling, Fyn activates the Erk MAPK pathway [49], but to our knowledge this is the first indication of Src-dependent induction of Erk MAPK by LMP1 in any cell type.
Discussion
EBV is associated with several lymphoid malignancies including Hodgkin's disease, Burkitt's lymphoma, and PTLD. Whereas the EBV protein LMP1 activates the oncogenic PI3K/Akt signaling pathway, the mechanism of PI3K/Akt activation is of interest for unveiling virally-controlled therapeutic targets for the treatment of EBV-associated malignancies. Using the inducible NGFR-LMP1 protein as a model system to study LMP1 signaling, we describe here the first evidence for non-canonical Syk activation in B cells by the EBV oncogene LMP1. Additionally, we provide the first evidence for activation of Src family kinases, in particular Fyn, as well as activation of c-Cbl, in B cells by LMP1. By demonstrating a requirement for Syk and Fyn in LMP1-induced PI3K/Akt, c-Cbl, and IL-10 induction, we also gained insight into the signaling mechanism by which LMP1 induces PI3K-dependent Akt phosphorylation in B cells.
Several lines of evidence indicate that the NGFR-LMP1 inducible chimeric protein is a useful model system to study LMP1 signaling. In particular, NGFR-LMP1 has replicated each signaling event and functional outcome associated with native LMP1 that we have tested. First, native LMP1 associates with TRAF1 and TRAF3 (Figure 2D and [7]–[9]). Similarly, TRAF1 and TRAF3 associate with NGFR-LMP1 after crosslinking (Figure 2C and [20]). Second, native LMP1 activates the following signaling pathways: PI3K/Akt [21], Erk [12], [13], p38 [10], [11], JNK [14], [15], and NF-kB [16], [17]. Similarly, each of these pathways is activated after NGFR-LMP1 crosslinking (Figures 2A, 3B, 4B, 4C, 5C and [20], [50]). Thirdly, native LMP1 induces the upregulation of ICAM (CD54) [17] and induces the production of IL-10 [11], a growth factor required for the autonomous proliferation of EBV+ PTLD-derived B cell lines [39]. Similarly, NGFR-LMP1 crosslinking induces the upregulation of ICAM (Figure S1B and [50]) and the production of IL-10 (Figures 3C, 4D, 5D and [20]). Finally, upon crosslinking, NGFR-LMP1 aggregates within the membrane in a manner similar to native LMP1 (Figures 1B, 1C). Together, these findings strongly support that the results obtained using the NGFR-LMP1 fusion protein are pertinent to native LMP1.
Our finding that LMP1 activates Syk sheds light on the mechanism of constitutive Syk activation observed in EBV-infected B cells, which had previously been attributed exclusively to LMP2a. Since LMP1 and LMP2a co-localize in the plasma membrane in latently-infected cells [43], they are spatially arranged to cooperate with each other. Indeed, there is precedence for cross-talk between these two proteins because LMP2a enhances LMP1-driven NF-κB and AP-1 activation [51]. Similarly, Syk activation in EBV-infected B cells may be a cooperative effort between LMP1 and LMP2a. Given that LMP1 and LMP2a are the viral homologs of CD40 and the BCR, respectively, our data suggests a more generalized function for non-canonical activation of Syk in B cells. Indeed, recent evidence suggests that CD40 can activate members of the BCR signaling complex, like BLNK, through Syk activation, resulting in enhanced BCR signaling [52]. Understanding the mechanism of Syk activation by LMP1 may yield more general insights into CD40 function and its modulation of BCR signaling.
