

Abstract
3-Iodobenzoylnaltrexamide 1 (IBNtxA) is a potent analgesic acting through a novel receptor target that lack many side-effects of traditional opiates composed, in part, of exon 11-associated truncated six transmembrane domain MOR-1 (6TM/E11) splice variants. To better understand the SAR of this drug target, a number of 4,5-epoxymorphinan analogs were synthesized. Results show the importance of a free 3-phenolic group, a phenyl ring at the 6 position, an iodine at the 3′ or 4′ position of the phenyl ring and an N-allyl or c-propylmethyl group to maintain high 6TM/E11 affinity and activity. 3-Iodobenzoylnaloxamide 15 (IBNalA) with a N-allyl group displayed lower delta opioid receptor affinity than its naltrexamine analog, was 10-fold more potent an analgesic than morphine, elicited no respiratory depression or physical dependence and only limited inhibition of gastrointestinal transit. Thus, the aryl-naloxamide scaffold can generate a potent analgesic acting through the 6TM/E11 sites with advantageous side-effect profile and greater selectivity.
INTRODUCTION
Opiates have been the subject of intense research since the isolation of morphine in the early 1800’s.1 Despite their efficacy and utility, side-effects greatly limit the usefulness of virtually all the clinically available opiates, including respiratory depression, constipation and physical dependence, reward and addiction. Attempts to separate these unwanted actions from analgesia have largely been unsuccessful. A number of studies exploring the pharmacology of naloxonazine dissociated morphine analgesia from respiratory depression,2, 3 inhibition of gastrointestinal transit,4, 5 and physical dependence.6 Yet, dissociation of these actions functionally failed to yield selective analgesics lacking these side-effects. The identification of delta and kappa1 receptors led to extensive efforts to generate alternative analgesics acting through different receptors to achieve a better pharmacological profile. Although highly selective agents were developed, they have yet to fill the clinical need. The kappa1 receptor drugs that were tested clinically revealed unacceptable psychotomimetic effects, as well a profound diuresis, while the utility of delta-selective agents have not yet been fully validated. Thus, there still remains a need for potent analgesics with safer side-effect profiles.
Clinicians have long appreciated differences among mu opioids, with some patients responding better to one agent than another.7–9 These clinical observations, along with preclinical pharmacological and binding studies, led to the initial suggestion of multiple mu receptors,10 a concept that has been confirmed with the cloning of a vast array of splice variants of the mu opioid receptor (MOR-1) gene Oprm1 in mice, rats and humans.11–20 Included in these variants is a set of truncated six transmembrane domain splice variants generated by an alternative promoter associated with exon 11 of the OPRM1 gene of mice,16 rats21 and humans.22 Although it was suggested that these variants might be active alone,23, 24 the poor affinity of the expressed variants for opiates and the very high doses of drug necessary to activate the receptors seems to make this unlikely.
Recently, we reported the synthesis25 and pharmacological characterization26 of a β-naltrexamine analog, 3′-iodobenzoylnaltrexamide 1 (IBNtxA, fig 1). Pharmacological studies revealed an analgesic with a potency 10-fold greater than morphine with no observable respiratory depression, physical dependence, cross tolerance to morphine or rewarding behavior in the conditioned place preference assay and limited inhibition of gastrointestinal transit. This compound displayed high affinity for a binding site in brain that was distinct from traditional mu, delta or kappa1 receptors, most clearly shown by its persistent analgesia and binding in a triple knockout mouse lacking the traditional mu, delta and kappa1 receptors. The loss of both its analgesia and its binding site in an exon 11 knockout mouse, further established a unique mechanism of action and implicated exon 11-associated 6 transmembrane domain splice variants (6TM/E11). Additional studies suggest that these truncated variants do not act alone. Rather, 1 appears to label with high affinity a heterodimer of a 6TM/E11 variant with an additional G-protein coupled receptor, although the partner is still unknown. We now describe the structure-activity relationships for this new drug target by synthesizing several analogs of 1 with substitutions on the 6, 17 and 3 positions of the 4,5-epoxymorphinan scaffold. All synthesized molecules were evaluated for their relative binding affinity for cloned mu (MOR-1), kappa1 (KOR-1) and delta (DOR-1) sites in cell lines and 6TM/E11 sites in mouse brain and for their analgesic activity in vivo with the radiant heat tail flick assay.
Figure 1.

Structures of selected opiates
RESULTS
Chemistry
The ketone at the 6-position of naloxone 4 and 3-methoxynaloxone 11 were transformed to an amine (Opiate-NH2) by reductive amination using sodium cyanoborohydride (NaBH3CN) and ammonium acetate (NH4OAc) to yield a mixture of beta and alpha isomers.27 The beta and alpha isomers were then purified by column chromatography. In a parallel synthesis, substituted carboxylic acids 5–7 were converted to their respective N-succinimidyl esters 8–10 by reacting them with N-hydroxysuccinimide (NHS) in the presence of DCC and THF. The corresponding activated esters were then reacted with the beta or alpha isomer of the opiate-NH2 in the presence of diisopropylethylamine (DIEA) and DCM. The corresponding aryl, alkyl and carbocyclic amido derivatives of opiates were then purified by column chromatography. Alternatively, the substituted carboxylic acids were directly coupled to the opiate-NH2 using benzotriazol-1yl-oxy-tris(dimethylamino)phosphonium hexafluorophosphate (BOP) and DIEA in DCM to give 3, 6-disubstituted derivatives. The 3,6-disubstituted opiate derivatives were then subjected to basic hydrolysis with potassium carbonate to yield 6-alkyl, aryl and carbocyclic derivatives of naloxamide and 3-methoxynaloxamide (Scheme 1; table 1). 1, 14–15 were synthesized as previously reported.25 The pharmacological characterization of 14 and 15 and all newly synthesized compounds (16–38) is summarized in table 1.
Scheme 1a. Synthesis of Aryl and alkyl amido derivatives of β-naloxamine and 3-methoxy-β-naloxamine.

aReagents: (a) NHS, DCC, dry THF, 24 h, rt, N2; (b) NH4OAc, NaBH3CN, dry MeOH, 24 h, rt, N2; (c) 8–10, DIEA, dry DCM, rt, N2, 2h; (d) R3COOH, BOP, DIEA, DCM, 2h; (e) K2CO3, MeOH, 3h.
Table 1.
