

Abstract
Hereditary spastic paraplegia (HSP) is a motor neuron disease caused by a progressive degeneration of the motor axons of the corticospinal tract. Point mutations or exon deletions in the microtubule-severing ATPase, spastin, are responsible for approximately 40% of cases of autosomal dominant HSP. Here, we report the 3.3 Å X-ray crystal structure of a hydrolysis- deficient mutant (E442Q) of the human spastin protein AAA domain. This structure is analyzed in the context of the existing Drosophila melanogaster spastin AAA domain structure and crystal structures of other closely related proteins in order to build a more unifying framework for understanding the structural features of this group of microtubule-severing ATPases.
Keywords: AAA ATPase, microtubule severing, HSP, Spastin
Introduction
Microtubules, an essential component of the cytoskeleton, can interconvert between periods of growth and periods of depolymerization. This process, termed dynamic instability, depends heavily upon specialized proteins that modulate microtubule dynamics (Mitchison & Kirschner, 1984). A number of diseases are attributed to mutations in proteins that regulate microtubule homeostasis. One such disease, hereditary spastic paraplegia (HSP), is commonly the result of mutations in the ATPase spastin. Point mutations and exon deletions in spastin account for the majority of cases of autosomal dominant hereditary spastic paraplegia (HSP) (Depienne et al., 2007). This disease, which primarily affects the lower limbs, is caused by a progressive degeneration of the motor axons of the corticospinal tract. Previous studies have shown that spastin has a microtubule severing activity (Evans, Gomes, Reisenweber, Gundersen, & Lauring, 2005), and that this activity is linked to the manifestation of neurological disorders in Drosophila (Roll-Mecak & Vale, 2005). Because of spastin’s significant role in the development of HSP, there have been an increasing number of studies on this protein and its role in axonal degeneration.
Spastin is a member of the AAA+ family of proteins. These proteins have a wide variety of cellular functions (hence the name AAA – ATPases Associated with various cellular Activities) (Vale, 2000). A structural hallmark of AAA+ proteins is that they generally form hexameric rings. It has been shown by sedimentation velocity and gel filtration assays (White, Evans, Lary, Cole, & Lauring, 2007), and more recently, by small angle X-ray scattering (SAXS) (Roll-Mecak & Vale, 2008) that spastin forms a hexamer in solution. By mutating a glutamic acid residue of the Walker B motif to glutamine (E442Q), spastin can be trapped in the hexameric form, even in the presence of ATP, the binding of which normally causes hexamer disassembly. This mutation has also been used to characterize the binding of the spastin hexamer to the C-terminal tail of tubulin (White et al., 2007). Although it is clear that spastin binds to the C-terminal tail of tubulin, the specific residues required for this activity are still unknown, and existing studies are not in complete agreement (White et al., 2007; Roll-Mecak & Vale, 2008). However, there does appear to be consensus that the binding of the negatively charged tubulin tail takes place within the positively charged, central pore of spastin, and that the pore loops of the AAA domain are involved.
Like all other AAA+ proteins, spastin contains an AAA domain. The AAA domain can be divided into a large / nucleotide-binding domain (NBD) and a smaller four-helix bundle domain (HBD)(White et al., 2007; Roll-Mecak & Vale, 2008). Within the large AAA domain are several motifs that are important for the catalytic ATPase activity, including a Walker A (P-loop) motif and a Walker B (Switch II) motif, which in ATPases are involved in both hydrolysis and in sensing the presence or absence of the nucleotide -phosphate. N-terminal to the AAA domain is the MIT (microtubule interacting and trafficking) domain (Ciccarelli et al., 2003). The crystal structure of this domain has been solved in complex with CHMP1B (Yang et al., 2008), an ESCRT-III protein that targets spastin to the midbody, where it is involved in microtubule organization during cytokinesis (Roll-Mecak & McNally, 2010; Schiel et al., 2011; Yang et al., 2008). The MIT domain is not needed for microtubule severing (White et al., 2007), nor does it bind strongly to microtubules on its own (Roll-Mecak & Vale, 2005). Connecting the MIT domain to the AAA domain is a flexible linker containing a putative microtubule binding domain (MTBD) that binds to tubulin in an ATP-independent manner (White et al., 2007). Although studies have shown that several residues at the C-terminal end of this linker are necessary for microtubule severing (White et al., 2007; Roll-Mecak & Vale, 2008), further studies are needed in order to fully characterize the function of this region.