We observed activation of Syk by LMP1 in the absence of physical interaction. Studies of Syk activation by integrins and P-selectin glycoprotein ligand 1 (PSGL1) have generated four potential models for non-canonical Syk activation [25]. The first, an ITAM-independent model, involves the interaction of Syk with the integrin β-chain, and the subsequent activation of Syk by Src family kinases. In the ITAM-mediated model, integrin ligation triggers the phosphorylation of ITAM-bearing signaling adaptors DAP12 or FcRγ by Src family kinases, resulting in the recruitment and activation of Syk. The third model involves a combination of the first two models, with Syk binding both integrins and the Src family-phosphorylated ITAMs of a signaling adaptor. In the fourth model, PSGL1 activation results in phosphorylation of the ITAMs exrin, radixin, and moesin (ERM) family proteins, which recruit and activate Syk [53]. Given that we observed no direct physical interaction between LMP1 and Syk, it is likely that the ITAM-independent model is not involved. Closer examination into the role of ITAM-containing signaling proteins, like ERM family proteins, and adaptor proteins, like DAP12 and FcRγ, is warranted to better understand the mechanism of Syk activation by LMP1.
Common among the potential mechanisms of non-canonical Syk activation is the involvement of Src family kinases. Indeed, our data shows activation of Src family kinases, specifically Fyn, by LMP1. Fyn also phosphorylates another viral protein reminiscent of LMP1 – K15 of Kaposi's sarcoma herpes virus [54]. Fyn is only one of multiple Src family kinases expressed in B lymphocytes. Depending on the receptor and signaling adaptor involved, different Src family kinases are activated. For example, while our data indicates LMP1 activates Fyn, the BCR and LMP2a primarily utilize Lyn, and DAP12/FcRγ-mediated PSGL1 signaling involves Fgr activation [55]. The unique contribution of different Src family kinases to Syk activation and the subsequent cellular response is of particular interest for further investigation. Fyn is more typically associated with function in T lymphocytes, where Fyn activation by the TCR results in recruitment and activation of Syk [56], [57]. Given the role of Fyn in Syk activation by the TCR, it is likely that Fyn activation by LMP1 results in the subsequent activation of Syk. However, further examination of the involvement of Fyn in Syk activation by LMP1 is warranted.
We show that LMP1 signals through PI3K in a manner at least partially dependent on Fyn and Syk. Both Syk and Fyn are reported c-Cbl kinases. Syk can phosphorylate c-Cbl at tyrosines 700, 731, and 774 with equal affinity [47] while Fyn phosphorylates c-Cbl preferentially at tyrosine 731 [58]. Our observation that LMP1 activation results in c-Cbl phosphorylation provides insight into the mechanism by which LMP1 induces PI3K-dependent Akt phosphorylation in B cells. Both Fyn- and Syk- phosphorylated c-Cbl strongly associate with the p85α subunit of PI3K to the same degree in vitro [47]. Fyn and Syk act together in T cells to phosphorylate c-Cbl and activate PI3K [56], [57]. In B cells, CD40, the cellular homolog of LMP1, is reported to activate PI3K through involvement of the Src family kinase Lyn, c-Cbl, and p85α [59]. More recent reports on c-Cbl suggest that it may also act as a regulator of PI3K activation through targeted ubiquitination and degradation of associated molecules. For instance, Syk has been reported as a target of c-Cbl mediated ubiquitination following BCR signaling [60], as well as following FcεRI signaling [61]. Whether the positive or the negative regulatory aspects of c-Cbl phosphorylation are necessary for LMP1 activation of the PI3K pathway is one that should be explored in future studies.
We also observed that Fyn and Syk activation by LMP1 had effects on MAPK pathways. Fyn kinases have been reported to stimulate the Erk pathway in TCR signaling [49]. In LMP1 signaling, we observed that both Fyn and Syk are involved in activating the Erk pathway, providing the first evidence for involvement of Src-family kinases in the activation of Erk by LMP1. Additionally, Fyn appears to be involved in activation of the JNK pathway. However, neither Syk nor Fyn plays any role in p38 activation. Such distinct activation requirements upstream of similar MAPK pathways may contribute to the bifurcation of signaling outcomes.
In summary, our data demonstrate the requirement for activation of Syk and Fyn by LMP1 upstream of PI3K. Understanding how an oncoprotein such as LMP1 activates Src family kinases, Syk and PI3K has implications for treatment of EBV+ B cell lymphomas.