Evaluation of receptor binding affinity and analgesic activity
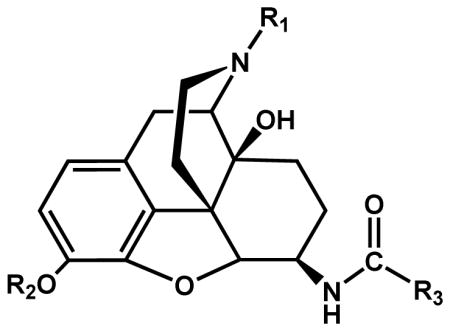
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Ki (nM) | Tail flick analgesia | |||||||
| Compound | R1 | R2 | R3 | MOR-1 | KOR-1 | DOR-1 | 6TM/E11 | ED50 mg/kgc (95%CL) |
| Naltrexone 3 | 32.7 ± 4.1a | |||||||
| Naloxone 4 | 53.2 ± 7.13a | |||||||
| Oxymorphone 2 | >1μM | |||||||
| 1 | -CPM | H | Ph-3′-I | 0.11 ± 0.02 | 0.03 ± 0.001 | 0.24 ± 0.05 | 0.16 ± 0.04a | 0.39 (0.15, 0.58)a |
| 14 | -CH3 | H | Ph-3′-I | 0.97 ± 0.2 | 47.22 ± 14.2 | 2.45 ± 1 | 41.22 ± 12.3 | >10 |
| 15 | -allyl | H | Ph-3′-I | 0.22 ± 0.12 | 0.08 ± 0.06 | 2.55 ± 0.18 | 0.25 ± 0.12 | 0.60 (0.42, 0.9) |
| 16 | -allyl | H | Ph-3′-Ib | 5.07 ± 1.9 | 12.16 ± 3.8 | 7.64 ± 2.56 | 8.46 ± 2.9 | 4.9 (4.47, 5.37) |
| 17 | -allyl | H | Ph-2′-I | 1.56 ± 0.4 | 1.00 ± 0.11 | 22.8 ± 10.3 | 29 ± 5.9 | >10 |
| 18 | -allyl | H | Ph-4′-I | 0.11 ± 0.04 | 0.28 ± 0.08 | 3.36 ± 2.57 | 0.64 ± 0.22 | 0.14 (0.08, 0.24) |
| 19 | -allyl | H | Ph-3′-F | 0.47 ± 0.03 | 2.05 ± 0.49 | 18.19 ± 3.81 | 8.09 ± 3.45 | 2.9 (2.4, 3.6) |
| 20 | -allyl | H | Ph-3′-Cl | 1.15 ± 0.11 | 0.52 ± 0.04 | 4.87 ± 3.32 | 5.49 ± 0.98 | 1.6 (0.8, 3.2) |
| 21 | -allyl | H | Ph-3′-Br | 3.85 ± 3.53 | 1.58 ± 0.89 | 23.37 ± 20.24 | 2.05 ± 1.3 | 0.71 (0.47, 1.1) |
| 22 | -allyl | H | Ph | 4.03± 1.04 | 14.27 ± 3.99 | 60.78 ± 11.44 | 5.82 ± 1.79 | 5.3 (4.3, 6.5) |
| 23 | -allyl | H | Ph-3′-CH3 | 0.29 ± 0.05 | 1.62 ± 0.28 | 8.24 ± 4.84 | 8.98 ± 0.1 | 2.25 (1.8, 2.9) |
| 24 | -allyl | H | Ph-3′-CF3 | 0.85 ± 0.24 | 0.22 ± 0.13 | 2.96 ± 2.09 | 9.32 ± 1.3 | 0.21 (0.14, 0.3) |
| 25 | -allyl | H | Ph-3′-OCH3 | 0.18 ± 0.1 | 4.97 ± 0.63 | 17.22 ± 3.53 | 1.64 ± 0.45 | 0.23 (0.13, 0.41) |
| 26 | -allyl | H | Ph-3′-NH2 | 0.43 ± 0.12 | 0.4 ± 0.1 | 36 ± 11 | 7.62 ± 3.94 | >10 |
| 27 | -allyl | H | Ph-3′-N(CH3)2 | 6.4 ± 2.3 | 34.91 ± 10.5 | 51.35 ± 15.6 | 10.79 ± 1.31 | >10 |
| 28 | -allyl | H | Ph-3′-OH | 0.23 ± 0.01 | 2.75 ± 0.87 | 11.25 ± 4.13 | 5.21 ± 1.51 | >10 |
| 29 | -allyl | H | Ph-3′-NO2 | 1.41 ± 0.12 | 1.51 ± 0.14 | 18.13 ± 6.86 | 4.53 ± 1.2 | 5.3 (4.22, 6.7) |
| 30 | -allyl | H | Ph-4′-OCF3 | 0.66 ± 0.04 | 3.16 ± 0.19 | 17.88 ± 3.25 | 7.43 ± 2.67 | 0.67 (0.52,0.88) |
| 31 | -allyl | H | Naphthalene | 0.74 ± 0.63 | 1.29 ± 0.45 | 5.51 ± 1.15 | 6.64 ± 1.82 | 1 (0.7,1.42) |
| 32 | -allyl | H | Biphenyl | 0.95 ± 0.2 | 25.79 ± 10.1 | 19.15 ± 5.6 | 7.17± 0.93 | >10 |
| 33 | -allyl | H | CH3 | 20.46 ± 1.4 | >100 | >100 | >100 | >10 |
| 34 | -allyl | H | C6H13 | 9.5 ± 26 | 9.15 ± 0.94 | 32.2 ± 9.45 | 29.65 ± 9.29 | >10 |
| 35 | -allyl | H | C12H25 | 0.61 ± 0.1 | 9.35 ± 1.55 | 15.25 ± 0.38 | 39.43 ± 1.54 | >10 |
| 36 | -allyl | H | cHexane | 11.54 ± 2.64 | 17.9 ± 3.59 | >100 | 30.17 ± 7.79 | >10 |
| 37 | -allyl | H | Adamantane | 6.5 ± 1.94 | 7.1 ± 3.15 | >100 | 30.27 ± 6.41 | >10 |
| 38 | -allyl | CH3 | Ph-3I | >100 | >100 | >100 | >100 | >10 |
Values from the literature.26
Alpha isomer
(95% confidence limits)
All analogs were the β-isomer at the 6 position, with the exception of 16, which was the α-isomer of 6-naloxamine. Competition studies were performed with the indicated compounds against 125I-BNtxA (0.1 nM) in membranes from CHO cells stably expressing the indicated cloned mouse opioid receptors or in mice brain membranes for 6TM/E11 sites with blockers to prevent binding to traditional mu, kappa1 and delta receptors as described in the Methods section. Ki values were calculated from the IC50values43 and represent the means ± SEM of at least three independent replications. 125IBNtxA KD values for MOR-1, KOR-1, DOR-1 and 6TM/E11 sites were 0.11, 0.03, 0.24 and 0.16 nM for respectively.
Pharmacology
1 is a potent analgesic lacking many of the problematic side-effects associated with traditional opiates. Our objective is to establish the criteria important for both potency and selectivity for the 6TM/E11 sites by exploring the structure-activity relationship (SAR).
The N-substituent is important for defining both affinity and analgesic activity. Receptor binding studies reveal that an N-methyl substituent dramatically lowers the affinity of the compound for the 6TM/E11 site, as shown with oxymorphone 2 (Ki >1 μM, compared to the cpropylmethyl group in naltrexone 3 (Ki 32 nM) or the allyl group in naloxone 4 (Ki 53 nM) (fig 1; table 1).
The increased affinity of 1 (Ki = 0.16 nM) illustrates the importance of substituents at the 6-position of β-naloxamine. To further explore the role of the 3′-iodophenyl moiety at the 6-position of the 4,5-epoxymorphinan, we synthesized 14 and 15 which incorporated a 3′-iodophenyl amido moiety into the oxymorphone and naloxone scaffold, respectively. 14 has a Ki of 41 nM for 6TM/E11 sites. Although the addition of the 3′-iodophenyl amido moiety increased the affinity by at least 50-fold, its affinity remained over 100-fold poorer for the 6TM/E11 site than either the naloxone analog (3-Iodobenzoylnaloxamide 15 (IBNalA)), Ki = 0.25 nM, fig 1) or the naltrexone analog (1, Ki 0.16 nM). The 3′-iodophenyl amido moiety also influenced the pharmacology of the compounds. Oxymorphone 2 is a potent analgesic. Yet, its analog, 14, showed no analgesia at doses as high as 10 mg/kg, s.c. despite a reasonable affinity for mu receptors. Conversely, the 3′-iodophenylamido substituent at the 6 position converted the naltrexone and naloxone analogs into potent analgesics, contrasting markedly with the antagonist character of naloxone and naltrexone themselves. It is important to note that 15 retained a similar affinity for the 6TM/E11 binding site and analgesic potency as 1 (fig 2a). Compared to 1, 15 had a 10-fold lower affinity for delta receptors and about 2 fold lower affinity for mu and kappa receptors. In view of this increased selectivity, we decided to pursue the naloxone series of analogs.
