

Abstract
To prevent excessive inflammatory responses to commensal microbes, intestinal macrophages unlike their systemic counterparts do not produce inflammatory cytokines in response to enteric bacteria. Consequently, loss of macrophage tolerance to the enteric microbiota plays a central role in the pathogenesis of the inflammatory bowel diseases. Therefore, we examined whether the hyporesponsive phenotype of intestinal macrophages is programmed by prior exposure to the microbiota. IL-10, but not in vivo exposure to the microbiota, programs intestinal macrophage tolerance, as wild-type (WT) colonic macrophages from germ free and specific-pathogen free (SPF) derived mice produce IL-10 but not IL-12 p40 when activated with enteric bacteria. Basal and activated IL-10 expression is mediated through a MyD88 dependent pathway. Conversely, colonic macrophages from germ free and SPF derived colitis-prone Il10−/− mice demonstrated robust production of IL-12 p40. Next, mechanisms through which IL-10 inhibits Il12b expression were investigated. While Il12b mRNA was transiently induced in LPS-activated WT bone marrow derived macrophages (BMDMs), expression persisted in Il10−/− BMDMs. There were no differences in nucleosome remodeling, mRNA stability, NF-κB activation or MAPK signaling to explain prolonged transcription of Il12b in Il10−/− BMDMs. However, acetylated histone H4 (AcH4) transiently associated with the Il12b promoter in WT BMDMs, whereas association of these factors was prolonged in Il10−/− BMDMs. Experiments utilizing histone deacetylase (HDAC) inhibitors and HDAC3 shRNA indicate that HDAC3 is involved in histone deacetylation of the Il12b promoter by IL-10. These results suggest that histone deacetylation on the Il12b promoter by HDAC3 mediates homeostatic effects of IL-10 in macrophages.
INTRODUCTION
The gastrointestinal tract represents a complex interface between the enteric microbiota and immune cell populations. A multitude of diverse microorganisms reside in the intestinal lumen separated from the body’s largest reservoir of macrophages by a single layer of epithelial cells. These macrophages serve as the first line of defense against the external environment. To prevent excessive inflammatory responses to commensal microbes, intestinal macrophages have acquired a unique phenotype. Intestinal macrophages, unlike their systemic counterparts, do not produce inflammatory cytokines in response to enteric bacteria (1). Consequently, loss of macrophage tolerance to the enteric microbiota plays a central role in the pathogenesis of the inflammatory bowel diseases (IBD) (2, 3).
The anti-inflammatory cytokine IL-10 is implicated in the maintenance of intestinal homeostasis. Mutations in genes encoding the IL-10 receptor subunit proteins IL10RA and IL10RB were reported in patients with early onset enterocolitis (4). Moreover, mice deficient in IL-10 or IL-10 receptors develop spontaneously occurring intestinal inflammation dependent on the presence of the enteric microbiota (5, 6). IL-10 is secreted by many cell types, including T cells, mast cells, epithelial cells, macrophages, and dendritic cells; however, a major source of IL-10 involved in the maintenance of intestinal homeostasis is lamina propria or mesenteric lymph node macrophages (7–9).
The IL-12 family members IL-12 and IL-23 expressed by macrophages are important inhibitory targets of IL-10 and are central mediators of chronic intestinal inflammation (10–13). IL-12/IL-23 p40 (encoded by the Il12b gene) is the common subunit of IL-12 and IL-23. Despite extensive investigation, molecular mechanisms through which IL-10 inhibits Il12b expression have not been fully elucidated (14–16).
Gene transcription is regulated at the chromatin level. DNA-binding factors cannot access DNA in closed chromatin. Therefore, chromatin structure needs to be altered to facilitate gene transcription (17). Histone acetylation induces an open chromatin conformation that allows the transcription machinery to access promoters, whereas histone deacetylation correlates with gene silencing. Inducible chromatin modifications serve as important restriction points in TLR-regulated gene expression. Recruitment of histone acetyltransferases (HATs) such as p300 and CREB-binding protein (CBP) to the Il12b promoter has been implicated in its transcriptional activation (18). TLR stimulation of macrophages results in rapid changes in chromatin remodeling at the Il12b locus via histone acetylation, enabling transcription factor recruitment (17). Accordingly, histone deacetylation on the Il12b promoter by histone deacetylase (HDAC) negatively regulates Il12b transcription (19). Therefore, epigenetic changes that inhibit and induce Il12b expression in macrophages are likely to be central determinants of intestinal homeostasis and inflammation, respectively.
Here, we report that the anti-inflammatory phenotype of resident colonic macrophages is programmed by IL-10 without requirement for exposure to the microbiota in vivo. In bone marrow derived macrophages (BMDM), IL-10 inhibits IL-12/IL-23 p40 expression through altered kinetics of histone acetylation on the Il12b promoter. Inhibition of HDAC3 results in decreased inhibition of Il12b by IL-10. These experiments suggest that histone deacetylation on the Il12b promoter by HDACs mediates homeostatic effects of IL-10 in macrophages. Consequently, the absence of IL-10 leads to prolonged histone acetylation with persistent transcription of Il12b.
MATERIALS AND METHODS
Mice
Wild-type (WT) and Il10−/− mice on 129/SvEv background were used to isolate colonic CD11b+ lamina propria mononuclear cells (LPMCs). IL-10–IRES-EGFP reporter (Vert-X) mice were created by insertion of a floxed neomycin–IRES-EGFP cassette between the endogenous stop site and the polyadenosine site of IL-10 (20). Germ-free (GF) mice were maintained in the Gnotobiotic Core Facility at the University of North Carolina at Chapel Hill. WT and Il10−/− mice on C57BL/6 background maintained in specific-pathogen free (SPF) condition were used for bone marrow derived macrophage (BMDM) derivation. All animal experiments were in accordance with protocols approved by the International Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Reagents
LPS was purchased from InvivoGen (San Diego, CA). M-CSF and IL-10 were obtained from PeproTech Inc (Rocky Hill, NJ). Heat-killed bacteria was prepared as described previously (1). Briefly, E. coli and E. faecalis in log-phase growth were harvested and washed twice with ice-cold PBS. Bacterial suspensions were heated at 80°C for 30 minutes, washed, resuspended in PBS, and stored at −80°C. Non-viability was confirmed by 72-hour incubation at 37°C on plate medium. Heat-killed bacteria were added at multiplicity of infection (MOI) 10 or 100 for cell stimulation. HDAC inhibitors, trichostatin A (TSA) and MS275 were obtained from Sigma (St. Louis, Mo) and Selleck Chemicals (Houston, TX), respectively.
Cell isolation
BMDMs were cultured as described previously (21). LPMCs were isolated from mouse colons by an enzymatic method as previously described (1). LPMCs were further separated into CD11b+ cells using anti-CD11b microbeads (Miltenyi Biotec, Auburn, CA).
Quantitative RT-PCR
Quantitative real-time RT-PCR was performed as described previously (22). Primer sequences are available upon request.
