

Abstract
Stroke pathology involves multifactorial pro-death responses, including inflammation, oxidative stress, vascular dysfunction, and activation of necrotic and apoptotic pathways. The interruption of a single specific pathway in defined stroke model systems has not been sufficient to address the multifactorial nature of stroke-induced injuries in the human population. CD36 is a class B scavenger receptor that functions in regulating normal physiological and pathological functions. CD36 pathways are activated by several distinct ligands. Convergence of these pathways results in inflammatory responses and endothelial dysfunction, which may be an underlying cause of cardio- and cerebrovascular diseases. The current review describes receptor CD36-ligand interactions relevant to endothelial function and discusses how targeting CD36 may have therapeutic utility in stroke.
Keywords: CD36, Stroke, Angiogenesis, inflammation, endothelial dysfunction
CD36 overview
CD36, an 88 kDa glycoprotein, was originally described as a platelet receptor glycoprotein IV that recognizes thrombospondins and plasmodium falciparum-parasitized erythrocytes [1–3]. The receptor belongs to the class B scavenger receptor family that also includes Lysosomal Integral Membrane Protein II (LIMP-II) and the receptor for selective cholesterol ester uptake from HDL, CD36-LIMP-II Analogous-I (CLA-1; known as SR-BI in rodents). CD36 consists of two transmembrane domains, a large extracellular domain, and two short cytoplasmic tails. Features of the extracellular domain include a hydrophobic region that may be buried within the plasma membrane, a proline-cysteine rich domain and ten potential N-linked glycosylation sites. Both the N- and C-terminal intracellular domains have a pair of cysteines that are palmitoylated [4], a process that may target the protein to lipid rafts within the membrane [5].
CD36 is expressed in many types of mammalian cells, including microvascular endothelial cells, microglia, monocytes/macrophages, platelets, cardiac and skeletal muscle, adipocytes, and epithelia of a number of tissues including retina, breast, kidney, and tongue [6–12]. As a result of its expression in multiple cell types and its ability to recognize a host of structurally distinct lipid and non-lipid based ligands, the activation of CD36 results in highly diverse cellular responses.
Acting either singly or in concert with other proteins as a multi-protein complex, CD36 elicits signaling cascades in response to diverse ligands and regulates cellular responses (Fig. 1). Also known as fatty acid translocase (FAT), CD36 is also involved in lipid metabolism. Its localization in the circumvallate papillae in the tongue was reported to be important for distinguishing food that contains lipids, indicating that CD36 functions as a taste receptor for free fatty acids [6, 13, 14]. CD36 also recognizes membranes of cells undergoing apoptosis [15, 16]. Although the ligand on lipoproteins and apoptotic cells recognized by CD36 was thought to be a phosphatidylserine [17], several studies show that the recognized ligand is a phosphatidylcholine/serine species whose specific esterified sn-2 fatty acid has been modified by reactive oxygen or nitrogen species to yield a γ-hydroxy, oxo, or α,β-unsaturated carbonyl moiety [18–20]. As a pattern recognition receptor, CD36 is involved in innate immunity, in conjunction with toll like receptors (TLRs) [21, 22]. CD36 is also able to recognize thrombospondin type I repeats (TSR) in thrombospondin-1 and -2, functioning as an angiostatic factor [23]. Interaction of CD36 with fibrillar β-amyloid (fAβ)/integrins induces inflammatory responses by elevating the expression of pro-inflammatory cytokines and chemokines [24, 25]. Inflammation associated with foam cell formation by excess uptake of lipids by macrophage CD36 is an underlying cause of atherosclerotic lesion development [26–28]. The diverse functionality of CD36 in the regulation of angiogenesis, inflammation, atherosclerosis, oxidative stress and lipid metabolism, innate immunity, and taste preference has been described in detail elsewhere (refer to reviews [29, 30]).
Figure 1. CD36 is a multi-functional receptor.

CD36 elicits diverse cellular responses by interacting with specific ligands. CD36 is involved in lipid metabolism, taste preference, and innate immunity. Several CD36 pathways activated by distinct ligands elicit inflammatory responses. The binding of thrombospondins produces pro-inflammatory factors and causes endothelial apoptosis. β-Amyloid fibrils bind CD36/integrin complex, producing ROS and pro-inflammatory mediators. The uptake of oxidatively modified LDL by macrophage CD36 contributes to the pro-inflammatory atherosclerotic milieu. Outline area indicates CD36 contribution to endothelial dysfunction. FFA, free fatty acid; TSPs, thrombospondins; EC, endothelial cells; fAβ, fibrillar Aβ; ROS, reactive oxygen species; MCP-1, monocyte chemoattractant protein-1;ox/mLDL, oxidized/modified low density lipoprotein.
The nature of multi-ligand recognition and the diverse expression of CD36 outside of the vasculature make difficult to render the receptor’s contribution to vascular dysfunction. Nevertheless, CD36 pathways that result in the production of inflammatory factors in endothelial cells or in non-endothelial cells including astrocytes, microglia, and macrophages suggest that the receptor is either directly or indirectly involved in negatively regulating vascular tone and integrity. This review describes potential roles for CD36 in vascular dysfunction and angiogenesis in cerebrovascular diseases with a particular focus on ischemic stroke.
Vascular Dysfunction
The responses elicited by the interactions between CD36 and its respective ligands are predicted on the basis of the specific ligand and the type of cell expressing CD36, both of which may be coupled to multiple downstream effectors. Although CD36 responses occur in a ligand-specific manner, the pathways triggered by different ligands often converge, generating free radicals and inflammatory mediators within the cell, both inside and outside of the vasculature. The resultant inflammatory responses compromise endothelial function and the integrity of the blood brain barrier (BBB) (Fig. 1 outlined area).
Loss of vascular tone and BBB integrity underlies the pathology of cardio- and cerebrovascular diseases such as stroke, Alzheimer’s disease, and atherosclerosis. Examining functional characteristics of CD36 in other diseases associated with vascular dysfunction helped to predict the potential roles of the receptor in cerebral ischemia because of commonly shared pathological mechanisms among these vascular diseases (Fig. 2). Overwhelming evidence also indicates that the expression and function of CD36 are altered in prevalent risk factors, including hypertension, dyslipidemia, and diabetes. Understanding and managing the risk factors that modulate CD36 expression suggest the importance of CD36 as a common mediator for vascular diseases. Hypertension, whether isolated systolic, isolated diastolic, or in combination, is a major modifiable risk factor for the increased incidence of stroke [31, 32]. Various clinical trials employing antihypertensive agents that reduce blood pressure have been effective in the management and prevention of stroke [33, 34]. A link between CD36 and hypertension is derived from the observation that changes in CD36 expression are related to the clustering of multiple cardiovascular risk factors. The Spontaneously Hypertensive Rat (SHR) is a widely used animal model for hypertension, and a defective cd36 gene in chromosome 4 has been implicated as a risk factor. Transferring a segment of chromosome 4 from Brown Norway rat onto the SHR background reduces blood pressure and ameliorate glucose intolerance, hyperinsulinemia, and hyperlipidemia [35]. Although the study suggested that cd36 deletion is not critical to the initial selection for hypertension in the SHR model, the linkage of cd36 to other genes within the differential segment of the SHR was implicated in influencing multiple cardiovascular risk factors, including hypertension. A recent study showing that the stroke-prone SHR rats display increased CD36 expression in vessels and BBB impairment in the brain, supports a role for CD36 in cerebral vascular injury [36].
Figure 2. CD36 is a common mediator for neurological and vascular diseases.

Cardiovascular and cerebrovascular diseases share common pathological features. Inflammation is one of the frequently observed pathologies in atherosclerosis, neurological, and neurodegenerative diseases. CD36 mediates inflammatory responses by generating free radicals, producing cytokines and chemokines. Risk factors such as hypertension, hyperlipidemia, and diabetes are associated with elevated expression of CD36 and higher incidence of vascular diseases.
Endothelin-1, a vasoactive peptide, is synthesized and secreted by vascular endothelial cells and is a potent endogenous vasoconstrictor [37]. By stimulating the proliferation of vascular smooth muscle cells (VSMCs), the mitogenic property of VSMCs results in the formation of lesions within the intima and leads to hypertension and atherosclerosis. Both CD36 and endothelin-1 are considered atherosclerosis-related genes. Haug and colleagues [38] reported the involvement of CD36 in the endothelin-1 mediated atherogenic effect of oxLDL. The study showed that LDL and oxLDL up-regulate endothelin-1 expression in smooth muscle cells and monocytes, and pre-incubation of antibodies against CD36 reduced the expression. Similarly, angiotensin II, another vasoconstrictor oligopeptide, has been shown to enhance oxLDL uptake, and CD36 is involved in the process [39, 40]. Hypertensive subjects show significant increase in CD36 expression in macrophages with an enhanced adhesion of monocytes to endothelial cells and greater production of ROS [41]. In contrast, Kwok and colleagues reported a regulatory effect of endothelin-1 that inhibits CD36 protein expression in VSMCs through endothelin receptor A [42], although the functional significance of this finding in vivo remains to be addressed. Investigations on the effects of the vasoactive peptides on CD36 expression, or vice versa, following cerebral ischemia would provide a contributing role for CD36 in the pathology of cerebral ischemia in hypertensive subjects.
