

Abstract
Current therapy for multiple myeloma is complex and prolonged. Antimyeloma drugs are combined in induction, consolidation and/or maintenance protocols to destroy bulky disease, then suppress or eradicate residual disease. Oncolytic viruses have the potential to mediate both tumor debulking and residual disease elimination, but this curative paradigm remains unproven. Here we engineered an oncolytic vesicular stomatitis virus to minimize its neurotoxicity, enhance induction of antimyeloma immunity, and facilitate noninvasive monitoring of its intratumoral spread. Using high resolution imaging, autoradiography and immunohistochemistry, we demonstrate that the intravenously administered virus extravasates from tumor blood vessels in immunocompetent myeloma-bearing mice, nucleating multiple intratumoral infectious centers which expand rapidly and necrose at their centers, ultimately coalescing to cause extensive tumor destruction. This oncolytic tumor debulking phase lasts only for 72 hours after virus administration, and is completed before antiviral antibodies become detectable in the bloodstream. Anti-myeloma T cells, cross-primed as the virus-infected cells provoke an antiviral immune response, then eliminate residual uninfected myeloma cells. The study establishes a curative oncolytic paradigm for multiple myeloma where direct tumor debulking and immune eradication of minimal disease are mediated by a single intravenous dose of a single therapeutic agent. Clinical translation is underway.
Keywords: Oncolytic Virotherapy, multiple myeloma, Vesicular Stomatitis Virus, Intravenous, Immunotherapy
Introduction
Multiple myeloma is a malignancy of antibody secreting plasma cells that accounts for 1% of all cancers and 10% of hematologic malignancies.1 Over 20,000 new cases are diagnosed each year in the USA.2. Various chemotherapeutic drugs including steroids, melphalan, cyclophosphamide, bortezomib, thalidomide and lenalidomide are used in various different schedules and combinations to treat patients with myeloma, typically resulting in a reasonable period of disease remission and significant extension of survival.3, 4 The standard approach to frontline myeloma therapy is to administer two to four cycles of induction therapy followed by high dose melphalan with autologous stem cell rescue or continued monthly cycles of multiagent chemotherapy as consolidation for up to a year or more from diagnosis. Subsequent to the induction/consolidation phase, many patients are now placed on long term bortezomib or lenalidomide maintenance therapy, or given experimental immunotherapy to suppress or eliminate residual myeloma cells.4, 5 However, despite this complex and protracted approach to myeloma therapy, remissions are not usually maintained indefinitely and eventually the multiply relapsed disease becomes refractory to further treatment.
Oncolytic virotherapy is a promising experimental approach to cancer therapy in which viruses with evolved or engineered cancer-specific tropisms are used to mediate tumor destruction.6, 7 Because of the virus-permissivity of neoplastic plasma cells and the disease-associated immune paresis, multiple myeloma is perhaps the ideal malignancy to target using oncolytic viruses.8 Theoretically, in favorable conditions, tumor destruction by oncolytic viruses is achieved through a combination of direct and indirect mechanisms. Direct destruction is a consequence of selective virus replication and spread through the cancerous tissue which damages the tumor cells and provokes both local inflammation and adaptive antiviral immune responses.9, 10 Assuming an adequate dose of the virus is administered, tumor debulking occurs through a combination of direct viral lysis and cytotoxic T cell-mediated killing of virus infected tumor cells, as well as collateral damage to the stromal elements of the tumor.11-13 Direct killing can be can be further increased by virally encoded transgenes that sensitize infected cells to pro-drug or radioisotope therapy.14, 15 Indirect killing of tumor cells after oncolytic virus administration is mediated by the host immune system and is a consequence of cross-priming of cytotoxic T cell responses specifically directed against antigens present on the surface of uninfected tumor cells. Cross-priming occurs in the context of the host antiviral immune response and generates immune effector cells that have the potential to eliminate residual disease.16-18 In contrast to other cancer therapies, the first dose of an oncolytic virus is the only exposure that is likely to be able to mediate tumor eradication. This is because neutralizing antiviral antibodies are rapidly generated, persist long after the oncolytic virus has been eliminated from the body, and greatly diminish the antitumor potency of subsequent exposures to the same agent.19, 20 This is especially the case when the virus is administered via the bloodstream.
Based on the above considerations, oncolytic virotherapy has the potential to be a simple, elegant, and potentially curative single-shot therapy capable of activating multiple mechanisms of tumor cell killing, to achieve both tumor debulking and immune eradication of minimal disease. Numerous studies in experimental rodent cancer models (including myeloma) have shown that oncolytic viruses have anticancer activity, either via intratumoral virus spread and direct oncolytic destruction of tumor cells (typically in immunocompromised mice)9, 11, 13 or via to the induction of antitumor immunity, which can even be curative if the tumor burden is low and the treatment is administered repeatedly.16-18, 21 But the paradigm of oncolytic debulking followed by curative immune mediated elimination of residual disease after a single systemic administration of an oncolytic virus has not previously been demonstrated in any cancer model.
VSV is a bullet-shaped rhabdovirus whose single stranded negative sense RNA genome contains only 5 genes. The virus causes a blistering disease (vesicular stomatitis) in ungulate species and is thought to be transmitted by flies. Human exposure is not uncommon on cattle ranches in endemic areas but typically causes no more than a flu-like illness. Lab-adapted strains of VSV have shown considerable promise as an oncolytic agents in preclinical myeloma models,22, 23 and in a broad spectrum of other cancer models,24-27 making VSV a logical platform from which to build an “ideal” oncolytic virus for single-shot curative myeloma therapy. Additional advantages of VSV include the very low prevalence of human seropositivity, a very broad species tropism (allowing preclinical validation in immunocompetent rodents), rapid kinetics of virus replication in susceptible tissues, suitability for large-scale manufacture and stability of clinical grade virus.
Based on previous work with oncolytic VSVs,23, 28 we determined that the optimal configuration of VSV for single-shot curative cancer therapy would require the insertion of two foreign transgenes into the virus genome, coding respectively for interferon-β (IFNβ) to confer myeloma specificity and to reduce neurotoxic potential, and the thyroidal sodium iodide symporter (NIS) to permit noninvasive in vivo monitoring of virus spread while allowing if necessary for further potency enhancement by combination with 131I radiotherapy, a particularly attractive option for the treatment of radiosensitive disease such as myeloma23. Since excessive NIS expression is toxic to infected cells (Russell, unpublished data), we placed the NIS transgene close to the end of the viral genome between the G and L cistrons, and moved the IFNβ gene upstream of G, resulting in higher IFNβ expression levels compared to previous IFNβ-expressing VSV recombinants.29
Using VSV-IFNβ-NIS as a single-shot intravenous therapy we found that the virus was able to cure advanced syngeneic myelomas in immunocompetent mice. We therefore performed serial SPECT/CT imaging studies to monitor the in vivo spread of the virus and subjected tumors at regular intervals to immunohistochemical (IHC) staining using antibody reagents that identify virus-infected cells at very high resolution. Our studies validate the one-shot curative virotherapy treatment paradigm and define the distinct mechanisms by which a single intravenous dose of VSV-mIFNβ-NIS is able to eradicate large myeloma tumors in fully immunocompetent mice.