Supporting Information
Inducible LMP1 Signaling. (A) Stable NGFR-LMP1 expressing lines and the parental BL41 line were stained with biotinylated anti-NGFR followed by streptavadin-PE to assess surface expression levels of the NGFR-LMP1 construct. (B) Cells were treated with anti-NGFR and goat-anti mouse Ig to induce NGFR-LMP1 signaling for 18 hours. After excess crosslinking antibody was neutralized by the addition of mouse IgG isotype control antibodies, cells were stained with ICAM-PE to assay for NGFR-LMP1 functionality.
(TIF)
Acknowledgments
We would like to thank Rigel, particularly Dr. P. Pine, Dr. A. Lowe, and Dr. Y. Hitoshi, and AstraZeneca for providing R406. The authors thank Dr. M. Vaysberg, Dr. K. Piard-Ruster, L.K. Phillips, and R. Frey for helpful discussions.
Funding Statement
This work was funded by an National Institutes of Health (NIH) National Institute of Allergy and Infectious Diseases (NIAID) grant ROI AI41769 (OMM) (nih.gov). O.H. was supported by a Stanford Graduate Fellowship (sgf.stanford.edu). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Young LS, Rickinson AB (2004) Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4: 757–768 doi:10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 2. Gottschalk S, Rooney CM, Heslop HE (2005) Post-transplant lymphoproliferative disorders. Annu Rev Med 56: 29–44 doi:10.1146/annurev.med.56.082103.104727. [DOI] [PubMed] [Google Scholar]
- 3. Caldwell RG, Brown RC, Longnecker R (2000) Epstein-Barr virus LMP2A-induced B-cell survival in two unique classes of EmuLMP2A transgenic mice. J Virol 74: 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mancao C, Hammerschmidt W (2007) Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 110: 3715–3721 doi:10.1182/blood-2007-05-090142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miller CL, Burkhardt AL, Lee JH, Stealey B, Longnecker R, et al. (1995) Integral membrane protein 2 of Epstein-Barr virus regulates reactivation from latency through dominant negative effects on protein-tyrosine kinases. Immunity 2: 155–166. [DOI] [PubMed] [Google Scholar]
- 6. Dykstra ML, Longnecker R, Pierce SK (2001) Epstein-Barr virus coopts lipid rafts to block the signaling and antigen transport functions of the BCR. Immunity 14: 57–67. [DOI] [PubMed] [Google Scholar]
- 7. Devergne O, Hatzivassiliou E, Izumi KM, Kaye KM, Kleijnen MF, et al. (1996) Association of TRAF1, TRAF2, and TRAF3 with an Epstein-Barr virus LMP1 domain important for B-lymphocyte transformation: role in NF-kappaB activation. Mol Cell Biol 16: 7098–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, et al. (1995) The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell 80: 389–399. [DOI] [PubMed] [Google Scholar]
- 9. Eliopoulos AG, Dawson CW, Mosialos G, Floettmann JE, Rowe M, et al. (1996) CD40-induced growth inhibition in epithelial cells is mimicked by Epstein-Barr Virus-encoded LMP1: involvement of TRAF3 as a common mediator. Oncogene 13: 2243–2254. [PubMed] [Google Scholar]
- 10. Eliopoulos AG, Gallagher NJ, Blake SM, Dawson CW, Young LS (1999) Activation of the p38 mitogen-activated protein kinase pathway by Epstein-Barr virus-encoded latent membrane protein 1 coregulates interleukin-6 and interleukin-8 production. J Biol Chem 274: 16085–16096. [DOI] [PubMed] [Google Scholar]
- 11. Vockerodt M, Haier B, Buttgereit P, Tesch H, Kube D (2001) The Epstein-Barr virus latent membrane protein 1 induces interleukin-10 in Burkitt's lymphoma cells but not in Hodgkin's cells involving the p38/SAPK2 pathway. Virology 280: 183–198 doi:10.1006/viro.2000.0768. [DOI] [PubMed] [Google Scholar]
- 12. Roberts ML, Cooper NR (1998) Activation of a ras-MAPK-dependent pathway by Epstein-Barr virus latent membrane protein 1 is essential for cellular transformation. Virology 240: 93–99 doi:10.1006/viro.1997.8901. [DOI] [PubMed] [Google Scholar]
