

Abstract
The cardiac electrical impulse depends on an orchestrated interplay of transmembrane ionic currents in myocardial cells. Two critical ionic current mechanisms are the inwardly rectifying potassium current (IK1), which is important for maintenance of the cell resting membrane potential, and the sodium current (INa), which provides a rapid depolarizing current during the upstroke of the action potential. By controlling the resting membrane potential, IK1 modifies sodium channel availability and therefore, cell excitability, action potential duration, and velocity of impulse propagation. Additionally, IK1–INa interactions are key determinants of electrical rotor frequency responsible for abnormal, often lethal, cardiac reentrant activity. Here, we have used a multidisciplinary approach based on molecular and biochemical techniques, acute gene transfer or silencing, and electrophysiology to show that IK1–INa interactions involve a reciprocal modulation of expression of their respective channel proteins (Kir2.1 and NaV1.5) within a macromolecular complex. Thus, an increase in functional expression of one channel reciprocally modulates the other to enhance cardiac excitability. The modulation is model-independent; it is demonstrable in myocytes isolated from mouse and rat hearts and with transgenic and adenoviral-mediated overexpression/silencing. We also show that the post synaptic density, discs large, and zonula occludens-1 (PDZ) domain protein SAP97 is a component of this macromolecular complex. We show that the interplay between Nav1.5 and Kir2.1 has electrophysiological consequences on the myocardium and that SAP97 may affect the integrity of this complex or the nature of Nav1.5–Kir2.1 interactions. The reciprocal modulation between Nav1.5 and Kir2.1 and the respective ionic currents should be important in the ability of the heart to undergo self-sustaining cardiac rhythm disturbances.
Keywords: reentry, scaffolding proteins, conduction velocity, protein trafficking
In the heart, the inward rectifying potassium current (IK1) is the major current responsible for the maintenance of the resting membrane potential (RMP), whereas the sodium current (INa) provides the largest fraction of the inward depolarizing current that flows during an action potential (1). It is well-known that a relationship exists between these two ionic currents that is crucial for proper cardiac electrical function; disruption of this balance results in changes in sodium channel availability, cell excitability, action potential duration, and conduction velocity (2). Accordingly, IK1–INa interactions are important in stabilizing and controlling the frequency of the electrical rotors that are responsible for the most dangerous cardiac arrhythmias, including ventricular tachycardia and fibrillation (3).
Post synaptic density, discs large, and zonula occludens-1 (PDZ) domain proteins link different and in many cases, multiple proteins to macromolecular complexes through interactions with their various domains. More than 70 PDZ domain-containing proteins have been identified that interact with different ion channels, receptors, and signaling molecules (4). In the heart, NaV1.5 and Kir2.1 interact independently with at least two distinct PDZ domain scaffolding proteins (SAP97 and α1-syntrophin) (5–7). Previous studies have shown that SAP97 regulates Kir2.x channels by multiple mechanisms and that IK1 may be regulated by this SAP97-mediated modulation of the Kir2 channels expression (7, 8). It has also been shown that NaV1.5 interacts with dystrophin through syntrophin (9) and that this association connects NaV1.5 to the nNOS-plasma membrane Ca-ATPase complex in the heart (10).
To our knowledge, there is no evidence to date for molecular interactions between NaV1.5 and Kir2.1 through a common partner(s) or in a macromolecular complex. Despite the pathophysiological implications of the (im)balance between INa and IK1, our extensive review of the relevant literature yielded no data on any molecular interactions (direct or indirect) between NaV1.5 and Kir2.1 other than interactions related to cardiac excitability and the control of the RMP and action potential duration (APD). We hypothesized that NaV1.5 and Kir2.1 interact with each other functionally and that this interaction involves and/or is mediated by one or more common partners in a macromolecular complex, with particular focus on the membrane-associated guanylate kinase (MAGUK; i.e., SAP97) family of proteins. We have, therefore, used a multidisciplinary approach based on molecular and biochemical techniques, immunofluorescence, patch clamp, acute gene transfer, RT-PCR, and gene silencing to show that IK1–INa interactions involve a reciprocal modulation of expression of their respective channel proteins (Kir2.1 and NaV1.5) within a macromolecular complex. This reciprocal modulation may involve SAP97, which we and others have shown affects IK1 (7, 8) and INa densities. Here, we also show that expression of NaV1.5 itself may affect the turnover and regulation of the biophysical properties of Kir2.1 channels. We also show that this reciprocal modulation has important implications for the ability of the heart to undergo self-sustaining cardiac rhythm disturbances.
Results
Reciprocal Regulation of NaV1.5 and Kir2.1 in Adult Rat Ventricular Myocytes.
We overexpressed NaV1.5 in adult rat ventricular myocytes (ARVMs) using an adenoviral construct (Ad-NaV1.5); control myocytes were infected with an adenoviral construct encoding GFP (Ad-GFP). Average INa-density voltage (I–V) plots from voltage clamp experiments are shown in Fig. 1A. There was a significant increase (P < 0.005) in INa density at several tested voltages compared with control. Fig. 1B represents the voltage-dependent activation and inactivation (m∞ and h∞) curves for the data presented in Fig. 1A. There was no significant difference for V1/2 values between control and Ad-NaV1.5-treated cells. Reversal potentials were calculated under both conditions, returning values of 5.01 and 0 mV for Ad-NaV1.5– and Ad-GFP–expressing cells, respectively. Overexpression of NaV1.5 also resulted in a significant increase in IK1 density in both the inward (P < 0.01) and outward (P < 0.05) directions (Fig. 1C). We next considered the possibility that the reverse scenario may also be true (i.e., that Kir2.1 overexpression would increase NaV1.5 functional expression). Infection with Ad-Kir2.1 resulted in an increase of IK1 in ARVMs (Fig. 1D). Peak inward (−100 mV) and outward (−60 mV) currents were significantly larger (P < 0.01) than in control (Ad-GFP). As hypothesized, peak INa density was also increased (P < 0.005) (Fig. 1E). These results suggest that common molecular mechanisms might be involved in the regulation of Kir2.1 and NaV1.5 functional expression. Voltage-dependent activation and inactivation (m∞ and h∞) curves were generated for this set of data. There was no significant difference in values of V1/2 (Fig. 1F).
Fig. 1.

Reciprocal regulation of NaV1.5 and Kir2.1 in ARVMs. (A and C) NaV1.5 overexpression increases both INa and IK1 densities. (A) Superimposed INa density/voltage relationships (5 mmol/L [Na+]o) for Ad-GFP– (black; N = 1, n = 7) and Ad-NaV1.5–infected (red; N = 2, n = 6) cells. (B) Voltage-dependent activation and inactivation (m∞ and h∞) curves for the data presented in A. (C) Superimposed IK1 density/voltage relationship for Ad-GFP– (black; N = 3, n = 8) and Ad-NaV1.5–infected (red; N = 3, n = 8) cells. (Inset) Magnification of the outward component of the IK1 I–V relationship. (D and E) Kir2.1 overexpression increases both IK1 and INa densities. (D) Superimposed IK1 density/voltage relationships for Ad-GFP (black; N = 3, n = 8) and Ad-Kir2.1 (red; N = 2, n = 5). (E) Superimposed INa density/voltage relationships (20 mmol/L [Na+]0) for Ad-GFP (black; N = 2, n = 6) and Ad-Kir2.1 (red; N = 4, n = 7). (F) Voltage-dependent activation and inactivation (m∞ and h∞) curves for the data presented in E. *P < 0.005; #P < 0.05; δP < 0.01 (unpaired t test with Welch’s correction). N represents the number of animals/litters, and n equals the number of experiments or cells.