Methods
Cell culture and Viruses
Cell lines were cultured in media supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100mg/ml streptomycin. BHK-21 and MPC-11 cells, obtained from American Type cell culture (ATCC), were grown in Dulbecco Modified eagles medium (DMEM). 5TGM1 cells were obtained from Dr. Babatunde Oyajobi (UT Health Sciences Center, San Antonio, TX) and grown in Iscove's modified Dulbecco medium (IMDM). B-16 murine melanoma cells were obtained from R. Vile (Mayo Clinic) and grown in DMEM. All cell lines tested negative for mycoplasma contamination.
Restriction sites were engineered into the previously constructed pVSV-XN2 plasmid, containing the VSV positive strand antigenome, at the M/G and the G/L gene junctions preceded by the putative VSV intergenic sequence [TATG(A)7CTAACAG] required for functional transgene expression56. Restriction site flanked cDNA coding for murine IFNβ, human IFNβ and NIS genes were generated by PCR. Murine or human IFNβ were incorporated into a single NotI site (M/G junction), while NIS was incorporated into XhoI and NheI sites (G/L junction) to generate VSV-IFN-NIS plasmid. VSV-IFNβ-NIS virus was rescued using previously described methods30. Viruses were subsequently amplified in BHK-21 cells, purified by filtration of cell supernatant and pelleted by centrifugation through 10% w/v sucrose. Viral titer was measured in BHK-21 cells following infection using serially diluted virus stock to measure Tissue culture infective dose (TCID50) determined using the Spearman and Karber equation.
In vitro viral characterization
Viral titer was measured in supernatant following infection of BHK-21 cells (MOI 1.0, 1h at 37°C). To measure in vitro radio-iodide uptake, cells were incubated in Hanks buffered salt solution (HBSS) with 10mM HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.3) in the presence of radio-labeled NaI (I125 at 1×105 cpm) +/- 100uM potassium perchlorate (KCl04). Interferon-β secretion in supernatant of infected cells was determined using enzyme-linked immunoadsorbent assay (ELISA) against murine or human IFNβ (PBL Interferon Source). To compare IFN responsiveness, cells were pre-incubated with 100 U/ml murine IFNβ for 12h, followed by infection with VSV-GFP. Proliferation of viable cells was assessed by MTT assay (ATCC). Killing of 5TGM1 and MPC-11 by VSV-IFN-NIS (MOI 1.0) was similarly monitored at specific time points following infection by MTT assay shown as a percentage of untreated cells.
In vivo studies
5×106 5TGM1 or MPC-11 murine myeloma cells were subcutaneously (SQ) implanted on the right flank of 6-10 week-old syngeneic female C57Bl6/KaLwRij (Harlan, Netherlands) or Balb/c mice (Taconic) respectively. Tumor burden was measured by serial caliper measurements. Mice were administered with a single, intravenous dose of 1×108/0.1ml VSV-IFN-NIS or equal volume PBS by tail vein injection. SPECT-CT imaging was carried out following Intra-peritoneal (IP) administration of 0.5mCi 99mTcO4 and quantified as previously described57.
High-resolution tumor analysis
Tumors harvested at 24h intervals were frozen in optimal cutting medium (OCT) for sectioning. Tumor sections were analyzed by autoradiography and immunofluorescence for (i) VSV antigens using polyclonal rabbit anti-VSV generated in-house by the Mayo Clinic Viral Vector Production Laboratory, followed by Alexa-labeled anti-rabbit IgG secondary antibody (Invitrogen, Molecular Probes), (ii) cell death by TUNEL staining (DeadEnd™ Fluorometric TUNEL kit, Promega) and (iii) cellular nuclei using Hoescht 33342 (Invitrogen). Image quantification was performed on four random images from three VSV-mIFNβ-NIS treated tumors (except n=2 tumors at 72h post treatment) using ImageJ software to obtain VSV or TUNEL positive regions as percentage of tumor area. Immunofluorescence analysis of tumors harvested at 6h intervals detected VSV antigens and tumor blood vessels using a rat anti-mouse CD31 antibody (BD Pharmingen). Intratumoral foci sizes were quantified by measuring 7-8 foci from 2 tumors and dividing diameters by average tumor cell size (based on diameter measurements of 50 individual cells) to obtain foci diameter in numbers of cells. Volume of approximately spherical foci was estimated using formula, v=4/3(π*r3). Average width of rim of viable, VSV-infected cells was similarly quantified from immunofluorescence images from n=3 tumors harvested at 48h post VSV-IFNβ-NIS administration.
Immune studies in immune competent mice
To measure generation of antiviral antibodies, serial 2-fold dilutions of heat-inactivated serum were pre-incubated with 500 TCID50 VSV-GFP, and subsequently used to infect BHK-21 cells. Minimum serum dilution allowing VSV induced CPE was plotted. In vivo IFNβ secretion was measured in serum by ELISA. 5TGM1 vaccinations were administered by injecting 1×107 VSV-mIFNβ-NIS infected cells (MOI 10.0) SQ in the left flank of syngeneic mice. T-cell depletion studies were performed in C57Bl6/KaLwRij mice by IP administration anti-CD4 and anti-CD8 antibodies (50ug each) administered 3 times/week, followed by a weekly maintenance dose.
Statistical methods
Visual displays of the data were used to assess for outliers or substantial departures from normality, and t-test was utilized where described. In all cases, two-tail P-values are provided which are not adjusted for multiple comparisons. Comparison of survival differences was performed using Log-rank test from Kaplan Meier survival curves. For comparing tumor relapse rates in animal studies, the Fischer exact test was utilized due to small sample size.
Results
Generation and characterization of VSV-IFNβ-NIS viruses
New oncolytic Vesicular stomatitis viruses (VSVs), VSV-hIFNβ-NIS and VSV-mIFNβ-NIS, were constructed by inserting a gene coding for human or mouse interferon-β upstream of the viral gene coding for the surface glycoproten (G), and a second gene coding for the human sodium iodide symporter (hNIS) downstream of G. The viral genomes are shown schematically in figure 1A. Each inserted gene is flanked by consensus VSV transcriptional start and stop signals and the viruses were rescued and amplified in BHK cells using established methods30. Interferon-β binds the widely expressed αβ IFN receptor, initiating a protective innate cellular antiviral antibody response, and amplifying the adaptive antiviral T cell response31. Interferon-β is species specific such that human IFNβ does not signal to mouse cells32, 33, making VSV-hIFNβ-NIS the ideal control virus for comparison with VSV-mIFNβ-NIS in mouse models. Purified stocks of the two new viruses were titrated on BHK (hamster) cells (Fig 1B), and cell supernatants were harvested to confirm the secretion of virally encoded IFNβ. As shown in Fig 1C, high concentrations of human or murine IFNβ were detected in supernatants of BHK cells infected with VSV-hIFNβ-NIS and VSV-mIFNβ-NIS respectively and radioiodine uptake studies confirmed perchlorate sensitive (i.e. NIS-mediated) concentration of radioactive iodine in virus-infected cells, maximal at 24 hours after high multiplicity infection (Fig 1D).
