

Abstract
In somatic cells, the length of the G1 phase of the cell cycle is tightly linked to differentiation, and its elongation can drive differentiation in many cases. Although it has been suggested that the situation is very similar in embryonic stem cells (ESCs), where a rapid cell cycle and a short G1 phase maintain the pluripotent state, evidence has been contradictory. Here we show that, in murine ESCs, elongation of the cell cycle and elongation of G1 are compatible with their pluripotent state. Multiple methods that lengthen the cell cycle and that target cyclin-dependent kinase, retinoblastoma protein, and E2F activity all fail to induce differentiation on their own or even to facilitate differentiation. The resistance of murine ESCs to differentiation induced by lengthening G1 and/or the cell cycle could allow for separate control of these events and provide new opportunities for investigation and application.
Keywords: stemness, proliferation, decoupling
In many somatic cell culture models, progression through the cell cycle is essential to maintain the undifferentiated state (1–4). If the cell cycle is lengthened or arrested, for example, in G1 by overexpression of G1 cyclin-dependent kinase (CDK) inhibitors (e.g., p21, p27, p16), differentiation will generally result. Several explanations have been proposed for this linkage of differentiation and proliferation. One of them is that G1 may be a period that is more susceptible to differentiation signals from the external environment (5). Continuous passage through G1 could, under these conditions, limit the total amount of differentiation signal received over time. The observation that an inhibitor of TGF-β signaling, SnoN, is a substrate of the anaphase-promoting complex/cyclosome (APC/C) in G1 indicates that different cell-cycle states may differ in their responsiveness to exogenous signals (6). In a similar way, passage through the cell cycle may also limit the opportunity for epigenetic changes to take place. Such changes associated with differentiation may include a remodeling of chromatin and activation or repression of lineage-specific gene expression. That G1 length may be limiting epigenetic changes is supported by the observation that many transcription factors are removed from condensed chromatin during mitosis (7). The next G1 would then be a critical period to initiate new transcriptional programs, which may not be feasible if the cell rapidly proceeds through this stage into S phase. Finally, it has been suggested that rapid progression through G1 limits accumulation of the activity of the G1/S checkpoint protein retinoblastoma (Rb) or Rb family members, which normally promote differentiation (8), because Rb is both phosphorylated and inactivated simultaneously by CDKs. In this view, when the cell cycle is lengthened or arrested in G1, more of the Rb protein will be dephosphorylated and activated, allowing activity of differentiation-inducing transcription factors.
Embryonic stem cells (ESCs) have a very short G1 phase and high proliferation rate. Like somatic cells, they may use this property to inhibit differentiation and preserve their pluripotent state (5, 9–16). Although plausible, direct evidence for this idea has been limited and equivocal. Early experiments treating D3 mouse ESCs with the CDK inhibitors roscovitine and Ro09-3033 found no effect on the pluripotency markers Oct4 and Rex-1 (17). Furthermore, it was later shown that knockout mice of CDK2, the main CDK thought to be responsible for G1/S progression in ESCs, were viable and therefore presumably did not prematurely differentiate their stem cells (18, 19). In contrast, it was recently reported that olomoucine II, another CDK inhibitor, could induce mRNA up-regulation of differentiation markers in mouse ESCs and that CDK2 siRNA could induce morphology changes characteristic of differentiation (20). In human ESCs exposure to roscovitine and other CDK inhibitors sometimes led to a loss of pluripotency markers but other times did not (21–24). We have now reexamined this question by using several methods to affect cell-cycle progression through the G1 phase. These results support the idea that the undifferentiated state of mouse ESCs can persist even with a highly elongated G1 phase and therefore that elongation of G1 does not in itself promote differentiation.
Results
Expression of p21 and p27 Lengthens G1 but Does Not Induce Differentiation.
To promote G1 elongation, we introduced the CDK inhibitors p21 and p27 into ESCs. The p21 and p27 proteins belong to a family of proteins that are normally induced during differentiation and after DNA damage. Overexpression of p21 or p27 is sufficient in many somatic cell culture models to induce both lengthening and differentiation of the G1 cell cycle through the inhibition of CDK1 and CDK2 activity (1).