Potential Role of SAP97 in NaV1.5–Kir2.1 Interactions.
We used adenoviral transfer technology to knock down SAP97 expression in ARVMs and studied properties of IK1 and INa under these conditions (Fig. 2). The SAP97 silencing construct contains an shRNA for SAP97 (Ad-shSAP97) (8, 11). Samples were collected 3 d postinfection for analysis (day 0 = day of cell isolation). Separate groups of ARVMs were infected with a control virus (Ad-515) previously generated by our laboratory (8, 12). Silencing of SAP97 expression was confirmed by Western blot analysis (Fig. 2A). Relative levels of SAP97 were reduced by ∼56% on day 3 postinfection (Fig. 2B). In ARVMs infected with Ad-shSAP97, IK1 density was reduced by ∼50% compared with control (Fig. 2C). Also, as we have recently shown (8), unitary IK1 properties (i.e., conductance and mean open and closed times) were unchanged in ARVMs infected with Ad-shSAP97. Furthermore, IK1 properties were unchanged as a function of days in culture (SI Materials and Methods and Fig. S1) (8). Overall, our results strongly suggest that a major mechanism for alterations in IK1 function is channel clustering and/or stabilization in the cell membrane (6, 8, 13). Similar to shSAP97-induced reduction of IK1 density, infection with shSAP97 also reduced (P < 0.005) whole-cell INa density at several tested voltages (Fig. 2D). Voltage-dependent activation and inactivation (m∞ and h∞) curves were generated for control and SAP97-silenced cells. No significant difference in values of V1/2 was shown (Fig. 2E).
Fig. 2.

Role of SAP97 in the reciprocal modulation of IK1 and INa. (A and B) Ad-shSAP97 infection silences SAP97 expression in adult rat ventricular myocytes (ARVMs) by day 3 in culture compared with control. (A) Immunoblot showing immunodetection of SAP97 expression (Top and Middle) and GAPDH (loading control; Bottom) from cells infected with either a control (Ad-515) or shSAP97 (Ad-shSAP97) adenovirus. Middle (SAP97) shows a longer exposure of the same immunoblot shown in Top; the overexposure confirms expression of SAP97 in the Ad-SAP97–silenced cells and shows the contrasting intensities and apparent protein levels between control and SAP97-silenced ARVMs. (B) Densitometric analysis of SAP97 expression normalized to GAPDH levels in control (black) and shSAP97-silenced (red) cells. Values represent data (mean ± SEM) from two different preparations harvested 3 d after infection. SAP97 expression was effectively knocked down by ∼56% on day 3. (C) IK1 density is reduced after SAP97 silencing in ARVMs. Peak inward current density at −100 mV (control: −7.67 ± 1.52 pA/pF; Ad-shSAP97: −4.55 ± 0.47 pA/pF) and peak outward current density at −60 mV (control: 1.03 ± 0.46 pA/pF; Ad-shSAP97: 0.34 ± 0.03 pA/pF) were significantly different (P = 0.02 and P = 0.04, respectively) between myocytes infected with shSAP97 (N = 4, n = 11) and myocytes infected with Ad-515 (control; N = 2, n = 6). Inset shows the protocol used to measure the current. (D) Effects of SAP97 knockdown on sodium channel current density in ARVMs. Superimposed INa density/voltage relationships in control (Ad-515) and SAP97-silenced (Ad-shSAP97) cells. A significant reduction in INa density was observed for SAP97-silenced cells at several tested voltages. For both control and silenced conditions, N = 2 and n = 11. (Inset) Voltage clamp step protocol. (E) Voltage-dependent activation and inactivation (m∞ and h∞) curves for the data presented in D. *P < 0.005; #P < 0.05; δP < 0.01 (unpaired t test with Welch’s correction).
SAP97 Interacts with NaV1.5 and Kir2.1.
Kir2.1–Kir2.3 channel proteins as well as NaV1.x have a class I PDZ binding motif and thus, are able to bind PDZ domain proteins such as SAP97 and syntrophin (9, 13, 14). Previous studies have indeed shown interactions of SAP97 with Kir2.1 (6–8). Fig. 3A, a–j show results of immunohistochemical analyses of SAP97 association with Kir2.1 or NaV1.5 channels in the adult rat ventricle; Fig. 3A, k–o show association between NaV1.5 and Kir2.1 channels. Tissue sections of 8- to 10-μm thickness were prepared from adult rat heart and immunostained with antibodies specific for SAP97 (Fig. 3A, a, d, f, and i) and either NaV1.5 (Fig. 3A, b, e, k, and n) or Kir2.1 (Fig. 3A, g, j, l, and o). Note that SAP97 exhibits a striated staining pattern reminiscent of a t-tubular distribution (Fig. 3A, double arrows) and also concentrates at the intercalated disk (Fig. 3A, asterisk). Overlapping areas of staining were observed for SAP97 with both NaV1.5 and Kir2.1 at each of these locales. A similar distribution at the t-tubules and intercalated disk was also observed between NaV1.5 (Fig. 3A, k) and Kir2.1 (Fig. 3A, l) when the staining patterns were directly compared for these two proteins. Fig. 3A, d and e, i and j, and n and o show magnifications of the boxed regions from Fig. 3A, c, h, and m, respectively; images are presented in black and white for clarity. Quantification of colocalization showed significant overlap between the fluorescent staining patterns and subcellular distribution of NaV1.5/Kir2.1 and SAP97. The average Pearson’s correlation coefficient was 0.86 ± 0.043 (n = 9) for NaV1.5 and SAP97, 0.9 ± 0.03 (n = 6) for Kir2.1 and SAP97, and 0.91 ± 0.010 (n = 3) for NaV1.5 and Kir2.1. These results strongly suggest that SAP97, NaV1.5, and Kir2.1 colocalize to the same discrete subcellular regions in the rat ventricle.
Fig. 3.

SAP97 immunolocalizes and immunoprecipitates with NaV1.5 and Kir2.1 in the rat ventricle. (A) Immunohistochemical analysis and localization for SAP97, NaV1.5, and Kir2.1 in the adult rat heart. SAP97 localization with NaV1.5 (a–c) and Kir2.1 (f–h) channels and NaV1.5 (k and n) localization with Kir2.1 channels (l and o). Double arrows denote a staining pattern reminiscent of a t-tubular distribution. *Intercalated disk. d and e, i and j, and n and o are magnifications of the boxed areas from the merged images shown in c, h, and m, respectively. SAP97 staining is presented in the enlarged d and i images, NaV1.5 is presented in e and n, and Kir2.1 is presented in j and o; images are shown in black and white for clarity. (Scale bars: 20 μm.) SAP97 (B, Top), NaV1.5 (C, Top), and Kir2.1 (D, Top) are detected by Western blot after immunoprecipitation with specific antibodies raised to the respective protein, which shows that each immunoprecipitation is specific for the desired protein. NaV1.5 coimmunoprecipitates with SAP97 (B, Middle), and SAP97 coimmunoprecipitates with NaV1.5 in the reverse reaction (C, Middle). Kir2.1 coimmunoprecipitates with SAP97 antibodies (B, Bottom), and SAP97 coimmunoprecipitates with Kir2.1 in the reverse reaction (D, Bottom). NaV1.5 and Kir2.1 each coimmunoprecipitate with the other ion channel protein (C, Bottom and D, Middle, respectively). All (co)immunoprecipitation reactions used membrane-enriched preparations generated from the ventricles of rat heart. IB, antibody used for immunoblotting; IP, antibody used for immunoprecipitation.