Figure 1. Generation and characterization of VSV expressing IFNβ and NIS.
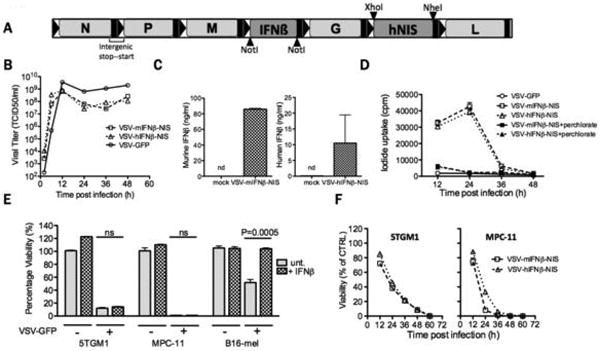
(A) Schematic of VSV-IFNβ-NIS. Two viruses were constructed, one encoding mouse IFNβ, the other human IFNβ. (B) One-step virus growth curves on BHK cells infected with VSV-GFP, VSV-mIFNβ-NIS or VSV-hIFNβ-NIS at MOI 1.0; (C) secretion of murine or human IFNβ by VSV-IFNβ-NIS-infected BHK cells, measured by ELISA. n.d. is not detectable. (D) Radioiodine uptakes by BHK cells infected with VSV-GFP, VSV-mIFNβ-NIS or VSV-hIFNβ-NIS at MOI 1.0. Uptakes were determined with (black symbols) or without (grey symbols) potassium perchlorate (KClO4), a specific inhibitor of NIS-mediated radioiodine uptake (E) Killing of myeloma cell lines by VSV. Viability of mouse IFNβ-treated (100U/ml for 12 hours) or untreated 5TGM1 and MPC-11 murine myeloma cells and B-16 murine melanoma cells was assessed by MTT assay at 48h after infection with VSV-GFP (MOI 1.0) and plotted as % viability compared to untreated cells. Significant differences were measured by t-test and P values are shown. (F) Timecourse of 5TGM1 and MPC-11 cell killing was monitored following infection with VSV-mIFNβ-NIS or VSV-hIFNβ-NIS (MOI 1.0) by measuring cell viability at 12h intervals by MTT assay. MPC-11 was killed more rapidly than 5TGM1. Error bars indicate Standard error of the mean (SEM)
Intratumoral spread of intravenously injected VSV-IFNβ-NIS in immunocompetent mice
We and others have previously shown that primary human myeloma cells as well as mouse and human myeloma cell lines are highly susceptible to oncolytic VSVs23, 34. To evaluate the in vivo activity of the new viruses, we chose the 5TGM1 and MPC-11 murine myeloma cell lines because they reliably form subcutaneous tumors in immunocompetent syngeneic mice35, 36. Both lines were confirmed susceptible to VSV-IFNβ-NIS infection (Fig 1E,F), resulting in functional NIS expression, IFNβ release (not shown) and subsequent cell killing. Neither cell line could be protected from the virus by interferon pretreatment, whereas the control interferon-responsive B16 melanoma cell line was protected (Fig. 1E). To determine whether intravenously administered VSV-IFNβ-NIS would localize specifically to sites of myeloma tumor growth, subcutaneous 5TGM1 or MPC-11 tumors were grown (∼5mm diameter) in syngeneic mice, a single intravenous dose of 108 TCID50 VSV-IFNβ-NIS was administered and the bio-distribution of virally encoded NIS expression was noninvasively monitored by daily SPECT/CT imaging using 99mTcO4 (6 hour half-life) as tracer (Fig 2A-B). Tracer uptake quantification suggested that the virus was efficiently infecting the tumors and that this was followed by rapid intratumoral viral propagation with NIS expression peaking at approximately 48h in the subcutaneous myeloma tumors (Fig C-D).
Figure 2. Monitoring intratumoral spread of intravenously administered VSV-IFN-NIS.

Female, 6-10 week old C57Bl6/KaLwRij mice bearing subcutaneous syngeneic 5TGM1 myeloma tumors or Balb/c mice bearing subcutaneous MPC-11 myeloma tumors were treated with a single intravenous (IV) dose of 100ul PBS, or 1×108TCID50 VSV-mIFNβ-NIS. SPECT-CT imaging was performed at 24h intervals, each image being obtained one hour after intraperitoneal administration of 99mTcO4 (500μCi). Serial day 1, 2, 3 and 4 SPECT/CT images from one representative animal (right panel) bearing (A) 5TGM1 myeloma or (B) MPC-11 myeloma show rapid radioiodine uptake following virus administration. Radioisotope uptake is seen in the thyroid gland (Th), and stomach (St), with slight excreted radioisotope visible in the bladder (Bl). Tumors from control PBS-treated animals (on left) show only background 99mTcO4 uptake. Semi-quantitative monitoring of intratumoral virus spread in subcutaneous (C) 5TGM1 and (D) MPC-11 tumor models. SPECT/CT images from n=5 VSV-mIFNβ-NIS-treated and n=2 control (PBS-treated) animals were analyzed to quantify 99mTcO4 radioisotope uptake by tumors days 1 through 5 following virus therapy. Mean group values are plotted for each timepoint (errors bars indicate SEM)
To confirm the impression that the virus was replicating and spreading in the tumor parenchyma, selected tumors were harvested immediately after SPECT/CT imaging at 24, 48 and 72 hours post VSV-IFNβ-NIS administration and subjected to (i) autoradiography to detect viable NIS-expressing tumor cells; (ii) immunofluorescence to detect VSV antigens, and (iii) TUNEL staining to identify dead or dying cells. Careful analysis of the data shown in figure 3A point to the existence of large, approximately spherical zones of VSV infection in which the tumor cells at the center are apoptotic and those at the periphery remain viable (see also supplemental Fig 1), express NIS and concentrate 99mTcO4 (supplemental Fig 2). Quantitative analysis of immunofluorescence and TUNEL data indicated a significant increase in the number of virus-infected and apoptotic cells between 24 and 48 hours post virus administration (Figure 3A). By 72 hours after infection the enlarging foci of VSV infection had largely coalesced, resulting in wholesale tumor destruction (Fig 3A,B and supplemental Fig 2). These studies indicate that SPECT/CT imaging correlates with intratumoral virus propagation, spread and tumor destruction and validates the use of NIS based imaging as a tool to monitor in vivo activity of systemically delivered VSV-IFNβ-NIS.