Figure 2. Pharmacology of 17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-iodo)benzamido]morphinan (15).

a) Analgesia: Cumulative dose-response curves were carried out on groups of mice (n=10) with 15 at the indicated doses of 15 (s.c.) and analgesia tested 30 min later at peak effect. ED50 values (and 95% confidence limits) were 0.6 mg/kg (0.42, 0.87) in CD1 mice by using the radiant heat tail-flick assay.
b) Respiratory rate: Animals are randomly assigned to receive saline (n=5, s.c.), or the opioid 15 (2.5 mg/kg, s.c.; n = 5), or morphine (20mg/kg, s.c.; ED50, n = 5) at 5-fold and 4-fold their respective ED50 and tested as described in Methods. Each animal’s baseline average breath rate was measured every 5 minutes for 25 minutes prior to drug injection, and breath rates following drug injection expressed as a percent of baseline. 15 did not depress respiratory rate and was not significantly different from saline at any time point while morphine decreases respiratory depression in comparison to both saline and 15 (p<0.001) as determined by repeated measures ANOVA followed by Bonferroni multiple comparison test.
c) Gastrointestinal transit: Groups of mice (n=10) received saline (s.c.), morphine aty its analgesic ED50 (5mg/kg, s.c.), or 15 at twice its analgesic ED50 (1 mg/kg, s.c.) before receiving a charcoal meal (0.2 ml; 2.5% gum tracaganth in 10% activated charcoal in water) by gavage and sacrificed 30 min later. The distance travelled by the charcoal was measured 30 min later. 15 lowered transit significantly compared to saline (p<0.05), but less than morphine (p<0.05) as determined by ANOVA followed by Tukeys multiple comparison test.
d) Physical dependence: Groups of mice (n ≥ 10) received either morphine (10 mg/kg, s.c.) or 15 (1 mg/kg, s.c.) at twice their analgesic ED50 for 5 days. They then were challenged with the indicated antagonist. Naloxone precipitated a profound withdrawal syndrome in the morphine-treated animals, as shown by the number of jumps/15 min, which was significantly greater than that in the morphine or 15 controls (i.e. given no antagonist) or in 15 mice given the indicated antagonist. Mice chronically administered 15 showed no significant difference from controls when challenged by naloxone or levallorphan (1 mg/kg, s.c.).
The stereochemistry of the 6-position is important. The alpha isomer, 16, showed a 33-fold decreased binding affinity for the 6TM/E11 site, a 23, 150 and 3 fold lower affinity for mu, kappa and delta receptors and a 10-fold higher ED50 in analgesic assays (Table 1). Thereafter, we focused upon β-naloxamine compounds.
Moving the position of the iodine on the aryl ring influences activity. Although an iodine at position 4′, 18, retains a very similar profile compared to the 3′ position, 15, placing it at position 2′, 17, lowered its binding affinity for the 6TM/E11 sites by 100-fold with approximately 10-fold lower affinities for mu, kappa and delta receptors and eliminated its analgesic activity at doses as high as 10 mg/kg.
We next examined a series of halogens at the 3′ position of the aryl ring (19–21, Table 1), and compared it to hydrogen in 22. We chose the 3′ position based on the higher affinity of the 3′-iodo-analog, 15 when compared to the 4′-iodo-analog, 18. 22 illustrates the importance of the iodine when compared to a hydrogen. Removal of the iodine at either the 3′ or the 4′-position lowered the affinity at the 6TM/E11 site by over 20-fold while the affinity across mu, kappa and delta receptors also decreased by about 20-, 180- and 25-fold, respectively. The 6TM/E11 affinity increased as the size of the halogen increased and electron withdrawing nature of the halogen atom decreased while no observable trends in affinity were seen with the traditional mu, kappa and delta receptors. The analgesic activity correlated with the 6TM/E11 site binding, increasing from F to I. The existence of halogen bonding in protein-ligand complexes has been shown for adenosine kinase inhibitors,28 HIV-RT inhibitors29, 30 and PD5 inhibitors.31 This raises the possibility that the ability of iodine to form weak halogen bonds32 in the protein pocket, rather than the electron-withdrawing nature of the halogen atom, maybe responsible for its higher affinity for the 6TM/E11 sites.
To further evaluate the role of electron releasing or electron withdrawing groups on the aryl ring on 6TM/E11 affinity we synthesized compounds with both electron releasing groups (CH3, OCH3, OH and NMe2) and electron withdrawing groups (NO2, OCF3 and CF3; 23–30, table 1). No clear trend was seen with either series, with these substituents lowering the affinities for the 6TM/E11 site and the traditional opioid receptors when compared to 15. However, the analgesic activity of 24 with a CF3 at the 3′position and 25 with OMe at the 3′position of the aryl ring stand out from the others with regards to their potent analgesic actions. 24 is 42, 4 and 10 fold more selective for kappa1 sites than 6TM/E11, mu or delta receptors. Similarly, 25 is 9, 27 and 100 fold more mu selective than 6TM/E11, kappa and delta receptors respectively, raising the question whether the strong analgesic actions of 24 and 25 might be due to traditional opioid receptors. If true, this would be unusual since the placement of an allyl group on the nitrogen in the 17-position typically yield antagonists for the traditional receptors.
An aryl ring may be sufficient to instill high affinity for the 6TM/E11 site in view of the similar affinity of analogs with either electron withdrawing or releasing groups. More complex aryl rings (31–32, table 1) maintained a modest binding affinity for the 6TM/E11 site. 31, with its naphthalene ring also shows modest analgesic activity. Despite its similar binding affinity for the 6TM/E11 site, 32 was not analgesic. Both 31 and 32 have higher affinity for the mu receptor over 6TM/E11 sites and offer no additional advantage in terms of 6TM/E11 selectivity over the traditional opioid receptors.
Lipophilicity alone appears to have little role in defining binding affinity for the 6TM/E11 sites or activity. Methyl, hexyl, dodecyl, cyclohexyl and adamantyl groups at the 6 position (33–37, table 1) all showed poor binding affinity for the 6TM/E11 sites and moderate to poor affinities across the traditional opioid receptors and no analgesic activity at doses as high as 10 mg/kg, s.c. This contrasts markedly with the aryl substituent’s. 22, with a simple phenyl ring (Ki 5nM; ED50 5 mg/kg, s.c.) is far superior to any of the above analogs, raising the question of a pipi stacking interaction between the aromatic ring at the 6-position and the 6TM/E11 protein binding site.
Finally, we examined the need for a free 3-OH group. It has long been known that traditional mu opiates of the 4,5-epoxymorphinan series require a free hydroxyl group at this position. Although codeine and oxycodone, which have a 3-methoxy group, are effective analgesics, many believe that the activity results from demethylation to morphine and oxymorphone, respectively. Like other opiates, the presence of a methyl group in 38 eliminates binding across the 6TM/E11 sites and the traditional opioid receptors and analgesia.