Given these immunolocalization data as well as the Ad-shSAP97 data in Fig. 2 C and D, we then investigated the possibility that NaV1.5 and Kir2.1 associate with SAP97. Coimmunoprecipitation results show that SAP97 does associate with Kir2.1 (Fig. 3 B, Bottom and D, Bottom) and NaV1.5 (Fig. 3 B, Middle and C, Middle). NaV1.5 and Kir2.1 were also found to coimmunoprecipitate together in both the forward and reverse coimmunoprecipitation reactions (Fig. 3 C, Middle and D, Middle). Control immunoprecipitation reactions were conducted for SAP97 (Fig. 3B, Top), NaV1.5 (Fig. 3C, Top), and Kir2.1 (Fig. 3D, Top) to show specificity of the immunoprecipitating antibody for the desired protein. In a different set of control experiments, we confirmed the specificity of the PDZ domain-mediated interaction(s). Kir6.2 was used as a negative control, because this protein sequence lacks a PDZ consensus sequence at its extreme C terminus (6–8). As expected, Kir6.2 failed to immunoprecipitate SAP97, and neither SAP97 nor Kir2.1 coimmunoprecipitated Kir6.2 (Fig. S2).
Kir2.1 Overexpression Increases NaV1.5 in the Mouse Heart.
To rule out the possibility that the above interactions might be model-dependent or an artifact of adenoviral infection, we conducted patch clamp experiments in ventricular myocytes isolated from a transgenic mouse model that overexpresses Kir2.1 and shows a subsequent up-regulation of IK1 (∼12×) (15). A comparison of INa density in WT and Kir2.1-overexpressing (OE) mice is shown in Fig. 4. Representative INa traces from each genotype are presented in Fig. 4A, Inset. The I–V relationship for INa in myocytes from Kir2.1 OE mice shows a significant increase in INa density at several tested voltages (Fig. 4A, Left) (P < 0.005). The m∞ and h∞ curves (Fig. 4A, Right) generated for the WT and Kir2.1 OE mice showed a significant hyperpolarizing shift in the inactivation profile (P = 0.041). Values for V1/2 were −86.44 and −81.18 mV for the Kir2.1 OE and WT myocytes, respectively. Furthermore, the activation profile also showed a hyperpolarizing shift in the V1/2 that tended towards significance (P = 0.056). Values for V1/2 were −60.76 and −55.55 mV for the Kir2.1 OE and WT myocytes, respectively. We speculate that the apparent shift in the activation profile, if any, could be because of altered stoichiometry of other necessary accessory proteins (i.e., β-subunits or ankyrin-G) as a consequence of the modification in the transgene.
Fig. 4.

Biophysical characterization of sodium current in Kir2.1 OE mice. (A) Superimposed INa density/voltage relationships in WT (black) and Kir2.1 OE (red) mice. (Inset) Representative examples of INa traces in each group. Dotted line denotes 0 pA. WT: N = 4, n = 10; Kir2.1 OE: N = 2, n = 6. *P < 0.005 (unpaired t test with Welch’s correction). (Right) Voltage-dependent activation and inactivation (m∞ and h∞) curves for the data presented in A. (B) The unitary conductance properties of sodium channels were characterized in ventricular myocytes isolated from the Kir2.1 OE mouse. Representative traces from control (littermate WT) and Kir2.1 OE mouse myocytes are illustrated in Inset. Histogram of the unitary events from WT littermates (N = 3, n = 240, seven patches) and Kir2.1 OE mice (N = 3, n = 231, eight patches) are superimposed and plotted.
To investigate the mechanism for the increase in the sodium current density from Kir2.1 OE mouse ventricular myocytes, the unitary conductance properties of sodium channels were characterized (Fig. 4B). Representative traces from control and Kir2.1 OE mouse myocytes are illustrated in Fig. 4B, Inset. Histogram of the unitary sodium channel events from WT littermates and Kir2.1 OE mice are superimposed. Average unitary conductance was 36.1 ± 0.39 pS in control and 37.46 ± 0.85 pS in cells isolated from OE mice. Also, mean open probability was 0.29 ± 0.05 (N = 2, n = 3) in control and 0.25 ± 0.03 (N = 2, n = 4) in the OE myocytes (16). These results show that the increase in INa was not because of changes in unitary conductance or open probability (P value was not significant) of the underlying channels. Thus, the likely explanation is an increased number of functional channels on the cell membrane. Consistent with this possibility, additional investigation showed that increased Kir2.1 expression leads to altered membrane protein expression of SAP97 and NaV1.5 in the transgenic Kir2.1 OE mouse heart (Fig. 5). Relative levels of SAP97 and NaV1.5 were significantly increased 1.6 and 2.2 times, respectively (Fig. 5B) compared with levels in WT littermates (P < 0.01). In addition, as shown in Fig. S3, RT-PCR analyses showed that mRNA levels for the genes encoding for SAP97 and NaV1.5 were not significantly different between WT and Kir2.1 OE mouse hearts. These results rule out the possibility that increased protein production could potentially lead to more channels at the cell membrane; rather, the observed increases in protein and current density seem to result from increased protein trafficking to and/or stabilization at the plasma membrane.
Fig. 5.

Relative levels of SAP97 and NaV1.5 are significantly increased in hearts of transgenic mice overexpressing Kir2.1. Crude membrane vesicles were prepared from the ventricles of control (WT) and Kir2.1 OE mice. Samples (16 μg/lane) were analyzed by SDS/PAGE and immunoblotted using specific antibodies for SAP97 or NaV1.5, as indicated. (A) Representative immunoblots after detection of protein immunoreactivity with HRP-conjugated secondary antibodies and chemiluminescence (Upper). The corresponding Amido Black nitrocellulose (protein stain) is shown in Lower to show analysis of equal total protein. Protein concentrations were also verified by Lowry assay. (B) Densitometric analysis of data shown in A comparing relative protein levels between WT and Kir2.1 OE mice. Results are expressed as mean signal intensity, and they represent data from three animals for each genotype (N = 3 per genotype, δP < 0.01, mean ± SEM).
To ensure that the interaction between reciprocal modulation of NaV1.5 and Kir2.1 was not simply a phenomenon caused by transgenic manipulation, we also investigated the effect of Kir2.1 overexpression on the protein, mRNA, and current density of another potassium ion channel, Kir6.2. This ATP-sensitive inward rectifier potassium channel lacks both a PDZ domain and a PDZ domain consensus binding motif. Transgenic overexpression of Kir2.1 in this mouse model leads to a significant increase in INa density (Fig. 4) and NaV1.5 relative protein levels (Fig. 5). Interestingly, peak IKATP density is not significantly different in ventricular myocytes isolated from WT and Kir2.1 OE mice (Fig. S4). Accordingly, mRNA and protein levels of Kir6.2 were not significantly altered as a result of Kir2.1 overexpression (Fig. S5).