Figure 3. Intratumoral extravasation, spread and cell killing by intravenously administered VSV-IFNβ-NIS.

(A) Intratumoral distribution of virus-infected cells was analyzed 1, 2 and 3 days after virus administration by harvesting tumors immediately following SPECT-CT imaging and subjecting adjacent tumor sections to autoradiography and immunofluorescence staining to detect VSV antigens (red) as well as TUNEL-positive dead and dying cells (green). Note the increase in VSV-infected cells and TUNEL positive cells at 72 hours, with associated reduction of radioisotope uptake due to loss of cell viability. (B) VSV and TUNEL positivity were quantified using Image J software to determine percent positive cells averaged for 4 sections from n=3 analyzed tumors at the 24 and 48 hour timepoints, or n=2 tumors at the 72 hour timepoint. There was a significant increase in both VSV and TUNEL positivity between 24 and 48h post treatment using t-test (P=0.0455 and P=0.0163 respectively). (C) 5TGM1 tumors from virus-treated mice were harvested at 6 hour intervals following intravenous administration of VSV-mIFNβ-NIS, and were analyzed by immunofluorescent staining to detect VSV-infected cells (green) and CD31-positive blood vessels (red). Representative images are shown at (i) 6h, (ii) 12h, (iii) 18h and (iv-v) 24h following virus administration, magnification 100×.
Additional experiments were conducted to characterize the kinetics of virus spread at very early time-points, during the first 24 hours after virus administration (Fig 3C). Analysis of tumor sections harvested 6h after IV virus administration and stained for both VSV and CD31-positive blood vessels show individual scattered VSV infected cells, mostly close to tumor blood vessels. By 12 hours, small clusters of virus infected cells are visible and by 18 hours they have grown significantly until by 24 hours they have the typical appearance described previously, apoptotic at the center and viable at the periphery (Fig 3C i-iv). To confirm that the virus was replicating in the expanding foci of virus-infected cells, tumor-bearing mice were injected with VSV-mIFNβ-NIS or VSV-ΔG, a non-replicating VSV vector.37 Immunohistochemical studies 24 hours after virus administration showed large foci of infection as previously observed in tumors from mice that were treated with VSV-IFNβ-NIS, compared to singly infected cells scattered throughout the tumor parenchyma following VSV-ΔG administration (supplemental Figure 3A-B). These images document the infection of tumor cells following extravasation of virus from tumor blood vessels, and indicate that subsequent expansion of the infectious centers is a result of intratumoral viral replication. Further analysis of dual CD31/VSV stained sections (Fig 3C i-iv) indicate that the endothelial cells lining tumor blood vessels do not succumb to VSV infection, even when completely surrounded by VSV-infected tumor cells (shown at high magnification in supplemental Fig 3C).
VSV-IFNβ-NIS-mediated tumor debulking in syngeneic immunocompetent mice
To determine whether efficient extravasation and rapid intratumoral spread of the virus is associated with tumor regression, additional groups of C57KaLwRij mice with subcutaneous 5TGM1 tumors were treated with a single intravenous dose of 108 TCID50 VSV-IFNβ-NIS and were followed longer term with daily health status checks and tumor measurements. Figure 4A shows that tumors regressed rapidly in the majority of VSV-mIFNβ-NIS and VSV-hIFNβ-NIS treated animals. Interestingly, two to three weeks after administration of the viral therapy, tumor recurrence was seen in a majority of animals treated with VSV-hIFNβ-NIS, but not in those treated with VSV-mIFNβ-NIS (Fig 4A,B), suggesting that the virally encoded mouse IFNβ (but not the human IFNβ) is capable of activating mechanisms that lead to the complete eradication of residual disease in this syngeneic immunocompetent mouse model. Retreatment of relapsing tumors with VSV-IFNβ-NIS was not attempted since all of the mice had by that time developed high titers of anti-VSV antibodies (Fig 4C) which completely neutralize the antitumor activity of intravenously administered oncolytic VSV38.
Figure 4. Therapeutic efficacy of systemically administered VSV-IFNβ-NIS.

Mice bearing subcutaneous 5TGM1 tumors were treated with a single intravenous dose of (i) PBS, (ii) VSV-mIFNβ-NIS or (iii) VSV-hIFNβ-NIS. (A) Tumor burden was measured by serial caliper measurements which were used to calculate tumor volume over time. (B) Tumor responses are categorized as tumor progression, early death (≤ 3 days post treatment), complete tumor regression, or regression followed by relapse. NI: no incidence. Statistical difference in rate of tumor relapse within mice with sustained tumor regression was measured by Fischer Exact test indicating significantly higher rate of tumor relapse in VSV-hIFNβ-NIS treated mice vs. VSV-mIFNβ-NIS treated mice (P=0.026). (C) Generation of VSV neutralizing antibodies is measured in serum of PBS treated (n=2) and VSV-IFNβ-NIS treated (n=3 for each virus) mice in the first 5 days post treatment and plotted as the minimum fold dilution that protects BHK cells from infection with 500 TCID50 VSV-GFP.
Immune mediated eradication of residual disease in VSV-mIFNβ-NIS treated mice
Measurement of serum IFNβ levels in virus treated animals indicate that this virally encoded cytokine is released into the bloodstream by virally infected tumor cells at early time-points after virus administration (Fig 5A). The known antitumor actions of interferon-β include the direct inhibition of tumor cell proliferation, natural killer cell activation, antiangiogenesis and the enhancement of antitumor T cell responses31, 39, 40. However, proliferation of 5TGM1 and MPC11 myeloma cells in vitro was not adversely affected even at high concentrations of IFNβ (Fig 1F). Moreover, analysis of CD31 or CD3 stained sections of virus treated tumors did not show any evidence for inhibition of angiogenic activity, nor for early tumor infiltration by host T lymphocytes (Fig 3B and data not shown). However, virus treated animals whose tumors did not recur were found to be resistant to re-challenge with 5TGM1 tumor cells (Fig 5B), indicating that mice had developed 5TGM1 specific antitumor immunity.
Figure 5. Immune mediated elimination of tumor cells prevents tumor relapse.