- 13. Chuang H-C, Lay J-D, Hsieh W-C, Wang H-C, Chang Y, et al. (2005) Epstein-Barr virus LMP1 inhibits the expression of SAP gene and upregulates Th1 cytokines in the pathogenesis of hemophagocytic syndrome. Blood 106: 3090–3096 doi:10.1182/blood-2005-04-1406. [DOI] [PubMed] [Google Scholar]
- 14. Eliopoulos AG, Blake SM, Floettmann JE, Rowe M, Young LS (1999) Epstein-Barr virus-encoded latent membrane protein 1 activates the JNK pathway through its extreme C terminus via a mechanism involving TRADD and TRAF2. J Virol 73: 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kieser A, Kilger E, Gires O, Ueffing M, Kolch W, et al. (1997) Epstein-Barr virus latent membrane protein-1 triggers AP-1 activity via the c-Jun N-terminal kinase cascade. EMBO J 16: 6478–6485 doi:10.1093/emboj/16.21.6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eliopoulos AG, Stack M, Dawson CW, Kaye KM, Hodgkin L, et al. (1997) Epstein-Barr virus-encoded LMP1 and CD40 mediate IL-6 production in epithelial cells via an NF-kappaB pathway involving TNF receptor-associated factors. Oncogene 14: 2899–2916 doi:10.1038/sj.onc.1201258. [DOI] [PubMed] [Google Scholar]
- 17. Huen DS, Henderson SA, Croom-Carter D, Rowe M (1995) The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 10: 549–560. [PubMed] [Google Scholar]
- 18. Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129: 1261–1274 doi:10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27: 5497–5510 doi:10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lambert SL, Martinez OM (2007) Latent membrane protein 1 of EBV activates phosphatidylinositol 3-kinase to induce production of IL-10. J Immunol 179: 8225–8234. [DOI] [PubMed] [Google Scholar]
- 21. Dawson CW, Tramountanis G, Eliopoulos AG, Young LS (2003) Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J Biol Chem 278: 3694–3704 doi:10.1074/jbc.M209840200. [DOI] [PubMed] [Google Scholar]
- 22. Hatton O, Phillips LK, Vaysberg M, Hurwich J, Krams SM, et al. (2011) Syk Activation of Phosphatidylinositol 3-Kinase/Akt Prevents HtrA2-dependent Loss of X-linked Inhibitor of Apoptosis Protein (XIAP) to Promote Survival of Epstein-Barr Virus+ (EBV+) B Cell Lymphomas. J Biol Chem 286: 37368–37378 doi:10.1074/jbc.M111.255125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mainou BA, Everly DNJ, Raab-Traub N (2005) Epstein-Barr virus latent membrane protein 1 CTAR1 mediates rodent and human fibroblast transformation through activation of PI3K. Oncogene 24: 6917–6924 doi:10.1038/sj.onc.1208846. [DOI] [PubMed] [Google Scholar]
- 24. Cooray S (2004) The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J Gen Virol 85: 1065–1076. [DOI] [PubMed] [Google Scholar]
- 25. Mócsai A, Ruland J, Tybulewicz VLJ (2010) The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol 10: 387–402 doi:10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reth M (1989) Antigen receptor tail clue. Nature 338: 383–384 doi:10.1038/338383b0. [PubMed] [Google Scholar]
- 27. Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, et al. (2009) Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458: 899–903 doi:10.1038/nature07750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abtahian F, Guerriero A, Sebzda E, Lu MM, Zhou R, et al. (2003) Regulation of blood and lymphatic vascular separation by signaling proteins SLP-76 and Syk. Science 299: 247–251 doi:10.1126/science.1079477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, et al. (2004) The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci U S A 101: 6158–6163 doi:10.1073/pnas.0401602101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kerrigan AM, Brown GD (2010) Syk-coupled C-type lectin receptors that mediate cellular activation via single tyrosine based activation motifs. Immunol Rev 234: 335–352 doi:10.1111/j.0105-2896.2009.00882.x. [DOI] [PubMed] [Google Scholar]