When we cotransfected p21 or p27 into J1 ESCs with a plasmid expressing EGFP to label the cells, we found that both proteins increased the percentage of G1 cells and decreased the percentage of cells in S phase, as determined by DNA content (+10.7% G1 and approximately −10.3% S phase) (Fig. 1A). This change in the DNA content is similar to the gradual change in the cell-cycle structure as cells undergo differentiation, where p21 and p27 levels similarly rise (16). To see how much of an effect the CDK inhibitors have on proliferation rate, we examined living cells in real time after tagging p27 with mCherry to construct a fusion protein and then expressing it in ESCs. We found that cells expressing p27 had much longer cell-cycle times compared with cells that contained background levels of mCherry fluorescence (Fig. 1B; p27+ cells, mean 27.3 h; p27− cells, mean 12.4 h). Thus, expression of p27 generated cell-cycle times in ESCs that are as long or longer than most cultured somatic cells (compare, for example, the 27.3-h generation time of ESCs expressing p27 to 25.3 h for mouse embryonic fibroblasts and 22.1 h for NIH 3T3 cells) (16).
Fig. 1.

Overexpression of p21 and p27 elongate the cell cycle in G1 but are insufficient to induce loss of pluripotency. (A) Flow cytometry analysis of p21 and p27 overexpression showing cell-cycle stage distribution of cells expressing high levels of cotransfected EGFP. Quantification of the cell-cycle elongation in G1 is shown below the histograms. (B) Real-time time-lapse microscopy measurements of cell-cycle times of WT cells vs. cells with G1 lengthened by CDK inhibitor overexpression. (C) Day 4 Oct4 reporter, Nanog reporter, and SSEA-1 levels remain constant in p21- and p27-overexpressing cells. The mCherry, p21, and p27 distributions are gated for expression of the fusion protein (i.e., mCherry+, p21+, p27+). The ESC lines used are Oct4-GiP, Nanog-GFP, and J1. In the combined retinoic acid (RA) plus p21/p27 (RA + p21 and RA + p27) treatments, RA was added 24 h after the addition of p21/p27. (D) Quantitative RT-PCR analysis of lineage markers for FACS-sorted cells expressing mCherry, p21, or p27. For reference, samples differentiated by LIF withdrawal for 4 d are provided.
We tested whether p21 and p27 overexpression could induce differentiation under standard self-renewing conditions [with leukemia inhibitory factor (LIF)] by assaying Oct4, Nanog, and stage-specific embryonic antigen 1 (SSEA-1) levels. We wanted to do this at the single-cell level so that we could choose cells with high levels of CDK inhibitor expression. To do so, we used reporter GFP lines for Oct4 and Nanog (called Oct4-GiP and Nanog-GFP, respectively) along with the J1 line for measuring SSEA-1 levels by immunostaining. By introducing mCherry-tagged p21 and p27 into these lines, we could specifically examine cells expressing high levels of the CDK inhibitors by flow cytometry. At 4 d after p21 and p27 addition, we found that cells that continued to express high levels of CDK inhibitor contained similar levels of Oct4, Nanog, and SSEA-1 as control cells expressing mCherry (Fig. 1C and Table S1). Furthermore, there was no significant decrease in pluripotency marker expression between cells expressing high levels of CDK inhibitors versus background levels, indicating that progressive elongation in G1 did not lead to differentiation. Similarly, we observed no drop in Oct4, Nanog, or SSEA-1 even at 10 d after p21/p27 addition (Table S1). As an additional control, we were able to reproduce the induction of differentiation by p27 in the somatic neuroblastoma differentiation model N1E-115, as previously reported (1).
In addition to observing no indication of a drop in pluripotency markers, we also observed no significant increases in genes that are used to characterize differentiated lineages and therefore might suggest differentiation. To compare the populations of cells expressing CDK inhibitor, we FACS-sorted the cells that were positive at 48 h posttransfection and assayed Fgf5 and Msx1 (ectoderm), Brachyury (mesoderm), GATA4 and GATA6 (endoderm), and Cdx2 (trophectoderm) by quantitative PCR. We noticed no significant increase (within approximately twofold) in any of these transcripts (Fig. 1D).
Knockdown of CDK2 or Cyclin A with siRNA Reduces the Proliferation Rate but Does Not Induce Differentiation.
Although some previous studies reported that a reduction of CDK2 protein is capable of inducing differentiation in human ESCs and producing morphological changes similar to differentiation in mouse ESCs (21–24), these results are inconsistent with the fact that CDK2 knockout mice are viable unless the knockout mice compensated by other functional changes. To reexamine the effect of reducing CDK2 under more acute conditions, we used siRNA to knock down CDK2 levels in our ESC lines. We were able to achieve a knockdown efficiency of ∼50% in the mean fluorescence of the population (Fig. S1). CDK2 knockdown slowed down the growth rate of ESCs by ∼14% but did not lead to any changes in cell-cycle structure (Fig. 2A) compared with a nonspecific control siRNA. Thus, each phase of the cell cycle was lengthened, including G1. With regard to morphology, we did not see the enlargement and flattening of the cells associated with differentiation, even at 4 d after the knockdown (Fig. S2). There was also no change in the levels of Oct4, Nanog, or SSEA-1 at 4 d after knockdown (Fig. 2B). These results support the conclusions about the dispensability of CDK2 that were suggested by the mouse knockout studies.