Despite extensive efforts, until recently very little structural information has been available for the spastin protein, and the detailed kinetic studies required to uncover the mechanism of severing are not possible without structural information. The crystal structure of the AAA domain of the Drosophila spastin protein has been determined (Roll-Mecak & Vale, 2008), and its structure closely resembles that of other AAA proteins. We report here the 3.3 Å X-ray crystal structure the AAA domain of human spastin (SPG4) and show that, despite amino acid differences in a number of key residues, the human and Drosophila spastin structures are highly conserved.
Materials and Method
Expression and purification
The human spastin gene was placed into the pGEX-6P-3 vector to express a glutathione-S-transferase fusion of amino acids 228-616 containing the E442Q mutation, as previously described (Evans et al., 2005). The spastin protein construct was expressed in E. coli BL21-CodonPlus (DE3)-RIPL cells, grown in LB with antibiotic selection to an optical density of 1.0, by induction with 0.5 mM IPTG. After overnight growth at 30°C, cells were pelleted and resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM MgCl2, 5 mM DTT, 1 mM ATP, and EDTA-free protease inhibitor tablet [Roche]) for sonication. Following addition of Triton to 1%, lysate was cleared by centrifugation at 45,000 rpm, 45 min, 4°C and applied to a 5 mL GSTrap column (GE Life Sciences) in equilibration buffer (lysis buffer with 500 mM NaCl, 1 mM DTT and no ATP), then bound proteins were washed with equilibration buffer containing 150 mM NaCl. Protein was eluted with a glutathione gradient, exchanged into 50 mM Hepes, pH 7.4, 150 mM NaCl, 5 mM MgCl2, and 1 mM DTT and the GST tag was cleaved overnight at 4°C with PreScission Protease (GE Life Sciences). The protease and tag were removed using GST- sepharose resin (GE Life Sciences), and, after addition of ATP and concentration of purified protein, hexameric spastin was isolated by gel filtration on a Superdex 200 column, resulting in a yield of up to 2 mg L−1 purified protein.
Crystallization
Diffracting crystals of the spastin construct were initially obtained by mixing equal parts 5.3 mg mL−1 spastin with a solution of 1.6 M (NH3)2SO4, 0.1 M MES monohydrate (pH 6.5) and 10% 1,4-dioxane (Hampton Research) in a hanging drop. The crystals were soaked in 35% glycerol and diffracted to 8 Å at the National Synchrotron Light Source beamline X6A at Brookhaven National Laboratory. Improved crystals, ranging from 60 to 180 M in length, were grown under similar conditions to those above, but with protein concentration at 3.4 mg mL−1 and in the presence of 4% benzamidine HCl (Hampton Research). Crystals were mounted and flash frozen in liquid nitrogen with 35% glycerol in mother liquor. A complete data set was collected to 3.3 Å resolution at the Advanced Photo Source beamline 23-ID-B at Argonne National Laboratory.
Structure determination
Data was processed in XDS (Kabsch, 2010) and structure determination was carried out using AutoMR in PHENIX (Adams, Afonine, & Bunkoczi, 2010). One copy of the search model, Drosophila spastin AAA domain (PDB: 3B9P), was placed in the unit cell. Using data to 3.3 Å, multiple rounds of refinement and rebuilding were carried out in PHENIX.refine (Afonine & Grosse-Kunstleve, 2005) and COOT (Emsley & Cowtan, 2004), respectively, resulting in a final Rwork of 20.7% and Rfree of 27.5%. Validation with MOLPROBITY placed this structure in the 90th percentile, among comparable structures. Coordinates for the human spastin AAA domain have been deposited in the Protein Databank with accession code 3VFD. Data collection, processing and refinement statistics are presented in Table 1. Structural representations rendered in MacPymol (DeLano, 2002).
Table 1.