To see if these compounds are acting in a manner similar to 1, we extended the pharmacological assessment of 15, the naloxone analog. The N-allyl analog had an advantage in that its affinity for delta receptors is approximately 10-fold lower than the CPM compound, 1. Despite its potent analgesic activity, 15 displayed no respiratory depression at doses approximately 4-fold greater than its analgesic activity, showed no evidence of physical dependence after repeated administration based upon the absence of withdrawal signs following the administration of naloxone or levallorphan and far less inhibition of GI transit than morphine (Fig. 2). Thus, the overall pharmacology of 15 mimics that of 1, consistent with activity through the 6TM/E11 target.
DISCUSSION
Our earlier work with 1 illustrated the ability to develop opioids capable of acting through a novel target, which is comprised, in part, by exon 11-associated splice variants of MOR-1.26 The overall profile of this class of analgesic was quite unusual in that the agents had no respiratory depression, physical dependence, or cross tolerance to morphine and only limited effects on gastrointestinal transit. Furthermore, 1 retained full analgesic activity in a MOR-1/DOR-1/KOR-1 triple knockout mouse lacking traditional mu, delta and kappa1 receptors while it was without activity in an E11 knockout mouse with a disruption of the exon 11-associated variants.26 Our current study explores a series of analogs. The pharmacological profile of the analogous naloxone derivative, 15, was quite similar. Again, it was a potent analgesic with no evidence of respiratory depression or physical dependence and limited inhibition of gastrointestinal transit.
The structure-activity relationships of these analogs are interesting. The N-substituent is important, as the analgesic activity is limited to the N-allyl, 15 or N-methylcyclopropyl 6β-naltrexamine derivatives, 1. No analgesia could be detected for the N-methyl derivative, 14. This was opposite to what might be expected since analgesia is seen only with oxymorphone and not naloxone and naltrexone. This might reflect a reversal of the effect of the N-substituent on the traditional agonist/antagonist character of the molecules, but the N-methyl group also was associated with a marked decrease in affinity for the 6TM/E11 sites. It is interesting to note that an aryl ring at the 6-position enhances affinity for the 6TM/E11 sites. All analogs lacking the aryl ring had poorer affinity with Ki >10 nM. Placement of an iodine on the aryl ring at the 3′ or 4′ position further enhances the affinity for the 6TM/E11 sites and as well as analgesic activity.
Structurally analogous aryl naltrexamides have been described by Ghirmai et al., as alcohol cessation agents33, 34 and by Zhang and co-workers as mu antagonists.35, 36 However, none of these molecules were noted to have analgesic activity. Similarly, 6-arylamido morphines have been reported as M6G analogues by McDougal et al.37 More recently, Portoghese described a naphthoyl analog of 6β-naltrexamine (NNTA) analogous to the naloxone version synthesized in our study 31 (Table 1) with analgesic activity and lacking physical dependence and rewarding behavior in the place preference assay that he ascribed to activation of a mu/kappa1 opioid receptor dimer.38 As with the 6β-naltrexamine, the 6β-naloxamine analog was active in the mouse radiant heat tailflick assay. However, it is unclear whether this compound is acting through the same target as 1 and 15. Compared to 15, the napthoyl derivative, 31, showed over a 26-fold lower binding affinity for the 6TM/E11 site while its analgesic activity was only 1.5-fold lower. The loss of NNTA analgesia as reported in their MOR-1 knockout mouse38 does not help since the disruption of exons 2 and 3 in their mouse leads to the loss of both the exon 11-associated 6TM variants and the full length MOR-1 ones and therefore would be expected to abolish the actions of compounds acting through any of the 6TM or 7 TM MOR-1 variants.
There is, in general, a reasonably good correlation between binding affinity for the 6TM/E11 sites and analgesic potency for most of the compounds, consistent with activity through the 6TM/E11 sites. However, there are some exceptions. For example, the 3-OCH3 analog, 25, shows an analgesic potency over 3-fold greater than 15, despite an affinity for the 6TM/E11 site that is 10-fold poorer. A similar dissociation of analgesic activity and binding affinity for the 6TM/E11 site is seen with 24 (4-CF3) and 30 (4-OCF3), raising questions about their targets as well. Thus, interpretation of the analgesic activity and correlating it with binding affinities must be done cautiously since a wide range of targets can all elicit the same pharmacological response. Finally, both 1 and 15 show a remarkable side-effect profile, confirming that it is possible to develop superior analgesics. However, it is premature to extrapolate these observations to the other compounds that may, or may not, have a similar side-effect profiles.
CONCLUSION
A series of C-6-substituted 4,5-epoxymorphinans (i.e. aryl, alkyl, and carbocycle naloxamides) were synthesized as analogs of 1 to probe the structure-activity relationships of both affinity and selectivity of ligands for the 6TM/E11 sites. These compounds were prepared by the reaction of 6β-naloxamine with alkyl, aryl and carbocyclic carboxylic acids or their corresponding NHS activated esters. This is the first report of the pharmacological properties of aryl naloxamides and their ability to demonstrate potent agonist activity, although structurally analogous aryl naltrexamides have been reported before. To our knowledge, this is the first study demonstrating aryl-naloxamide analgesics. Although many of the derivatives displayed high affinity for other opiate receptor targets, particularly mu and kappa1, there were clear predictors for the 6TM/E11 target. The most important was the aryl ring at the C-6 position of the 4,5-epoxymorphinan scaffold. All derivatives lacking a phenyl ring were completely inactive in analgesia assays and had lower affinity in 6TM/E11 binding assays. The presence of an iodine at either the 3′ or 4′position, but not the 2′position, of the phenyl ring also increased the 6TM/E11 affinity and activity. Replacing the N-CPMof 1 with an N-allyl substituent, 15, produced an analog with similar affinity for the 6TM/E11 site and a similar pharmacological profile, as well as increased selectivity over the delta opioid receptor. The superior pharmacological profile of 15 compared to traditional mu analgesics suggested that the 6TM/E11 site may provide a useful target in developing useful analgesics.
EXPERIMENTAL PROCEDURES
Materials
Naltrexone, oxymorphone, levallorphan, morphine, U50,488H, [D-Pen2,DPen 5]enkephalin, [D-Ala2,MePhe4,Gly(ol)5]enkephalin and naloxone were a generous gift from the Research Technology Branch of the National Institute of Drug Abuse (Rockville, MD) and other chemicals were purchased from Fischer Scientific (Pittsburgh, PA, USA) and Sigma (St Louis, MO, USA). Na125I was bought from Perkin-Elmer (Waltham, MA, USA).
Synthesis of compounds (15–38)
General Procedures
All reactions were carried out under positive nitrogen atmosphere with a magnetic stirrer at ambient temperatures using oven-dried glassware. 1H-NMR were taken on a 500 MHz Bruker Advance III instrument using CDCl3 as solvent and calibrated using an internal reference. Chemical shifts are expressed in parts per million (ppm) and coupling constants (J) in hertz. High resolution mass spectra were obtained with a Waters LCT premiere spectrometer while a Waters SQD mass spectrometer instrument operating in the ESI mode was also used. A reversed-phase HPLC using a Perkin Elmer LC pump series 200 and a 785A uv/vis detector (214 nM) was used. A Varian microsorb MV 100-5 reversed-phase column (5 μm × 4.6 mm × 250 mm) with the mobile phases being 0.1% TFA in water and 0.1% TFA in ACN with a gradient elution at a flow rate of 1 mL/min was used. Silica gel (230–400 mesh) was used in column chromatography. C18 sep paks were purchased from Waters (Milford, MA). Radioiodinated samples were counted on a Wizard 1470 automatic gamma counter from Perkin Elmer (Waltham, MA, USA). All chemicals and were either purchased from Fischer Scientific (Pittsburgh, PA, USA) or Sigma (St Louis, MO). All synthesized compounds were characterized and purity determined by 1H-NMR, 13C-NMR, HPLC, MS and HRMS. Analytical data confirmed the purity of the products ≥95%.