Fig. 2.

Effects of CDK2 and cyclin A knockdown on the cell cycle and pluripotency. (A) Effect of knockdown on proliferation rate and cell-cycle structure by DNA content. (B) Flow cytometry analysis of Oct4 reporter, Nanog reporter, and SSEA-1 levels at 4 d after siRNA treatment.
To further examine whether loss of CDK2 activity could induce differentiation, we knocked down cyclin A, the principal partner of CDK2 at the G1/S transition. This experiment is difficult to perform because complete removal of cyclin A is lethal (25). We therefore tested whether a partial knockdown of cyclin A might lead to some loss of self-renewal. Cyclin A siRNA, which achieved a population mean knockdown of ∼75% (Fig. S1), did not generate any significant changes to ESC morphology (Fig. S2). The knockdown did, however, lead to an accumulation of cells in G2/M, as previously reported (26), and a 77% reduction in growth rate (Fig. 2A). Again there was no detectable change in pluripotency marker levels at 4 d after the knockdown (Fig. 2B).
Effects of Mutant CDK2, E2F1, and Rb Overexpression.
Any given method for elongating G1 could affect pluripotency by indirect means, so we attempted some alternative approaches to affect CDK, Rb, or E2F activity. We overexpressed dominant-negative CDK2 (dnCDK2) and dominant-negative E2F1 (dnE2F1) as well as both WT and constitutively active forms of Rb. The dnCDK2 consists of the human CDK2 with an aspartyl residue replacing an asparaginyl residue in the ATP binding site (D145N). This mutant presumably acts by competitively inhibiting the binding of the WT CDK2 to its cyclin binding partners. It was previously shown to induce G1 arrest in human U2OS osteosarcoma cells (27). To generate the dnE2F1 mutant, we used the DNA binding domain of mouse E2F1 (amino acids 90–186). This region is homologous to the 95–191 region of human E2F1, which was previously shown to block S-phase entry in 3T3 cells (28). The dnE2F1 mutant presumably acts to block binding of WT E2F1 to its DNA targets. Finally, we constructed a WT (WTLP) and a mutant (7LP) version of the human Rb large pocket (i.e., the region of Rb that binds E2Fs basic helix–loop–helix factors such as MyoD, amino acids 37–928). The putative dominant-negative mutant contains inactivating mutations in seven phosphorylation sites, rendering it uninhibitable by CDK phosphorylation and, hence, constitutively active. Introduction of the 7LP mutant into Rat-1 cells was previously shown to induce growth arrest in G1 (29). Each of these constructs was tagged with mCherry.
Overexpression of these constructs led to clear cell-cycle effects but not differentiation. The dnCDK2 led to a similar percentage of G1 cells but decreased S phase and increased G2/M percentage of cells (−9.4% S phase, +11% G2/M) (Fig. 3A). The dnCDK2 also led to a 20% slower proliferation rate, which implies a longer time spent in G1 (Fig. 3B). The WT Rb construct had a small effect on increasing S-phase percentage and decreasing G2/M percentage, and this effect was more pronounced in the mutant Rb construct (WT: +3.5% S phase, −3.8% G2/M; mutant: +10.2% S phase, −9.2% G2/M). However, both Rb constructs had little or no effect on proliferation rate (3% and 6% decrease for WT and mutant, respectively). The dnE2F1 construct led to both an increase in G1 cells (+26.7% G1) and a 28% slowdown in proliferation rate. We found no significant changes in the pluripotency markers Oct4, Nanog, and SSEA-1 after introducing these constructs into our ESC lines (Fig. 3C).
Fig. 3.

Treatment of ESCs with mCherry-tagged dnCDK2, dnE2F1, and Rb constructs induce cell-cycle effects but not differentiation. (A) Effect of overexpressing mutants on cell-cycle structure by DNA content. (B) Growth curves of treated cells over a 3-d period. (C) Analysis of Oct4 reporter, Nanog reporter, and SSEA-1 levels by flow cytometry after 4 d of overexpression.