- 31. Jakus Z, Fodor S, Abram CL, Lowell CA, Mocsai A (2007) Immunoreceptor-like signaling by beta 2 and beta 3 integrins. Trends Cell Biol 17: 493–501 doi:10.1016/j.tcb.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 32. Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O, et al. (2005) Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 22: 507–517 doi:10.1016/j.immuni.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 33. Vines CM, Potter JW, Xu Y, Geahlen RL, Costello PS, et al. (2001) Inhibition of beta 2 integrin receptor and Syk kinase signaling in monocytes by the Src family kinase Fgr. Immunity 15: 507–519. [DOI] [PubMed] [Google Scholar]
- 34. Mócsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, et al. (2006) Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol 7: 1326–1333 doi:10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zou W, Reeve JL, Liu Y, Teitelbaum SL, Ross FP (2008) DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Mol. Cell 31: 422–431 doi:10.1016/j.molcel.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Obergfell A, Eto K, Mócsai A, Buensuceso C, Moores SL, et al. (2002) Coordinate interactions of Csk, Src, and Syk kinases with [alpha]IIb[beta]3 initiate integrin signaling to the cytoskeleton. J Cell Biol 157: 265–275 doi:10.1083/jcb.200112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Calender A, Billaud M, Aubry JP, Banchereau J, Vuillaume M, et al. (1987) Epstein-Barr virus (EBV) induces expression of B-cell activation markers on in vitro infection of EBV-negative B-lymphoma cells. Proc Natl Acad Sci U S A 84: 8060–8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ghosh D, Kieff E (1990) cis-acting regulatory elements near the Epstein-Barr virus latent-infection membrane protein transcriptional start site. J Virol 64: 1855–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Beatty PR, Krams SM, Martinez OM (1997) Involvement of IL-10 in the autonomous growth of EBV-transformed B cell lines. J Immunol 158: 4045–4051. [PubMed] [Google Scholar]
- 40. Lam N, Sugden B (2003) LMP1, a viral relative of the TNF receptor family, signals principally from intracellular compartments. EMBO J 22: 3027–3038 doi:10.1093/emboj/cdg284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palmer TD, Willhoite AR, Gage FH (2000) Vascular niche for adult hippocampal neurogenesis. J Comp Neurol 425: 479–494. [DOI] [PubMed] [Google Scholar]
- 42. Mann KP, Staunton D, Thorley-Lawson DA (1985) Epstein-Barr virus-encoded protein found in plasma membranes of transformed cells. J Virol 55: 710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Longnecker R, Kieff E (1990) A second Epstein-Barr virus membrane protein (LMP2) is expressed in latent infection and colocalizes with LMP1. J Virol 64: 2319–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fu C, Turck CW, Kurosaki T, Chan AC (1998) BLNK: a central linker protein in B cell activation. Immunity 9: 93–103. [DOI] [PubMed] [Google Scholar]
- 45. Wienands J, Schweikert J, Wollscheid B, Jumaa H, Nielsen PJ, et al. (1998) SLP-65: a new signaling component in B lymphocytes which requires expression of the antigen receptor for phosphorylation. J Exp Med 188: 791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shao Y, Yang C, Elly C, Liu YC (2004) Differential regulation of the B cell receptor-mediated signaling by the E3 ubiquitin ligase Cbl. J Biol Chem 279: 43646–43653 doi:10.1074/jbc.M404082200. [DOI] [PubMed] [Google Scholar]
- 47. Grossmann AH, Kolibaba KS, Willis SG, Corbin AS, Langdon WS, et al. (2004) Catalytic domains of tyrosine kinases determine the phosphorylation sites within c-Cbl. FEBS Lett 577: 555–562 doi:10.1016/j.febslet.2004.10.054. [DOI] [PubMed] [Google Scholar]
- 48. Panchamoorthy G, Fukazawa T, Miyake S, Soltoff S, Reedquist K, et al. (1996) p120cbl is a major substrate of tyrosine phosphorylation upon B cell antigen receptor stimulation and interacts in vivo with Fyn and Syk tyrosine kinases, Grb2 and Shc adaptors, and the p85 subunit of phosphatidylinositol 3-kinase. J Biol Chem 271: 3187–3194. [DOI] [PubMed] [Google Scholar]