Kir2.1 Down-Regulation Decreases Nav1.5 in the Mouse Heart.
Data presented in Fig. S6 show the effects of KCNJ2 gene reduction on mRNA levels of NaV1.5 and SAP97 in heterozygous Kir2.1 KO (Kir2.1−/+) mice. RT-PCR experiments showed that the expression levels of the genes encoding for NaV1.5 and SAP97 mRNA are unchanged in ventricles of Kir2.1−/+ mice compared with WT littermates. However, 50% allelic reduction of KCNJ2 gene expression by homologous recombination resulted in a significant decrease in relative membrane protein levels of NaV1.5 and SAP97 (P < 0.05) (Fig. 6). Moreover, similar to what we showed for transgenic Kir2.1 overexpression, altered expression of Kir2.1 protein in Kir2.1−/+ mouse ventricles had no significant effect on the protein or mRNA levels of Kir6.2 (Fig. S7).
Fig. 6.

Transgenic reduction of Kir2.1 gene expression leads to a significant decrease in relative protein levels of NaV1.5 and SAP97. Crude membrane vesicles were prepared from the ventricles of control (WT) and Kir2.1−/+ mice. Fig. 5 has methods describing SDS/PAGE and immunoblotting. (A) Representative immunoblots after detection of protein immunoreactivity with HRP-conjugated secondary antibodies and chemiluminescence (Upper). The corresponding Amido Black nitrocellulose (protein stain) is shown in Lower to show analysis of equal total protein. Protein concentrations were also verified by Lowry assay. (B) Densitometric analysis comparing relative protein levels between WT and Kir2.1−/+ mice. Results are expressed as mean signal intensity (N = 7 per genotype, #P < 0.05, mean ± SEM).
Altogether, the data presented in Results, Kir2.1 Overexpression Increases NaV1.5 in the Mouse Heart and this section show that reciprocal regulation between NaV1.5 and Kir2.1 also occurs in the mouse heart, and it is, thus, not model-specific. The results also provide assurance that the reciprocal regulation observed in ARVMs was not a virally mediated phenomenon. Furthermore, they show that, whether these reciprocal changes lead to down- or up-regulation of channel proteins, they seem to involve posttranscriptional and/or posttranslational mechanisms.
NaV1.5 Promotes Cell Surface Expression of Kir2.1.
It is now well-established that the balance between trafficking pathways to and from the cell membrane is an important determinant of the steady-state cell surface density of membrane proteins. Therefore, to test the hypothesis that reciprocal NaV1.5–Kir2.1 interactions involve protein trafficking, we measured steady-state levels of cell surface Kir2.1-pHluorin in the presence or absence of NaV1.5, which is shown in Fig. 7. Coexpression with NaV1.5 led to an increase in steady-state surface Kir2.1-pHluorin compared with control in neonatal (Fig. 7A) and adult (Fig. 7B) rat ventricular myocytes as well as HL-1 cells (Fig. 7C) (neonatal rat: n > 58 myocytes, P < 0.005; adult rat ventricular myocytes: n > 75 cells, P < 0.005; HL-1 cells: n > 56 cells, P < 0.005). We then investigated constitutive internalization of Kir2.1 channels in HL-1 cells transiently expressing Kir2.1-pHluorin (SI Materials and Methods). Cells were live cell labeled with anti-GFP antibody before incubation for 0, 10, 20, 30, 60, or 120 min at 37 °C. Fig. S8 shows the time course of constitutive internalization of Kir2.1 channels, with representative images showing the increase in internalized Kir2.1-pHluorin with time (t1/2 = 23.7 min). Based on these data, we examined the effect of NaV1.5 coexpression on Kir2.1 stability with respect to membrane expression. Surface (Fig. 8A) and internalized (Fig. 8B) Kir2.1-pHluorin were quantified after 20 min of internalization for both transfection conditions. Compared with control at the same time point, concomitant NaV1.5 expression increased the surface level of Kir2.1-pHluorin (30.9 ± 6.9%, n > 30 cells, P = 0.0147); accordingly, this finding led to a decrease in internalized Kir2.1-pHluorin channels (17.2 ± 3.4%, n > 30 cells, P = 0.0025). Altogether, these data show that NaV1.5 promotes cell surface expression of Kir2.1, at least in part, by reducing its internalization.
Fig. 7.
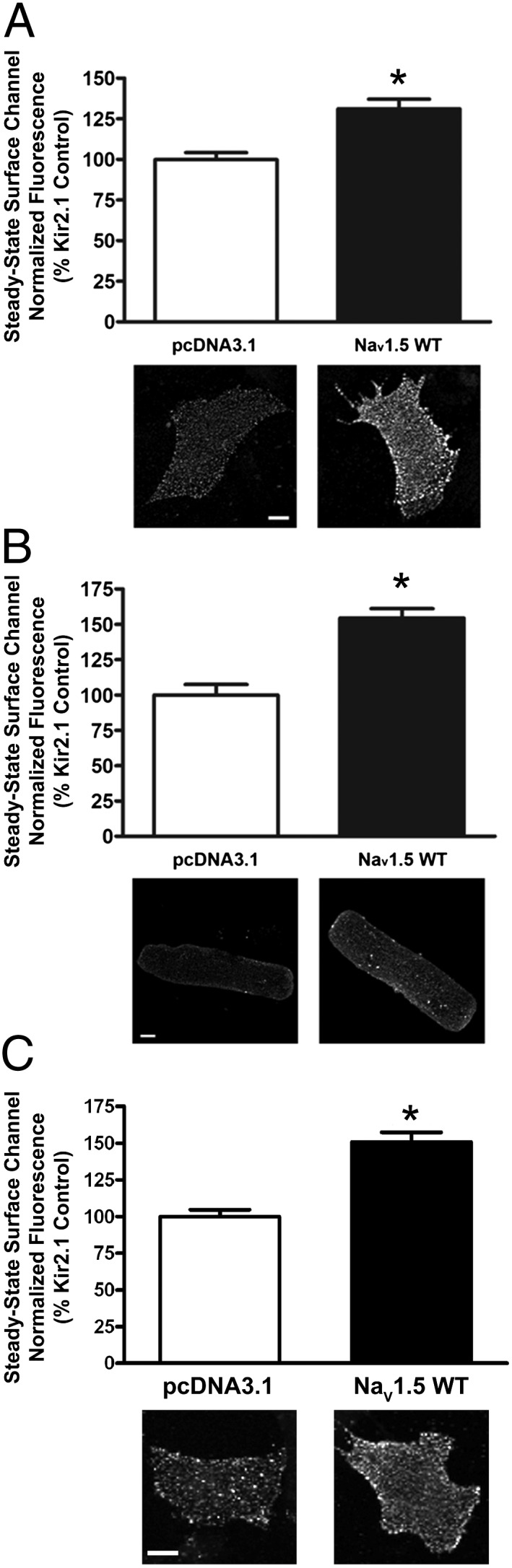
Coexpression of Kir2.1-pHluorin with NaV1.5 promotes cell surface expression of Kir2.1-pHluorin. Quantification of steady-state cell surface Kir2.1-pHluorin in cells cotransfected/coinfected with either empty pcDNA3.1 and DsRed vectors or plasmids/adenoviruses encoding NaV1.5 and DsRed. Cells were live cell labeled with anti-GFP antibody before incubation for 20 min at 37 °C. Cell surface channels were then labeled as described in SI Materials and Methods. Representative images of surface Kir2.1-pHluorin channels for each condition and cell type are shown. (Scale bars: 10 μm.) *P < 0.005 as determined by unpaired Student t test. (A) Rat neonatal ventricular myocytes (n > 58 myocytes). (B) ARVMs (n > 75 cells). (C) HL-1 cells (n > 56 cells). DNA plasmid transfection was used for rat neonatal ventricular myocytes and HL-1 cells; adenoviral-mediated overexpression was used for ARVMs.