Aberrant Effects Caused by Small-Molecule CDK Inhibitors.
Probably the most available and common approach to perturb cell-cycle progression is the use of small-molecule kinase inhibitors. Although potent kinase inhibitors are now widely available, each has a range of potential enzyme targets, and some have a very broad range. Nevertheless, they are potentially useful because they produce acute effects, and the dose–response range can easily be explored. Several groups have previously used small molecules (e.g., roscovitine) targeted to CDKs but with a host of off-target effects on ESCs and found drastically different results on promoting differentiation (17, 20). To explore the effects of these small molecule inhibitors further, we applied both roscovitine and CVT-313, a more specific CDK2 inhibitor, to ESCs. Titrations of both drugs showed that they affect the rate of cell proliferation at low micromolar concentrations (Fig. 4A). Furthermore, both exhibited changes on cell-cycle structure (Fig. S3). CVT-313 induced a decrease in S phase percentage and an increase in G2/M percentage (e.g., at 8 μM: −11.6% S phase, +14.6% G2/M). Roscovitine induced fluctuations in S and G2/M percentages across the range of concentrations tested (for 5, 10, 15, and 20 μM: −8.3%, +3.4%, +6.7%, and −6.5% S phase; +7.9%, −7.6%, −1.7%, and +10.6% G2/M). In both cases, the estimated G1 fraction did not change dramatically over the range of concentrations.
Fig. 4.

Pharmacological CDK inhibitors can induce multiple effects at sublethal concentrations. (A Left) Proliferative dose–response curves of two small-molecule inhibitors: CVT-313, a CDK2 inhibitor, and roscovitine, a broader CDK inhibitor. (Right) Percentage of cells in G1, as determined by FACS cell-cycle profile. (B) Flow cytometry analysis of pluripotency marker levels after 10 d of treatment with sublethal concentrations of inhibitors. The CDK inhibitors induced a minor decrease in fluorescence marker levels (demarcated by a shift in arrow position over the major peak). Reductions in marker levels of magnitude suggestive of differentiation could be found only in a fraction of cells in roscovitine-treated samples. (C) Morphologies of ESCs treated with sublethal concentrations of inhibitors for an extended period (10 d). CVT-313 did not induce noticeable changes, but roscovitine did in some cells.
To examine their effect on pluripotency, we applied a chronic dose of drugs at concentrations that reduced the proliferation rate to approximately a third of normal (Fig. 4A, 4 μM CVT-313 and 10 μM roscovitine). At the end of 10 d of treatment, we observed small decreases in Oct4, Nanog, and SSEA-1 levels for both drugs in the majority of the population (Fig. 4B). The decrease was minor (−33% Oct4, −60% Nanog, and −43% SSEA-1) compared with the level normally observed for differentiated cells (−90% Oct4, −98.5% Nanog, and −98.5% SSEA-1). Thus, in cases of differentiation, the Oct4, Nanog, and SSEA-1 levels drop to almost undetectable levels while, in response to these two drugs, appreciable levels of Oct4, Nanog, and SSEA-1 remain. These results suggest to us that this drop may not be recording differentiation but rather some other generally toxic effect of the drug. On top of this general effect, a small fraction of roscovitine-treated cells had further reduced levels of Oct4, Nanog, and SSEA-1, indicating that differentiation could be induced by roscovitine in some cells. Because this effect was not observed with CVT-313 cells, the effect appeared to be roscovitine-specific and not attributable simply to the reduction in proliferation rate, which was similar for both drugs. Cell morphology correlated with the molecular data. CVT-313-treated cells were unchanged compared with untreated ESCs (Fig. S4). The majority of cells also maintained an undifferentiated colony morphology in response to roscovitine. A distinct minority exhibited morphological signs of differentiation in both the Oct4 reporter line and the J1 line; they were larger, flatter, and generally resembled fibroblasts.
G1-Lengthened ESCs Contain Hypophosphorylated, Active Rb.
One way the short cell cycle of ESCs is thought to protect against differentiation is by constitutively phosphorylating the Rb protein (9). The Rb protein is a substrate of CDKs in G1 and has 16 Ser/Thr-Pro sites that potentially can be phosphorylated (27). In somatic cells, Rb is normally dephosphorylated exiting mitosis and progressively becomes rephosphorylated during the G1/S transition (Fig. 5A).
Fig. 5.