(A) Quantification of murine IFNβ in serum of mice bearing subcutaneous 5TGM1 tumors treated intravenously with PBS, VSV-mIFNβ-NIS or VSV-hIFNβ-NIS, measured by ELISA. nd: not detectable (B) C57Bl6/KaLwRij mice that had complete tumor regression after intravenous therapy with VSV-mIFNβ-NIS treatment (n=6) and naïve age-matched syngeneic mice (n=6) were subsequently challenged subcutaneously with 1×107 5TGM1 cells. Tumor occurrence was recorded on day 21 post-challenge. NI is no incidence. (C) Immunotherapeutic efficacy of a single subcutaneous immunization with VSV-infected 5TGM1 cells. 1×107 5TGM1 cells were infected at MOI 10.0 with VSV-mIFNβ-NIS and 12 hours later implantated on the left flank. One day or five days later, 5×106 uninfected 5TGM1 tumor cells were implanted subcutaneously on the right flank. Log rank survival analysis comparison shows that day(-5) vaccination prolongs survival of mice following tumor implantation compared to unvaccinated mice (P=0.0253). (D) Mice bearing subcutaneous 5TGM1 tumors were treated with a single intravenous dose of (i) PBS, (ii) VSV-mIFNβ-NIS or (iii) VSV-mIFNβ-NIS combined with antibodies to deplete CD4+ and CD8+ T cells. Tumor burden was measured by serial caliper measurements. (E) Relapse rates between T-depleted and control groups were compared by Fischer Exact test indicating a higher rate of tumor relapse when VSV-mIFNβ-NIS was combined with T-cell depletion (P=0.0498).
To determine whether syngeneic VSV-infected myeloma cells could provoke a specific anti-myeloma immune response we immunized syngeneic mice with a single subcutaneous injection of 107 VSV-infected 5TGM1 cells, either one day after or five days prior to subcutaneous tumor cell implantation. Tumor growth was delayed resulting in a significant enhancement of survival in mice that were immunized 5 days prior to tumor challenge (Fig 5C), indicating that the VSV-infected tumor cells do provoke a modest antitumor immune response. However, the VSV-infected tumor cell vaccine had no detectable antitumor activity in mice bearing even small, established tumors, suggesting that antitumor immunity was effective only in the context of minimal disease burden.
To formally determine whether the lower tumor relapse rates in VSV-mIFNβ-NIS treated mice could be attributed to virally encoded IFNβ enhancing the antitumor T-cell response, we used a cocktail of anti-CD4 and anti-CD8 antibodies to deplete T-cells. As shown in Figures 5D and 5E, tumors responded equally well to the intravenous VSV-mIFNβ-NIS therapy regardless of T cell depletion status, but the rate of tumor recurrence was significantly higher in T-cell depleted mice. We conclude from these studies that eradication of residual tumor cells after oncolytic debulking by VSV-mIFNβ-NIS is mediated by tumor-specific T cells whose amplification is stimulated by the virally encoded murine IFNβ.
Discussion
Here we have demonstrated that a single intravenous dose of an appropriately engineered vesicular stomatitis virus (VSV-mIFNβ-NIS) can mediate both direct oncolytic destruction of bulky disease and subsequent indirect immune eradication of minimal residual disease in immune competent myeloma bearing mice. Having demonstrated that recombinant VSVs encoding both IFNβ and NIS were potently oncolytic in two different syngeneic myeloma models, we used high-resolution CT/SPECT and autoradiographic imaging of NIS expression with immunofluorescence analysis of serially explanted tumors to demonstrate that wholesale tumor destruction was due to rapid intratumoral virus spread leading to direct killing of virus infected tumor cells (the oncolytic paradigm). By comparing the rates of early tumor recurrence after treatment with viruses coding for either mouse or human IFNβ or in animals with intact or depleted T cells, we could demonstrate that residual tumor cells persisting after direct oncolytic tumor debulking could be eradicated by IFNβ-stimulated antimyeloma T cells which were presumably activated and amplified in the milieu of the initial debulking oncolytic infection, rich in tumor antigens as well as virally encoded IFNβ.
The rationale for generating a new recombinant VSV incorporating both IFNβ and NIS transgenes was based on a substantial body of published and unpublished work23, 29, 31. Several previous studies have shown that VSV neurotoxicity can be ameliorated and the virus targeted for cancer therapy by disabling its interferon combat machinery. This has been achieved either by mutating the matrix protein (VSV-δM51), or by inserting an interferon transgene (VSV-IFNβ) into the viral genome29, 41. Viruses that contain the IFNβ transgene have superior replication kinetics and drive higher expression levels of IFNβ compared to those with the δM51 mutation (Russell, unpublished data) which, by virtue of the antiproliferative, antiangiogenic and immune enhancing actions of IFNβ, may significantly enhance their antitumor potency. Our preliminary unpublished data shows that the safety profile of the newly created VSV-mIFNβ-NIS virus is superior to that of VSV-hIFNβ-NIS, indicating that the virally encoded IFNβ, when biologically active, does indeed ameliorate its neurotoxic potential.
The thyroidal sodium iodide symporter (NIS) powerfully concentrates radioactive iodide and has been expressed from various oncolytic viruses to facilitate noninvasive imaging of virus spread, as well as potency enhancement with concurrent radioiodine therapy.23 The potential use of NIS based therapy is especially attractive for the treatment of Multiple myeloma, a particularly radiosensitive malignancy.42 Inserting the NIS gene into VSV-δM51, however, produced a virus with severely retarded replication kinetics and with limited antitumor potency although noninvasive radioiodine imaging was convincingly demonstrated in immune compromised tumor bearing mice, as well as efficacy boosting by concurrent administration of therapeutic 131I23. The VSV-IFNβ-NIS viruses demonstrated greatly superior replication kinetics compared to VSV-δM51-NIS (unpublished data), possibly a consequence of the specific design of the new virus. The expression levels of VSVs genes are determined by their position in the genome with a well-characterized 3′ to 5′ transcriptional gradient43. Previous studies from our laboratory have taught us that viral fitness can be adversely affected by very high levels of NIS gene expression, probably due to protein misfolding resulting in premature death of infected cells (Russell, unpublished data). The NIS gene was therefore inserted downstream of G achieving NIS expression sufficiently low to maintain cell viability, but also sufficiently high for effective viral monitoring. The IFNβ gene was inserted upstream of G inducing higher IFNβ expression levels (compared to VSV-IFNβ and VSV-δM51), but not high enough to debilitate viral propagation and spread in myeloma cells and tumors. The configuration of VSV-IFNβ-NIS was compatible with efficient virus propagation and preparation of high titer virus stocks in BHK cells, without loss, mutation or compromised expression of either of the inserted transgenes. High level IFNβ secretion and robust radioiodine concentration by VSV-IFNβ-NIS-infected cells were confirmed before proceeding with in vivo studies and the stability of the virus was tested in multiple substrates to ensure that the virus could feasibly be manufactured for clinical applications (not shown).