CD36 ligands to elicit inflammatory responses
CD36 is involved in free radical production and contributes to brain injury associated with post-ischemic inflammation [43]. Reduced free radical production and injury size in CD36 KO mice further substantiate the role of CD36 in stroke-induced brain injury. Several CD36 ligands including fibrillar β amyloid (fAβ), modified/oxidized low density lipoproteins (mLDL, oxLDL), TSPs, long chain fatty acids, and membrane components of apoptotic cells are elevated following stroke [44–48]. Depending on prevalent clinical co-morbid conditions, differential sets of CD36 ligands may prevail and affect vascular integrity following stroke. Because CD36 expression occurs in a feed forward manner [49], the presence of CD36 ligands in the infarct territory suggests excess activation of CD36 pathways. CD36 ligands that influence vascular dysfunction are amyloid-β, oxLDLs and lipid based ligands, and advanced glycation end products (AGE)-LDL, which are closely associated with AD, dyslipidemia, and diabetes.
a. Fibrillar β amyloid (fAβ)
Accumulation of amyloid β (Aβ) in the vicinity of plaques is one of the hallmarks of AD. The deposition of Aβ in the microvasculature contributes to oxidative stress and compromises BBB integrity [50, 51]. Due to the nature of the ligand, many of the studies on CD36 relevant to fAβ were focused on innate host response and inflammation associated with AD. Aβ binds to a number of biomolecules such as proteins and membrane lipids. Binding of membrane lipids facilitates fibrillation of Aβ (fAβ) which, in turn, alters the structure and function of the membrane [52].
Receptors that interact with fAβ include scavenger receptor (SR-A) [53], SR-B1[54], the receptor for advanced glycation end products (RAGE) [55], and CD36 [25, 56]. In response to fAβ, CD36 activates signaling cascades to trigger innate host responses [24, 25, 56]. A key role of CD36 in innate pro-inflammatory events was demonstrated. CD36 deficiency attenuated fAβ-induced secretion of cytokines, chemokines, and reactive oxygen species in microglia. Intraperitoneal and intracerebral injection of fAβ in CD36 KO mice attenuated macrophage and microglia recruitment into peritoneum and brain [24]. Bamberger and colleagues further identified a multi-receptor complex comprising the B-class scavenger receptor CD36, α6β1-integrin, and the integrin-associated protein/CD47 [57]. This cell surface receptor complex stimulates intracellular Tyr kinase-based signaling cascades and cellular activation, which leads to the secretion of pro-inflammatory molecules. A mechanistic study revealed that intracellular signaling by the receptor complex and fAβ involves Vav guanine nucleotide exchanging factor phosphorylation and Rac1, an essential component of NADPH oxidase, demonstrating a direct link of Vav phosphorylation to free radical production through the fAβ-receptor complex [58]. In the animal model of AD, interaction of Aβ with CD36 causes cerebrovascular oxidative stress and neurovascular dysfunction. The dysfunction was abrogated in the absence of CD36, suggesting that a strategy of CD36 inhibition to normalize cerebrovascular dysfunction might be effective [59]. Although literature regarding Aβ-induced vascular dysfunction following stroke is limited, cerebral amyloid angiopathy, in which amyloid is deposited in the vessel walls of the brain, is a risk factor for hemorrhagic stroke [60, 61]. Lee and colleagues showed an increased level of circulating Aβ in patients with acute ischemic stroke and suggested that the ligand is derived from brain as a consequence of ischemic insult [62].
b. Oxidized/modified LDLs
High cholesterol in conjunction with elevated inflammatory mediators in plasma is positively correlated with increased incidence of stroke in humans [63]. The episode occurs either singly or in association with other risk factors including obesity, hypertension, and insulin/glucose intolerance [63, 64]. These clinical conditions may influence the progression and outcomes of stroke-induced injury. Podrez and colleagues reported that an excess amount of lipid-based CD36 ligands are generated in hyperlipidemia [65]. This study demonstrated a profound up-regulation of structurally defined oxidized choline glycerophospholipid species (oxPCCD36) that serve as high affinity ligands for CD36 in the plasma of hypercholesterolemic mice and also in humans with low HDL levels [20]. Although the study did not investigate the potential up-regulation in non-lipid based ligands such as TSPs, the abundance of oxPCCD36 (oxidized or modified lipid moiety on lipoproteins) in hyperlipidemic plasma suggests the relevance of the lipid-based ligands in intensifying CD36-mediated function. Uptake of excess lipid ligands by monocyte/macrophage CD36 leads to foam cell formation, a key event for atherosclerotic lesion development in vessels [27].
High fat diet fed ApoE KO mice that were subjected to ischemic stroke exhibited increased infarct size as well as increased CCR2 and MCP-1 levels [66]. The hyperlipidemic ApoE KO mice showed increased IL-1β, TNF-α, and IL-6 mRNA levels, increased lipid content in the infarct, and displayed an increased number of lipid laden foam cells in the peri-infarct area. The involvement of CD36 in hyperlipidemia-induced exacerbation in ischemic outcome was further supported by the reversal of the hyperlipidemic phenotype in mice that lack CD36. CD36/ApoE double KO mice that were subjected to ischemic stroke exhibited smaller infarct size, reduced expression of the pro-inflammatory cytokines MCP-1 and CCR2, and fewer foam cells in the ischemic brain [66]. Whereas these findings suggest the importance of CD36 in stroke pathology in hyperlipidemic conditions, several other studies showed the effect of hyperlipidemia on endothelial dysfunction. In carotid arteries of ApoE-deficient mice, hypercholesterolemia causes loss of endothelial-dependent relaxation in response to acetylcholine. The deficit occurs via impairment of activation of endothelial nitric oxide synthase (eNOS) via superoxide anion [67, 68].
Specialized micro domains in the plasma membrane, caveolae, are enriched with cholesterol, sphingomyelin, caveolin and eNOS, and they are involved in signal transduction [5, 69]. Hypercholesterolemia was shown to deplete eNOS in the caveolae [70, 71]. CD36 is enriched in this cholesterol-rich lipid raft. A role of CD36 on eNOS-mediated vascular dysfunction in hyperlipidemic conditions was reported. Using ApoE KO and ApoE/CD36 double KO mice, the study showed reduced agonist-stimulated NO generation and vasodilation in ApoE KO. The eNOS-mediated vascular dysfunction was reversed in the absence of CD36 in the hyperlipidemic condition, thereby providing a direct link between CD36 function and eNOS activation in the presence of excess CD36 ligands [72]. Additionally, exposure of oxLDL, a major CD36 ligand in the development of atherosclerosis, to endothelial cells increases endothelial cell stiffness, supporting the influence of the CD36 lipid ligands in vascular diseases [73].
c. Advanced glycation end-product (AGE)-LDL
Evidence suggests an involvement of CD36 in insulin resistance [74, 75]. The receptor was identified as a novel marker of insulin resistance, and soluble CD36 level in plasma was positively correlated with type 2 diabetes risks [76, 77]. There is a strong association between diabetes and CD36 expression. The expression of CD36 occurs in the types of cells where diabetic complications prevail, including microvascular endothelium, retinal pigment epithelium, and proximal renal tubular epithelial cells, respectively resulting in vascular neuropathy, retinopathy, and nephropathy. It has been shown that diabetic conditions upregulate CD36 expression in several types of cells and tissue [78, 79].
Hyperglycemia increases the formation and accumulation of AGEs, which are recognized by CD36 [80]. In elevated levels of plasma AGE, CD36 protein expression is increased in VSMCs [81]. The advanced glycation end product-modified LDL (AGE-LDL) also has high affinity toward CD36. The study by Sima and colleagues showed that exposure of AGE-LDL induced a pro-oxidant and pro-inflammatory state in human smooth muscle cells, increasing lipid accumulation [82]. These studies indicate that increasing the burden of CD36 ligands in diabetic conditions promotes a pro-inflammatory state and compromises endothelial functions. Because post-ischemic inflammation is a contributing component of ischemic brain injury, inhibition of CD36 pathways by reducing the burden of diabetes-relevant CD36 ligands may be a potential strategy to attenuate vascular inflammation and dysfunction following stroke.
Angiogenesis in stroke
The survival of ischemic tissue heavily depends on the level of oxygen and substrate from collateral blood flow. Injury-induced angiogenesis plays a critical role in stroke outcome and recovery [83, 84]. Unlike vasculogenesis during development, angiogenesis in ischemic tissue involves a sprouting of new vessels from pre-existing vessels. The process requires endothelial cell replication, degradation of surroundings, and migration of endothelial cells to form lumen vessels to revascularize the injured area [85]. Enhanced angiogenesis in the peri-infarct area was correlated with longer survival in stroke patients [84]. Thus, the promotion of ischemia-induced angiogenesis, especially within the ischemic boundary, has been suggested as a therapeutic strategy to improve stroke outcome [83, 86].
The infarct typically begins to develop only hours after ischemic insult. However, studies indicate that endothelial cell proliferation occurs over several days post-ischemia [45, 87, 88]. Although a time point for the cessation of angiogenesis is not known, it was reported that new vessels are formed up to 92 days after stroke in human [89] and up to 21 days in an animal model of stroke [45]. In line with a potential benefit of promoting angiogenesis in the post-ischemic brain, several pro- and anti-angiogenic factors relevant to CD36 activation have been identified.
CD36, an angiostatic receptor
The relevance of CD36 function in vasculature is its anti-angiogenic nature, which offsets compensatory angiogenesis-promoting cascades in response to tissue ischemia. Binding of thrombospondin repeat (TSR) type 1 in TSP-1 to CD36 activates a signaling cascade that leads to endothelial cell apoptosis [23, 90, 91]. The ligand/receptor-mediated angiostatic effect requires the C-terminal cytoplasmic tail of CD36, since a point mutation in the region blocks CD36-mediated intracellular signaling [92].