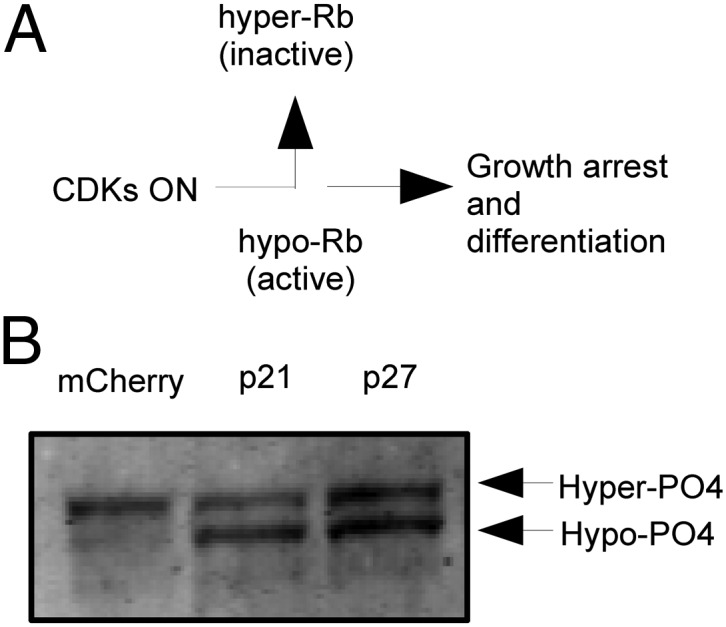
Rb dephosphorylation generated by p21 and p27 overexpression. (A) Schematic of Rb inactivation during the G1/S transition in somatic cells. In mouse ESCs, CDK activity is high, and Rb appears to be largely in the hyperphosphorylated form. (B) Gel-shift analysis shows that the active, hypophosphorylated form of Rb accumulates from p21/p27 overexpression.
We examined the effect of G1 lengthening on Rb phosphorylation by mobility shift on a Western blot. In J1 cells, Rb ran as the hyperphosphorylated species (Fig. 5B), which is the only detectable form of Rb so far in mouse ESCs (30). Addition of p21 or p27 led to the appearance of a second faster-running form, which was the hypophosphorylated form (here detected with anti–pan-Rb-phospho form pMG3-245 antibody). That this form was in fact the hypophosphorylated form of Rb was confirmed by λ phosphatase treatment, which showed a same size gel shift. Previous studies have shown that the hypophosphorylated form in cycling cells is the active form that binds and suppresses E2F function (31). Thus, addition of p21 or p27 led to the expected accumulation of the active, hypophosphorylated form of Rb that can bind E2F, but it did not lead to differentiation.
G1 Extension Does Not Potentiate Differentiation by LIF Withdrawal.
Although a lengthening of G1 might not be sufficient on its own to undermine pluripotency and promote differentiation, it might potentiate differentiation driven by LIF withdrawal. To test this possibility, we manipulated the cell cycle in G1 by introducing p21 and p27 into our Nanog reporter line and monitored the effect of removing LIF from the media. If lengthening the cell cycle promoted differentiation, we would expect a more rapid loss of pluripotency markers on LIF withdrawal. In the opposite condition, shortening the G1 period by expression of proteins that promote the G1/S transition would be expected to delay or block differentiation induced by LIF withdrawal. There was no indication that lengthening G1 by overexpression of p21 or p27 facilitated differentiation under G1 elongation (Fig. 6). In p21- and p27-expressing cells, LIF withdrawal was followed by identical kinetics of Nanog loss. In an attempt to shorten G1, we expressed cyclin A, cyclin E, and cyclin D individually (and tagged with mCherry). The introduction of the new cyclin constructs all led to a decrease in the percentage of G1 cells by DNA content (Fig. S5), consistent with their role as S-phase–promoting cyclins (G1: −10.4% cyclin A, −5.6% cyclin E, −6.9% cyclin D). Unlike p21 and p27, however, some of the cyclins led to a short temporary delay in the kinetics of Nanog loss. The extent and duration of the delay were unique to the different cyclins, with cyclin E having almost no loss of Nanog during the first 2 d, cyclin D having a comparatively milder effect (∼25% loss of signal by day 2), and cyclin A having little if any effect. Also, the effect of cyclin E appeared immediately (difference with mCherry control was detectable by day 1), whereas the effect of cyclin D only appeared later (detectable at day 2). Thus, there was no facilitating effect of lengthening G1, but shortening G1 by overexpressing specific cyclins did slow down the rate of differentiation as measured by Nanog loss.
Fig. 6.