Through immunofluorescence staining and microscopic study of tumor blood vessels, TUNEL-positive cells and VSV-infected cells, and autoradiographic analysis of NIS-mediated pertechnetate uptake, we gained important insights into the mechanism of VSV-mediated tumor destruction in these myeloma models. Early after infusion, intratumoral extravasation of blood-borne virus leads to preferential infection of myeloma cells in close proximity to the leaky tumor blood vessels. Each infected cell is able to transmit the virus infection outward to its immediate neighboring cells resulting in an expanding focus of infection. Outward viral transmission to successive layers of cells in each expanding focus of infection continues at a steady rate until the enlarging infectious centers come into contact with each other and coalesce, resulting in wholesale tumor destruction. Tumor cell destruction at this point is not complete as evidenced by the high rate of tumor relapse in animals whose T cell responses are inhibited by monoclonal antibody therapy, or in animals treated with a recombinant VSV coding for the human IFNβ protein which lacks biological activity in mice. But treatment with VSV-mIFNβ-NIS, coding for murine IFNβ, greatly decreases the tumor relapse frequency, indicating that the virally encoded murine IFNβ is able to recruit additional tumor killing mechanisms to effectively control or eliminate residual myeloma cells.
It has long been known that type I interferons (alpha or beta) released by virus infected cells can signal to adjacent cells, protecting them from virus infection44, 45. The VSV matrix protein (M) negates this host defense by inhibiting cytoplasmic export of messenger RNAs and hence IFN production by infected cells46. This is dramatically overridden in cells infected with VSV-IFNβ-NIS, which drives high level expression of the virally encoded IFNβ, thereby constraining virus spread in normal tissues and reducing neurotoxicity. While high level IFNβ production by virus-infected cells is known to diminish viral toxicity in normal host tissues with intact IFN response machinery29, 47, its effect on antitumor activity is more complex48. Many cancers are partially or fully resistant to the antiproliferative and antiviral activities of IFNβ48, 49, and the cell lines providing a basis for the in vivo myeloma models used in the current study were completely unresponsive to IFNβ, allowing unimpeded propagation in 5TGM1 and MPC11 cells, and through their corresponding tumors in vivo.
Aside from its anti-proliferative actions, IFNβ has the potential to inhibit tumor growth through its antiangiogenic activity40, 50-52 but immunofluorescence analysis of blood vessels in virus-treated 5TGM1 tumors indicate that antiangiogenic activity of the virally encoded IFNβ was not a significant factor contributing to the tumor responses seen in the current study, probably because the kinetics of virus propagation and tumor destruction were so extraordinarily rapid relative to the speed with which inhibition of angiogenesis can mediate antitumor activity.
A third and indirect way in which IFNβ can mediate antitumor activity is through the modulation of adaptive antitumor immune responses. Thus IFNβ has been shown to directly enhance the generation of antigen-specific T cells or facilitate dendritic cell presentation of antigens to T cells, and has been used successfully to stimulate antitumor immunity in cancer therapy models50-52. Our histology and immunofluorescence studies did not show detectable immune cell infiltration in tumors responding to VSV-IFNβ-NIS infection (data not shown), supporting the view that antitumor immunity was not a significant mechanism contributing to the rapid tumor debulking. However, the comparison of early tumor recurrence rates between mice treated with VSVs coding for human and mouse IFNβ, and of mice depleted of T lymphocytes, provides clear proof of the importance of tumor-specific T lymphocytes for the eradication of minimal residual disease.
The intratumoral extravasation and subsequent intratumoral spread of VSV-IFNβ-NIS in both of the myeloma tumor models examined was extremely efficient and rapid. Presumably this is a reflection of several favorable factors in these models including the absence of pre-existing immunity to VSV, the high levels of VEGF secreted by myeloma cells resulting in highly permeable tumor blood vessels53, the absence of stromal barriers to intratumoral virus dispersion in multiple myeloma54, and the nonresponsiveness of myeloma cells to IFNβ55. In addition, the potency of VSV in various myeloma cell lines and patient samples23 suggests VSV-IFNβ-NIS will be a potent therapeutic for even relapsed or refractory myeloma. It remains to be determined whether these favorable factors can be replicated or created in other haematologic malignancies or tumor types, and this will be an important area for future study.
Intratumoral spread of the recombinant viruses was easily followed in living animals by serial noninvasive SPECT/CT imaging of NIS-mediated radioisotope uptake. SPECT/CT image intensities were maximal 24 to 48 hours after initiation of therapy indicating that the total number of viable, virus infected cells was also peaking at this time. Autoradiographic and immunoshistochemical analysis of tissue sections show that only the viable infected cells (VSV-positive, TUNEL-negative) efficiently concentrate the 99mTcO4, and that uptake is no longer possible once they die and become TUNEL-positive. Future studies will be conducted to determine whether lower, subtherapeutic doses of the NIS-expressing viruses will be fully curative when combined with 131I as radiovirotherapy.
To summarize, the work described in the paper establishes a curative single-shot myeloma treatment paradigm in which a targeted, trackable oncolytic virus is able to mediate both direct oncolytic tumor debulking and subsequent immune mediated eradication of minimal disease in an immune competent murine myeloma model. A manufacturing process for clinical grade VSV-IFNβ-NIS has been developed, preliminary intravenous rodent toxicology studies have been completed with no unexpected findings and efforts are underway to move this work forward to clinical testing.
Supplementary Material
Acknowledgments
This work was supported by funds from Mayo Clinic, NIH/NCI (R01 CA100634 and R01 CA129966), the Richard M Schulze Family Foundation and a gift from Al and Mary Agnes McQuinn. The authors are grateful to Guy Griesmann and Sharon Stephan in the Viral Vector Production Laboratory (Mayo Clinic), Theresa Decklever (Department of Nuclear Medicine) and Wendy Ferguson (Mayo Clinic) for their technical assistance.
Footnotes
There are no conflicts of interest to disclose.
Supplemental information is available at Leukemia's website.