Synthesis of β-naloxamine 12 and β-3-methoxynaloxamine 13
Reductive amination of naloxone 4 and 3-methoxynaloxone 11 was carried out using a literature protocol published by Jiang et al.27 Typically, 10 g of opiate (30 mmol), was stirred with NH4OAc (22g, 0.3 mol, 10 eqv) in 40 mL dry methanol for 10 minutes at room temperature. NaBH3CN (1.31 g, 21 mmol, 0.7 eqv) in 5 mL dry methanol was then added to the reaction mixture and contents stirred overnight. The reaction was quenched by addition of 10 mL 1N NaOH, the solvents were evaporated on a rotavapor at 40°C. The residue was then extracted with 30 mL DCM three times; the organic extracts were combined and washed with 25 mL water. The organic extracts were dried over Na2SO4 and concentrated to a white solid, which was purified by silica gel column chromatography. The reaction gave a mixture of alpha and beta isomers. The respective isomers were isolated by column chromatography using 87:10:3 of EtOAc: MeOH: NH4OH as the eluent. The beta isomer had a higher Rf than the alpha isomer on a TLC plate and eluted first when the mixture was subjected to column chromatography. Yields for beta isomer were about 2.5–3 g (25–30%). The stereochemistry of the diastereomers at the C-6 position were determined on the basis of the size of the NMR coupling constant, J 5, 6. For the α-isomer, the chemical shift of the C-5 H is δ 4.58 with 3.5 Hz as the J 5,6 coupling constant, compared to the literature value 27 of δ 4.55 and 4.0 Hz; for the β-isomer, the chemical shift of the C-5 H is δ 4.24 with 7.1 Hz as the J 5,6 coupling constant, comparing to the literature value25, 27 of δ 4.18 and 7.4 Hz.
Synthesis of N-hydroxysuccinimide (NHS) esters of carboxylic acids 8–10
Substituted carboxylic acid (5–7, 7.8 mmol), NHS (1g, 8.6 mmol, 1.1 eqv), DCC (1.79 g, 8.6 mmol, 1.1 eqv) in 20 mL dry THF were stirred overnight. The white suspension was filtered and the clear filterate was evaporated on a rotavapor at 40°C. The white solid seen was purified by column chromatography using EtOAc/hexanes as eluents. A singlet at δ2.9 integrating to 4 protons in 1H-NMR and corresponding to four protons of succinimide was seen in all NHS esters of substituted carboxylic acids. Crude yields were about 80–100%. The crude activated ester was used in the next step without any purification.
Synthesis of aryl-α-naloxamide, aryl-β-naloxamides and β-3-methoxynaloxamides 15–38
Coupling of α-naloxamine, β-naloxamine 12 or β-3-methoxynaloxamine 13 (Opiate-NH2) to the corresponding carboxylic acid was carried out by either reacting the amine to the NHS-activated carboxylic acid ester or by direct coupling of the amine to the carboxylic acid in presence of BOP.
Procedure I
Opiate-NH2 (12–13, 200 mg, 0.6 mmol) was reacted with DIEA (116 ul, 0.66 mmol, 1.1 eqv) and NHS esters of substituted carboxylic acids (8–10, 0.66 mmol, 1.1 eqv) in dry DCM (5 mL) for 2 h. The reaction was diluted to 20 mL with DCM and washed with 5 mL water. The organic extracts were dried over Na2SO4 and then concentrated to a white solid, which was purified by silica gel flash column chromatography using a gradient run of 1–5% MeOH:DCM as eluents.
Alternate procedure II
Opiate-NH2 (12–13, 200 mg, 0.6 mmol) was reacted with BOP (271 mg, 1.2 mmol, 2 eqv), DIEA (313 ul, 1.8 mmol, 3 eqv) and substituted carboxylic acid (1.2 mmol, 2 eqv) in dry DCM (5 mL) for 2 h. The reaction mixture poured into a small silica gel column and eluted with 100 mL EtOAc. The ethyl acetate fraction was evaporated and a white solid was obtained. The solid obtained was hydrolyzed in K2CO3 and MeOH. Briefly, the contents, usually a white suspension were stirred with K2CO3 (622 mg, 4.22 mmol, 7 eqv) and MeOH for 3h. The white suspension seen was filtered and the filterate concentrated to a yellowish oil or a white solid. The oily residue or white solid obtained was then purified by flash column chromatography using a gradient run of 1–5% MeOH: DCM as the eluent.
Synthesis of individual embodiments
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-iodo)benzamido]morphinan (15)
Compound 15 was synthesized according to the general procedure (I) described above using β-naloxamine, NHS ester of 3-iodobenzoic acid 8 and DIEA in DCM. A white solid was obtained. Yield: 255 mg (75%); mp 203–207 °C dec; [α]20D −161.2 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 8.16 (s, 1H), 7.8 (d, J=8.9 Hz, 1H), 7.76 (d, J=8.9 Hz, 1H), 7.15-7.11 (m, 1H), 6.69 (d, J=10.6 Hz, 1H), 6.57 (d, J=10.6 Hz, 1H), 5.8 (m, 1H), 5.23-5.16 (m, 2H), 4.57 (d, J=8.85 Hz, 1H), 4.13 (m, 1H), 3.14-1.2 (m, 14H). 13C NMR (150 MHz, CDCl3) δ: 165.4, 142.9, 140.3, 139.2, 136.4, 136.2, 135.2, 130.6, 130.1, 126.1, 124.7, 119.3, 118.1, 117.6, 94.3, 92.9, 70.2, 62.4, 57.8, 50.5, 47.3, 43.6, 31.5, 29.0, 23.2, 22.7 ppm. ESI-MS m/z: 559.1 (MH+). HRMS calcd for C26H28N2O4I (MH+), 559.1094; found, 559.1099. HPLC purity: 96.5%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6α-[(3′-iodo)benzamido]morphinan (16)