Effects of modulating G1 length on the kinetics of Nanog reporter loss during LIF withdrawal. Nanog-GFP reporter ESCs were first transfected for 24 h, then LIF was removed to begin kinetics measurement. Values indicated are the means of GFP fluorescence in the Nanog-GFP line for the construct-expressing population after background correction. The mCherry curve is reproduced in light blue in each subsequent graph for reference.
Discussion
We have reexamined the notion that the short G1 of mouse ESCs actively maintains their stem cell state. Our results support the conclusions of some previous reports (17–19) and dispute those of others (20–24). The conflict may partially reflect differing criteria for assessing pluripotency. The criteria we used is a drop in pluripotency factors such as Oct4, Nanog, and SSEA-1. The experiments were performed in single cells, where the potential heterogeneity of the experimental treatment can be recognized. By themselves, assessments of cell morphology or the expression of lineage-specific transcription factors may be misleading because morphology is hard to assess objectively and quantitatively, and lineage-specific genes can often be expressed promiscuously in ESCs without affecting self-renewal (32). Given these criteria, several previously contradictory studies would not be in conflict with our conclusions (20, 24). Furthermore, any particular method used to elongate G1 and shorten the cell cycle may individually harbor potential artifacts, which may be a reason why some previous studies have reached contradictory conclusions. We addressed this issue by using a total of 10 different methods involving the perturbation of G1 CDK activity, Rb, and E2F. Perhaps the most natural method for lengthening G1 was the overexpression of p21 and p27 because these genes are thought to be highly specific for their targets. Expression of these genes induced a cell-cycle length beyond typical somatic cells and produced a cell-cycle structure that was elongated in G1. Some of the other methods generated effects that were more complicated and not just limited to lengthening the G1 phase. Given the potential off-target effects of small-molecule CDK inhibitors and their strong toxicity at slightly higher doses, it may be that their effects on G1 are uninterpretable.
Lengthening G1 by p21 and p27 overexpression did not accelerate differentiation induced by LIF withdrawal (as measured by Nanog loss) (Fig. 6). However, shortening G1 by overexpressing some G1 cyclins did produce a delay in differentiation. These results suggest that, although the natural lengthening of G1 during ESC differentiation may be important for the differentiation process to proceed on schedule, it is not itself capable of driving the process as it does in many somatic cell models.
Provided that the short G1 is not involved in maintaining the ESC state, an interesting question is whether a basal rate of passage through the cell cycle is required for the pluripotent state. This scenario would suggest an alternative model in which there is a threshold of G1 length above which pluripotency cannot be maintained. Such a model may be possible, but it would need to be reconciled with the fact that, upon exit from the pluripotent state, G1 lengthens gradually and not in a discrete step-wise fashion. At the moment, we believe our data cannot exclude the possibility of the threshold model because we may not have hit this threshold. With CDK inhibitor overexpression, we have observed cell-cycle lengths elongated beyond that of typical somatic cells (mean cell-cycle time of p27-expressing cells of 27.3 h vs. 22.1 h for NIH 3T3 cells and 25.3 h for mouse embryonic fibroblasts) (Fig. 1B). In the future, it will be interesting to try to induce even longer cell cycles.
Why is the length of the cell cycle tightly tied to differentiation of somatic cells but not to ESCs? What is the nature of the block to differentiation in both? At present, the mechanisms of both are unknown, but one possibility raised by these experiments is that features unique to ESCs dictate their strong resistance to differentiation under circumstances where G1 length or cell-cycle length is perturbed. One such mechanism is the dependence on LIF signaling or the requirement of blocking MAPK and FGF signaling to maintain pluripotent status. In those specific cases, the unique combination of external signals could override the normal cell-cycle trigger for differentiation. Another possibility is that the normal triggers for differentiation exerted by slowing of the cell cycle in G1 only take effect later during embryogenesis rather than in the ESC stage. Indeed, most of the documented somatic cell culture models in which G1 phase length has an effect have been models of late-stage, terminal differentiation (1). Much less is known about the role of the cell cycle and Rb in differentiation in these early embryonic stages. Surprisingly, our results from G1 lengthening and Rb overexpression indicate that both may have little to do with the regulation of differentiation in the ESC context.