References
- 1.Rajkumar SV. Multiple myeloma: 2012 update on diagnosis, risk-stratification, and management. Am J Hematol. 2012 Jan;87(1):78–88. doi: 10.1002/ajh.22237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010 Sep-Oct;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.Zhou Y, Barlogie B, Shaughnessy JD., Jr The molecular characterization and clinical management of multiple myeloma in the post-genome era. Leukemia. 2009 Nov;23(11):1941–1956. doi: 10.1038/leu.2009.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar SK, Mikhael JR, Buadi FK, Dingli D, Dispenzieri A, Fonseca R, et al. Management of newly diagnosed symptomatic multiple myeloma: updated Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) consensus guidelines. Mayo Clin Proc. 2009 Dec;84(12):1095–1110. doi: 10.4065/mcp.2009.0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlude C, Beckhove P. Immunology and immunotherapeutic approaches in multiple myeloma. Recent Results Cancer Res. 2011;183:97–109. doi: 10.1007/978-3-540-85772-3_5. [DOI] [PubMed] [Google Scholar]
- 6.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007 Feb;4(2):101–117. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 7.Russell SJ, Peng KW. Viruses as anticancer drugs. Trends Pharmacol Sci. 2007 Jul;28(7):326–333. doi: 10.1016/j.tips.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thirukkumaran CM, Morris DG. Oncolytic virotherapy for multiple myeloma: past, present, and future. Bone Marrow Res. 2011;2011:632948. doi: 10.1155/2011/632948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007 Apr;15(4):651–659. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 10.Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. 2011 Jun;19(6):1008–1016. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shafren DR, Au GG, Nguyen T, Newcombe NG, Haley ES, Beagley L, et al. Systemic therapy of malignant human melanoma tumors by a common cold-producing enterovirus, coxsackievirus a21. Clin Cancer Res. 2004 Jan 1;10(1 Pt 1):53–60. doi: 10.1158/1078-0432.ccr-0690-3. [DOI] [PubMed] [Google Scholar]
- 12.Wojton J, Kaur B. Impact of tumor microenvironment on oncolytic viral therapy. Cytokine Growth Factor Rev. 2010 Apr-Jun;21(2-3):127–134. doi: 10.1016/j.cytogfr.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu YA, Galanis C, Woo Y, Chen N, Zhang Q, Fong Y, et al. Regression of human pancreatic tumor xenografts in mice after a single systemic injection of recombinant vaccinia virus GLV-1h68. Mol Cancer Ther. 2009 Jan;8(1):141–151. doi: 10.1158/1535-7163.MCT-08-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dingli D, Peng KW, Harvey ME, Greipp PR, O'Connor MK, Cattaneo R, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004 Mar 1;103(5):1641–1646. doi: 10.1182/blood-2003-07-2233. [DOI] [PubMed] [Google Scholar]
- 15.Tai CK, Kasahara N. Replication-competent retrovirus vectors for cancer gene therapy. Front Biosci. 2008;13:3083–3095. doi: 10.2741/2910. [DOI] [PubMed] [Google Scholar]
- 16.Bridle BW, Hanson S, Lichty BD. Combining oncolytic virotherapy and tumour vaccination. Cytokine Growth Factor Rev. 2010 Apr-Jun;21(2-3):143–148. doi: 10.1016/j.cytogfr.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 17.Prestwich RJ, Harrington KJ, Pandha HS, Vile RG, Melcher AA, Errington F. Oncolytic viruses: a novel form of immunotherapy. Expert Rev Anticancer Ther. 2008 Oct;8(10):1581–1588. doi: 10.1586/14737140.8.10.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tong AW, Senzer N, Cerullo V, Templeton NS, Hemminki A, Nemunaitis J. Oncolytic Viruses for Induction, of Anti-Tumor Immunity. Curr Pharm Biotechnol. 2011 Jul 8; doi: 10.2174/138920112800958913. [DOI] [PubMed] [Google Scholar]
- 19.Chiocca EA. The host response to cancer virotherapy. Curr Opin Mol Ther. 2008 Feb;10(1):38–45. [PubMed] [Google Scholar]
- 20.Guo ZS, Parimi V, O'Malley ME, Thirunavukarasu P, Sathaiah M, Austin F, et al. The combination of immunosuppression and carrier cells significantly enhances the efficacy of oncolytic poxvirus in the pre-immunized host. Gene Ther. 2010 Dec;17(12):1465–1475. doi: 10.1038/gt.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castelo-Branco P, Passer BJ, Buhrman JS, Antoszczyk S, Marinelli M, Zaupa C, et al. Oncolytic herpes simplex virus armed with xenogeneic homologue of prostatic acid phosphatase enhances antitumor efficacy in prostate cancer. Gene Ther. 2010 Jun;17(6):805–810. doi: 10.1038/gt.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lichty BD, Stojdl DF, Taylor RA, Miller L, Frenkel I, Atkins H, et al. Vesicular stomatitis virus: a potential therapeutic virus for the treatment of hematologic malignancy. Hum Gene Ther. 2004 Sep;15(9):821–831. doi: 10.1089/hum.2004.15.821. [DOI] [PubMed] [Google Scholar]
- 23.Goel A, Carlson SK, Classic KL, Greiner S, Naik S, Power AT, et al. Radioiodide imaging and radiovirotherapy of multiple myeloma using VSV(Delta51)-NIS, an attenuated vesicular stomatitis virus encoding the sodium iodide symporter gene. Blood. 2007 Oct 1;110(7):2342–2350. doi: 10.1182/blood-2007-01-065573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hadaschik BA, Zhang K, So AI, Fazli L, Jia W, Bell JC, et al. Oncolytic vesicular stomatitis viruses are potent agents for intravesical treatment of high-risk bladder cancer. Cancer Res. 2008 Jun 15;68(12):4506–4510. doi: 10.1158/0008-5472.CAN-08-0238. [DOI] [PubMed] [Google Scholar]
- 25.Wu L, Huang TG, Meseck M, Altomonte J, Ebert O, Shinozaki K, et al. rVSV(M Delta 51)-M3 is an effective and safe oncolytic virus for cancer therapy. Hum Gene Ther. 2008 Jun;19(6):635–647. doi: 10.1089/hum.2007.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wollmann G, Rogulin V, Simon I, Rose JK, van den Pol AN. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J Virol. 2010 Feb;84(3):1563–1573. doi: 10.1128/JVI.02040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stewart JH, Ahmed M, Northrup SA, Willingham M, Lyles DS. Vesicular stomatitis virus as a treatment for colorectal cancer. Cancer Gene Ther. 2011 Dec;18(12):837–849. doi: 10.1038/cgt.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Obuchi M, Fernandez M, Barber GN. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J Virol. 2003 Aug;77(16):8843–8856. doi: 10.1128/JVI.77.16.8843-8856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Obuchi M, Fernandez M, Barber GN. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J Virol. 2003 Aug;77(16):8843–8856. doi: 10.1128/JVI.77.16.8843-8856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whelan SP, Ball LA, Barr JN, Wertz GT. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc Natl Acad Sci U S A. 1995 Aug 29;92(18):8388–8392. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Bon A, Tough DF. Type I interferon as a stimulus for cross-priming. Cytokine Growth Factor Rev. 2008 Feb;19(1):33–40. doi: 10.1016/j.cytogfr.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 32.Bodo G, Palese P, Lindner J. Activity of mouse interferon in human cells. Proc Soc Exp Biol Med. 1971 Sep;137(4):1392–1395. doi: 10.3181/00379727-137-35796. [DOI] [PubMed] [Google Scholar]