Chromatin immunoprecipitation assays (ChIP)
ChIP was performed with ChIP-IT Express kit (Active Motif, Carlsbad, CA) according to manufacturer’s instruction and as previously reported (23, 24). Briefly, 2×106 (for AcH4, me3H3K4) or 5×106 (for RNA polymerase II (RNA Pol II)) BMDMs were stimulated, washed with PBS, and fixed with 1% formaldehyde for 10 minutes at room temperature. Fixed cells were harvested, lysed, and sonicated for 10 cycles of 20-second on/20-second off with Sonic Dismembrator 60 (Thermo Fisher Scientific, Waltham, MA). For AcH4 ChIP, sodium butyrate (20 mM) was added to all the solutions to preserve histone acetylation. Antibodies for AcH4 and RNA pol II were obtained from Millipore (Billerica, MA), and me3H3K4 was from Abcam (Cambridge, MA). Primer pairs for monitoring binding to Il12b promoter were as follows: acetylated histone H4 (AcH4), forward 5′-ATGCACTCAGGGAGGCAAG-3′, reverse 5′-TCTGATGGAAACCCAAAGTAGAAAC-3′, RNA polymerase II (RNA Pol II), Forward 5′-GAAGGAACAGTGGGTGTCCAG-3′, Reverse 5′-AGGGAGTTAGCGACAGGGAAG-3′.
Restriction enzyme accessibility assay
To monitor nucleosome remodeling, chromatin accessibility was measured by real-time PCR as previously described (25, 26). Purified DNA was amplified by three sets of primers. PCR-based analysis was validated by Southern blot, as described (25) (Supplemental Fig. 1).
Lentivirus-mediated gene transduction
Lentiviral transduction was optimized based on manufacturer’s instructions for FuGENE Transfection Reagent (Roche, Indianapolis, IN) and as previously described (23). Lentivirus for HDAC3-specific shRNA was obtained from Open Biosystems (Huntsville, AL) and transduced to BMDMs from Il10−/− mice in 12-well plates. Transduced cells were selected by puromycin, and transduction efficiency was confirmed by Western blot and RT-PCR.
Enzyme-linked immunosorbent assay (ELISA)
IL-12 p40 and IL-10 concentrations were determined by sandwich ELISA according to the manufacturer’s instructions (BD Biosciences, San Jose, CA).
Western immunoblots
Western blot analyses were performed on whole cell extracts as described previously (21). NF-κB p65, IκBα, phospho-p38, and phospho-ERK antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), phospho-NF-κB p65, phospho-IκBα, and phospho-JNK antibodies were obtained from Cell Signaling (Danvers, MA).
Statistical analysis
Statistical significance for data subsets were assessed by the two-tailed Student’s t test. p values < 0.05 were considered to be significant. All data are expressed as mean ± standard error (SEM).
RESULTS
IL-10 but not the enteric microbiota programs the anti-inflammatory phenotype of colonic macrophages
Germ free (GF) Il10−/− mice, but not wild type (WT) mice, develop colitis when colonized with the enteric microbiota (27). Colitis in Il10−/− mice is associated with increased colonic production of IL-12/23 p40 (24). As peripheral macrophages become tolerized to activation of inflammatory pathways upon prolonged or repeated exposure to pathogen associated molecular patterns (28, 29), we reasoned that colonic macrophages may require exposure to the enteric microbiota in vivo to develop a tolerant, anti-inflammatory phenotype. To address this question, colonic CD11b+ lamina propria mononuclear cells (LPMCs) were isolated from GF and specific pathogen free (SPF) colonized WT mice (Fig. 1A and B). Surprisingly, CD11b+ LPMCs from GF WT mice activated with heat killed E. coli or E. faecalis did not express IL-12/23 p40 (Fig. 1A), but secrete basal and activated IL-10 (Fig. 1B). This suggests that the anti-inflammatory phenotype is programmed by factors in the local microenvironment and does not require in vivo exposure to the enteric microbiota. CD11b+ LPMCs isolated from SPF colonized WT mice demonstrated patterns of IL-12/23 p40 and IL-10 expression identical to that of CD11b+ LPMCs from GF WT mice (Fig. 1A and B). Interestingly, CD11b+ LPMCs from GF Il10−/− mice demonstrate IL-12/23 p40 production upon activation with E. coli and E. faecalis (Fig. 1A). Indeed, IL-12 p40 activation was as robust in GF CD11b+ LPMCs as in CD11b+ LPMCs from SPF colonized Il10−/− mice (Fig. 1A). No difference was observed in expression of surface markers (F4/80, CD11c, CD40, MHC II, CD80, CD86) between WT and Il10−/− CD11b+ LPMCs (from GF or SPF colons). CD40, CD86 and MHC II expression appeared to be higher in GF compared to SPF CD11b+ LPMCs, although these differences did not impact Il10 or Il12b expression (Supplemental Fig. 2). To substantiate the role of IL-10 in determining colonic macrophage phenotype, CD11b+ LPMCs from GF WT colons were cultured with blocking antibodies to IL-10 and IL-10 receptor prior to activation with heat-killed E. coli and E. faecalis. By blocking IL-10 signaling, activated WT colonic CD11b+ LPMCs demonstrate robust production of IL-12/23 p40 compared to CD11b+ LPMCs cultured with isotype control antibodies (Fig. 1C). These results indicate that endogenous IL-10 production is essential for the regulation of IL-12 p40 and consequently, the anti-inflammatory phenotype of colonic macrophages. Interestingly, IL-10 production by colonic CD11b+ LPMCs was MyD88-dependent (Fig. 1D): Both basal and enteric bacteria-activated IL-10 expression is absent in colonic CD11b+ LPMC from SPF colonized Myd88−/− but not Trif−/− mice, suggesting that endogenous signals through MyD88 control homeostatic macrophage function. The in vivo expression of IL-10 in different subsets of CD11b+ cells was further elucidated using IL-10 transcriptional GFP reporter mice, Vert-X mice (20). First, IL-10 expressing cells were quantitated and compared in colonic CD11b+CD11c− macrophage and CD11b+ CD11c+ dendritic cell populations from SPF-raised Vert-X mice. CD11b+CD11c− macrophages demonstrated greater numbers of IL-10 producing cells compared to CD11b+CD11c+ dendritic cells, as reported previously (30) (Fig. 1E). We confirmed that the presence and abundance of IL-10 producing CD11b+CD11c− macrophages was independent of colonization status by the enteric microbiota in Vert-X mice raised in GF, SPF or transferred from GF to SPF microbiota (Fig. 1F). Overall, these findings implicate locally produced IL-10, not exposure to the microbiota, as a requisite factor determining colonic macrophage phenotype through attenuated expression of IL-12/23 p40 upon subsequent exposure to enteric microbial products.
Figure 1. IL-10 but not the enteric microbiota programs the anti-inflammatory phenotype of colonic macrophages.