Crystallographic Statistics
| Native spastin AAA domain | |
|---|---|
| Data collection | |
| Space group | P65 |
| Cell dimensions | |
| a, b, c (Å) | 88.5, 88.5, 88.6 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 44.3 −3.3 (3.3-3.49) |
| R meas | 0.116(0.598) |
| I/σI | 24.8(4.7) |
| Completeness (%) | 99.7 (98.3) |
| Redundancy | 1.3 (11.1) |
| Refinement | |
| Resolution (Å) | 44.3-3.3 |
| No. reflections | 5983 |
| Rwork/ Rfree | 0.207/0.275 |
| No. atoms | |
| Protein | 2063 |
| Ligand | 5 |
| Wilson B-factor | 64.4 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.0078 |
| Bond angles (°) | 1.181 |
Highest resolution shell is shown in parenthesis.
Silver staining
Two crystals of spastin that had been previously rinsed, mounted on loops and flash-frozen were prepared as a 20 μL SDS-PAGE sample and loaded onto a 10% SDS-PAGE gel with protein standards. The samples were visualized with the ProteoSilver kit (Sigma Aldrich), following manufacturer’s directions.
Results and Discussion
Human spastin structure
We set out to solve the structure of a 42.5 kDa (389 amino acid) construct encompassing the MTBD, N-terminal linker residues and AAA domain of human spastin; however, the MTBD and linker residues were unresolved in the crystal structure. The resulting structure of human spastin AAA domain is remarkably similar to the Drosophila protein, with an overall C -RMSD of 1.7 Å over 261 atoms (Fig. 1A). Despite addition of 1 mM ATP to spastin protein samples prior to crystallization and the presence of the E442Q substitution, which is known to block ATP hydrolysis, the crystal structure is in the nucleotide-free form, with an open nucleotide-binding pocket, very similar to that described previously for the wild-type Drosophila spastin AAA domain (Roll-Mecak & Vale, 2008). A region of positive electron density was observed near the P-loop, which is likely due to the presence of a sulfate ion. In Drosophila spastin, a chloride ion was observed in this position (Roll-Mecak & Vale, 2008). However, placement of a chloride into the human spastin resulted in clear difference density, supporting the placement of a sulfate ion.
Figure 1.

Comparison of the two existing spastin structures. A) Superposition of human spastin (deep teal) and Drosophila spastin (wheat) AAA domains. (B) The loop between a1 and a2 (shown as ribbon representation) is held in the open configuration by a number of interactions with the ATPase domain (shown as surface), including the hydrophobic and ionic interactions seen above. (C) Similar interactions are seen in the nucleotide-free Drosophila spastin structure.
In human spastin, the “linchpin” residue W482 that was observed Drosophila spastin (Roll-Mecak & Vale, 2008) is replaced by a conserved phenylalanine residue (F341). Despite the substitution at this position, the key functional role of this side chain appears to be conserved (Fig. 1B). The extended loop between 1 and 2 is affixed to the NBD by hydrophobic interactions between F341 and a conserved hydrophobic pocket, as well as a salt bridge between K393 and D334. Hydrophobic interactions at the 1- 5 interface, previously reported for the Drosophila protein (Roll-Mecak & Vale, 2008), are also observed in the human spastin structure, with the exception of the substitution of M329 in place of L470, which again appears to play the same functional role. Therefore, despite a number of differences in the amino acid sequences of Drosophila and human spastin, the overall structural and functional characteristics appear to be highly conserved.
Spastins are highly homologous to the proteins VPS4 (Scott et al., 2005), fidgetin (Cox, Mahaffey, Nystuen, Letts, & Frankel, 2000) and katanin p60 (Hartman et al., 1998) within the conserved AAA domain, and together these proteins form the meiotic clade of the AAA+ superfamily. The peripheral N- and C-terminal helices, 1 and 11, have been observed in the structures of Drosophila spastin (Roll-Mecak & Vale, 2008), human fidgetin-like protein 1 [PDB: 3D8B (Karlberg et al. 2008)], and in PSIPRED secondary structure predictions of katanin (Buchan et al. 2010, Jones 1999). In the crystal structure of VPS4 (PDB: 1XWI), the only meiotic AAA+ ATPase that does not sever microtubules, 1 is not present. These findings suggest that 1 is a defining structural characteristic of the microtubule-severing ATPases and its importance is underscored by previous studies showing that non-conservative mutations in 1 severely impair ATPase activity (Roll-Mecak & Vale, 2008).