Stroke up-regulates CD36 expression in the ipsilateral side of the brain. CD36-deficient mice exhibited smaller injuries and the neuroprotection was associated with reduced free radical generation in these mice [43]. Our lab investigated whether an underlying mechanism for this neuroprotection was partly mediated by enhanced angiogenesis in the absence of this negative regulator. To exclude the possible effect of infarct size between the genotypes on the degree of angiogenesis, infarct size between wild type and CD36 KO was normalized by increasing the ischemic duration to 45 min in CD36 KO mice in contrast to 30 min for WT mice. In spite of similar infarct size, CD36 KO mice showed profoundly increased vessel densities in the post-ischemic brain (Fig. 3). The observation supports the necessary role of CD36 in antagonizing injury-induced vessel formation. Additionally, reported down-regulation of VEGFR-2 phosphorylation by CD36 suggests a mechanism by which CD36 negatively modulates the classical mitogen signaling pathway [92]. Whether the increased vessel density in the absence of CD36 is due to the absence of the negative regulator alone, an increased pro-angiogenic cue, or a shifted balance toward a pro-angiogenic state still remains unknown.
Figure 3. Absence of CD36 increases stroke-induced vessel density.
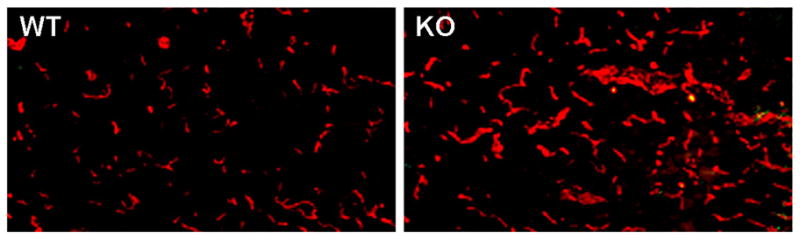
Fluorescence Immunohistochemistry of CD31, an endothelial marker, in CD36 WT and CD36 KO mice 3 days after ischemia. Note the profound increase in vessel density in CD36 KO mice in the infarct territory.
TSPs, inhibitors of angiogenesis
TSPs are extracellular matrix proteins that mediate cell-cell interaction via several membrane receptors including integrins, MMPs, fibronectin, CD36, other extracellular matrix proteins, and cytokines [93]. Among the five known TSPs, each coded by a separate gene, TSP-1 and-2 are involved in platelet aggregation, inflammation and angiogenesis [94]. TSPs are synthesized and secreted by several different cell types in response to injury and injury-induced growth factors. The importance of TSP-1 in inhibiting angiogenesis was demonstrated by antagonizing NO-stimulated angiogenic pathways. NO-stimulated angiogenesis is significantly increased in the absence of TSP-1, and exogenous TSP blocks NO-stimulated increase in endothelial cell proliferation, adhesion and migration [95]. Produced in vascular cells, TSP-1 also modulates cellular functions through a number of cell surface receptor complexes. CD36 was shown to be involved in the anti-angiogenic action of TSP-1 and 2 [96–99]. Additionally, the necessity of CD47, an integrin associated protein that forms a surface receptor complex with CD36, was reported in the inhibition of NO-stimulated vascular cell responses by TSP-1 [100].
Although ischemia increases the expression of TSP-1 and TSP-2 in the post-ischemic brain [45, 94], their expression profiles show distinct temporal and spatial expressions with an early rise of TSP-1 and delayed TSP-2 expression after stroke [94]. My lab determined the temporal expression of TSP-1 and TSP-2 following stroke. Temporal CD36 expression overlaps with that of TSP-1, but not TSP-2, during acute cerebral ischemia (Fig. 4). The observation suggests that the early CD36/TSP-1 interactions contribute to the angiogenic deficit associated with acute stroke pathology. Complete profiles of TSP and CD36 expression during a long-term stroke recovery phase will identify whether there is a secondary rise of CD36 and TSP expression and how their expression may regulate functions that are relevant to late angiogenesis.
Figure 4. Expression of angiostatic factors, CD36 and TSP-1, overlaps following stroke.
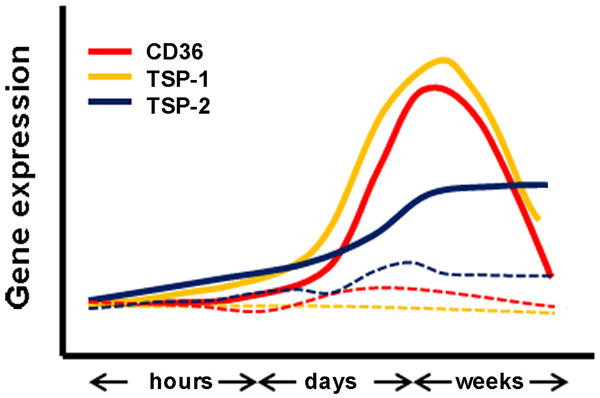
Relative CD36, TSP-1, and TSP-2 mRNA levels in the ipsilateral side (solid lines) of the brain hours, days, and weeks after ischemia. Expression of these factors in the contralateral side (dotted lines) was relatively unchanged.
BDNF, a pro-angiogenic factor
Vascular endothelial growth factors (VEGF), basic fibroblast growth factor (FGF), and hepatocyte growth factor, among others, promote stroke-induced angiogenesis [101–104]. Besides these mitogens, brain derived neurotrophic factor (BDNF) was also shown to be essential in maintaining vessel stability through direct angiogenic actions on endothelial cells [105]. Numerous reports have uncovered critical roles for BDNF and receptors in non-neuronal cells, such as endothelial cells, smooth muscle cells, immune cells, and epithelial cells in different organs [106–108]. Unlike VEGF-A that activates receptors that are expressed mostly in endothelial cell populations and are critical for early stages of vascular development, BDNF is expressed in an organ-specific manner in perinatal and adult vasculature [105]. BDNF deficiency causes reduced endothelial cell-cell contacts and endothelial cell apoptosis leading to hypo-contractility of the heart and perinatal lethality. Conversely, BDNF overexpression in the mid-gestational mouse heart results in an increase in capillary density, establishing an essential role for BDNF in modulating cardiac microvascular endothelial cells during development [105].
BDNF has been described as a newly identified angiogenic mediator in ischemic tissue that acts through TrkB receptor tyrosine kinase [109, 110]. Given the critical role of BDNF in perinatal vessel stabilization and continued expression of TrkB in the adult vasculature, Kermani and colleagues studied direct angiogenic actions of exogenous delivery of BDNF in normal and ischemic conditions in the adult mouse [110]. In a femoral artery ligation model, BDNF protein is significantly induced in ischemic tissue in comparison to non-ischemic tissue. Local overexpression of BDNF promotes blood flow recovery and capillary density comparable to that elicited by VEGF. Importantly, these effects appear to be mediated by TrkB, as the effects of exogenous BDNF are attenuated in haplodeficient animals (TrkB+/−), as assessed by blood flow and capillary density.
In the normal brain, BDNF is localized in neurons with the highest expression in the hippocampus and lower expression in the cortex [111, 112]. Transient focal ischemia induces BDNF mRNA and increase the number of BDNF immunoreactive neurons in the ipsilateral cingulate and frontal cortices outside the infarct, but less within the infarct core, suggesting a role for BDNF in survival and plasticity of cortical neurons after focal ischemia [113, 114]. The role of BDNF in stroke outcome and recovery is controversial. While some studies indicate that enhanced BDNF availability and signaling protect against ischemic brain injury and promote functional recovery [115–118], others argue against the beneficial effect of BDNF [119, 120]. The reason for the discrepancy needs further investigation.
Effect of BDNF SNP on CD36/TSP expression and angiogenesis
A single nucleotide polymorphism (SNP) of the bdnf gene occurs only in humans with high frequency. The BDNF SNP results in a substitution of a valine (Val) with a methionine (Met) in the prodomain of the BDNF protein at codon 66 and represents the first genetic alteration in the neurotrophin system that has been linked to human disease [121–125]. This alteration of the BDNF protein has been implicated in conferring susceptibility to depression, bipolar disorder, as well as altered episodic memory in patients with psychiatric disease [122, 126, 127]. Using mice with humanized BDNF variant in both allele (BDNFMet/Met) and wild type (BDNF+/+), Chen and others reported that the BDNF SNP is associated with the impairment of activity-regulated, but not constitutive, BDNF secretion [128].
From the observation that BDNF is critical for post-stroke outcome and recovery in rodents, we and others have investigated the effect of this BDNF SNP on stroke outcome. Although limited, clinical studies showed that the BDNFMet allele is correlated to poor ischemic outcome and low memory performance among survivors of subarachnoid hemorrhage [129, 130]. In contrast, the BDNFMet allele, not BDNFVal allele, is responsible for the promotion of executive function recovery in individuals with traumatic brain injury [131], suggesting a controversy in the effect of the BDNF SNP in post-injury outcome. Our lab investigated the effect of the BDNF SNP on infarct size and post-stroke angiogenesis. The study showed that BDNFMet/Met mice exhibited reduced BDNF levels in the post-ischemic brain compared to BDNF+/+ mice. In spite of the reduced BDNF levels in the post-ischemic brain of BDNFMet/Met, infarct size did not differ between the genotypes, indicating that stroke-induced BDNF secretion is not critical for acute infarct development [132]. However, BDNFMet/Met mice displayed reduced angiogenesis in the peri-infarct. The angiogenic deficit was associated with elevated expression of CD36 and TSP-1, suggesting the functional role of CD36 in antagonizing angiogenic response in Met homozygosity at the BDNF locus (Fig. 5). The observation in BDNFMet/Met mice - a shift toward angiostatic state with reduced BDNF and increased TSP-1/CD36 - suggests that CD36 inhibition may be a viable strategy to enhance angiogenesis and possible recovery in human stroke victims who have the BDNF SNP.