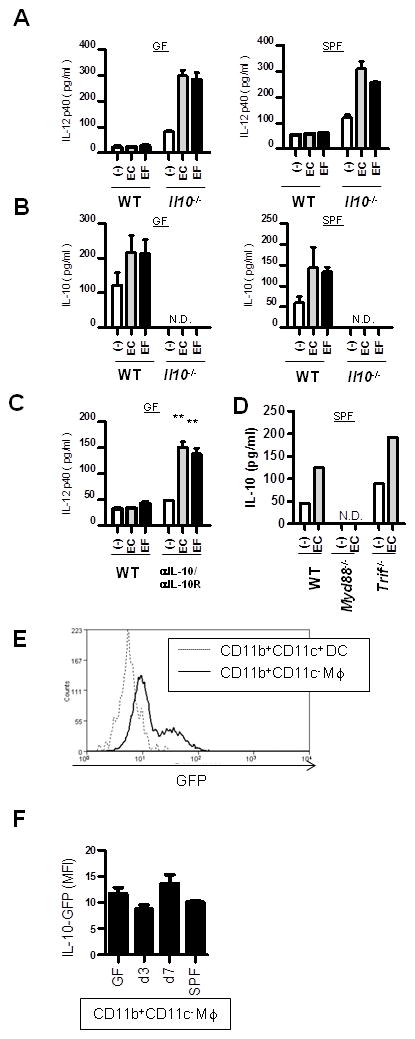
WT and Il10−/− colonic CD11b+ LPMCs from GF (left panel) and SPF microbiota colonized (right panel) mice were stimulated with heat-killed E. coli (EC) or E. faecalis (EF) (MOI = 10) for 24 hours. (A) IL-12/23 p40 and (B) IL-10 secretion was determined by ELISA. (C) Colonic CD11b+ LPMCs from WT GF mice were stimulated with heat-killed EC or EF (MOI = 10) ± neutralizing anti-IL-10 (10 μg/ml), IL-10 receptor (anti-IL-10R) antibodies (10 μg/ml) or isotype control antibodies for 24 hours. IL12/IL-23 p40 secretion was determined by ELISA. **p < 0.01 relative to isotype stimulated WT CD11b+ LPMCs. Results represent mean ± SEM from three independent experiments. N.D., not detected. (D) Colonic CD11b+ LPMCs from WT, Myd88−/−, and Trif−/− SPF mice were stimulated with heat-killed EC (MOI = 10). IL-10 secretion was determined by ELISA. Representative result from 2 independent experiment is shown. N.D., not detected. (E) Colonic lamina propria mononuclear cells from IL-10 transcriptional reporter, Vert-X mice, are analyzed by flow cytometry. A representative histogram from 3 independent experiments is shown for GFP, representing IL-10 expression in gated CD11b+CD11c+ dendritic cells and CD11b+CD11c− macrophages in SPF-raised Vert-X mice. (F) Germ-free raised (GF) mice were colonized with the SPF microbiota and cells were isolated 3 and 7 days post-colonization. Results are shown as mean fluorescence intensity (MFI) for GFP representing IL-10 expression in gated CD11b+CD11c− macrophages, at each time point of colonization. Results represent mean ± SEM from three independent experiments.
IL-10 regulates Il12b in macrophages through epigenetic mechanisms
M-CSF-derived bone marrow derived macrophages (BMDMs) produce more IL-10 and less IL-12 p40 compared to GM-CSF-derived BMDM (1). Therefore, we utilized M-CSF-derived BMDMs as a model to explore molecular mechanisms through which IL-10 attenuates Il12b activation. Indeed, kinetics of Il12b and Il10 expression was similar between LPS-stimulated BMDMs and heat-killed bacteria-stimulated colonic macrophages from WT and Il10−/− mice (Fig. 2A, B). LPS-induced Il12b mRNA was transient in WT BMDMs with peak expression at 3 hours. However, Il12b expression from Il10−/− BMDMs was still increasing at 12 hours (Fig. 2A). In the presence of anti-IL-10, the kinetics of Il12b expression in WT BMDMs was identical to Il10−/− BMDMs (Fig. 2C). There was no detectable difference in Il12b mRNA stability between WT and Il10−/− BMDMs (Fig. 2D). NF-κB (IκBα phosphorylation and degradation, RelA phosphorylation) and MAPK kinase activation kinetics were also identical in WT and Il10−/− BMDMs (Fig. 2E), with peak activation between 0.5 and 3 hours. Interestingly, when exogenous IL-10 was added to Il10−/− BMDMs 3 hours post-LPS stimulation, Il12b expression was still attenuated at later time points (Fig. 2F). These results suggest that IL-10 inhibits IL-12 p40 not through altered induction of downstream signal transduction pathways, but through other mechanisms that affect gene transcription.
Figure 2. IL-10 inhibition of Il12b expression does not involve mRNA stability, NF-κB activation, MAPK signaling, or nucleosome remodeling.
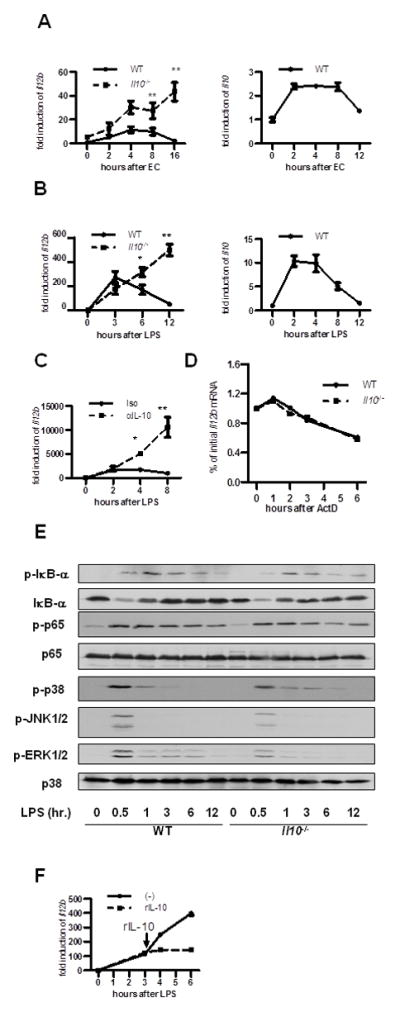
(A) WT and Il10−/− colonic CD11b+ LPMCs were stimulated with heat-killed E. coli (EC) (MOI=100). Cells were harvested at indicated time points post-EC stimulation and Il12b and Il10 expression analyzed by real-time RT-PCR. ** p < 0.01 versus WT. (B) WT and Il10−/− BMDMs were stimulated with LPS (10 ng/ml). Cells were harvested at indicated time points post-LPS stimulation and Il12b and Il10 expression analyzed by real-time RT-PCR. *p < 0.05; ** p < 0.01 versus LPS stimulated WT BMDMs. (C) WT BMDMs were stimulated with LPS (10 ng/ml) ± anti-IL-10 or isotype control (iso) antibodies for 2, 4, and 8 hours. Kinetics of Il12b mRNA expression was analyzed by real-time RT-PCR. ** p < 0.01 versus LPS stimulated WT BMDMs incubated with isotype control antibody. For real time RT-PCR experiments, results are expressed as fold induction versus unstimulated control BMDMs normalized to β-actin (mean ± SEM from three independent experiments). (D) WT and Il10−/− BMDMs were stimulated with LPS (10 ng/ml) for 2 hours + actinomycin D (ActD, 5 μg/ml). Cells were harvested at the indicated time points and Il12b expression analyzed. Results are representative of three independent experiments. (E) Kinetics of NF-κB and MAPK pathway activation was analyzed by Western immunoblot for indicated proteins in WT and Il10−/− BMDMs stimulated with LPS (10 ng/ml). Representative results from three independent experiments are shown. (F) Il10−/− BMDMs were stimulated with LPS (10 ng/ml) for 3 hours and recombinant IL-10 (20 ng/ml) was added. Cells were harvested at the indicated time points for mRNA purification and Il12b mRNA expression was analyzed by real-time PCR. Results are expressed as fold induction versus unstimulated WT BMDMs normalized to β-actin (mean ± SEM from three independent experiments).