N-terminal linker and microtubule binding domain
The N-terminal 96 residues of the construct used to obtain the crystals in this study were not visible in the final structure. As this region of the protein is predicted to have very little secondary structure (Kabsch, 1983), it was unclear whether the entire construct had been crystallized and simply contained a disordered N-terminus, or the N- terminal amino acids were actually absent. Previous evidence exists for a large, disordered N- terminus in the Vsp4 crystal structure (Scott et al., 2005) and for cleavage of the N-terminus in the structure of Rca, another AAA+ ATPase involved in activation of Rubisco (Stotz et al., 2011). To determine the size of the human spastin domain that was actually crystallized, crystals of spastin were thoroughly washed and the protein was visualized on a silver-stained SDS-PAGE gel (Fig. 2). The major band migrates at approximately the size of the AAA domain of spastin (32 kDa). Additionally, in a sample of purified spastin protein, several low molecular weight bands were detected, indicating that the N-terminus of this construct is degraded or cleaved. It appears, therefore, that the N-terminal residues were clipped off from the AAA domain in the 5-7 days between the setting up of crystallization drops and the formation of crystals.
Figure 2.
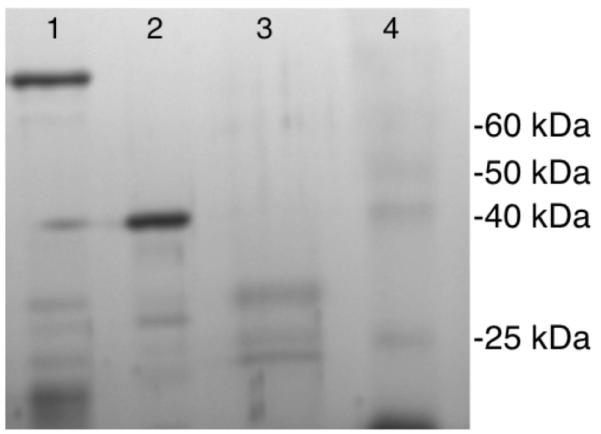
Molecular weight determination of the crystallized spastin protein. Silver-stained gel showing size of crystallized protein in comparison to size of freshly purified spastin. Lane 1 contains purified GST-spastin, lane 2 contains purified spastin after cleavage and removal of GST tag, lane 3 contains crystallized spastin protein, lane 4 contain molecular weight marker.
Comparison to hexameric AAA+ structures
In addition to the structures of spastin, a number of diversely-functioning AAA+ crystal structures have been solved in the P65 space group, including human Vps4 (PDB:1XWI), Rca (PDB:3T15) and the helicase, RuvB, from Thermotoga maritima (PDB:1IN4) (Putnam et al., 2001; Scott et al., 2005; Stotz et al., 2011). Rather than forming a hexameric ring, this crystal-packing symmetry results in a helical, six-fold spiral of monomers in which any six consecutive monomers in a given turn resemble a hexamer when viewed down the six-fold axis. Observation of this helical six-fold arrangement, which is closely related a hexameric ring (in an manner analogous to the way a metal flat washer is related to that of a lock washer), raises the intriguing question of whether the helical arrangement observed in these crystal structures could resemble an actual physiologically relevant conformation that spastin might assume during its mechanical cycle. Some support for this idea comes from the arrangement of monomers in the crystal, which appear to be only slightly distorted in the helical arrangement from that predicted for the hexameric ring (Roll-Mecak & Vale, 2008). For example, an interaction between the conserved arginine finger in the ATPase domain and the nucleotide of the adjacent subunit in the hexamer has been observed in hexameric AAA+ structures, including the bacterial protease FtsH (PDB:3CF1)(Langklotz, Baumann, & Narberhaus, 2012) in its ADP-bound state. We observed the arginine finger residue in human spastin (R459) from an adjacent monomer in our helical structure to be in a similar position and orientation with respect to the ATP binding domain. To visualize this, the ADP nucleotide in FtsH was modeled into the spastin nucleotide- binding pocket by aligning the ATPase domain of spastin onto that of FtsH (C RMSD for 141 alpha carbons = 3.3), while fixing the relative orientations of two symmetry-related spastin monomers (Fig 3). The arginine finger of FtsH interacts with the ADP- -phosphate at a distance of 2.5 Å, and although the analogous R459 in spastin is in a similar position, it is too distant to interact with the modeled