Fig. 8.

Deletion of the PDZ domain of Kir2.1-pHluorin abolishes the NaV1.5-mediated increase in cell surface channel. (A and B) Coexpression with NaV1.5 reduces internalization of Kir2.1-pHluorin in HL-1 cells. HL-1 cells expressing Kir2.1-pHluorin were cotransfected with either empty pcDNA3.1 and DsRed vectors or plasmids encoding NaV1.5 and DsRed. Representative images of surface channels for each of these conditions are shown. Quantification of surface (A) and internalized (B) Kir2.1-pHluorin in HL-1 cells coexpressing either pcDNA3.1 + DsRed or NaV1.5 + DsRed. (C) Quantification of steady-state cell surface Kir2.1 in HL-1 cells expressing WT Kir2.1, Kir2.1ΔPDZ, or Kir2.1ΔPDZ + NaV1.5. The absence of an intact PDZ binding consensus sequence significantly decreased cell surface expression of Kir2.1ΔPDZ compared with WT Kir2.1-pHluorin. The reduced surface expression of Kir2.1ΔPDZ was not rescued by concomitant coexpression with NaV1.5. Cell surface labeling (Materials and Methods and Fig. 7) was conducted 48 h posttransfection (n ≥ 55 cells). Representative images of surface channel for each experimental condition are shown in the indicated panels. (Scale bars: 10 μm.) *P < 0.005; #P < 0.05 as determined by unpaired Student t test.
To confirm specificity and further investigate the role of the PDZ domain in mediating the interaction between Kir2.1 and NaV1.5, we measured steady-state levels of surface Kir2.1-pHluorin in the presence and absence of a functional PDZ consensus sequence. Kir2.1ΔPDZ was generated by deleting the 3 aa at the C terminus that are known to be necessary for PDZ domain binding interactions from the Kir2.1 sequence. In the absence of an intact PDZ domain binding consensus sequence, there was a significant decrease in the cell surface expression of Kir2.1 compared with WT Kir2.1 (Fig. 8C). In contrast to what was observed with WT Kir2.1 (Fig. 7C), cooverexpression with NaV1.5 was not able to rescue this effect, because cell surface expression of Kir2.1ΔPDZ was significantly reduced compared with WT Kir2.1 with or without concomitant overexpression of NaV1.5 (n > 55 cells, P < 0.005). These data support that the interaction between Kir2.1 and NaV1.5 is specific and that it is mediated, at least in part, by PDZ domain-mediated binding.
Electrophysiological Consequences of NaV1.5–Kir2.1 Interactions.
Data in Kir2.1 OE mice have provided strong evidence that the electrophysiological interplay between INa and IK1 is essential in controlling the stability and frequency of reentry and ventricular tachycardia/ventricular fibrillation (3). In addition, those results provided direct evidence that IK1 up-regulation is a substrate for very fast electrical rotors in structurally normal ventricles. Here, we show the electrophysiological consequences of the reciprocal regulation of NaV1.5 and Kir2.1 on the frequency and dynamics of rotors in neonatal rat ventricular myocyte (NRVM) monolayers. First, we investigated the changes in dV/dt and APD resulting from the molecular interplay between NaV1.5 and Kir2.1 when one or both protein channels are overexpressed in isolated NRVMs. Fig. 9 A–D shows current clamp recordings obtained from four different infection conditions where cells were paced at a cycle length of 500 ms. Fig. 9A has examples of control action potentials. In Fig. 9B, infection of the cell with Ad-NaV1.5 alone hyperpolarized the RMP (Fig. S9), increased the dV/dt (Ad-GFP: 125.5 ± 16.8 V/s, n = 6 vs. Ad-NaV1.5: 185.7 ± 19.0 V/s, n = 4; P < 0.05), and prolonged the APD. In Fig. 9C, infection with Ad-Kir2.1 alone also hyperpolarized the RMP and increased dV/dt (Ad-GFP: 125.5 ± 16.8 V/s, n = 6 vs. Ad-Kir2.1: 210.3 ± 13.4 V/s, n = 3; P < 0.05). However, in this case, the APD was greatly abbreviated. Finally, in contrast with the effects of Ad-NaV1.5 infection alone, infection with Ad-Kir2.1 + NaV1.5 resulted in a significant reduction of the APD (Fig. 9D). Composite results (mean ± SEM) of APD at 80% repolarization (APD80) are presented in Fig. 9E.
Fig. 9.

NaV1.5 and Kir2.1 cooverexpression significantly reduces the APD in single NRVMs. (A–D) Action potential recordings from NRVMs infected with adenoviruses encoding (A) GFP (n = 5), (B) NaV1.5 + GFP (n = 4), (C) Kir2.1 + GFP (n = 4), or (D) Kir2.1 + NaV1.5 (n = 5) paced at 2 Hz. (E) Summary plots of mean ± SEM. APD80 for each group. Ad-GFP was used as an adenoviral control for single infections to account for the higher viral load expected as a result of Kir2.1 and NaV1.5 coinfection (red bar). #P < 0.05; δP < 0.01.
Fig. 10 illustrates phase maps (Fig. 10A) and rotation frequency plots (Fig. 10B) for NRVM monolayers infected with Ad-GFP, Ad-NaV1.5 alone, Ad-Kir2.1 alone, or a combination of Ad-NaV1.5 + Ad-Kir2.1. Color-coded signals in Fig. 10A indicate the phase in the excitation–recovery cycle at each pixel location (3, 17, 18). Stable reentry was present in all monolayers, and in each, there was a single stationary rotor. However, in single NRVMs, Ad-NaV1.5 infection alone prolonged the APD (Fig. 9). As shown in Fig. 10A, APD prolongation during reentry in the monolayer was manifest as a lengthening of the depolarization phases in the reentry cycle. Consequently, Ad-NaV1.5 infection alone reduced the rotation frequency (Fig. 10B). However, the shortened APD produced by Ad-Kir2.1 in neonatal cells (Fig. S9) likely contributed to significantly decreased wavelength and increased rotation frequency (Fig. 10). Most importantly, the combined infection (Ad-NaV1.5 + Ad-Kir2.1) hyperpolarized the RMP (Fig. S9), resulting in a shortened APD (Fig. 9) and a faster conduction velocity (CV) (Fig. S9). Consequently, the frequency of reentry was even higher with cooverexpression of NaV1.5 and Kir2.1 than Ad-Kir2.1 infection alone (Fig. 10B). These results are also consistent with the idea of molecular interplay between NaV1.5 and Kir2.1 channel proteins, and they also support that the underlying mechanism of this interplay is important in determining the frequency and stability of reentry in the well-polarized heart.
Fig. 10.