IL-10 alters histone H4 acetylation kinetics on the Il12b proximal promoter
Gene expression is regulated at the chromatin level through nucleosome remodeling and covalent histone modifications. Histone acetylation is associated with transcriptionally active chromatin, whereas deacetylation correlates with gene repression. Upon LPS stimulation, Il12b promoter activation is accompanied by selective remodeling of a nucleosome (referred to as Nuc1) in the proximal promoter and a DNase I hypersensitive site (HSS1) approximately 10 kb upstream of the transcription start site (16). As previously described (25), restriction enzyme accessibility assays revealed that Nuc1 and HSS1 were remodeled upon LPS stimulation of WT BMDMs (Fig. 3A and B; Confirmation by Southern blot shown in Supplemental Fig. 1). Il10−/− BMDMs demonstrated identical kinetics of nucleosome remodeling, despite the marked difference in Il12b expression kinetics between WT and Il10−/− BMDMs. Therefore, we next examined histone modifications on the Il12b promoter by chromatin immunoprecipitation (ChIP) using acetylated histone H4 as an indicator of open chromatin. Histone H4 on the Il12b promoter was transiently acetylated in WT BMDMs, peaking at 1.5 hours after LPS stimulation and decreasing to baseline by 3 hours, whereas histone H4 acetylation on the Il12b promoter persisted for 6 hours in Il10−/− BMDMs (Fig. 3C). Recombinant IL-10 inhibited LPS-induced histone acetylation on the Il12b promoter in Il10−/− BMDMs (Fig. 3D). In contrast, trimethylation of histone H3 at lysine 4 (H3K4me3), another marker of transcriptionally active promoters, was induced upon LPS stimulation and persisted at 6 hours in both WT and Il10−/− BMDMs (Supplemental Fig. 3A), whereas Il12b mRNA is decreasing at 6 hours post-LPS in WT BMDMs (Fig. 2A). This suggests that IL-10 specifically induces histone deacetylation on Il12b promoter.
Figure 3. IL-10 alters histone H4 acetylation kinetics on the Il12b proximal promoter.
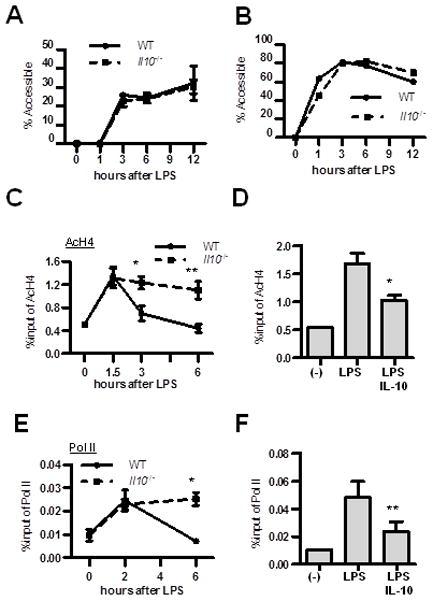
(A) Effects of IL-10 on nucleosome remodeling at nucleosome 1 (Nuc1) position and (B) an upstream DNase I hypersensitivity site 1 (HSS1) was monitored by restriction enzyme accessibility assays. Quantification of SpeI and PstI cleavage products was analyzed by real-time RT-PCR using primers spanning the Nuc1 and HSS1 regions in the Il12b promoter. Results are expressed as a percentage of Nuc1 and HSS1 accessibility observed in LPS stimulated BMDMs relative to unstimulated DNA (mean ± SEM from three independent experiments). (C) WT and Il10−/− BMDMs were stimulated with LPS (10 ng/ml) for 1.5, 3, and 6 hours and kinetics of acetylation of histone H4 (AcH4) on the Il12b promoter was analyzed by ChIP. Results are presented as enrichment (percentage of input DNA) of AcH4 associated with the Il12b promoter. (D) Il10−/− BMDMs were stimulated with LPS (10 ng/ml) ± recombinant IL-10. Acetylation of histone H4 on the Il12b promoter was analyzed by ChIP 3 hours following LPS stimulation. Results are presented as enrichment (percentage of input DNA) of AcH4 associated with the Il12b promoter. (E) WT and Il10−/− BMDMs were stimulated with LPS (10 ng/ml) for 2 and 6 hours and recruitment of RNA polymerase II (RNA pol II) to the Il12b promoter was assessed by ChIP. (F) Il10−/− BMDMs were stimulated with LPS (10 ng/ml) ± recombinant IL-10 for 3 hours and RNA pol II binding on the Il12b promoter was assessed by ChIP. Results are presented as enrichment (percentage of input DNA) of RNA pol II promoter occupancy. All ChIP assays are presented as mean ± SEM of chromatin preparations from three independent experiments. *p < 0.05, **p < 0.01 versus LPS-stimulated BMDMs.
Histone deacetylation decreases the accessibility of chromatin to the basal transcriptional machinery. Therefore, we next determined whether IL-10-mediated histone deacetylation correlates with decreased occupancy of RNA polymerase II (RNA pol II) on the Il12b promoter. LPS-stimulated WT BMDMs demonstrated transient RNA pol II occupancy on the Il12b promoter. In Il10−/− BMDMs, RNA pol II occupancy persisted for 6 hours following LPS stimulation (Fig. 3E). Recombinant IL-10 inhibited RNA pol II binding in Il10−/− BMDMs (Fig. 3F). Kinetics of NF-κB p65 recruitment to the Il12b promoter was also similar to RNA pol II in WT and Il10−/− BMDMs and binding of p65 was also inhibited by exogenous IL-10 (Supplemental Fig. 3B and C), similar to findings in bone marrow derived dendritic cells described previously (31). Taken together, these results demonstrate that IL-10 limits transcriptional activity of the Il12b promoter, likely through alterations in histone acetylation kinetics.
Inhibition of LPS-induced IL-12 p40 by IL-10 involves histone deacetylation by HDAC3 in bone marrow derived and colonic macrophages
The altered kinetics of histone acetylation led us to study the role of HDACs in IL-10-mediated Il12b inhibition. We first used an inhibitor of class I and II HDACs, trichostatin A (TSA). Pretreatment of BMDMs with TSA prior to LPS stimulation significantly inhibit Il12b transcription induction, as previously described (32) (Supplemental Fig. 4A), greatly affecting our ability to detect IL-10 mediated inhibition of Il12b transcription. Therefore, Il12b transcription was first induced with LPS for one hour before treating the BMDMs with TSA. Interestingly, TSA prolonged the expression of Il12b in WT BMDMs resulting in kinetics similar to that in Il10−/− BMDMs (Fig. 4A). However, TSA also affected the kinetics of other cytokines, including IL-10 (Supplemental Fig. 4B). Next, exogenous IL-10 was added back to Il10−/− BMDMs in the presence or absence of TSA to compare IL-10-induced deacetylation on the Il12b promoter without potential confounding effects mediated by endogenous IL-10 production (Fig. 4B). As predicted, inhibition of IL-12 p40 by IL-10 was significantly impaired by TSA(Fig. 4C). Quantitative PCR also showed marked decrease of Il12b inhibition by IL-10 in the presence of TSA (Fig. 4C), suggesting IL-10 mediated inhibition of LPS-induced IL-12 p40 is partially dependent on class I or II HDACs.
Figure 4. HDACs are involved in inhibition of Il12b by IL-10.