nucleotide, the closest rotamer still being 4.5 Å from the -phosphate. This suggests that a relatively small movement between adjacent subunits must occur upon binding of nucleotide, and that the interactions seen in the crystal packing (buried surface area between monomers of 749 Å2) could represent an intermediate state in which nucleotide-free subunits interact weakly. More extensive interactions, such as those between homologous AAA domain subunits of ADP-bound FtsH (average buried surface area, 1450 Å2) and ADP-bound ClpX (average buried surface area, 1319 Å2) hexamers, could form upon interdomain motions driven by ATP binding. These motions would cause the twist that is required to transition from the spiral, as in the crystal, to a more planar hexamer. The relatively small conformation change required to cause this transition can be visualized by comparing the difference in the relative orientation of the AAA subdomains on spastin and FtsH (Fig. 3B).
Figure 3.

Comparison of spastin monomer with hexameric AAA+ protein, FtsH. (A) Spastin (cyan) nucleotide binding pocket with modeled ADP molecule from FtsH. Arginine finger side chain shown as sticks and distance between arginine amine and beta phosphate of ADP shown as dashed line. (B) Interaction between the arginine finger and nucleotide of FtsH (C) Superposition of human spastin (cyan) and FtsH (grey) four-helix bundle domains, highlighting the difference in relative orientation of the HBD and NBDs of these structures.
In summary, the crystal structure of the human spastin AAA domain presented here provides an atomic level foundation for future studies investigating the mechanism utilized by human spastin to sever microtubules, as well as setting a structural context in the human protein for many of the mutations resulting in hereditary spastic paraplegia. Comparison with the Drosophila spastin AAA domain shows a very high degree of structural similarity, as well as conservation of key structural and functional elements. Finally, the observation that both human and Drosophila spastin AAA domains, as well as those of other AAA proteins, form a helical arrangement in crystals, suggests that future studies should explore the possibility that these proteins might utilize a structural transition from a hexameric ring to a helical spiral during their mechanochemical cycle.
Acknowledgements
We would like to thank Dr. Anita Prasad for data collection at APS, as well as Dr. Robert Fischetti and the GM-CA/CAT team at the APS, Argonne National Laboratory, for their assistance in X-ray data collection. Research carried out at the X6A beam line was funded by the NIH (NIGMS) under agreement GM-0080. The GM/CA CAT beamline has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104). The use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under contract DE- AC02-06CH11357. SRW was supported by NIH/NINDS predoctoral fellowship F31NS056781.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams P, Afonine P, Bunkoczi G. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P, Grosse-Kunstleve R. The Phenix refinement framework. CCP4 newsletter. 2005 [Google Scholar]
- Buchan DW, Ward SM, Lobley AE, Nugent TC, Bryson K, et al. Protein annotation and modelling servers at University College London. Nucl. Acids Res. 2010;38(Suppl):W563–W568. doi: 10.1093/nar/gkq427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli FD, Proukakis C, Patel H, Cross H, Azam, et al. The identification of a conserved domain in both spartin and spastin, mutated in hereditary spastic paraplegia. Genomics. 2003;81(4):437–441. doi: 10.1016/s0888-7543(03)00011-9. [DOI] [PubMed] [Google Scholar]
- Cox GA, Mahaffey CL, Nystuen A, Letts VA, Frankel WN. The mouse fidgetin gene defines a new role for AAA family proteins in mammalian development. Nature genetics. 2000;26(2):198–202. doi: 10.1038/79923. [DOI] [PubMed] [Google Scholar]
- DeLano W. The PyMOL molecular graphics system. 2002.