Compound 16 was synthesized according to the general procedure (I) described above using α-naloxamine, NHS ester of 3-iodobenzoic acid 8 and DIEA in DCM. A white solid was obtained. Yield: 248 mg (73%); mp 134–136 °C; [α]20D −225.7 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 8.01 (s, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.11 (t, J = 7.8 Hz, 1H), 6.70 (d, J = 8.1 Hz, 1H), 6.56 (d, J = 8.1 Hz, 1H), 6.37 (d, J = 8.2 Hz, 1H), 5.80 (m, 1H), 5.18 (d, J = 18.5 Hz, 1H,), 5.15 (d, J = 10.9 Hz, 1H), 4.74 (m, 2H), 3.50-1.00 (m, 15 H) ppm. 13C NMR (150 MHz, CDCl3) δ: 165.5, 145.1, 140.3, 137.2, 136.6, 136.0, 135.2, 130.8, 130.1, 126.3, 125.9, 119.4, 118.0, 117.3, 94.2, 90.1, 69.7, 62.3, 58.1, 47.2, 46.7, 42.9, 33.3, 28.9, 23.0, 21.0 ppm. MS (ESI) m/z (%) 559 (MH+). HRMS calcd for C26H28N2O4 I (MH+), 559.1094; found, 559.1107. HPLC purity: 97.5%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(2′-iodo)benzamido]morphinan (17)
Compound 17 was synthesized according to the general procedure (II) described above using β-naloxamine, 2-iodobenzoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 204 mg (60%); mp 208–211 °C dec; [α]20D −180.0 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 7.87 (d, J=8.35 Hz, 1H), 7.42 (d, J=8.35, 1H), 7.38-7.36 (m, 1H), 7.11-7.08 (m, 1H), 6.75 (d, J=8.35, 1H), 6.6 (d, J=8.35, 1H), 6.41 (m, 1H), 5.78 (m, 1H), 5.14 (m, 2H), 4.51 (d, J=8.35, 1H), 4.17 (m, 1H), 3.49-1.26 (m, 14H). 13C NMR (150 MHz, CDCl3) δ: 169.2, 142.9, 142.2, 139.9, 139.6, 135.2, 131.1, 130.8, 128.3, 128.2, 124.8, 119.3, 118.0, 117.6, 93.2, 92.4, 70.2, 62.4, 57.7, 50.8, 47.5, 43.6, 31.0, 29.5, 23.5, 22.7 ppm. MS(ESI) m/z (%) 559 (MH+). HRMS calcd for C26H28N2O4 I (MH+), 559.1094; found, 559.1115. HPLC purity: 96.5%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4′-iodo)benzamido]morphinan (18)
Compound 18 was synthesized according to the general procedure (II) described above using β-naloxamine, 4-iodobenzoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 146 mg (43%); mp 210–213 °C dec; [α]20D −163.2 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 7.78 (d, J=9.8 Hz, 2H), 7.53 (d, J=9.8 Hz, 2H), 6.7 (d, J=9.8 Hz, 1H), 6.57 (d, J=9.8 Hz, 1H), 5.82 (m, 1H), 5.23-5.2 (m, 2H), 4.51 (d, J=8.2 Hz, 1H), 4.23 (m, 1H), 3.19-1.5 (m, 14H). 13C NMR (150 MHz, CDCl3) δ 166.1, 143.2, 139.0, 137.7, 135.2, 133.9, 130.5, 128.7, 124.9, 119.3, 118.1, 117.5, 98.4, 92.7, 70.2, 62.4, 57.8, 49.9, 47.1, 43.5, 31.9, 28.7, 22.9, 22.7 ppm. MS(ESI) m/z (%) 559 (MH+). HRMS calcd for C26H28N2O4 I (MH+), 559.1094; found, 559.1099. HPLC purity: 90.1%, α-isomer: 8.6%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-fluoro)benzamido]morphinan (19)
Compound 19 was synthesized according to the general procedure (II) described above using β-naloxamine, 3-fluorobenzoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 192 mg (70%); mp 193–196 °C dec; [α]20D −135.5 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 7.59 (d, J=9.2 Hz, 1H), 7.55 (d, J=9.2Hz, 1H), 7.41-7.36 (m, 2H), 7.21-7.17 (m, 1H), 6.73 (d, J=9.2 Hz, 1H), 6.59 (d, J=10Hz, 1H), 5.81 (m, 1H), 5.23-5.16 (m, 2H), 4.51 (d, J=9.2 Hz, 1H), 4.25 (m, 1H), 3.14-1.28 (m, 14H). 13C NMR (150 MHz, CDCl3) δ 166.6, 143.0, 140.1, 136.4, 130.3, 130.2, 122.8, 119.2, 118.8, 118.7, 118.6, 114.6, 114.4, 93.1, 70.6, 62.4, 57.7, 50.8, 47.4, 43.7, 31.2, 29.3, 23.9, 22.7 ppm. MS(ESI) m/z (%) 451 (MH+). HRMS calcd for C26H28N2O4F(MH+), 451.2033; found, 451.2031. HPLC purity: 94.3%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-chloro)benzamido]morphinan (20)
Compound 20 was synthesized according to the general procedure (II) described above using β-naloxamine, 3-chlorobenzoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 205 mg (72%); mp 207–211 °C dec; [α]20D −181.7 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 7.82 (s, 1H), 7.69 (d, J=7.85 Hz, 1H), 7.47 (d, J=7.85Hz, 1H), 7.39-7.35 (m, 1H), 6.73 (d, J=8.05 Hz, 1H), 6.59 (d, J=8.05 Hz, 1H), 5.82-5.81 (m, 1H), 5.2-5.17 (m, 2H), 4.51-4.5 (d, J=5 Hz, 1H), 4.25 (m, 1H), 3.14-1.28 (m, 14H). 13C NMR (150 MHz, CDCl3) δ 165.7, 142.9, 139.2, 136.1, 135.2, 134.6, 131.5, 130.5, 129.8, 127.5, 125.1, 124.7, 119.3, 118.1, 117.6, 92.7, 70.3, 62.4, 57.8, 50.5, 47.2, 43.6, 31.6, 29.0, 23.2, 22.7 ppm. MS(ESI) m/z (%) 467 (MH+). HRMS calcd for C26H28N2O4Cl(MH+), 467.1738; found, 467.1737. HPLC purity: 97.4%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-bromo)benzamido]morphinan (21)
Compound 21 was synthesized according to the general procedure (II) described above using β-naloxamine, 3-bromobenzoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 218 mg (70%); mp 152–154 °C; [α]20D −193.1 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 7.96 (s, 1H), 7.72 (d, J=8.75 Hz, 1H), 7.61 (d, J=8.75 Hz, 1H), 7.31-7.28 (m, 1H), 7.24-7.22 (m, 1H), 6.72 (d, J=8.75 Hz, 1H), 6.58 (d, J=8.75 Hz, 1H), 5.8 (m, 1H), 5.23-5.16 (m, 2H), 4.52 (d, J=8.75 Hz, 1H), 4.18 (m, 1H), 3.14-1.5 (m, 14H). 13C NMR (150 MHz, CDCl3) δ 165.6, 143.2, 139.4, 136.4, 135.4, 135.2, 134.5, 130.4, 130.1, 125.6, 124.5, 122.7, 119.3, 118.1, 117.8, 92.5, 70.2, 62.4, 57.8, 50.2, 47.1, 43.5, 31.9, 28.7, 23.0, 22.7 ppm. MS (ESI) m/z (%) 511 (MH+). HRMS calcd for C26H28N2O4Br (MH+), 511.1232; found, 511.1250. HPLC purity: 94.4%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-(benzamido)morphinan (22)