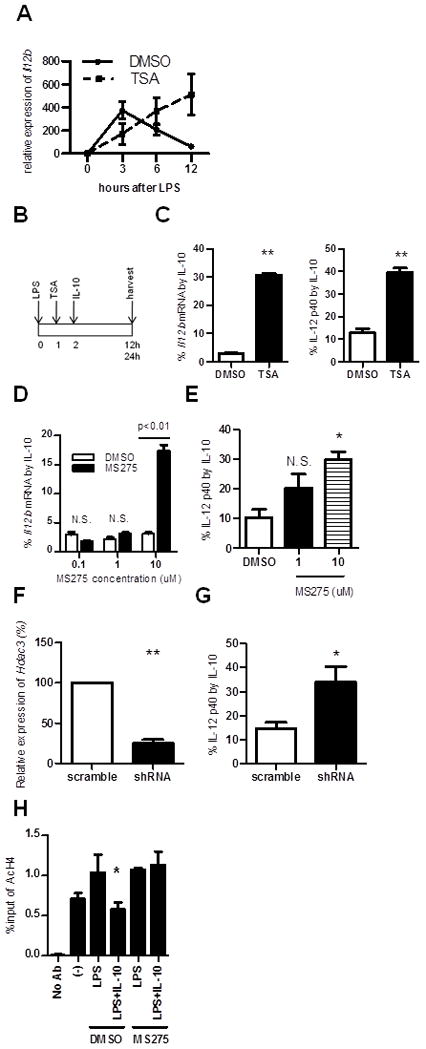
(A) WT BMDMs were stimulated with LPS (10 ng/ml) with or without trichostatin A (TSA) (100 nM) 1 hour after LPS. Cells were harvested at 3, 6, and 12 hours post-LPS stimulation and Il12b expression analyzed by real-time RT-PCR. (B, C) Il10−/− BMDMs were stimulated with LPS (10 ng/ml) and treated with TSA (100 nM) or DMSO 1h post-LPS. IL-10 (1 ng/ml) was added 2h-post LPS. Il12b expression was determined by real-time RT-PCR 12 h post-LPS and IL-12 p40 protein by ELISA after 24 hours. Results are presented as percent Il12b (4C, left) and IL-12 p40 (4C, right) expression by LPS in the presence of IL-10 relative to LPS alone. (D, E) Il10−/− BMDMs were stimulated with LPS (10 ng/ml) and treated with MS275, an inhibitor of HDAC1 (IC50=0.3 μM) and HDAC3 (IC50=8 μM), or DMSO 1 h post-LPS. IL-10 was added 2h-post LPS. Inhibition of Il12b expression (D) and IL-12 p40 (E) was examined as above. (F) Il10−/− BMDMs transduced HDAC3 shRNA or control scrambled shRNA were analyzed for Hdac3 mRNA expression. Results are expressed as relative expression (%) versus BMDMs transduced with scrambled shRNA normalized to β-actin. (G) Il10−/− BMDMs transduced with HDAC3 shRNA or control scrambled shRNA were stimulated with LPS (10 ng/ml), treated with IL-10 2 hours post-LPS, and harvested 24 hours post-LPS. Inhibition of IL-12 p40 is presented as above. (H) Il10−/− BMDMs were stimulated with LPS (10 ng/ml) ± IL-10 in the presence or absence of MS275 (10 μM). Acetylation of histone H4 on the Il12b promoter was analyzed by ChIP four hours following LPS stimulation. Results are presented as enrichment (percentage of input DNA) of AcH4 associated with the Il12b promoter. *p < 0.05 versus LPS + DMSO.
Since HDAC1 is reported to be associated with histone deacetylation on the Il12b and Il6 promoters in macrophages (19, 33), we next utilized the HDAC1- and 3- specific inhibitor MS275 (IC50 0.3 μM for HDAC1 and 8 μM for HDAC3). Unexpectedly, loss of IL-10 mediated inhibition of Il12b was observed only at the highest dose of MS275 (10 μM), suggesting HDAC3 is more important to this process (Fig. 4D and E). HDAC3-specific lentiviral shRNA(Fig. 4F) was used to confirm this role of HDAC3 in the inhibitory effect of IL-10 on LPS-induced IL-12 p40. IL-12 p40 inhibition by IL-10 was significantly diminished in LPS and HDAC3 shRNA-treated cells compared to control cells (scrambled HDAC3 shRNA) (Fig. 4G). Furthermore, inhibition of HDAC3 prevented histone H4 deacetylation on the Il12b promoter by IL-10 (Fig. 4H).
HDAC3 is a homeostatic factor in IL-10-mediated intestinal immunity
To determine whether our findings in BMDMs are relevant for colonic macrophage function, IL-12 p40 production by colonic CD11b+ LPMCs from Il10−/− mice was determined in the presence of HDAC inhibitors. Il10−/− colonic CD11b+ LPMCs were activated with heat killed E.coli prior to the addition of MS275 or TSA and IL-10. As demonstrated in BMDMs, IL-12 p40 inhibition by IL-10 was significantly diminished by blocking HDAC3 compared to the control (Fig. 5A). Next, colonic Hdac3 expression was characterized before and after transition of GF mice to an SPF microbiota (34). Interestingly, colonic Hdac3 expression was significantly induced after colonization of WT mice, but not colitis-prone Il10−/− mice (Fig. 5B). Colonic Il12b expression was significantly induced in Il10−/− but not WT mice upon exposure to SPF microbiota, inversely correlating with Hdac3 induction. As a control, no significant induction of colonic Hdac1 was observed (Fig. 5B).
Figure 5. HDAC3 is a homeostatic factor in IL-10-mediated intestinal immunity.
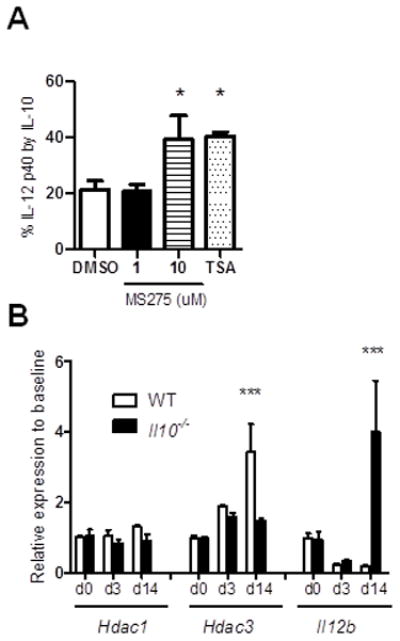
(A)Il10−/− colonic CD11b+ LPMCs were stimulated with heat-killed E. coli (EC, 100 MOI) and treated with MS275 (1 or 10 μM), TSA (100 nM) or DMSO 1h post-EC. Recombinant IL-10 (1 ng/ml) was added 2h post-EC. Inhibition of IL-12 p40 was examined as above. All results represent mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01 versus controls. (B) GF WT and Il10−/− mice were transitioned to an SPF enteric microbiota. Colonic mucosal Hdac1, Hdac3, and Il12b expression were analyzed before, 3 and 14 days post-colonization. Results are expressed as fold induction versus WT GF colons normalized to β-actin (mean ± SEM from four mice per each time point). ***p < 0.005 versus Il10−/−.