- 33.Stewart WE, 2nd, Havell EA. Characterization of a subspecies of mouse interferon cross-reactive on human cells and antigenically related to human leukocyte interferon. Virology. 1980 Feb;101(1):315–318. doi: 10.1016/0042-6822(80)90512-7. [DOI] [PubMed] [Google Scholar]
- 34.Lichty BD, Stojdl DF, Taylor RA, Miller L, Frenkel I, Atkins H, et al. Vesicular stomatitis virus: a potential therapeutic virus for the treatment of hematologic malignancy. Hum Gene Ther. 2004 Sep;15(9):821–831. doi: 10.1089/hum.2004.15.821. [DOI] [PubMed] [Google Scholar]
- 35.Oyajobi BO, Franchin G, Williams PJ, Pulkrabek D, Gupta A, Munoz S, et al. Dual effects of macrophage inflammatory protein-1alpha on osteolysis and tumor burden in the murine 5TGM1 model of myeloma bone disease. Blood. 2003 Jul 1;102(1):311–319. doi: 10.1182/blood-2002-12-3905. [DOI] [PubMed] [Google Scholar]
- 36.Turner J, Tan J, Crucian B, Sullivan D, Ballester O, Dalton W, et al. Broadened clinical utility of gene gun-mediated, granulocyte-macrophage colon-stimulating factor cDNA-based tumor cell vaccines as demonstrated with a mouse myeloma model. Human Gene Therapy. 1998;9:1121–1130. doi: 10.1089/hum.1998.9.8-1121. [DOI] [PubMed] [Google Scholar]
- 37.Majid AM, Ezelle H, Shah S, Barber GN. Evaluating replication-defective vesicular stomatitis virus as a vaccine vehicle. J Virol. 2006 Jul;80(14):6993–7008. doi: 10.1128/JVI.00365-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Power AT, Wang J, Falls TJ, Paterson JM, Parato KA, Lichty BD, et al. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol Ther. 2007 Jan;15(1):123–130. doi: 10.1038/sj.mt.6300039. [DOI] [PubMed] [Google Scholar]
- 39.Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clin Cancer Res. 2011 May 1;17(9):2619–2627. doi: 10.1158/1078-0432.CCR-10-1114. [DOI] [PubMed] [Google Scholar]
- 40.Taylor KL, Leaman DW, Grane R, Mechti N, Borden EC, Lindner DJ. Identification of interferon-beta-stimulated genes that inhibit angiogenesis in vitro. J Interferon Cytokine Res. 2008 Dec;28(12):733–740. doi: 10.1089/jir.2008.0030. [DOI] [PubMed] [Google Scholar]
- 41.Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003 Oct;4(4):263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 42.Dingli D, Diaz RM, Bergert ER, O'Connor MK, Morris JC, Russell SJ. Genetically targeted radiotherapy for multiple myeloma. Blood. 2003 Jul 15;102(2):489–496. doi: 10.1182/blood-2002-11-3390. [DOI] [PubMed] [Google Scholar]
- 43.Conzelmann KK. Nonsegmented negative-strand RNA viruses: genetics and manipulation of viral genomes. Annu Rev Genet. 1998;32:123–162. doi: 10.1146/annurev.genet.32.1.123. [DOI] [PubMed] [Google Scholar]
- 44.Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006 May 12;312(5775):879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 45.Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 46.von Kobbe C, van Deursen JM, Rodrigues JP, Sitterlin D, Bachi A, Wu X, et al. Vesicular stomatitis virus matrix protein inhibits host cell gene expression by targeting the nucleoporin Nup98. Mol Cell. 2000 Nov;6(5):1243–1252. doi: 10.1016/s1097-2765(00)00120-9. [DOI] [PubMed] [Google Scholar]
- 47.Jenks N, Myers R, Greiner SM, Thompson J, Mader EK, Greenslade A, et al. Safety studies on intrahepatic or intratumoral injection of oncolytic vesicular stomatitis virus expressing interferon-beta in rodents and nonhuman primates. Hum Gene Ther. 2010 Apr;21(4):451–462. doi: 10.1089/hum.2009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saloura V, Wang LC, Fridlender ZG, Sun J, Cheng G, Kapoor V, et al. Evaluation of an attenuated vesicular stomatitis virus vector expressing interferon-beta for use in malignant pleural mesothelioma: heterogeneity in interferon responsiveness defines potential efficacy. Hum Gene Ther. 2010 Jan;21(1):51–64. doi: 10.1089/hum.2009.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Balachandran S, Barber GN. Defective translational control facilitates vesicular stomatitis virus oncolysis. Cancer Cell. 2004 Jan;5(1):51–65. doi: 10.1016/s1535-6108(03)00330-1. [DOI] [PubMed] [Google Scholar]
- 50.Natsume A, Mizuno M, Ryuke Y, Yoshida J. Antitumor effect and cellular immunity activation by murine interferon-beta gene transfer against intracerebral glioma in mouse. Gene Ther. 1999 Sep;6(9):1626–1633. doi: 10.1038/sj.gt.3300990. [DOI] [PubMed] [Google Scholar]
- 51.Saito R, Mizuno M, Nakahara N, Tsuno T, Kumabe T, Yoshimoto T, et al. Vaccination with tumor cell lysate-pulsed dendritic cells augments the effect of IFN-beta gene therapy for malignant glioma in an experimental mouse intracranial glioma. Int J Cancer. 2004 Sep 20;111(5):777–782. doi: 10.1002/ijc.20331. [DOI] [PubMed] [Google Scholar]
- 52.Shibata S, Okano S, Yonemitsu Y, Onimaru M, Sata S, Nagata-Takeshita H, et al. Induction of efficient antitumor immunity using dendritic cells activated by recombinant Sendai virus and its modulation by exogenous IFN-beta gene. Journal of Immunology. 2006 Sep 15;177(6):3564–3576. doi: 10.4049/jimmunol.177.6.3564. [DOI] [PubMed] [Google Scholar]
- 53.Ria R, Roccaro AM, Merchionne F, Vacca A, Dammacco F, Ribatti D. Vascular endothelial growth factor and its receptors in multiple myeloma. Leukemia. 2003 Oct;17(10):1961–1966. doi: 10.1038/sj.leu.2403076. [DOI] [PubMed] [Google Scholar]
- 54.Peng KW, Dogan A, Vrana J, Liu C, Ong HT, Kumar S, et al. Tumor-associated macrophages infiltrate plasmacytomas and can serve as cell carriers for oncolytic measles virotherapy of disseminated myeloma. Am J Hematol. 2009 Jul;84(7):401–407. doi: 10.1002/ajh.21444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liberati AM, Cinieri S, Senatore MG, Portuesi MG, De Angelis V, Di Clemente F, et al. Phase I-II trial on natural beta interferon in chemoresistant and relapsing multiple myeloma. Haematologica. 1990 Sep-Oct;75(5):436–442. [PubMed] [Google Scholar]
- 56.Schnell MJ, Buonocore L, Whitt MA, Rose JK. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J Virol. 1996 Apr;70(4):2318–2323. doi: 10.1128/jvi.70.4.2318-2323.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Penheiter AR, Wegman TR, Classic KL, Dingli D, Bender CE, Russell SJ, et al. Sodium iodide symporter (NIS)-mediated radiovirotherapy for pancreatic cancer. AJR Am J Roentgenol. 2010 Aug;195(2):341–349. doi: 10.2214/AJR.09.3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.