Figure 5. BDNF SNP increases CD36/TSP expression and reduces angiogenesis.

The diagram depicts the effect of BDNF SNP on the expression of CD36 and TSP-1 in the post-ischemic brain. Compared to wild type (BDNF+/+), BDNF SNP (BDNFM/M) impairs activity-regulated BDNF secretions, elevates angiostatic factors CD36 and TSP-1, and attenuates stroke-induced angiogenesis. Arrows indicate pathways that promote (blue) or inhibit (red) angiogenesis.
Targeting CD36 to promote endothelial function
It has been suggested that down-regulation of CD36 pathways attenuates post-ischemic inflammation and injury [133, 134]. Genetic ablation of CD36 is associated with a less inflammatory state and delays in vascular disease progression. In light of the receptor’s anti-angiogenic property, our lab observed enhanced stroke-induced angiogenesis in the absence of CD36 in C57BL/6 (Fig. 3) and in BDNFMet/Met genetic background [132].
Apart from genetic approaches, several pharmacological agents have been identified to reduce CD36 expression and functions. Synthetically engineered nanoblockers were targeted against scavenger receptors CD36 and SR-A. These nanoparticles block the uptake of oxLDLs and the subsequent formation of foam cells, thereby effectively attenuating the atherogenicity of inflammatory cells [135]. Other promising CD36 antagonists that block oxLDL uptake include hexarelin and its analogues. Hexarelin is a member of the hexapeptide growth hormone-releasing peptides (GHRPs) family that possesses growth hormone releasing activity and acts though G-protein-coupled ghrelin receptors in the hypothalamic-pituitary region [136]. In addition to binding to ghrelin receptors, it also binds to CD36 receptors [137]. Mice treated with hexarelin or a structurally related analogue, EP80317, demonstrated a marked decrease in atherosclerotic lesions [138]. The protective effect of hexarelin was mediated partly by negative modulation of CD36 expression [139]. A photo-affinity cross linking study showed that the binding domain of hexarelin overlaps with that of oxLDLs in CD36 [137], suggesting that the effect of hexarelin occurs through competition of the oxLDL binding site. Using a high-throughput screening assay, Wang and others reported several compounds that inhibit oxLDL uptake, including sodium danshensu, rosmaric acid, and salvianolic acid B [140]. While its efficacy in vivo remains to be investigated, treatment with salvianolic acid B in rat was associated with improved motor function following stroke [141].
Other approaches have been used to modulate CD36 expression or its downstream effects. Statins have been used to reduce cardiovascular events, not only by lowering cholesterol but also through other beneficial effects. The beneficial effects of statins may be related to the reduction of CD36 expression and function because they down-regulate CD36 expression and also suppress oxLDL uptake [142–144]. For antagonism that is specific to amyloid β, a recent study identified ursolic acid as a novel inhibitor that blocks Aβ-CD36 interaction [145].
Increased expression of CD36 has been associated with pro-oxidative conditions. Antioxidants, such as α-tocopherol, can reduce expression of CD36 and the uptake of oxLDLs by macrophages [146–148]. In addition, a pro-inflammatory response triggered by oxidative stress is abrogated in macrophages from CD36-deficient patients [149], indicating a direct association between manifestations of oxidative stress and CD36 expression. Another new class of antioxidants, SS peptides, have been shown to protect against myocardial ischemia-reperfusion injury in isolated hearts and to significantly reduce reperfusion arrhythmia, myocardial stunning, and infarct size in vivo [150, 151]. The potency of the SS peptide antioxidants is due to their ability to penetrate cell membranes and concentrate in the inner mitochondrial membrane, the primary site of intracellular ROS generation [150]. In addition to their scavenging action, the SS peptides inhibit the mitochondrial permeability transition, mitochondrial swelling, cytochrome c release, and apoptosis induced by lipid peroxides [152]. The efficacy of SS31 as an antioxidant in stroke was investigated in our laboratory. Treating mice with SS31 peptide attenuated ischemia-induced glutathione (GSH) depletion in the cortex and reduced infarct size [153]. The protective effect of SS31 was absent in CD36 KO mice, suggesting that SS31-induced neuroprotection occurs through antagonizing CD36-mediated activity and indicates that anti-oxidative approaches that target CD36 may be a useful strategy to treat ischemic stroke.
Conclusions
Cardiovascular and cerebrovascular diseases share common pathological features. CD36, a multi-ligand and multi-functional inflammatory receptor, has been implicated in the pathogenesis of cerebro/cardiovascular diseases, including stroke. Because the expression of the receptor in the post-ischemic brain occurs in cells within vessels as well as in cells outside of the vasculature, including microglia, monocytes/macrophages infiltrating into injury sites, and astrocytes [43], summarizing a role for CD36 in vascular dysfunction is a complicated issue. While stroke-induced TSP expression suggests a direct inhibition of angiogenesis in the endothelial cells, heightened inflammation by CD36 in other types of cells also alters vascular tone and integrity. Several promising pharmacological agents that counteract CD36 expression and function are available. Some have shown efficacy in reducing atherosclerotic lesion size and infarct size in stroke. Rigorous characterization of a new class of CD36 antagonists and testing their efficacy in experimental animal models of stroke are essential to develop a potential stroke therapy that aims at restoring vascular dysfunction.
Acknowledgments
NIH grants HL82511 and HL82511-04S1 (SC) and Burke Foundation.
Footnotes
Conflict of interests: Cornell Center for Technology Enterprise & Commercialization filed patents on SS31 and Salvianolic acid B as methods to antagonize CD36
References
- 1.Silverstein RL, Asch AS, Nachman RL. Glycoprotein IV mediates thrombospondin-dependent platelet-monocyte and platelet-U937 cell adhesion. J Clin Invest. 1989;84:546–52. doi: 10.1172/JCI114197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tandon NN, Kralisz U, Jamieson GA. Identification of glycoprotein IV (CD36) as a primary receptor for platelet-collagen adhesion. J Biol Chem. 1989;264:7576–83. [PubMed] [Google Scholar]
- 3.Oquendo P, Hundt E, Lawler J, Seed B. CD36 directly mediates cytoadherence of Plasmodium falciparum parasitized erythrocytes. Cell. 1989;58:95–101. doi: 10.1016/0092-8674(89)90406-6. [DOI] [PubMed] [Google Scholar]
- 4.Tao N, Wagner SJ, Lublin DM. CD36 is palmitoylated on both N- and C-terminal cytoplasmic tails. J Biol Chem. 1996;271:22315–20. doi: 10.1074/jbc.271.37.22315. [DOI] [PubMed] [Google Scholar]
- 5.Lisanti MP, Scherer PE, Vidugiriene J, Tang Z, Hermanowski-Vosatka A, Tu YH, Cook RF, Sargiacomo M. Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J Cell Biol. 1994;126:111–26. doi: 10.1083/jcb.126.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abumrad NA. CD36 may determine our desire for dietary fats. J Clin Invest. 2005;115:2965–7. doi: 10.1172/JCI26955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swerlick RA, Lee KH, Wick TM, Lawley TJ. Human dermal microvascular endothelial but not human umbilical vein endothelial cells express CD36 in vivo and in vitro. J Immunol. 1992;148:78–83. [PubMed] [Google Scholar]
- 8.Abumrad NA, el-Maghrabi MR, Amri EZ, Lopez E, Grimaldi PA. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J Biol Chem. 1993;268:17665–8. [PubMed] [Google Scholar]
- 9.Van Nieuwenhoven FA, Verstijnen CP, Abumrad NA, Willemsen PH, Van Eys GJ, Van der Vusse GJ, Glatz JF. Putative membrane fatty acid translocase and cytoplasmic fatty acid-binding protein are co-expressed in rat heart and skeletal muscles. Biochem Biophys Res Commun. 1995;207:747–52. doi: 10.1006/bbrc.1995.1250. [DOI] [PubMed] [Google Scholar]
- 10.Clezardin P, Frappart L, Clerget M, Pechoux C, Delmas PD. Expression of thrombospondin (TSP1) and its receptors (CD36 and CD51) in normal, hyperplastic, and neoplastic human breast. Cancer Res. 1993;53:1421–30. [PubMed] [Google Scholar]
- 11.Talle MA, Rao PE, Westberg E, Allegar N, Makowski M, Mittler RS, Goldstein G. Patterns of antigenic expression on human monocytes as defined by monoclonal antibodies. Cell Immunol. 1983;78:83–99. doi: 10.1016/0008-8749(83)90262-9. [DOI] [PubMed] [Google Scholar]
- 12.Ryeom SW, Silverstein RL, Scotto A, Sparrow JR. Binding of anionic phospholipids to retinal pigment epithelium may be mediated by the scavenger receptor CD36. J Biol Chem. 1996;271:20536–9. doi: 10.1074/jbc.271.34.20536. [DOI] [PubMed] [Google Scholar]