DISCUSSION
Mechanisms operative in programming an anti-inflammatory phenotype in intestinal macrophages are incompletely understood. We initially speculated that a tolerant colonic macrophage phenotype might be acquired upon exposure to the enteric microbiota. Given this unique intestinal environment where macrophages intimately coexist with the enteric microbiota, we hypothesized that a phenomenon similar to the induction of endotoxin tolerance in peripheral macrophages may occur (28, 29). However, colonic macrophages isolated from GF WT mice were phenotypically identical to macrophages derived from colonized mice, and failed to produce Il12b upon stimulation with enteric bacteria. Moreover, both GF and SPF WT colonic macrophages produced abundant IL-10. Colonic macrophages from GF Il10−/− mice and WT macrophages treated with anti-IL-10/IL-10 receptor antibodies produced abundant IL-12 p40 upon activation by enteric bacteria, demonstrates that locally produced IL-10 is a requisite factor for maintaining anti-inflammatory responses in colonic macrophages. It is also intriguing that MyD88-deficient intestinal macrophages lack both basal and inducible IL-10. Further studies are needed to unravel precise mechanisms by which intestinal macrophages acquire the anti-inflammatory phenotype to produce IL-10 through MyD88. Speculatively, endogenous factors that activate the MyD88 signaling pathway in the colonic microenvironment may shape the colonic macrophage phenotype and mediate tolerance to the enteric microbiota. However, given inherent limitations of the germ free mouse model system, we also cannot exclude exogenous microbial products and other exogenous substance present in small amounts in the mouse diet as contributing to this process. Nonetheless, these results further define the unique immune environment in the gastrointestinal tract, focusing on IL-12 p40 regulation by endogenous IL-10 production as a well established prototype for a mucosal innate inflammatory response.
Utilizing BMDMs as a model to understand molecular mechanisms through which IL-10 inhibits Il12b expression, IL-10 was found to mediate histone deactelyation of the Il12b promoter with the consequence of attenuated transcription of Il12b. In Il10−/− BMDMs, prolonged kinetics of Il12b mRNA and protein expression correlated with prolonged histone H4 acetylation on the proximal promoter and prolonged occupancy by RNA pol II. Zhou et al. previously demonstrated that exogenous IL-10 abolishes RNA pol II binding to the Il12b promoter partly through inhibition of nucleosome remodeling of the Il12b promoter (16). While our results also demonstrated altered RNA pol II recruitment, we did not observe differences in nucleosome remodeling between WT and Il10−/− BMDMs. An important difference between our studies is that they used peritoneal macrophages. Additionally, alterations of nucleosome remodeling demonstrated in the prior study were relatively small in magnitude compared with profound IL-12 p40 inhibition by exogenous addition of recombinant IL-10. Indeed, the authors speculated that alterations in nucleosome remodeling induced by IL-10 were likely to be a consequence of transcription inhibition rather than the cause. Furthermore, comparisons between WT and Il10−/− BMDMs may be more relevant to dissect molecular differences given that we demonstrate the importance of autocrine regulation of IL-12 p40 by endogenous IL-10 (Fig. 1C and 2C).
Although our studies clearly implicate HDAC3 in the inhibitory effect of IL-10 on Il12b, it is important to note that the inhibitory effect was not complete. Indeed, HDAC inhibitors reverse approximately 30% of the inhibitory effect of IL-10. Based on many other studies looking at mechanisms of IL-10 inhibition, this is not surprising. IL-10 is such an important homeostatic factor that multiple independent mechanisms must mediate its inhibitory effect even on a single gene such as IL-12 p40, because the consequences of loss of IL-10 regulation are so significant biologically. Indeed, IL-10 induces many genes in macrophages at the same time as it inhibits others (35) and exerts its potent anti-inflammatory function in innate immunity through multiple mechanisms. Transcription elongation (36), miR-155 (37), and induction of transcriptional repressors such as tristetraprolin (38), ETV3, and Strawberry notch homologue 2 (SBNO2) (39) have been suggested as mechanisms for IL-10-mediated innate immune regulation, although they have not been explicitly implicated in IL-12 p40 inhibition. Accordingly, multiple redundant mechanisms for IL-10-mediated IL-12 p40 regulation have been described, including nuclear factor, interleukin-3 regulated (NFIL3) (40), interferon regulatory factor-8 (IRF-8) (41), nucleosome remodeling (16) all of which seem to have incremental but biologically significant effects.
We have utilized CD11b+ LPMCs as representative of colonic macrophages since these cells are the main sources of IL-12 p40 and IL-10 in the intestinal lamina propria (23, 24). IL-10 and IL-12 p40 production in response to heat-killed enteric bacteria from these cells are identical in both GF and SPF-raised WT mice. Although this population also includes CD11b+CD11c+ dendritic cells, CD11b+ CD11c− macrophages are more abundant than CD11b+CD11c+ dendritic cells in number. Furthermore, we also showed that vast majority of IL-10 producing cells belongs to CD11b+ CD11c− macrophages, as reported previously (30).
Histone acetylation and deacetylation are regulated by HATs and HDACs, respectively. We demonstrated altered histone acetylation on Il12b promoter by IL-10, suggesting that IL-10 represses Il12b transcription by this mechanism. Currently, eighteen HDACs have been identified in mammalian cells and are classified into 4 classes, of which class I and II are the major and best characterized groups. Class I HDACs are widely expressed in most cell types, whereas class II HDACs demonstrate more restricted expression and have roles in cell differentiation (42). Accordingly, HDAC inhibition has many overarching consequences in immune cells, including apoptosis (43), differentiation (42, 44), signal transduction (45, 46), and cytokine production (47–49). Indeed, the class I and II HDAC inhibitor, TSA was previously described to inhibit LPS-induced pro-inflammatory cytokine expression in macrophages, including IL-12 p40, when cells were treated with TSA prior to LPS activation (32). In contrast, treatment with TSA following activation of macrophages leads to an increased pro-inflammatory response (42, 50). Therefore, to specifically address the role of HDACs in IL-10 inhibition of Il12b, we treated macrophages with HDAC inhibitors post-LPS stimulation. Although the magnitude of LPS-induced IL-12 p40 production varied, both TSA and MS275 significantly diminished IL-12 p40 inhibition by IL-10, indicating that inhibition is HDAC dependent. Among class I and II HDACs, we have shown that HDAC3 is likely to be involved in this process by using the class I specific HDAC inhibitor, MS275. Moreover, HDAC3-specific knockdown resulted in impaired inhibition of IL-12 p40 by IL-10, and IL-10-mediated histone deacetylation of Il12b promoter was blocked by MS275. Furthermore, GF mice colonized with SPF microbiota were used as an in vivo model for characterizing colonic HDAC induction upon the exposure to commensal bacteria. Hdac3 expression was induced in WT mice but not in Il10−/− mice after colonization. Hdac3 expression inversely correlated with Il12b expression. This finding implicates HDAC3 in IL-10 mediated Il12b regulation, with colonic Hdac3 induction requiring IL-10 and the enteric microbiota in vivo. Hence, we provide multiple lines of evidence that HDAC3 contributes to IL-10-mediated Il12b inhibition. However, precise mechanisms for IL-10 control of HDAC3 function on the Il12b promoter await clarification. HDAC3 may function directly on the Il12b promoter or indirectly through expression of other genes involved in Il12b regulation. There are multiple factors controlling HDAC activity including its expression, nuclear translocation and binding to DNA. Additionally, co-repressors such as SMRT (silencing mediator for retinoid and thyroid receptors), and N-CoR (nuclear receptor co-repressor) (51) may participate in an HDAC3 regulation. Involvement of STAT3 in this process is also of interest. STAT3 is both necessary and sufficient for inhibitory effect of IL-10 on many target pro-inflammatory genes. In fact, Stat3−/− cells and mice show a phenotype similar to those of Il10−/− (52). Furthermore, Hoentjen et al. demonstrated that IL-10 regulates p65 recruitment to the Il12b promoter through STAT3 phosphorylation in bone marrow derived dendritic cells (31). Therefore, it is likely that control of HDAC3 function by IL-10 also involves STAT3.