Several authors have previously speculated on the relationship of ESCs to adult stem cells and cancer on the basis of cycling and the G1 phase. Specifically, it has been postulated previously that blocking G1 phase-induced differentiation could be a common strategy underlying self-renewal in ESCs and adult stem cells, either by constitutively cycling or entering quiescent G1, respectively (9). Our results argue against the particular mechanism of blocking differentiation by constitutive cycling but suggest that alternative mechanisms continue to block G1 phase-induced differentiation in ESCs. In this sense, ESCs may still share an underlying similarity with adult stem cells in their ability to proliferate slowly in G1 in an undifferentiated state. It has also been suggested that the short G1 and fast cycling in ESCs may be a mechanism by which oncogenic factors in ESCs, such as PI3K signaling and Myc, maintain pluripotency (5). We believe that our findings do not contradict this idea. Instead, they imply that these oncogenic factors may have cell-cycle–independent functions in maintaining pluripotency in addition to promoting fast cycling. Thus, the short G1 may be irrelevant or it may facilitate, but it cannot be sufficient for maintaining the pluripotent state. Finally, our results suggest that, because it is possible to separate proliferation from pluripotency in ESCs, additional opportunities for investigation and/or therapeutic application will be available.
Materials and Methods
ESCs were cultured under standard conditions with Knockout DMEM (Invitrogen) and 15% (vol/vol) ESC-qualified FBS (Invitrogen) supplemented with glutamine (Invitrogen), penicillin/streptomycin (Invitrogen), β-mercaptoethanol (Sigma), nonessential amino acids (Invitrogen), and LIF (Millipore). Cells were grown feeder-free on gelatin. Oct4-GiP cells were kindly provided by Austin Smith (University of Cambridge, Cambridge, United Kingdom). Nanog-GFP cells were purchased from Millipore.
Detailed methods for differentiation assays, time-lapse imaging, RNAi transfections, construction of fusion constructs and cotransfection experiments, flow cytometry and staining, quantitative RT-PCR, growth curve/drug response curve measurements, and Western blotting are available in SI Materials and Methods.
Supplementary Material
Acknowledgments
V.C.L. thanks William Kaelin, Andrew Murray, Paola Arlotta, Nicholas Dyson, Jodene Moore, the Nikon Imaging Center at Harvard Medical School, and members of M.W.K.’s laboratory for their advice, suggestions, and reagents. M.W.K. was supported by National Institute of General Medical Sciences Grant GM26875.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1206740109/-/DCSupplemental.
References
- 1.Muñoz-Alonso M, León J. G1 phase control and cell differentiation. In: Boonstra J, editor. G1 Phase Progression. Georgetown, TX: Landes Bioscience; 2003. pp. 236–264. [Google Scholar]
- 2.Zhu L, Skoultchi AI. Coordinating cell proliferation and differentiation. Curr Opin Genet Dev. 2001;11:91–97. doi: 10.1016/s0959-437x(00)00162-3. [DOI] [PubMed] [Google Scholar]
- 3.Miller JP, Yeh N, Vidal A, Koff A. Interweaving the cell cycle machinery with cell differentiation. Cell Cycle. 2007;6:2932–2938. doi: 10.4161/cc.6.23.5042. [DOI] [PubMed] [Google Scholar]
- 4.Myster DL, Duronio RJ. To differentiate or not to differentiate? Curr Biol. 2000;10:R302–R304. doi: 10.1016/s0960-9822(00)00435-8. [DOI] [PubMed] [Google Scholar]
- 5.Singh AM, Dalton S. The cell cycle and Myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell. 2009;5:141–149. doi: 10.1016/j.stem.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wan Y, Liu X, Kirschner MW. The anaphase-promoting complex mediates TGF-β signaling by targeting SnoN for destruction. Mol Cell. 2001;8:1027–1039. doi: 10.1016/s1097-2765(01)00382-3. [DOI] [PubMed] [Google Scholar]
- 7.Egli D, Birkhoff G, Eggan K. Mediators of reprogramming: Transcription factors and transitions through mitosis. Nat Rev Mol Cell Biol. 2008;9:505–516. doi: 10.1038/nrm2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khidr L, Chen PL. RB, the conductor that orchestrates life, death and differentiation. Oncogene. 2006;25:5210–5219. doi: 10.1038/sj.onc.1209612. [DOI] [PubMed] [Google Scholar]
- 9.Orford KW, Scadden DT. Deconstructing stem cell self-renewal: Genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9:115–128. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- 10.He S, Nakada D, Morrison SJ. Mechanisms of stem cell self-renewal. Annu Rev Cell Dev Biol. 2009;25:377–406. doi: 10.1146/annurev.cellbio.042308.113248. [DOI] [PubMed] [Google Scholar]