- 13.Laugerette F, Passilly-Degrace P, Patris B, Niot I, Febbraio M, Montmayeur JP, Besnard P. CD36 involvement in orosensory detection of dietary lipids, spontaneous fat preference, and digestive secretions. J Clin Invest. 2005;115:3177–84. doi: 10.1172/JCI25299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laugerette F, Passilly-Degrace P, Patris B, Niot I, Montmayeur JP, Besnard P. CD36, a major landmark on the trail of the taste of fat. Med Sci (Paris) 2006;22:357–9. doi: 10.1051/medsci/2006224357. [DOI] [PubMed] [Google Scholar]
- 15.Ren Y, Silverstein RL, Allen J, Savill J. CD36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. J Exp Med. 1995;181:1857–62. doi: 10.1084/jem.181.5.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savill J. Recognition and phagocytosis of cells undergoing apoptosis. Br Med Bull. 1997;53:491–508. doi: 10.1093/oxfordjournals.bmb.a011626. [DOI] [PubMed] [Google Scholar]
- 17.Fadok VA, Bratton DL, Frasch SC, Warner ML, Henson PM. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 1998;5:551–62. doi: 10.1038/sj.cdd.4400404. [DOI] [PubMed] [Google Scholar]
- 18.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, Silverstein RL, Hoff HF, Salomon RG, Hazen SL. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J Biol Chem. 2002a;277:38517–23. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- 19.Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. 2006;203:2613–25. doi: 10.1084/jem.20060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Gugiu B, Fox PL, Hoff HF, Salomon RG, Hazen SL. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J Biol Chem. 2002b;277:38503–16. doi: 10.1074/jbc.M203318200. [DOI] [PubMed] [Google Scholar]
- 21.Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, Sovath S, Shamel L, Hartung T, Zahringer U, Beutler B. CD36 is a sensor of diacylglycerides. Nature. 2005;433:523–7. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 22.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–61. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–8. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 24.El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–66. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, Freeman MW. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J Biol Chem. 2002;277:47373–9. doi: 10.1074/jbc.M208788200. [DOI] [PubMed] [Google Scholar]
- 26.Silverstein RL, Febbraio M. CD36 and atherosclerosis. Curr Opin Lipidol. 2000;11:483–91. doi: 10.1097/00041433-200010000-00006. [DOI] [PubMed] [Google Scholar]
- 27.Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, Silverstein RL. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105:1049–56. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahaman SO, Lennon DJ, Febbraio M, Podrez EA, Hazen SL, Silverstein RL. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006;4:211–21. doi: 10.1016/j.cmet.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108:785–91. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Febbraio M, Silverstein RL. CD36: implications in cardiovascular disease. Int J Biochem Cell Biol. 2007;39:2012–30. doi: 10.1016/j.biocel.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Droste DW, Ritter MA, Dittrich R, Heidenreich S, Wichter T, Freund M, Ringelstein EB. Arterial hypertension and ischaemic stroke. Acta Neurol Scand. 2003;107:241–51. doi: 10.1034/j.1600-0404.2003.00098.x. [DOI] [PubMed] [Google Scholar]
- 32.Amenta F, Di Tullio MA, Tomassoni D. Arterial hypertension and brain damage--evidence from animal models (review) Clin Exp Hypertens. 2003;25:359–80. doi: 10.1081/ceh-120023545. [DOI] [PubMed] [Google Scholar]
- 33.Bornstein N, Silvestrelli G, Caso V, Parnetti L. Arterial hypertension and stroke prevention: an update. Clin Exp Hypertens. 2006;28:317–26. doi: 10.1080/10641960600549405. [DOI] [PubMed] [Google Scholar]
- 34.Amenta F, Mignini F, Rabbia F, Tomassoni D, Veglio F. Protective effect of anti-hypertensive treatment on cognitive function in essential hypertension: analysis of published clinical data. J Neurol Sci. 2002;203–204:147–51. doi: 10.1016/s0022-510x(02)00281-2. [DOI] [PubMed] [Google Scholar]
- 35.Pravenec M, Zidek V, Simakova M, Kren V, Krenova D, Horky K, Jachymova M, Mikova B, Kazdova L, Aitman TJ, Churchill PC, Webb RC, Hingarh NH, Yang Y, Wang JM, Lezin EM, Kurtz TW. Genetics of Cd36 and the clustering of multiple cardiovascular risk factors in spontaneous hypertension. J Clin Invest. 1999;103:1651–7. doi: 10.1172/JCI6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ueno M, Nakagawa T, Nagai Y, Nishi N, Kusaka T, Kanenishi K, Onodera M, Hosomi N, Huang C, Yokomise H, Tomimoto H, Sakamoto H. The expression of CD36 in vessels with blood-brain barrier impairment in a stroke-prone hypertensive model. Neuropathol Appl Neurobiol. 2011;37:727–37. doi: 10.1111/j.1365-2990.2011.01172.x. [DOI] [PubMed] [Google Scholar]
- 37.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–5. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 38.Haug C, Schmid-Kotsas A, Zorn U, Schuett S, Gross HJ, Gruenert A, Bachem MG. Endothelin-1 synthesis and endothelin B receptor expression in human coronary artery smooth muscle cells and monocyte-derived macrophages is up-regulated by low density lipoproteins. J Mol Cell Cardiol. 2001;33:1701–12. doi: 10.1006/jmcc.2001.1421. [DOI] [PubMed] [Google Scholar]
- 39.Keidar S, Attias J. Angiotensin II injection into mice increases the uptake of oxidized LDL by their macrophages via a proteoglycan-mediated pathway. Biochem Biophys Res Commun. 1997;239:63–7. doi: 10.1006/bbrc.1997.7428. [DOI] [PubMed] [Google Scholar]
- 40.Keidar S, Heinrich R, Kaplan M, Hayek T, Aviram M. Angiotensin II administration to atherosclerotic mice increases macrophage uptake of oxidized ldl: a possible role for interleukin-6. Arterioscler Thromb Vasc Biol. 2001;21:1464–9. doi: 10.1161/hq0901.095547. [DOI] [PubMed] [Google Scholar]
- 41.Zapolska-Downar D, Siennicka A, Chelstowski K, Widecka K, Goracy I, Halasa M, Machalinski B, Naruszewicz M. Is there an association between angiotensin-converting enzyme gene polymorphism and functional activation of monocytes and macrophage in young patients with essential hypertension? J Hypertens. 2006;24:1565–73. doi: 10.1097/01.hjh.0000239292.32883.38. [DOI] [PubMed] [Google Scholar]
- 42.Kwok CF, Juan CC, Ho LT. Endothelin-1 decreases CD36 protein expression in vascular smooth muscle cells. Am J Physiol Endocrinol Metab. 2007;292:E648–52. doi: 10.1152/ajpendo.00084.2006. [DOI] [PubMed] [Google Scholar]
- 43.Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G, Silverstein RL, Iadecola C. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci. 2005;25:2504–12. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nihashi T, Inao S, Kajita Y, Kawai T, Sugimoto T, Niwa M, Kabeya R, Hata N, Hayashi S, Yoshida J. Expression and distribution of beta amyloid precursor protein and beta amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion. Acta Neurochir (Wien) 2001;143:287–95. doi: 10.1007/s007010170109. [DOI] [PubMed] [Google Scholar]
- 45.Hayashi T, Noshita N, Sugawara T, Chan PH. Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J Cereb Blood Flow Metab. 2003;23:166–80. doi: 10.1097/01.WCB.0000041283.53351.CB. [DOI] [PubMed] [Google Scholar]
- 46.Pilitsis JG, Coplin WM, O’Regan MH, Wellwood JM, Diaz FG, Fairfax MR, Michael DB, Phillis JW. Measurement of free fatty acids in cerebrospinal fluid from patients with hemorrhagic and ischemic stroke. Brain Res. 2003;985:198–201. doi: 10.1016/s0006-8993(03)03044-0. [DOI] [PubMed] [Google Scholar]
- 47.Uno M, Kitazato KT, Nishi K, Itabe H, Nagahiro S. Raised plasma oxidised LDL in acute cerebral infarction. J Neurol Neurosurg Psychiatry. 2003;74:312–6. doi: 10.1136/jnnp.74.3.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shie FS, Neely MD, Maezawa I, Wu H, Olson SJ, Jurgens G, Montine KS, Montine TJ. Oxidized low-density lipoprotein is present in astrocytes surrounding cerebral infarcts and stimulates astrocyte interleukin-6 secretion. Am J Pathol. 2004;164:1173–81. doi: 10.1016/S0002-9440(10)63205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–52. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 50.Hartz AM, Bauer B, Soldner EL, Wolf A, Boy S, Backhaus R, Mihaljevic I, Bogdahn U, Klunemann HH, Schuierer G, Schlachetzki F. Amyloid-beta Contributes to Blood-Brain Barrier Leakage in Transgenic Human Amyloid Precursor Protein Mice and in Humans With Cerebral Amyloid Angiopathy. Stroke. 2011 doi: 10.1161/STROKEAHA.111.627562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carrano A, Hoozemans JJ, van der Vies SM, Rozemuller AJ, van Horssen J, de Vries HE. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal. 2011;15:1167–78. doi: 10.1089/ars.2011.3895. [DOI] [PubMed] [Google Scholar]