In conclusion, we provide evidence that IL-10 is a necessary homeostatic factor for maintaining the anti-inflammatory phenotype of colonic macrophages, without requirement for in vivo exposure to the enteric microbiota. IL-10 altered the kinetics of histone H4 acetylation on the Il12b promoter. This finding led to the identification of a novel epigenetic mechanism of IL-10-mediated IL-12 p40 regulation through histone deacetylation by HDAC3 which is operative in colonic macrophages.
Supplementary Material
Acknowledgments
Grant support: This work was supported by grants from the National Institutes of Health (NIH): RO1 DK54452 (SEP), Gastroenterology Research Training Grant T32 DK007737 (SZS), NRSA F32 DK083186 (SZS), P40 RR018603 (National Gnotobiotic Rodent Resource Center), P30 DK034987 (Center for Gastrointestinal Biology and Disease; Immunotechnologies, Gnotobiotic and Histology Cores), and a Crohn’s and Colitis Foundation of America Research Fellowship Award (TK and KM), and the Uehara Memorial Foundation (TK).
Abbreviations
- AcH4
acetylated histone H4
- BMDMs
bone marrow derived macrophages
- ChIP
chromatin immunoprecipitation
- CBP
CREB-binding protein
- EF
Enterococcus feacalis
- EC
Escherichia coli
- GF
germ free
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HSS1
DNase I hypersensitive site
- H3K4me3
histone H3 trimethylated at lysine 4
- IBD
inflammatory bowel disease
- LPMCs
lamina propria mononuclear cells
- MOI
multiplicity of infection
- NFIL3
nuclear factor, interleukin-3 regulated
- N-CoR
nuclear receptor co-repressor
- RNA pol II
RNA polymerase II
- SBNO2
strawberry notch homologue 2
- shRNA
small hairpin ribonucleic acid
- SMRT
silencing mediator for retinoid and thyroid receptors
- SPF
specific pathogen free
- TSA
trichostatin A
- WT
wild-type
References
- 1.Kamada N, Hisamatsu T, Okamoto S, Sato T, Matsuoka K, Arai K, Nakai T, Hasegawa A, Inoue N, Watanabe N, Akagawa KS, Hibi T. Abnormally differentiated subsets of intestinal macrophage play a key role in Th1-dominant chronic colitis through excess production of IL-12 and IL-23 in response to bacteria. J Immunol. 2005;175:6900–6908. doi: 10.4049/jimmunol.175.10.6900. [DOI] [PubMed] [Google Scholar]
- 2.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 3.Plevy S. The immunology of inflammatory bowel disease. Gastroenterol Clin North Am. 2002;31:77–92. doi: 10.1016/s0889-8553(01)00006-1. [DOI] [PubMed] [Google Scholar]
- 4.Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, Perro M, Diestelhorst J, Allroth A, Murugan D, Hatscher N, Pfeifer D, Sykora KW, Sauer M, Kreipe H, Lacher M, Nustede R, Woellner C, Baumann U, Salzer U, Koletzko S, Shah N, Segal AW, Sauerbrey A, Buderus S, Snapper SB, Grimbacher B, Klein C. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 6.Spencer SD, Di Marco F, Hooley J, Pitts-Meek S, Bauer M, Ryan AM, Sordat B, Gibbs VC, Aguet M. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J Exp Med. 1998;187:571–578. doi: 10.1084/jem.187.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, Kronenberg M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009;10:1178–1184. doi: 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albright CA, Sartor RB, Tonkonogy SL. Endogenous antigen presenting cell-derived IL-10 inhibits T lymphocyte responses to commensal enteric bacteria. Immunol Lett. 2009;123:77–87. doi: 10.1016/j.imlet.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu B, Tonkonogy SL, Sartor RB. Antigen-presenting cell production of IL-10 inhibits T-helper 1 and 17 cell responses and suppresses colitis in mice. Gastroenterology. 2011;141:653–662. 662 e651–654. doi: 10.1053/j.gastro.2011.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davidson NJ, Hudak SA, Lesley RE, Menon S, Leach MW, Rennick DM. IL-12, but not IFN-gamma, plays a major role in sustaining the chronic phase of colitis in IL-10-deficient mice. J Immunol. 1998;161:3143–3149. [PubMed] [Google Scholar]
- 12.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du C, Sriram S. Mechanism of inhibition of LPS-induced IL-12p40 production by IL-10 and TGF-beta in ANA-1 cells. J Leukoc Biol. 1998;64:92–97. doi: 10.1002/jlb.64.1.92. [DOI] [PubMed] [Google Scholar]
- 15.Aste-Amezaga M, Ma X, Sartori A, Trinchieri G. Molecular mechanisms of the induction of IL-12 and its inhibition by IL-10. J Immunol. 1998;160:5936–5944. [PubMed] [Google Scholar]
- 16.Zhou L, Nazarian AA, Smale ST. Interleukin-10 inhibits interleukin-12 p40 gene transcription by targeting a late event in the activation pathway. Mol Cell Biol. 2004;24:2385–2396. doi: 10.1128/MCB.24.6.2385-2396.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smale ST, Fisher AG. Chromatin structure and gene regulation in the immune system. Annu Rev Immunol. 2002;20:427–462. doi: 10.1146/annurev.immunol.20.100301.064739. [DOI] [PubMed] [Google Scholar]
- 18.Sun H, Lu J, Wei L, Wang X, Xu X, Dong M, Huang B. Histone acetyltransferase activity of p300 enhances the activation of IL-12 p40 promoter. Mol Immunol. 2004;41:1241–1246. doi: 10.1016/j.molimm.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 19.Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Roach JC, Kennedy K, Hai T, Bolouri H, Aderem A. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 20.Madan R, Demircik F, Surianarayanan S, Allen JL, Divanovic S, Trompette A, Yogev N, Gu Y, Khodoun M, Hildeman D, Boespflug N, Fogolin MB, Grobe L, Greweling M, Finkelman FD, Cardin R, Mohrs M, Muller W, Waisman A, Roers A, Karp CL. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J Immunol. 2009;183:2312–2320. doi: 10.4049/jimmunol.0900185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiong H, Zhu C, Li F, Hegazi R, He K, Babyatsky M, Bauer AJ, Plevy SE. Inhibition of interleukin-12 p40 transcription and NF-kappaB activation by nitric oxide in murine macrophages and dendritic cells. J Biol Chem. 2004;279:10776–10783. doi: 10.1074/jbc.M313416200. [DOI] [PubMed] [Google Scholar]
- 22.Hegazi RA, Rao KN, Mayle A, Sepulveda AR, Otterbein LE, Plevy SE. Carbon monoxide ameliorates chronic murine colitis through a heme oxygenase 1-dependent pathway. J Exp Med. 2005;202:1703–1713. doi: 10.1084/jem.20051047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi T, Matsuoka K, Sheikh SZ, Elloumi HZ, Kamada N, Hisamatsu T, Hansen JJ, Doty KR, Pope SD, Smale ST, Hibi T, Rothman PB, Kashiwada M, Plevy SE. NFIL3 is a regulator of IL-12 p40 in macrophages and mucosal immunity. J Immunol. 2011;186:4649–4655. doi: 10.4049/jimmunol.1003888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheikh SZ, Matsuoka K, Kobayashi T, Li F, Rubinas T, Plevy SE. Cutting Edge: IFN-{gamma} Is a Negative Regulator of IL-23 in Murine Macrophages and Experimental Colitis. J Immunol. 2010;184:4069–4073. doi: 10.4049/jimmunol.0903600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinmann AS, Plevy SE, Smale ST. Rapid and selective remodeling of a positioned nucleosome during the induction of IL-12 p40 transcription. Immunity. 1999;11:665–675. doi: 10.1016/s1074-7613(00)80141-7. [DOI] [PubMed] [Google Scholar]