Molecular NaV1.5–Kir2.1 interactions modulate reentry frequency in NRVM monolayers. (A) Phase maps (3, 17, 18) for single rotations from representative optical mapping movies of monolayers infected with Ad-GFP, Ad-NaV1.5, Ad-Kir2.1, or Ad-Kir2.1 + Ad-NaV1.5. The color bar indicates the phase in the excitation–recovery cycle. (B) Reentry frequencies in monolayers infected with Ad-GFP (black; n = 11), Ad-NaV1.5 (blue; n = 13), Ad-Kir2.1 (yellow; n = 11), or Ad-Kir2.1 + Ad-Nav1.5 (red; n = 13). δP < 0.01 (ANOVA).
Discussion
Our results provide a demonstration that a change in functional expression of Kir2.1 modulates the expression of NaV1.5 and vice versa to alter cardiac excitability. We also show that SAP97 is a component of a macromolecular complex involved with these NaV1.5–Kir2.1 interactions. Furthermore, we provide evidence that the modulation is model-independent and demonstrable in myocytes isolated from adult rat and mouse hearts as well as NRVMs. Moreover, we show that NaV1.5 reduces internalization and promotes cell surface expression of Kir2.1 channels in rat ventricular myocytes (neonatal and adult) and HL-1 cells. The interaction between NaV1.5 and Kir2.1 is specific, and mediated, at least in part, by PDZ domain-mediated binding. Finally, we show that molecular interplay between NaV1.5 and Kir2.1 channel proteins is a crucial determinant of cardiac excitability and APD and that combined NaV1.5 + Kir2.1 overexpression in NRVM monolayers hyperpolarizes the RMP, shortens the APD, and increases the CV. Consequently, the frequency of reentry increases, and the rotating activity becomes more persistent than when each channel protein is overexpressed alone.
MAGUK proteins act as scaffolding proteins involved in intermolecular interactions and protein trafficking to the cell membrane of a number of ion channels. The last 3 aa of the NaV1.5 C terminus form a PDZ domain binding motif that is known to interact with PDZ domains found in the protein sequence of the syntrophin (14) and MAGUK family members (19). Like other ion channels, NaV1.5 has been reported to be part of the dystrophin multiprotein complex (14). The work by Gavillet et al. (9) showed that NaV1.5 interacts with dystrophin via syntrophin adaptor proteins through the PDZ domain binding motif on the NaV1.5 C terminus (14). Dystrophin-deficient mice (mdx5cv) have reduced levels of NaV1.5 protein in ventricular lysates, and this reduction is associated with reduced INa in isolated ventricular myocytes and conduction defects documented on ECG (9). The work by Petitprez et al. (20) detailed pull-down experiments using NaV1.5 C-terminal fusion proteins and human and mouse heart protein extracts to show that the association between SAP97 and NaV1.5 depended on the PDZ domain binding motif of NaV1.5. In patch clamp experiments, the work by Petitprez et al. (20) also suggested that silencing SAP97 reduced the whole-cell sodium current measured in HEK293 cells stably expressing NaV1.5 channels without decreasing the total protein content. These findings support the existence of an interaction between NaV1.5 and SAP97 in cardiac tissue, and they also led to the suggestion that there might be two pools of NaV1.5 channels: one pool targeted to lateral membranes by the syntrophin–dystrophin complex and another pool targeted to intercalated discs by SAP97 (20). Before that study (20), colocalization of NaV1.5 and SAP97 at the level of intercalated discs had not been shown. The results presented in Fig. 3A, a–e strongly support the hypothesis proposed in the work by Petitprez et al. (20), and they also show colocalization of NaV1.5 channels with SAP97 and Kir2.1 at the t-tubules.
A number of accessory proteins have also been shown to interact and form a multiprotein complex with NaV1.5 (21, 22). Ankyrin-G is a known accessory protein of NaV1.5. Both proteins exhibit a similar subcellular localization and localize at the intercalated disk and t-tubule membranes in cardiomyocytes; additionally, NaV1.5 coimmunoprecipitates with the 190-kDa ankyrin-G from detergent-soluble lysates of rat heart (22). In 2004, the work by Mohler et al. (22) identified the NaV1.5–ankyrin-G interaction as being important in determining cell surface localization of NaV1.5 in cardiomyocytes and maintaining normal cardiac function. The E1053K mutation associated with Brugada syndrome is the result of a point mutation in the ankyrin binding motif of NaV1.5. Disruption of this site abolishes binding of NaV1.5 to ankyrin-G and prevents accumulation of NaV1.5 at cell surface sites in ventricular cardiomyocytes (23). Such data suggest that association of NaV1.5 with accessory proteins (i.e., ankyrin-G, SAP97, and syntrophin) or other protein partners (i.e., Kir2.1) is required for NaV1.5 localization at excitable membranes in cardiomyocytes.
Members of Kir2.x (Kir2.1–2.3) have a C-terminal motif that enables interaction with PDZ domain proteins. In 2004, the work by Leonoudakis et al. (24) used a proteomics approach to identify proteins associated with Kir2 channels through the channel’s C-terminal PDZ binding motif. Based on immunoaffinity purification and affinity chromatography from skeletal and cardiac muscle as well as the brain, they showed that, in addition to MAGUK-type proteins, components of the dystrophin-associated protein complex, including α1-, β1-, and β2-syntrophin, dystrophin, and dystrobrevin, interact with Kir2 channels. Their affinity pull-down experiments revealed that Kir2.1, Kir2.2, Kir2.3, and Kir4.1 all bind to scaffolding proteins but with different affinities for the dystrophin-associated protein complex, including dystrophin and dystrobrevin as well as MAGUK proteins such as SAP97, CASK, and Veli (13). Moreover, their immunofluorescent localization studies showed that Kir2.2 colocalizes with syntrophin, dystrophin, and dystrobrevin at skeletal muscle neuromuscular junctions. Overall, the results of the work by Leonoudakis et al. (13, 24) suggest that Kir2 channels associate with protein complexes that may be important in trafficking and targeting channels to specific subcellular locations as well as anchoring and stabilizing the channels in the plasma membrane. Additionally, in cardiac myocytes, there is now evidence that these protein–protein interactions are important in the assembly of signaling complexes involved in the autonomic regulation of cardiac electrical activity (8). However, until recently, this hypothesis had not been directly tested, with the exception of the interaction of PSD-95 (a neuronal scaffolding protein) with Kir2.1 (25).
Sodium channel availability is a major determinant of CV and APD (1). However, recent studies in mice have shown that, rather than peak INa alone, it is the interplay between INa availability and the outward component of IK1 at the subthreshold levels of membrane potential that controls both excitability and CV (3). Moreover, in our optical mapping studies with NVRM monolayers, NaV1.5 overexpression facilitated an increase in IK1. This increase in IK1 forced the RMP to more negative levels, thereby enhancing the availability of sodium channels during sustained reentry. This result contributed to the observed increase in the frequency of rotors showed by the combined overexpression of NaV1.5 and Kir2.1. Our results are consistent with the idea that the molecular interplay between NaV1.5 and Kir2.1 channel proteins is an important determinant of the frequency and stability of reentrant tachyarrhythmias.