- 52.Verdier Y, Zarandi M, Penke B. Amyloid beta-peptide interactions with neuronal and glial cell plasma membrane: binding sites and implications for Alzheimer’s disease. J Pept Sci. 2004;10:229–48. doi: 10.1002/psc.573. [DOI] [PubMed] [Google Scholar]
- 53.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature. 1996;382:716–9. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 54.Husemann J, Loike JD, Kodama T, Silverstein SC. Scavenger receptor class B type I (SR-BI) mediates adhesion of neonatal murine microglia to fibrillar beta-amyloid. J Neuroimmunol. 2001;114:142–50. doi: 10.1016/s0165-5728(01)00239-9. [DOI] [PubMed] [Google Scholar]
- 55.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 56.Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El-Khoury JB. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160:101–12. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–74. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilkinson B, Koenigsknecht-Talboo J, Grommes C, Lee CY, Landreth G. Fibrillar beta-amyloid-stimulated intracellular signaling cascades require Vav for induction of respiratory burst and phagocytosis in monocytes and microglia. J Biol Chem. 2006;281:20842–50. doi: 10.1074/jbc.M600627200. [DOI] [PubMed] [Google Scholar]
- 59.Park L, Wang G, Zhou P, Zhou J, Pitstick R, Previti ML, Younkin L, Younkin SG, Van Nostrand WE, Cho S, Anrather J, Carlson GA, Iadecola C. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-beta. Proc Natl Acad Sci U S A. 2011;108:5063–8. doi: 10.1073/pnas.1015413108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold KH, Tolnay M, Staufenbiel M, Jucker M. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J Neurosci. 2001;21:1619–27. doi: 10.1523/JNEUROSCI.21-05-01619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hernandez-Guillamon M, Martinez-Saez E, Delgado P, Domingues-Montanari S, Boada C, Penalba A, Boada M, Pagola J, Maisterra O, Rodriguez-Luna D, Molina CA, Rovira A, Alvarez-Sabin J, Ortega-Aznar A, Montaner J. MMP-2/MMP-9 Plasma Level and Brain Expression in Cerebral Amyloid Angiopathy-Associated Hemorrhagic Stroke. Brain Pathol. 2011 doi: 10.1111/j.1750-3639.2011.00512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee PH, Bang OY, Hwang EM, Lee JS, Joo US, Mook-Jung I, Huh K. Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J Neural Transm. 2005;112:1371–9. doi: 10.1007/s00702-004-0274-0. [DOI] [PubMed] [Google Scholar]
- 63.Engstrom G, Stavenow L, Hedblad B, Lind P, Eriksson KF, Janzon L, Lindgarde F. Inflammation-sensitive plasma proteins, diabetes, and mortality and incidence of myocardial infarction and stroke: a population-based study. Diabetes. 2003;52:442–7. doi: 10.2337/diabetes.52.2.442. [DOI] [PubMed] [Google Scholar]
- 64.Pinto A, Tuttolomondo A, Di Raimondo D, Fernandez P, Licata G. Cerebrovascular risk factors and clinical classification of strokes. Semin Vasc Med. 2004;4:287–303. doi: 10.1055/s-2004-861497. [DOI] [PubMed] [Google Scholar]
- 65.Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, Chen J, Zhang R, Silverstein RL, Hazen SL. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–95. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim E, Tolhurst AT, Qin LY, Chen XY, Febbraio M, Cho S. CD36/fatty acid translocase, an inflammatory mediator, is involved in hyperlipidemia-induced exacerbation in ischemic brain injury. J Neurosci. 2008;28:4661–70. doi: 10.1523/JNEUROSCI.0982-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.d’Uscio LV, Baker TA, Mantilla CB, Smith L, Weiler D, Sieck GC, Katusic ZS. Mechanism of endothelial dysfunction in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2001a;21:1017–22. doi: 10.1161/01.atv.21.6.1017. [DOI] [PubMed] [Google Scholar]
- 68.d’Uscio LV, Smith LA, Katusic ZS. Hypercholesterolemia impairs endothelium-dependent relaxations in common carotid arteries of apolipoprotein e-deficient mice. Stroke. 2001b;32:2658–64. doi: 10.1161/hs1101.097393. [DOI] [PubMed] [Google Scholar]
- 69.Smart EJ, Graf GA, McNiven MA, Sessa WC, Engelman JA, Scherer PE, Okamoto T, Lisanti MP. Caveolins, liquid-ordered domains, and signal transduction. Mol Cell Biol. 1999;19:7289–304. doi: 10.1128/mcb.19.11.7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Uittenbogaard A, Shaul PW, Yuhanna IS, Blair A, Smart EJ. High density lipoprotein prevents oxidized low density lipoprotein-induced inhibition of endothelial nitric-oxide synthase localization and activation in caveolae. J Biol Chem. 2000;275:11278–83. doi: 10.1074/jbc.275.15.11278. [DOI] [PubMed] [Google Scholar]
- 71.Blair A, Shaul PW, Yuhanna IS, Conrad PA, Smart EJ. Oxidized low density lipoprotein displaces endothelial nitric-oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation. J Biol Chem. 1999;274:32512–9. doi: 10.1074/jbc.274.45.32512. [DOI] [PubMed] [Google Scholar]
- 72.Kincer JF, Uittenbogaard A, Dressman J, Guerin TM, Febbraio M, Guo L, Smart EJ. Hypercholesterolemia promotes a CD36-dependent and endothelial nitric-oxide synthase-mediated vascular dysfunction. J Biol Chem. 2002;277:23525–33. doi: 10.1074/jbc.M202465200. [DOI] [PubMed] [Google Scholar]
- 73.Shentu TP, Titushkin I, Singh DK, Gooch KJ, Subbaiah PV, Cho M, Levitan I. oxLDL-induced decrease in lipid order of membrane domains is inversely correlated with endothelial stiffness and network formation. Am J Physiol Cell Physiol. 2010;299:C218–29. doi: 10.1152/ajpcell.00383.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aitman TJ, Glazier AM, Wallace CA, Cooper LD, Norsworthy PJ, Wahid FN, Al-Majali KM, Trembling PM, Mann CJ, Shoulders CC, Graf D, St Lezin E, Kurtz TW, Kren V, Pravenec M, Ibrahimi A, Abumrad NA, Stanton LW, Scott J. Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat Genet. 1999;21:76–83. doi: 10.1038/5013. [DOI] [PubMed] [Google Scholar]
- 75.Griffin E, Re A, Hamel N, Fu C, Bush H, McCaffrey T, Asch AS. A link between diabetes and atherosclerosis: Glucose regulates expression of CD36 at the level of translation. Nat Med. 2001;7:840–6. doi: 10.1038/89969. [DOI] [PubMed] [Google Scholar]
- 76.Handberg A, Hojlund K, Gastaldelli A, Flyvbjerg A, Dekker JM, Petrie J, Piatti P, Beck-Nielsen H. Plasma sCD36 is associated with markers of atherosclerosis, insulin resistance and fatty liver in a nondiabetic healthy population. J Intern Med. 2011 doi: 10.1111/j.1365-2796.2011.02442.x. [DOI] [PubMed] [Google Scholar]
- 77.Handberg A, Levin K, Hojlund K, Beck-Nielsen H. Identification of the oxidized low-density lipoprotein scavenger receptor CD36 in plasma: a novel marker of insulin resistance. Circulation. 2006;114:1169–76. doi: 10.1161/CIRCULATIONAHA.106.626135. [DOI] [PubMed] [Google Scholar]
- 78.Sampson MJ, Davies IR, Braschi S, Ivory K, Hughes DA. Increased expression of a scavenger receptor (CD36) in monocytes from subjects with Type 2 diabetes. Atherosclerosis. 2003;167:129–34. doi: 10.1016/s0021-9150(02)00421-5. [DOI] [PubMed] [Google Scholar]
- 79.Susztak K, Ciccone E, McCue P, Sharma K, Bottinger EP. Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2005;2:e45. doi: 10.1371/journal.pmed.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ohgami N, Nagai R, Ikemoto M, Arai H, Kuniyasu A, Horiuchi S, Nakayama H. CD36, a member of class B scavenger receptor family, is a receptor for advanced glycation end products. Ann N Y Acad Sci. 2001;947:350–5. doi: 10.1111/j.1749-6632.2001.tb03961.x. [DOI] [PubMed] [Google Scholar]
- 81.de Oliveira Silva C, Delbosc S, Arais C, Monnier L, Cristol JP, Pares-Herbute N. Modulation of CD36 protein expression by AGEs and insulin in aortic VSMCs from diabetic and non-diabetic rats. Nutr Metab Cardiovasc Dis. 2006 doi: 10.1016/j.numecd.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 82.Sima AV, Botez GM, Stancu CS, Manea A, Raicu M, Simionescu M. Effect of irreversibly glycated LDL in human vascular smooth muscle cells: lipid loading, oxidative and inflammatory stress. J Cell Mol Med. 2010;14:2790–802. doi: 10.1111/j.1582-4934.2009.00933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Slevin M, Kumar P, Gaffney J, Kumar S, Krupinski J. Can angiogenesis be exploited to improve stroke outcome? Mechanisms and therapeutic potential. Clin Sci (Lond) 2006;111:171–83. doi: 10.1042/CS20060049. [DOI] [PubMed] [Google Scholar]
- 84.Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke. 1994;25:1794–8. doi: 10.1161/01.str.25.9.1794. [DOI] [PubMed] [Google Scholar]
- 85.Hayashi T, Deguchi K, Nagotani S, Zhang H, Sehara Y, Tsuchiya A, Abe K. Cerebral ischemia and angiogenesis. Curr Neurovasc Res. 2006;3:119–29. doi: 10.2174/156720206776875902. [DOI] [PubMed] [Google Scholar]
- 86.Rosell-Novel A, Montaner J, Alvarez-Sabin J. Angiogenesis in human cerebral ischemia. Rev Neurol. 2004;38:1076–82. [PubMed] [Google Scholar]
- 87.Chen HH, Chien CH, Liu HM. Correlation between angiogenesis and basic fibroblast growth factor expression in experimental brain infarct. Stroke. 1994;25:1651–7. doi: 10.1161/01.str.25.8.1651. [DOI] [PubMed] [Google Scholar]