Compound 22 was synthesized according to the general procedure (II) described above using β-naloxamine, benzoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 84 mg (32%); mp 177–181 °C; [α]20D −58.4 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 7.82 (d, J=9.2 Hz, 2H), 7.51-7.42 (m, 3H), 7.20 (m, 1H), 6.74 (d, J=9.2Hz, 1H), 6.59 (d, J=9.2 Hz, 1H), 5.82 (m, 1H), 5.23-5.17 (m, 2H), 4.5 (d, J=7.65 Hz, 1H), 4.26 (m, 1H), 3.13-1.25 (m, 14H). 13C NMR (150 MHz, CDCl3) δ 166.9, 143.3, 139.2, 135.2, 134.5, 131.5, 130.7, 128.6, 127.0, 125.0, 119.2, 118.1, 117.5, 93.3, 70.2, 62.5, 57.8, 49.8, 47.2, 43.6, 31.7, 28.9, 23.2, 22.7 ppm. MS (ESI) m/z (%) 433 (MH+). HRMS calcd for C26H29N2O4 (MH+), 433.2127; found, 433.2125. HPLC purity: 90.4%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-methyl)benzamido]morphinan (23)
Compound 23 was synthesized according to the general procedure (II) described above using β-naloxamine, 3-toluic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 133 mg (49%); mp 218–221 °C dec; [α]20D −163.4 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 7.67 (m, 2H), 7.51 (s, 1H), 7.35 (d, J=8.1 Hz, 1H), 6.71 (d, J=8.1Hz, 1H), 6.61 (d, J=8.1Hz, 1H), 5.82 (m, 1H), 5.23-5.17 (m, 2H), 4.55 (d, J=7.05 Hz, 1H), 4.06 (m, 1H), 3.36-1.5 (m, 16H). 13C NMR (150 MHz, CDCl3) δ 167.3, 143.1, 139.3, 138.4, 135.2, 134.4, 132.3, 130.7, 128.4, 127.8, 124.8, 123.9, 119.2, 118.1, 117.6, 93.3, 70.2, 62.5, 57.8, 50.2, 47.3, 43.6, 31.5, 29.1, 23.5, 22.7, 21.4 ppm. MS(ESI) m/z (%) 447 (MH+). HRMS calcd for C27H31N2O4 (MH+), 447.2284; found, 447.2290. HPLC purity: 98.6%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-trifluoromethyl)benzamido]morphinan (24)
Compound 24 was synthesized according to the general procedure (II) described above using β-naloxamine, 3-trifluorotoluic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 210 mg (69%); mp 88–91 °C; [α]20D −126.7 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 8.01 (s, 3H), 8.0 (m, 1H), 7.89-7.88 (m, 1H), 7.65 (m, 1H), 7.45 (m, 1H), 6.62 (d, J=8.15 Hz, 1H), 6.5 (d, J=8.15 Hz, 1H), 5.78-5.74 (m, 1H), 5.2-5.13 (m, 2H), 4.67 (d, J=6.15 Hz, 1H), 4.11-4.02 (m, 1H), 3.54-1.24 (m, 14H). 13C NMR (150 MHz, CDCl3) δ 165.5, 143.1, 139.2, 135.2, 130.5, 130.3, 129.1, 128.1, 124.8, 124.2, 119.3, 118.1, 117.5, 92.8, 70.2, 62.4, 57.8, 50.2, 47.2, 43.5, 31.8, 28.8, 23.0, 22.7 ppm. MS (ESI) m/z (%) 501 (MH+). HRMS calcd for C27H28N2O4F3 (MH+), 501.2001; found, 501.2004. HPLC purity: 96.4%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-methoxy)benzamido]morphinan (25)
Compound 25 was synthesized according to the general procedure (II) described above using β-naloxamine, 3-anisic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 169 mg (60%); mp 113–115 °C; [α]20D −124.4 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 7.56 (d, J=9 Hz, 1H), 7.39-7.26 (m, 3H), 7.0 (m, 1H), 6.72 (d, J=8.1 Hz, 1H), 6.55 (d, J=8.1 Hz, 1H), 5.78-5.74 (m, 1H), 5.24-5.17 (m, 2H), 4.52 (d, J=6.2 Hz, 1H), 4.12-4.11 (m, 1H), 3.78 (s, 3H), 3.72-1.25 (m, 14H). 13C NMR (150 MHz, CDCl3) δ 166.9, 159.8, 143.1, 139.3, 135.9, 134.9, 130.6, 129.5, 124.6, 119.2, 118.8, 117.8, 117.7, 112.4, 93.0, 70.2, 62.5, 57.8, 55.4, 50.2, 47.2, 43.7, 31.5, 29.0, 23.3, 22.7 ppm. MS (ESI) m/z (%) 463 (MH+). HRMS calcd for C27H31N2O5 (MH+), 463.2233; found, 463.2232. HPLC purity: 97.0%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-amino)benzamido]morphinan (26)
Compound 26 was synthesized according to the general procedure (II) described above using β-naloxamine, 3-amino benzoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 82 mg (30%); mp 191–193 °C dec; [α]20D −148.7 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 7.2-7.16 (m, 1H), 7.1 (d, J=7.95 Hz, 1H), 6.90 (d, J=7.95 Hz, 1H), 6.8 (d, J=7.95 Hz, 1H), 6.75 (d, J=8.1 Hz, 1H), 6.69 (d, J=8.1 Hz, 1H), 5.81-5.8 (m, 1H), 5.19-5.16 (m, 2H), 4.46 (d, J=5.85 Hz, 1H), 4.21-4.19 (m, 1H), 3.48-1.22 (m, 16H). 13C NMR (150 MHz, CDCl3) δ 167.6, 146.6, 143.0, 139.6, 135.5, 135.3, 130.7, 129.3, 124.4, 119.1, 118.3, 118.0, 117.9,116.8, 114.5, 92.8, 70.4, 62.5, 57.7, 50.7, 47.3, 43.6, 31.2, 29.2, 23.7, 22.7 ppm. MS (ESI) m/z (%) 448 (MH+). HRMS calcd for C26H30N3O4 (MH+), 448.2236; found, 448.2230. HPLC purity: 95.4%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-dimethylamino)benzamido]morphinan (27)
Compound 27 was synthesized according to the general procedure (II) described above using β-naloxamine, 3-dimethylamino benzoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 174 mg (60%); mp 170–173 °C; [α]20D −127.3 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 7.21 (s, 1H), 7.02 (d, J=10 Hz, 1H), 6.89-6.85 (m, 1H), 6.75 (d, J=10 Hz, 1H), 6.59 (m, 1H), 5.79 (m, 1H), 5.22-5.16 (m, 2H), 4.48 (d, J=10 Hz, 1H), 4.22 (m, 1H), 3.14-1.5 (m, 20H). 13C NMR (150 MHz, CDCl3) δ 167.8, 150.7, 143.2, 139.4, 135.3, 130.7, 129.1, 119.1, 118.0, 117.6, 115.3, 114.2, 111.6, 93.1, 70.2, 62.5, 57.8, 53.4, 50.2,47.2, 43.6, 40.5, 31.5, 29.1, 23.4, 22.7 ppm. MS (ESI) m/z (%) 476 (MH+). HRMS calcd for C28H34N3O4 (MH+), 476.2549; found, 476.2544. HPLC purity: 96.5%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-hydroxy)benzamido]morphinan (28)
Compound 28 was synthesized according to the general procedure (II) described above using β-naloxamine, 3-hydroxy benzoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 107 mg (39%); mp 201–204 °C dec; [α]20D −170.7 (c 0.1, CHCl3); 1HNMR (500 MHz, CDCl3) δ: 7.44 (m, 3H), 7.3-7.28 (m, 2H), 6.99 (d, J=7.75 Hz, 1H), 6.71 (d, J=7.75 Hz, 1H), 6.6 (d, J=7.75 Hz, 1H), 5.82-5.8 (m, 1H), 5.22-5.17 (m, 2H), 4.51 (d, J=7.75 Hz, 1H), 4.062 (m, 1H), 3.51-1.51 (m, 14H). 13C NMR (150 MHz, CDCl3) δ 168.0, 156.7, 142.7, 139.8, 135.1, 134.8, 130.6, 129.7, 124.1, 119.5, 119.2, 118.2, 118.1, 115.1, 93.3, 70.6, 62.4, 57.7, 51.3, 47.4, 43.7, 30.9, 29.3, 24.2, 22.7 ppm. MS (ESI) m/z (%) 449 (MH+). HRMS calcd for C26H29N2O5 (MH+), 449.2076; found, 449.2080. HPLC purity: 95.5%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(3′-nitro)benzamido]morphinan (29)