- 26.Albrecht I, Tapmeier T, Zimmermann S, Frey M, Heeg K, Dalpke A. Toll-like receptors differentially induce nucleosome remodelling at the IL-12p40 promoter. EMBO Rep. 2004;5:172–177. doi: 10.1038/sj.embor.7400078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shnyra A, Brewington R, Alipio A, Amura C, Morrison DC. Reprogramming of lipopolysaccharide-primed macrophages is controlled by a counterbalanced production of IL-10 and IL-12. J Immunol. 1998;160:3729–3736. [PubMed] [Google Scholar]
- 29.Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and toll-like receptor 2 and 4 gene expression. J Immunol. 2000;164:5564–5574. doi: 10.4049/jimmunol.164.11.5564. [DOI] [PubMed] [Google Scholar]
- 30.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 31.Hoentjen F, Sartor RB, Ozaki M, Jobin C. STAT3 regulates NF-kappaB recruitment to the IL-12p40 promoter in dendritic cells. Blood. 2005;105:689–696. doi: 10.1182/blood-2004-04-1309. [DOI] [PubMed] [Google Scholar]
- 32.Halili MA, Andrews MR, Labzin LI, Schroder K, Matthias G, Cao C, Lovelace E, Reid RC, Le GT, Hume DA, Irvine KM, Matthias P, Fairlie DP, Sweet MJ. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J Leukoc Biol. 2010;87:1103–1114. doi: 10.1189/jlb.0509363. [DOI] [PubMed] [Google Scholar]
- 33.Lu J, Sun H, Wang X, Liu C, Xu X, Li F, Huang B. Interleukin-12 p40 promoter activity is regulated by the reversible acetylation mediated by HDAC1 and p300. Cytokine. 2005;31:46–51. doi: 10.1016/j.cyto.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 34.Hansen JJ, Holt L, Sartor RB. Gene expression patterns in experimental colitis in IL-10-deficient mice. Inflamm Bowel Dis. 2009;15:890–899. doi: 10.1002/ibd.20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- 36.Smallie T, Ricchetti G, Horwood NJ, Feldmann M, Clark AR, Williams LM. IL-10 inhibits transcription elongation of the human TNF gene in primary macrophages. J Exp Med. 2010;207:2081–2088. doi: 10.1084/jem.20100414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCoy CE, Sheedy FJ, Qualls JE, Doyle SL, Quinn SR, Murray PJ, O’Neill LA. IL-10 inhibits miR-155 induction by toll-like receptors. J Biol Chem. 2010;285:20492–20498. doi: 10.1074/jbc.M110.102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaljo B, Kratochvill F, Gratz N, Sadzak I, Sauer I, Hammer M, Vogl C, Strobl B, Muller M, Blackshear PJ, Poli V, Lang R, Murray PJ, Kovarik P. Tristetraprolin is required for full anti-inflammatory response of murine macrophages to IL-10. J Immunol. 2009;183:1197–1206. doi: 10.4049/jimmunol.0803883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El Kasmi KC, Smith AM, Williams L, Neale G, Panopoulos AD, Watowich SS, Hacker H, Foxwell BM, Murray PJ. Cutting edge: A transcriptional repressor and corepressor induced by the STAT3-regulated anti-inflammatory signaling pathway. J Immunol. 2007;179:7215–7219. doi: 10.4049/jimmunol.179.11.7215. [DOI] [PubMed] [Google Scholar]
- 40.Smith AM, Qualls JE, O’Brien K, Balouzian L, Johnson PF, Schultz-Cherry S, Smale ST, Murray PJ. A distal enhancer in Il12b is the target of transcriptional repression by the STAT3 pathway and requires the basic leucine zipper (B-ZIP) protein NFIL3. J Biol Chem. 2011;286:23582–23590. doi: 10.1074/jbc.M111.249235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu C, Rao K, Xiong H, Gagnidze K, Li F, Horvath C, Plevy S. Activation of the murine interleukin-12 p40 promoter by functional interactions between NFAT and ICSBP. J Biol Chem. 2003;278:39372–39382. doi: 10.1074/jbc.M306441200. [DOI] [PubMed] [Google Scholar]
- 42.Barnes PJ, I, Adcock M, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. 2005;25:552–563. doi: 10.1183/09031936.05.00117504. [DOI] [PubMed] [Google Scholar]
- 43.Singh TR, Shankar S, Srivastava RK. HDAC inhibitors enhance the apoptosis-inducing potential of TRAIL in breast carcinoma. Oncogene. 2005;24:4609–4623. doi: 10.1038/sj.onc.1208585. [DOI] [PubMed] [Google Scholar]
- 44.Rahman MM, Kukita A, Kukita T, Shobuike T, Nakamura T, Kohashi O. Two histone deacetylase inhibitors, trichostatin A and sodium butyrate, suppress differentiation into osteoclasts but not into macrophages. Blood. 2003;101:3451–3459. doi: 10.1182/blood-2002-08-2622. [DOI] [PubMed] [Google Scholar]
- 45.Kao HY, Ordentlich P, Koyano-Nakagawa N, Tang Z, Downes M, Kintner CR, Evans RM, Kadesch T. A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev. 1998;12:2269–2277. doi: 10.1101/gad.12.15.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao W, Bao C, Padalko E, Lowenstein CJ. Acetylation of mitogen-activated protein kinase phosphatase-1 inhibits Toll-like receptor signaling. J Exp Med. 2008;205:1491–1503. doi: 10.1084/jem.20071728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, Dona G, Fossati G, Sozzani S, Azam T, Bufler P, Fantuzzi G, Goncharov I, Kim SH, Pomerantz BJ, Reznikov LL, Siegmund B, Dinarello CA, Mascagni P. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci U S A. 2002;99:2995–3000. doi: 10.1073/pnas.052702999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bode KA, Schroder K, Hume DA, Ravasi T, Heeg K, Sweet MJ, Dalpke AH. Histone deacetylase inhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment. Immunology. 2007;122:596–606. doi: 10.1111/j.1365-2567.2007.02678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, Ding XC, Chanson AL, Reymond MK, Miconnet I, Schrenzel J, Francois P, Calandra T. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. 2010;117:1205–1217. doi: 10.1182/blood-2010-05-284711. [DOI] [PubMed] [Google Scholar]
- 50.Cosio BG, Tsaprouni L, Ito K, Jazrawi E, Adcock IM, Barnes PJ. Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med. 2004;200:689–695. doi: 10.1084/jem.20040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wen YD, Perissi V, Staszewski LM, Yang WM, Krones A, Glass CK, Rosenfeld MG, Seto E. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc Natl Acad Sci U S A. 2000;97:7202–7207. doi: 10.1073/pnas.97.13.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.