- 88.Szpak GM, Lechowicz W, Lewandowska E, Bertrand E, Wierzba-Bobrowicz T, Dymecki J. Border zone neovascularization in cerebral ischemic infarct. Folia Neuropathol. 1999;37:264–8. [PubMed] [Google Scholar]
- 89.Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM. Some remarks on the growth-rate and angiogenesis of microvessels in ischemic stroke. Morphometric and immunocytochemical studies. Patol Pol. 1993;44:203–9. [PubMed] [Google Scholar]
- 90.Iruela-Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD. Inhibition of angiogenesis by thrombospondin-1 is mediated by 2 independent regions within the type 1 repeats. Circulation. 1999;100:1423–31. doi: 10.1161/01.cir.100.13.1423. [DOI] [PubMed] [Google Scholar]
- 91.Armstrong LC, Bornstein P. Thrombospondins 1 and 2 function as inhibitors of angiogenesis. Matrix Biol. 2003;22:63–71. doi: 10.1016/s0945-053x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 92.Primo L, Ferrandi C, Roca C, Marchio S, di Blasio L, Alessio M, Bussolino F. Identification of CD36 molecular features required for its in vitro angiostatic activity. FASEB J. 2005;19:1713–5. doi: 10.1096/fj.05-3697fje. [DOI] [PubMed] [Google Scholar]
- 93.Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004;36:961–8. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin TN, Kim GM, Chen JJ, Cheung WM, He YY, Hsu CY. Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion. Stroke. 2003;34:177–86. doi: 10.1161/01.str.0000047100.84604.ba. [DOI] [PubMed] [Google Scholar]
- 95.Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, Roberts DD. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc Natl Acad Sci U S A. 2005;102:13141–6. doi: 10.1073/pnas.0502977102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Isenberg JS, Hyodo F, Ridnour LA, Shannon CS, Wink DA, Krishna MC, Roberts DD. Thrombospondin 1 and vasoactive agents indirectly alter tumor blood flow. Neoplasia. 2008;10:886–96. doi: 10.1593/neo.08264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Simantov R, Febbraio M, Silverstein RL. The antiangiogenic effect of thrombospondin-2 is mediated by CD36 and modulated by histidine-rich glycoprotein. Matrix Biol. 2005;24:27–34. doi: 10.1016/j.matbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 98.Asch AS, Barnwell J, Silverstein RL, Nachman RL. Isolation of the thrombospondin membrane receptor. J Clin Invest. 1987;79:1054–61. doi: 10.1172/JCI112918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Silverstein RL, Baird M, Lo SK, Yesner LM. Sense and antisense cDNA transfection of CD36 (glycoprotein IV) in melanoma cells. Role of CD36 as a thrombospondin receptor. J Biol Chem. 1992;267:16607–12. [PubMed] [Google Scholar]
- 100.Isenberg JS, Shiva S, Gladwin M. Thrombospondin-1-CD47 blockade and exogenous nitrite enhance ischemic tissue survival, blood flow and angiogenesis via coupled NO-cGMP pathway activation. Nitric Oxide. 2009;21:52–62. doi: 10.1016/j.niox.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang JP, Liu HJ, Liu XF. VEGF promotes angiogenesis and functional recovery in stroke rats. J Invest Surg. 2010;23:149–55. doi: 10.3109/08941930903469482. [DOI] [PubMed] [Google Scholar]
- 102.Lee HJ, Kim KS, Park IH, Kim SU. Human neural stem cells over-expressing VEGF provide neuroprotection, angiogenesis and functional recovery in mouse stroke model. PLoS One. 2007;2:e156. doi: 10.1371/journal.pone.0000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zacharek A, Chen J, Cui X, Li A, Li Y, Roberts C, Feng Y, Gao Q, Chopp M. Angiopoietin1/Tie2 and VEGF/Flk1 induced by MSC treatment amplifies angiogenesis and vascular stabilization after stroke. J Cereb Blood Flow Metab. 2007;27:1684–91. doi: 10.1038/sj.jcbfm.9600475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Navarro-Sobrino M, Rosell A, Hernandez-Guillamon M, Penalba A, Boada C, Domingues-Montanari S, Ribo M, Alvarez-Sabin J, Montaner J. A large screening of angiogenesis biomarkers and their association with neurological outcome after ischemic stroke. Atherosclerosis. 2011;216:205–11. doi: 10.1016/j.atherosclerosis.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 105.Donovan MJ, Lin MI, Wiegn P, Ringstedt T, Kraemer R, Hahn R, Wang S, Ibanez CF, Rafii S, Hempstead BL. Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development. 2000;127:4531–40. doi: 10.1242/dev.127.21.4531. [DOI] [PubMed] [Google Scholar]
- 106.Coppola V, Barrick CA, Southon EA, Celeste A, Wang K, Chen B, Haddad el B, Yin J, Nussenzweig A, Subramaniam A, Tessarollo L. Ablation of TrkA function in the immune system causes B cell abnormalities. Development. 2004;131:5185–95. doi: 10.1242/dev.01383. [DOI] [PubMed] [Google Scholar]
- 107.Nemoto K, Fukamachi K, Nemoto F, Miyata S, Hamada M, Nakamura Y, Senba E, Ueyama T. Gene expression of neurotrophins and their receptors in cultured rat vascular smooth muscle cells. Biochem Biophys Res Commun. 1998;245:284–8. doi: 10.1006/bbrc.1998.8418. [DOI] [PubMed] [Google Scholar]
- 108.Botchkarev VA, Metz M, Botchkareva NV, Welker P, Lommatzsch M, Renz H, Paus R. Brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4 act as “epitheliotrophins” in murine skin. Lab Invest. 1999;79:557–72. [PubMed] [Google Scholar]
- 109.Kermani P, Hempstead B. Brain-derived neurotrophic factor: a newly described mediator of angiogenesis. Trends Cardiovasc Med. 2007;17:140–3. doi: 10.1016/j.tcm.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kermani P, Rafii D, Jin DK, Whitlock P, Schaffer W, Chiang A, Vincent L, Friedrich M, Shido K, Hackett NR, Crystal RG, Rafii S, Hempstead BL. Neurotrophins promote revascularization by local recruitment of TrkB+ endothelial cells and systemic mobilization of hematopoietic progenitors. J Clin Invest. 2005;115:653–63. doi: 10.1172/JCI200522655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde YA. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. Embo J. 1990;9:2459–64. doi: 10.1002/j.1460-2075.1990.tb07423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Phillips HS, Hains JM, Laramee GR, Rosenthal A, Winslow JW. Widespread expression of BDNF but not NT3 by target areas of basal forebrain cholinergic neurons. Science. 1990;250:290–4. doi: 10.1126/science.1688328. [DOI] [PubMed] [Google Scholar]
- 113.Kokaia Z, Andsberg G, Yan Q, Lindvall O. Rapid alterations of BDNF protein levels in the rat brain after focal ischemia: evidence for increased synthesis and anterograde axonal transport. Exp Neurol. 1998;154:289–301. doi: 10.1006/exnr.1998.6888. [DOI] [PubMed] [Google Scholar]
- 114.Kokaia Z, Zhao Q, Kokaia M, Elmer E, Metsis M, Smith ML, Siesjo BK, Lindvall O. Regulation of brain-derived neurotrophic factor gene expression after transient middle cerebral artery occlusion with and without brain damage. Exp Neurol. 1995;136:73–88. doi: 10.1006/exnr.1995.1085. [DOI] [PubMed] [Google Scholar]
- 115.Chen J, Li Y, Chopp M. Intracerebral transplantation of bone marrow with BDNF after MCAo in rat. Neuropharmacology. 2000;39:711–6. doi: 10.1016/s0028-3908(00)00006-x. [DOI] [PubMed] [Google Scholar]
- 116.Muller HD, Hanumanthiah KM, Diederich K, Schwab S, Schabitz WR, Sommer C. Brain-derived neurotrophic factor but not forced arm use improves long-term outcome after photothrombotic stroke and transiently upregulates binding densities of excitatory glutamate receptors in the rat brain. Stroke. 2008;39:1012–21. doi: 10.1161/STROKEAHA.107.495069. [DOI] [PubMed] [Google Scholar]
- 117.Schabitz WR, Berger C, Kollmar R, Seitz M, Tanay E, Kiessling M, Schwab S, Sommer C. Effect of brain-derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke. 2004;35:992–7. doi: 10.1161/01.STR.0000119754.85848.0D. [DOI] [PubMed] [Google Scholar]
- 118.Kurozumi K, Nakamura K, Tamiya T, Kawano Y, Kobune M, Hirai S, Uchida H, Sasaki K, Ito Y, Kato K, Honmou O, Houkin K, Date I, Hamada H. BDNF gene-modified mesenchymal stem cells promote functional recovery and reduce infarct size in the rat middle cerebral artery occlusion model. Mol Ther. 2004;9:189–97. doi: 10.1016/j.ymthe.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 119.Nygren J, Kokaia M, Wieloch T. Decreased expression of brain-derived neurotrophic factor in BDNF(+/−) mice is associated with enhanced recovery of motor performance and increased neuroblast number following experimental stroke. J Neurosci Res. 2006;84:626–31. doi: 10.1002/jnr.20956. [DOI] [PubMed] [Google Scholar]
- 120.Gustafsson E, Andsberg G, Darsalia V, Mohapel P, Mandel RJ, Kirik D, Lindvall O, Kokaia Z. Anterograde delivery of brain-derived neurotrophic factor to striatum via nigral transduction of recombinant adeno-associated virus increases neuronal death but promotes neurogenic response following stroke. Eur J Neurosci. 2003;17:2667–78. doi: 10.1046/j.1460-9568.2003.02713.x. [DOI] [PubMed] [Google Scholar]