At this time, it cannot be excluded that the observed effects could be indirectly mediated through other or additional ion channel accessory proteins (e.g., β-subunits) or proteins associating with PDZ domain-containing proteins. However, we propose that NaV1.5 and Kir2.1 are part of the same macromolecular complex in a given microdomain based on the following essentials. First, the subcellular localization and channel activity of both NaV1.5 and Kir2.1 are regulated by protein–protein interactions through their respective C-terminal PDZ binding motif (9, 10, 13, 24). Second, our voltage and current clamp data show reciprocal increases in the current densities, RMP and dV/dtmax, suggesting that common molecular mechanisms are involved in the regulation of NaV1.5 and Kir2.1 functional expression. Third, our coimmunoprecipitation data are consistent with Kir2.1 and SAP97 existing in a complex from crude membrane preparations of rat ventricle; the same holds true for NaV1.5 and SAP97 (Fig. 3) (9, 10, 24). Fourth, surface expression of Kir2.1ΔPDZ in HL-1 cells was significantly reduced compared with WT Kir2.1 with or without concomitant overexpression of NaV1.5. Fifth, although the stoichiometry of the interactions showed here has not been elucidated, there are reports of specific relations involving the PDZ domain region of SAP97 PDZ1–3 (6, 26–28). There are clear indications that NaV1.5 binds to PDZ1 (26). In addition, structural analyses using small-angle X-ray scattering and NMR spectroscopy have suggested that, although the Kir2.1 cytoplasmic domain interacts with all three PDZ domains, it has a clear preference for PDZ2 (29), which in principle, would allow PDZ1 to bind to NaV1.5 in sufficient proximity to yield a productive channelosome (29). In our SAP97 silencing experiments (Fig. 2), a 56% reduction in SAP97 protein was accompanied by a 49% decrease in INa peak density, a 40% reduction in the density of the inward component of IK1, and a 67% reduction in the outward IK1. Although these results are by no means a measure of stoichiometry, they allow us to speculate that there may indeed be proportionality in the NaV1.5 and Kir2.1 functional expression and the available SAP97 protein. Given these possible interactions, changes in the expression of NaV1.5 or Kir2.1 should modify functional expression of its counterpart on the cell membrane, which has been demonstrated here.
This study shows protein–protein interactions specifically for NaV1.5 and Kir2.1. Importantly, our results suggest the presence of a potential molecular program in the cell that enables it to monitor and adjust accordingly expression levels of proteins involved in generating and maintaining the cardiac impulse. These protein–protein interactions may involve (i) increased production or stability of proteins on the cell membrane or (ii) increased RNA levels of NaV1.5 and/or Kir2.1, which might suggest increased protein production and could potentially lead to more channels at the cell membrane. However, our RT-PCR analyses in both transgenic Kir2.1 OE and Kir2.1−/+ mouse hearts conclusively show that the latter is not the likely mechanism underlying the observed functional effects caused by Kir2.1 overexpression. Rather, our results suggest that such interactions involve dynamic trafficking at the plasma membrane through changes in internalization and recycling (30) of the channel proteins. Quantification of the steady-state cell surface Kir2.1-pHluorin in ARVM, NRVM, and HL-1 cells overexpressing NaV1.5 indicates that the presence of NaV1.5 promotes cell surface expression of Kir2.1 by reducing protein internalization and increasing its stability. Nonetheless, the molecular mechanisms underlying the reciprocal regulation of NaV1.5 and Kir2.1 may also involve additional proteins and/or processes and therefore, warrant future investigation.
In conclusion, we have provided evidence suggesting that the molecular correlates of INa and IK1 are part of a common macromolecular complex and that direct or indirect interactions therein provide a means for their reciprocal regulation, with important functional consequences for myocardial excitation, conduction velocity, and arrhythmogenesis. Our study opens a pathway in understanding the molecular mechanisms underlying abnormal cardiac electrical activity as well as a variety of heart diseases, including heart failure, where a defect in the functional expression of Kir2.1 has been clearly shown (23). Indeed, a mechanistic link between Kir2.1–NaV1.5 interactions, arrhythmogenesis, and heart failure is underscored by the demonstration that INa density decreases in chronic heart failure (31).
Materials and Methods
All experiments were approved by the University Committee on the Use and Care of Animals at the University of Michigan. All data are expressed as mean ± SEM. In all experiments, N represents the number of animals/litters, and n equals the number of experiments or cells. Unpaired t test with Welch’s correction or one-way ANOVA was used to compare an experimental group with its appropriate control unless specified otherwise; P < 0.05 was considered significant for all experiments.
SI Materials and Methods has a detailed description of the procedures used in this study.
ARVMs and Adult Mouse Ventricular Myocytes.
Calcium-tolerant ARVMs were isolated from hearts of Sprague–Dawley rats (200 g) as previously described (32). Rod-shaped Ca2+-tolerant cells were maintained in M199 media for viral/nonviral transfer and patch clamp experiments. Cells were viable for up to 8 d in culture (Fig. S1). The method for isolating adult ventricular myocytes from the mouse is a modification of the method previously described (17). Rod-shaped mouse myocytes were used for electrophysiological recording within 8 h of isolation.
NRVM.
NRVM monolayers were obtained from 1- to 2-d-old Sprague–Dawley rat hearts as described in detail in the work by Muñoz et al. (18) and SI Materials and Methods. Monolayers were maintained in an incubator (VWR) at 37 °C with 5% CO2; all experiments were performed after 4 d in culture.
SDS/PAGE and Immunoblotting.
SDS/PAGE was carried out according to the method in the work by Laemmli (33).
Cell Electrophysiology.
INa and IK1 properties were studied using standard patch clamp techniques. Sodium currents were recorded at room temperature (21–22 °C). To assess the INa density, cells were held at −120 mV and stepped to various test potentials (−100–30 mV in 5-mV increments, 200-ms duration, 2,800-ms interpulse intervals). IK1 recordings were conducted at 37 °C. BaCl2 (0.4–1 mmol/L) was used to isolate IK1 from other background currents. IK1 was recorded using a step protocol with a holding potential of −50 mV and stepping from −100 to +20 mV in 10-mV increments of 500-ms duration at each potential. Action potentials were recorded at 37 °C. Cells were paced with 2-ms depolarizing pulses at 1.5–2 times threshold to measure action potential parameters (RMP and APD80).
Optical Mapping.
We used the potentiometric dye di-8-ANEPPS (40 μM) and a CCD camera (80 × 80 pixels, 438 μm per pixel, 200 frames/s) (3, 18, 34). Acquired signals (37 ± 0.5 °C) were amplified, filtered, and digitalized (Lab-Windows). Analysis was carried out using customized tools (3, 18, 34).
Supplementary Material
Acknowledgments
We thank Nulang Wang for her technical support and Robin M. Shaw (University of California, San Francisco, CA) for insight and discussion of this study. Funding was provided by National Heart, Lung, and Blood Institute Award T32HL007853 (to M.L.M.), National Institutes of Health Grants R01 GM076608 (to J.M.B.A.), P01 HL39707 (to J.J.), and HL60843 (to J.J.), and the Leducq Foundation (J.J.). R.V. was supported by a predoctoral fellowship from the American Heart Association and Robert and Eileen Hutton.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Author Summary on page 12284 (volume 109, number 31).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1109370109/-/DCSupplemental.