- 11.Lange C, Calegari F. Cdks and cyclins link G1 length and differentiation of embryonic, neural and hematopoietic stem cells. Cell Cycle. 2010;9:1893–1900. doi: 10.4161/cc.9.10.11598. [DOI] [PubMed] [Google Scholar]
- 12.Edel MJ, Izpisua Belmonte JC. The cell cycle and pluripotency: Is there a direct link? Cell Cycle. 2010;9:2694–2695. doi: 10.4161/cc.9.14.12456. [DOI] [PubMed] [Google Scholar]
- 13.Neganova I, Lako M. G1 to S phase cell cycle transition in somatic and embryonic stem cells. J Anat. 2008;213:30–44. doi: 10.1111/j.1469-7580.2008.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savatier P, Malashicheva A. Cell-cycle control in embryonic stem cells. In: Lanza R, et al., editors. Handbook of Stem Cells. Vol 1. Burlington, MA: Elsevier Academic; 2004. pp. 53–62. [Google Scholar]
- 15.Burdon T, Smith A, Savatier P. Signalling, cell cycle and pluripotency in embryonic stem cells. Trends Cell Biol. 2002;12:432–438. doi: 10.1016/s0962-8924(02)02352-8. [DOI] [PubMed] [Google Scholar]
- 16.White J, Dalton S. Cell cycle control of embryonic stem cells. Stem Cell Rev. 2005;1:131–138. doi: 10.1385/SCR:1:2:131. [DOI] [PubMed] [Google Scholar]
- 17.Stead E, et al. Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene. 2002;21:8320–8333. doi: 10.1038/sj.onc.1206015. [DOI] [PubMed] [Google Scholar]
- 18.Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775–1785. doi: 10.1016/j.cub.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 19.Ortega S, et al. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet. 2003;35:25–31. doi: 10.1038/ng1232. [DOI] [PubMed] [Google Scholar]
- 20.Koledova Z, et al. Cdk2 inhibition prolongs G1 phase progression in mouse embryonic stem cells. Stem Cells Dev. 2010;19:181–194. doi: 10.1089/scd.2009.0065. [DOI] [PubMed] [Google Scholar]
- 21.Filipczyk AA, Laslett AL, Mummery C, Pera MF. Differentiation is coupled to changes in the cell cycle regulatory apparatus of human embryonic stem cells. Stem Cell Res (Amst) 2007;1:45–60. doi: 10.1016/j.scr.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Neganova I, Zhang X, Atkinson S, Lako M. Expression and functional analysis of G1 to S regulatory components reveals an important role for CDK2 in cell cycle regulation in human embryonic stem cells. Oncogene. 2009;28:20–30. doi: 10.1038/onc.2008.358. [DOI] [PubMed] [Google Scholar]
- 23.Ruiz S, et al. A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. Curr Biol. 2011;21:45–52. doi: 10.1016/j.cub.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menchón C, Edel MJ, Izpisua Belmonte JC. The cell cycle inhibitor p27Kip1 controls self-renewal and pluripotency of human embryonic stem cells by regulating the cell cycle, Brachyury and Twist. Cell Cycle. 2011;10:1435–1447. doi: 10.4161/cc.10.9.15421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalaszczynska I, et al. Cyclin A is redundant in fibroblasts but essential in hematopoietic and embryonic stem cells. Cell. 2009;138:352–365. doi: 10.1016/j.cell.2009.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ballabeni A, et al. Cell cycle adaptations of embryonic stem cells. Proc Natl Acad Sci USA. 2011;108:19252–19257. doi: 10.1073/pnas.1116794108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 28.Dobrowolski SF, Stacey DW, Harter ML, Stine JT, Hiebert SW. An E2F dominant negative mutant blocks E1A induced cell cycle progression. Oncogene. 1994;9:2605–2612. [PubMed] [Google Scholar]
- 29.Knudsen ES, Wang JY. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. 1997;17:5771–5783. doi: 10.1128/mcb.17.10.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savatier P, Huang S, Szekely L, Wiman KG, Samarut J. Contrasting patterns of retinoblastoma protein expression in mouse embryonic stem cells and embryonic fibroblasts. Oncogene. 1994;9:809–818. [PubMed] [Google Scholar]
- 31.Ezhevsky SA, Ho A, Becker-Hapak M, Davis PK, Dowdy SF. Differential regulation of retinoblastoma tumor suppressor protein by G1 cyclin-dependent kinase complexes in vivo. Mol Cell Biol. 2001;21:4773–4784. doi: 10.1128/MCB.21.14.4773-4784.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith AG. Embryo-derived stem cells: Of mice and men. Annu Rev Cell Dev Biol. 2001;17:435–462. doi: 10.1146/annurev.cellbio.17.1.435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.