- 121.He XM, Zhang ZX, Zhang JW, Zhou YT, Tang MN, Wu CB, Hong Z. Lack of association between the BDNF gene Val66Met polymorphism and Alzheimer disease in a Chinese Han population. Neuropsychobiology. 2007;55:151–5. doi: 10.1159/000106473. [DOI] [PubMed] [Google Scholar]
- 122.Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–69. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 123.Egan MF, Weinberger DR, Lu B. Schizophrenia, III: brain-derived neurotropic factor and genetic risk. Am J Psychiatry. 2003;160:1242. doi: 10.1176/appi.ajp.160.7.1242. [DOI] [PubMed] [Google Scholar]
- 124.Ventriglia M, Bocchio Chiavetto L, Benussi L, Binetti G, Zanetti O, Riva MA, Gennarelli M. Association between the BDNF 196 A/G polymorphism and sporadic Alzheimer’s disease. Mol Psychiatry. 2002;7:136–7. doi: 10.1038/sj.mp.4000952. [DOI] [PubMed] [Google Scholar]
- 125.Itoh K, Hashimoto K, Kumakiri C, Shimizu E, Iyo M. Association between brain-derived neurotrophic factor 196 G/A polymorphism and personality traits in healthy subjects. Am J Med Genet B Neuropsychiatr Genet. 2004;124B:61–3. doi: 10.1002/ajmg.b.20078. [DOI] [PubMed] [Google Scholar]
- 126.Sen S, Nesse RM, Stoltenberg SF, Li S, Gleiberman L, Chakravarti A, Weder AB, Burmeister M. A BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depression. Neuropsychopharmacology. 2003;28:397–401. doi: 10.1038/sj.npp.1300053. [DOI] [PubMed] [Google Scholar]
- 127.Sklar P, Gabriel SB, McInnis MG, Bennett P, Lim YM, Tsan G, Schaffner S, Kirov G, Jones I, Owen M, Craddock N, DePaulo JR, Lander ES. Family-based association study of 76 candidate genes in bipolar disorder: BDNF is a potential risk locus. Brain-derived neutrophic factor. Mol Psychiatry. 2002;7:579–93. doi: 10.1038/sj.mp.4001058. [DOI] [PubMed] [Google Scholar]
- 128.Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL, Lee FS. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–3. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Siironen J, Juvela S, Kanarek K, Vilkki J, Hernesniemi J, Lappalainen J. The Met allele of the BDNF Val66Met polymorphism predicts poor outcome among survivors of aneurysmal subarachnoid hemorrhage. Stroke. 2007;38:2858–60. doi: 10.1161/STROKEAHA.107.485441. [DOI] [PubMed] [Google Scholar]
- 130.Vilkki J, Lappalainen J, Juvela S, Kanarek K, Hernesniemi JA, Siironen J. Relationship of the met allele of the brain-derived neurotrophic factor Val66Met polymorphism to memory after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2008;63:198–203. doi: 10.1227/01.NEU.0000320382.21577.8E. discussion 203. [DOI] [PubMed] [Google Scholar]
- 131.Krueger F, Pardini M, Huey ED, Raymont V, Solomon J, Lipsky RH, Hodgkinson CA, Goldman D, Grafman J. The role of the Met66 brain-derived neurotrophic factor allele in the recovery of executive functioning after combat-related traumatic brain injury. J Neurosci. 2011;31:598–606. doi: 10.1523/JNEUROSCI.1399-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Qin L, Kim E, Ratan R, Lee FS, Cho S. Genetic variant of BDNF (Val66Met) polymorphism attenuates stroke-induced angiogenic responses by enhancing anti-angiogenic mediator CD36 expression. J Neurosci. 2011;31:775–83. doi: 10.1523/JNEUROSCI.4547-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A Novel Cell-permeable Antioxidant Peptide, SS31, Attenuates Ischemic Brain Injury by Down-regulating CD36. J Biol Chem. 2007;282:4634–42. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- 134.Cho S, Kim E. CD36: a multi-modal target for acute stroke therapy. J Neurochem. 2009;109 (Suppl 1):126–32. doi: 10.1111/j.1471-4159.2009.05801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chnari E, Nikitczuk JS, Wang J, Uhrich KE, Moghe PV. Engineered polymeric nanoparticles for receptor-targeted blockage of oxidized low density lipoprotein uptake and atherogenesis in macrophages. Biomacromolecules. 2006;7:1796–805. doi: 10.1021/bm0600872. [DOI] [PubMed] [Google Scholar]
- 136.Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, Hamelin M, Hreniuk DL, Palyha OC, Anderson J, Paress PS, Diaz C, Chou M, Liu KK, McKee KK, Pong SS, Chaung LY, Elbrecht A, Dashkevicz M, Heavens R, Rigby M, Sirinathsinghji DJ, Dean DC, Melillo DG, Van der Ploeg LH, et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science. 1996;273:974–7. doi: 10.1126/science.273.5277.974. [DOI] [PubMed] [Google Scholar]
- 137.Demers A, McNicoll N, Febbraio M, Servant M, Marleau S, Silverstein R, Ong H. Identification of the growth hormone-releasing peptide binding site in CD36: a photoaffinity cross-linking study. Biochem J. 2004;382:417–24. doi: 10.1042/BJ20040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Marleau S, Harb D, Bujold K, Avallone R, Iken K, Wang Y, Demers A, Sirois MG, Febbraio M, Silverstein RL, Tremblay A, Ong H. EP 80317, a ligand of the CD36 scavenger receptor, protects apolipoprotein E-deficient mice from developing atherosclerotic lesions. Faseb J. 2005;19:1869–71. doi: 10.1096/fj.04-3253fje. [DOI] [PubMed] [Google Scholar]
- 139.Ong H, Tremblay A, Febbriao M, Silverstein RL, Avallone R, Iken K, Wang Y, Bujoid K, Demers A, Sirois MG. Growth hormone-releasing peptides as inhibitors of fatty streaks formation: A new therapy for ahterosclerosis. In programs and abstracts fo the 85th Annual Meeting of the endocrine Socienty; 2003. p. 223. [Google Scholar]
- 140.Wang L, Bao Y, Yang Y, Wu Y, Chen X, Si S, Hong B. Discovery of antagonists for human scavenger receptor CD36 via an ELISA-like high-throughput screening assay. J Biomol Screen. 2010;15:239–50. doi: 10.1177/1087057109359686. [DOI] [PubMed] [Google Scholar]
- 141.Tang M, Feng W, Zhang Y, Zhong J, Zhang J. Salvianolic acid B improves motor function after cerebral ischemia in rats. Behav Pharmacol. 2006;17:493–8. doi: 10.1097/00008877-200609000-00015. [DOI] [PubMed] [Google Scholar]
- 142.Han J, Zhou X, Yokoyama T, Hajjar DP, Gotto AM, Jr, Nicholson AC. Pitavastatin downregulates expression of the macrophage type B scavenger receptor, CD36. Circulation. 2004;109:790–6. doi: 10.1161/01.CIR.0000112576.40815.13. [DOI] [PubMed] [Google Scholar]
- 143.Fuhrman B, Volkova N, Aviram M. Oxidative stress increases the expression of the CD36 scavenger receptor and the cellular uptake of oxidized low-density lipoprotein in macrophages from atherosclerotic mice: protective role of antioxidants and of paraoxonase. Atherosclerosis. 2002;161:307–16. doi: 10.1016/s0021-9150(01)00646-3. [DOI] [PubMed] [Google Scholar]
- 144.Puccetti L, Sawamura T, Pasqui AL, Pastorelli M, Auteri A, Bruni F. Atorvastatin reduces platelet-oxidized-LDL receptor expression in hypercholesterolaemic patients. Eur J Clin Invest. 2005;35:47–51. doi: 10.1111/j.1365-2362.2005.01446.x. [DOI] [PubMed] [Google Scholar]
- 145.Wilkinson K, Boyd JD, Glicksman M, Moore KJ, El Khoury J. A high content drug screen identifies ursolic acid as an inhibitor of amyloid beta protein interactions with its receptor CD36. J Biol Chem. 2011;286:34914–22. doi: 10.1074/jbc.M111.232116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Venugopal SK, Devaraj S, Jialal I. RRR-alpha-tocopherol decreases the expression of the major scavenger receptor, CD36, in human macrophages via inhibition of tyrosine kinase (Tyk2) Atherosclerosis. 2004;175:213–20. doi: 10.1016/j.atherosclerosis.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 147.Ricciarelli R, Zingg JM, Azzi A. Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation. 2000;102:82–7. doi: 10.1161/01.cir.102.1.82. [DOI] [PubMed] [Google Scholar]
- 148.Munteanu A, Zingg JM, Ogru E, Libinaki R, Gianello R, West S, Negis Y, Azzi A. Modulation of cell proliferation and gene expression by alpha-tocopheryl phosphates: relevance to atherosclerosis and inflammation. Biochem Biophys Res Commun. 2004;318:311–6. doi: 10.1016/j.bbrc.2004.04.028. [DOI] [PubMed] [Google Scholar]
- 149.Janabi M, Yamashita S, Hirano K, Sakai N, Hiraoka H, Matsumoto K, Zhang Z, Nozaki S, Matsuzawa Y. Oxidized LDL-induced NF-kappa B activation and subsequent expression of proinflammatory genes are defective in monocyte-derived macrophages from CD36-deficient patients. Arterioscler Thromb Vasc Biol. 2000;20:1953–60. doi: 10.1161/01.atv.20.8.1953. [DOI] [PubMed] [Google Scholar]
- 150.Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–90. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 151.Szeto HH. Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J. 2006;8:E277–83. doi: 10.1007/BF02854898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhao K, Luo G, Giannelli S, Szeto HH. Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochem Pharmacol. 2005;70:1796–806. doi: 10.1016/j.bcp.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 153.Cho S, Szeto HH, Kin E, Kim H, ATT, JPP A novel cell permeable antioxidant peptide, SS31, attneuates ischemic brain injury by down-regulating CD36. 2007 doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]