References
- 1.Fozzard HA, Hanck DA. Structure and function of voltage-dependent sodium channels: Comparison of brain II and cardiac isoforms. Physiol Rev. 1996;76:887–926. doi: 10.1152/physrev.1996.76.3.887. [DOI] [PubMed] [Google Scholar]
- 2.Lopatin AN, Nichols CG. Inward rectifiers in the heart: An update on I(K1) J Mol Cell Cardiol. 2001;33:625–638. doi: 10.1006/jmcc.2001.1344. [DOI] [PubMed] [Google Scholar]
- 3.Noujaim SF, et al. Up-regulation of the inward rectifier K+ current (I K1) in the mouse heart accelerates and stabilizes rotors. J Physiol. 2007;578:315–326. doi: 10.1113/jphysiol.2006.121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garner CC, Nash J, Huganir RL. PDZ domains in synapse assembly and signalling. Trends Cell Biol. 2000;10:274–280. doi: 10.1016/s0962-8924(00)01783-9. [DOI] [PubMed] [Google Scholar]
- 5.Nehring RB, et al. Neuronal inwardly rectifying K(+) channels differentially couple to PDZ proteins of the PSD-95/SAP90 family. J Neurosci. 2000;20:156–162. doi: 10.1523/JNEUROSCI.20-01-00156.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leonoudakis D, Mailliard W, Wingerd K, Clegg D, Vandenberg C. Inward rectifier potassium channel Kir2.2 is associated with synapse-associated protein SAP97. J Cell Sci. 2001;114:987–998. doi: 10.1242/jcs.114.5.987. [DOI] [PubMed] [Google Scholar]
- 7.Vikstrom KL, et al. SAP97 regulates Kir2.3 channels by multiple mechanisms. Am J Physiol Heart Circ Physiol. 2009;297:H1387–H1397. doi: 10.1152/ajpheart.00638.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaidyanathan R, Taffet SM, Vikstrom KL, Anumonwo JM. Regulation of cardiac inward rectifier potassium current (IK1) by synapse associated protein-97. J Biol Chem. 2010;285:28000–28009. doi: 10.1074/jbc.M110.110858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gavillet B, et al. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006;99:407–414. doi: 10.1161/01.RES.0000237466.13252.5e. [DOI] [PubMed] [Google Scholar]
- 10.Ueda K, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci USA. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakagawa T, et al. Quaternary structure, protein dynamics, and synaptic function of SAP97 controlled by L27 domain interactions. Neuron. 2004;44:453–467. doi: 10.1016/j.neuron.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 12.Sato PY, et al. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009;105:523–526. doi: 10.1161/CIRCRESAHA.109.201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leonoudakis D, Conti LR, Radeke CM, McGuire LMM, Vandenberg CA. A multiprotein trafficking complex composed of SAP97, CASK, Veli, and Mint1 is associated with inward rectifier Kir2 potassium channels. J Biol Chem. 2004;279:19051–19063. doi: 10.1074/jbc.M400284200. [DOI] [PubMed] [Google Scholar]
- 14.Gee SH, et al. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998;18:128–137. doi: 10.1523/JNEUROSCI.18-01-00128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piao L, Li J, McLerie M, Lopatin AN. Transgenic upregulation of IK1 in the mouse heart is proarrhythmic. Basic Res Cardiol. 2007;102:416–428. doi: 10.1007/s00395-007-0659-y. [DOI] [PubMed] [Google Scholar]
- 16.Camacho JA, et al. Modulation of Nav1.5 channel function by an alternatively spliced sequence in the DII/DIII linker region. J Biol Chem. 2006;281:9498–9506. doi: 10.1074/jbc.M509716200. [DOI] [PubMed] [Google Scholar]
- 17.Cerrone M, et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101:1039–1048. doi: 10.1161/CIRCRESAHA.107.148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muñoz V, et al. Adenoviral expression of IKs contributes to wavebreak and fibrillatory conduction in neonatal rat ventricular cardiomyocyte monolayers. Circ Res. 2007;101:475–483. doi: 10.1161/CIRCRESAHA.107.149617. [DOI] [PubMed] [Google Scholar]
- 19.Gardoni F, Marcello E, Di Luca M. Postsynaptic density-membrane associated guanylate kinase proteins (PSD-MAGUKs) and their role in CNS disorders. Neuroscience. 2009;158:324–333. doi: 10.1016/j.neuroscience.2008.07.068. [DOI] [PubMed] [Google Scholar]
- 20.Petitprez S, et al. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res. 2011;108:294–304. doi: 10.1161/CIRCRESAHA.110.228312. [DOI] [PubMed] [Google Scholar]
- 21.Abriel H. Roles and regulation of the cardiac sodium channel Na v 1.5: Recent insights from experimental studies. Cardiovasc Res. 2007;76:381–389. doi: 10.1016/j.cardiores.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 22.Mohler PJ, et al. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci USA. 2004;101:17533–17538. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li GR, Lau CP, Leung TK, Nattel S. Ionic current abnormalities associated with prolonged action potentials in cardiomyocytes from diseased human right ventricles. Heart Rhythm. 2004;1:460–468. doi: 10.1016/j.hrthm.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Leonoudakis D, et al. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2.x)-associated proteins. J Biol Chem. 2004;279:22331–22346. doi: 10.1074/jbc.M400285200. [DOI] [PubMed] [Google Scholar]
- 25.Leyland ML, Dart C. An alternatively spliced isoform of PSD-93/chapsyn 110 binds to the inwardly rectifying potassium channel, Kir2.1. J Biol Chem. 2004;279:43427–43436. doi: 10.1074/jbc.M407575200. [DOI] [PubMed] [Google Scholar]
- 26.Stiffler MA, et al. PDZ domain binding selectivity is optimized across the mouse proteome. Science. 2007;317:364–369. doi: 10.1126/science.1144592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peiretti F, et al. Identification of SAP97 as an intracellular binding partner of TACE. J Cell Sci. 2003;116:1949–1957. doi: 10.1242/jcs.00415. [DOI] [PubMed] [Google Scholar]
- 28.Sabio G, et al. p38gamma regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. EMBO J. 2005;24:1134–1145. doi: 10.1038/sj.emboj.7600578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goult BT, et al. Small-angle X-ray scattering and NMR studies of the conformation of the PDZ region of SAP97 and its interactions with Kir2.1. Biochemistry. 2007;46:14117–14128. doi: 10.1021/bi701257z. [DOI] [PubMed] [Google Scholar]
- 30.Schumacher SM, et al. Antiarrhythmic drug-induced internalization of the atrial-specific k+ channel kv1.5. Circ Res. 2009;104:1390–1398. doi: 10.1161/CIRCRESAHA.108.192773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maltsev VA, Sabbab HN, Undrovinas AI. Down-regulation of sodium current in chronic heart failure: Effect of long-term therapy with carvedilol. Cell Mol Life Sci. 2002;59:1561–1568. doi: 10.1007/s00018-002-8529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westfall MV, Rust EM, Metzger JM. Slow skeletal troponin I gene transfer, expression, and myofilament incorporation enhances adult cardiac myocyte contractile function. Proc Natl Acad Sci USA. 1997;94:5444–5449. doi: 10.1073/pnas.94.10.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Noujaim SF, et al. Universal scaling law of electrical turbulence in the mammalian heart. Proc Natl Acad Sci USA. 2007;104:20985–20989. doi: 10.1073/pnas.0709758104. [DOI] [PMC free article] [PubMed] [Google Scholar]