- 49. Lovatt M, Filby A, Parravicini V, Werlen G, Palmer E, et al. (2006) Lck regulates the threshold of activation in primary T cells, while both Lck and Fyn contribute to the magnitude of the extracellular signal-related kinase response. Mol Cell Biol 26: 8655–8665 doi:10.1128/MCB.00168-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vaysberg M, Hatton O, Lambert SL, Snow AL, Wong B, et al. (2008) Tumor-derived variants of Epstein-Barr virus latent membrane protein 1 induce sustained Erk activation and c-Fos. J Biol Chem 283: 36573–36585 doi:10.1074/jbc.M802968200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dawson CW, George JH, Blake SM, Longnecker R, Young LS (2001) The Epstein-Barr virus encoded latent membrane protein 2A augments signaling from latent membrane protein 1. Virology 289: 192–207 doi:10.1006/viro.2001.1142. [DOI] [PubMed] [Google Scholar]
- 52. Ying H, Li Z, Yang L, Zhang J (2011) Syk mediates BCR- and CD40-signaling integration during B cell activation. Immunobiology 216: 566–570 doi:10.1016/j.imbio.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Urzainqui A, Serrador JM, Viedma F, Yanez-Mo M, Rodriguez A, et al. (2002) ITAM-based interaction of ERM proteins with Syk mediates signaling by the leukocyte adhesion receptor PSGL-1. Immunity 17: 401–412. [DOI] [PubMed] [Google Scholar]
- 54. Brinkmann MM, Glenn M, Rainbow L, Kieser A, Henke-Gendo C, et al. (2003) Activation of mitogen-activated protein kinase and NF-kappaB pathways by a Kaposi's sarcoma-associated herpesvirus K15 membrane protein. J Virol 77: 9346–9358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zarbock A, Abram CL, Hundt M, Altman A, Lowell CA, et al. (2008) PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcR gamma to induce slow leukocyte rolling. J Exp Med 205: 2339–2347 doi:10.1084/jem.20072660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Deckert M, Elly C, Altman A, Liu YC (1998) Coordinated regulation of the tyrosine phosphorylation of Cbl by Fyn and Syk tyrosine kinases. J Biol Chem 273: 8867–8874. [DOI] [PubMed] [Google Scholar]
- 57. Palacios EH, Weiss A (2004) Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 23: 7990–8000 doi:10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- 58. Hunter S, Burton EA, Wu SC, Anderson SM (1999) Fyn associates with Cbl and phosphorylates tyrosine 731 in Cbl, a binding site for phosphatidylinositol 3-kinase. J Biol Chem 274: 2097–2106. [DOI] [PubMed] [Google Scholar]
- 59. Arron JR, Vologodskaia M, Wong BR, Naramura M, Kim N, et al. (2001) A positive regulatory role for Cbl family proteins in tumor necrosis factor-related activation-induced cytokine (trance) and CD40L-mediated Akt activation. J Biol Chem 276: 30011–30017 doi:10.1074/jbc.M100414200. [DOI] [PubMed] [Google Scholar]
- 60. Rao N, Ghosh AK, Ota S, Zhou P, Reddi AL, et al. (2001) The non-receptor tyrosine kinase Syk is a target of Cbl-mediated ubiquitylation upon B-cell receptor stimulation. EMBO J 20: 7085–7095 doi:10.1093/emboj/20.24.7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Macglashan D, Miura K (2004) Loss of syk kinase during IgE-mediated stimulation of human basophils. J Allergy Clin Immunol 114: 1317–1324 doi:10.1016/j.jaci.2004.08.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Inducible LMP1 Signaling. (A) Stable NGFR-LMP1 expressing lines and the parental BL41 line were stained with biotinylated anti-NGFR followed by streptavadin-PE to assess surface expression levels of the NGFR-LMP1 construct. (B) Cells were treated with anti-NGFR and goat-anti mouse Ig to induce NGFR-LMP1 signaling for 18 hours. After excess crosslinking antibody was neutralized by the addition of mouse IgG isotype control antibodies, cells were stained with ICAM-PE to assay for NGFR-LMP1 functionality.
(TIF)