- Depienne C, Fedirko E, Forlani S, Cazeneuve C, Ribaï P, et al. Exon deletions of SPG4 are a frequent cause of hereditary spastic paraplegia. J. Med. Genet. 2007;44(4):281–284. doi: 10.1136/jmg.2006.046425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Evans KJ, Gomes ER, Reisenweber SM, Gundersen GG, Lauring BP. Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing. J. Cell Biol. 2005;168(4):599–606. doi: 10.1083/jcb.200409058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman JJ, Mahr J, McNally K, Okawa K, Iwamatsu A, et al. Katanin, a microtubule-severing protein, is a novel AAA ATPase that targets to the centrosome using a WD40-containing subunit. Cell. 1998;93(2):277–287. doi: 10.1016/s0092-8674(00)81578-0. [DOI] [PubMed] [Google Scholar]
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Crystallogr. D Biol. Crystallogr. 2010;D66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlberg T, Wisniewska M, Andersson J, Arrowsmith CH, Berglund H, et al. Crystal structure of human fidgetin-like protein 1 in complex with ADP. 2008.
- Langklotz S, Baumann U, Narberhaus F. Structure and function of the bacterial AAA protease FtsH. Biochim. Biophys. Acta. 2012;1823(1):40–48. doi: 10.1016/j.bbamcr.2011.08.015. [DOI] [PubMed] [Google Scholar]
- Mitchison T, Kirschner M. Dynamic instability of microtubule growth. Nature. 1984;312(5991):237–242. doi: 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- Putnam CD, Clancy SB, Tsuruta H, Gonzalez S, Wetmur JG, et al. Structure and mechanism of the RuvB Holliday junction branch migration motor. J. Mol. Biol. 2001;311(2):297–310. doi: 10.1006/jmbi.2001.4852. [DOI] [PubMed] [Google Scholar]
- Roll-Mecak A, McNally FJ. Microtubule-severing enzymes. Curr. Opin. Cell Biol. 2010;22(1):96–103. doi: 10.1016/j.ceb.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roll-Mecak A, Vale RD. The Drosophila homologue of the hereditary spastic paraplegia protein, spastin, severs and disassembles microtubules. Curr. Biol. 2005;15(7):650–655. doi: 10.1016/j.cub.2005.02.029. [DOI] [PubMed] [Google Scholar]
- Roll-Mecak A, Vale RD. Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature. 2008;451(7176):363–367. doi: 10.1038/nature06482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiel JA, Park K, Morphew MK, Reid E, Hoenger A, et al. Endocytic membrane fusion and buckling-induced microtubule severing mediate cell abscission. J Cell Sci. 2011;124(9):1411–1424. doi: 10.1242/jcs.081448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A, Chung H-Y, Gonciarz-Swiatek M, Hill GC, Whitby FG, et al. Structural and mechanistic studies of VPS4 proteins. EMBO Journal. 2005;24(20):3658–3669. doi: 10.1038/sj.emboj.7600818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz M, Mueller-Cajar O, Ciniawsky S, Wendler P, Hartl FU, et al. Structure of green-type Rubisco activase from tobacco. Nat. Struct. Mol. Biol. 2011;18(12):1366–1370. doi: 10.1038/nsmb.2171. [DOI] [PubMed] [Google Scholar]
- Vale RD. AAA proteins. Lords of the ring. J. Cell Biol. 2000;150(1):F13–9. doi: 10.1083/jcb.150.1.f13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SR, Evans KJ, Lary J, Cole JL, Lauring BP. Recognition of C-terminal amino acids in tubulin by pore loops in Spastin is important for microtubule severing. J. Cell Biol. 2007;176(7):995–1005. doi: 10.1083/jcb.200610072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Rismanchi N, Renvoisé B, Lippincott-Schwartz J, Blackstone C, et al. Structural basis for midbody targeting of spastin by the ESCRT-III protein CHMP1B. Nat. Struct. Mol. Biol. 2008;15(12):1278–1286. doi: 10.1038/nsmb.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]