Compound 29 was synthesized according to the general procedure (II) described above using β-naloxamine, 3-nitro benzoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 172 mg (59%); mp 228–231 °C dec; [α]20D −128.8 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 8.68 (s, 1H), 8.36-8.34 (m, 1H), 8.22 (d, J=11.8 Hz, 1H), 7.67-7.63 (m, 2H), 6.69 (d, J=11.8 Hz, 1H), 6.58 (d, J=11.8 Hz, 1H), 5.81 (m, 1H), 5.2-5.17 (m, 2H), 4.59 (d, J=9.8 Hz, 1H), 4.27 (m, 1H), 3.14-1.25 (m, 14H). 13C NMR (150 MHz, CDCl3) δ 164.6, 148.1, 141.6, 139.4, 135.5, 135.2, 133.7, 130.6, 129.4, 125.8, 124.6, 121.9, 119.4, 118.1, 117.3, 92.1, 70.6, 62.4, 57.9, 51.9, 47.4, 43.4, 38.6, 31.0, 29.5, 23.7,22.7 ppm. MS(ESI) m/z (%) 478 (MH+). HRMS calcd for C26H28N3O6 (MH+), 479.1978; found, 478.1967. HPLC purity: 97.0%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4′-trifluoromethoxy)benzamido]morphinan (30)
Compound 30 was synthesized according to the general procedure (II) described above using β-naloxamine, 4-(trifluoromethoxy)benzoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 248 mg (79%); mp 138–140 °C; [α]20D −136.0 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 7.87 (d, J=11.75 Hz, 1H), 7.44 (d, J=11.75 Hz, 1H), 7.24 (m, 2H), 6.72 (d, J=11.75 Hz, 1H), 6.58 (d, J=11.75 Hz, 1H), 5.81 (m, 1H), 5.23-5.16 (m, 2H), 4.53 (d, J=9.8 Hz, 1H), 4.24 (m, 1H), 3.33-1.28 (m, 14H). 13C NMR (150 MHz, CDCl3) δ 165.7, 151.4, 143.1, 139.1, 135.1, 132.8, 130.5, 129.0, 124.7, 120.5, 119.3, 118.2, 117.6, 92.6, 70.3, 62.4, 57.8, 50.2, 47.1, 43.5, 31.8, 28.8, 23.0, 22.6 ppm. MS (ESI) m/z (%) 517 (MH+). HRMS calcd for C27H28N2O5F3 (MH+), 517.1950; found, 517.1956. HPLC purity: 99.9%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(2′-naphthyl)acetamido]morphinan (31)
Compound 31 was synthesized according to the general procedure (I) described above using β-naloxamine, NHS ester of naphthalene-2-carboxylic acid 9, and DIEA in DCM. A white solid was obtained. Yield: 261 mg (89%); mp 210–214 °C dec; [α]20D −260.2 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 8.17 (s, 1H), 7.78-7.70 (m, 4H), 7.47 (t, J= 7.5 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H), 6.91 (d, J = 8.8 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.51 (d, J = 8.1 Hz, 1H), 5.79 (m, 1H), 5.16 (m, 2H), 4.80 (m, 1H), 4.73 (d, J = 4.3 Hz, 1H), 3.10-1.05 (m, 15 H) ppm. 13C NMR (150 MHz, CDCl3) δ 167.3, 145.3, 137.4, 134.6, 132.5, 131.7, 128.9, 128.1, 127.6, 127.5, 127.4, 126.4, 123.9, 119.2, 117.4, 93.5, 90.2, 70.2, 69.7, 62.4, 58.1, 47.1, 46.6, 42.9, 33.3, 29.0, 22.9, 21.0 ppm. MS (ESI) m/z (%) 483 (MH+). HRMS calcd for C30H31N2O4 (MH+), 483.2284; found, 483.2293. HPLC purity: 97.3%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4′-phenyl)benzamido]morphinan (32)
Compound 32 was synthesized according to the general procedure (I) described above using β-naloxamine, NHS ester of biphenyl-4-carboxylic acid 10, and DIEA in DCM. A white solid was obtained. Yield: 263 mg (85%); mp 210–214 °C dec; [α]20D −145.8 (c 0.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 7.89 (d, J=8.15 Hz, 2H), 7.66-7.61 (m, 4H), 7.46 (m, 3H), 7.38 (m, 1H), 6.74 (d, J=8.15 Hz, 1H), 6.61 (d, J=8.15 Hz, 1H), 5.82-5.79 (m, 1H), 5.23-5.17 (m, 2H), 4.53-4.52 (d, J=5.15 Hz, 1H), 4.31-4.29 (m, 1H), 3.15-1.25 (m, 14H). 13C NMR (150 MHz, CDCl3) δ 166.7, 144.2, 143.2, 140.0, 139.2, 135.2, 133.1, 130.6, 128.9, 128.0, 127.6, 127.2, 124.8, 119.2,118.1,117.6, 92.9, 70.2, 62.5, 57.8, 50.1, 47.2, 43.6, 31.7, 28.9, 23.2, 22.7 ppm. ESI-MS m/z: 509.09 (MH+). HPLC purity: 95.9%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-(acetylamido)morphinan (33)
Compound 33 was synthesized according to the general procedure (II) described above using β-naloxamine, acetic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 120 mg (53%); mp 204–207 °C dec; [α]20D −157.0 (c 0.1, CHCl3); 1H NMR (500 MHz, CDCl3): δ 6.70 (d, J = 8.2 Hz, 1H,), 6.56 (d, J = 8.2 Hz, 1H), 5.96 (d, J = 9.2 Hz, 1H), 5.76 (m, 1H,), 5.18 (d, J = 17.8 Hz, 1H,), 5.14 (d, J = 10.5 Hz, 1H,), 4.33 (d, J = 6.5 Hz, 1H), 3.89 (m, 1H), 3.15-0.80 (m, 18 H) ppm. 13C NMR (150 MHz, CDCl3) δ 170.6, 142.8, 139.8, 135.2, 130.8, 124.3, 119.1, 118.0, 117.8, 93.0, 70.4, 62.4, 57.7, 50.7, 47.4, 43.7, 30.9, 29.5, 23.9, 23.6, 22.7 ppm. MS(ESI) m/z (%) 371 (MH+). HRMS calcd for C21H27N2O4 (MH+), 371.1971; found, 371.1965. HPLC purity: 97.0%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-(hexylamido)morphinan (34)
Compound 34 was synthesized according to the general procedure (II) described above using β-naloxamine, hexanoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 130 mg (50%); mp 113–116 °C; [α]20D −101.6 (c 0.1, CHCl3); 1H NMR (500 MHz, CDCl3): δ 6.71 (d, J = 8.2 Hz, 1H,), 6.55 (d, J = 8.2 Hz, 1H), 6.07 (d, J = 9.2 Hz, 1H), 5.77 (m, 1H), 5.18 (d, J = 17.4 Hz, 1H), 5.14 (d, J = 10.1 Hz, 1H,), 4.34 (d, J = 6.4 Hz, 1H), 3.91 (m, 1H), 3.15-0.80 (m, 26 H) ppm. 13C NMR (150 MHz, CDCl3) δ 173.0, 142.9, 139.6, 135.3, 130.8, 124.7, 119.2, 118.0, 117.6, 93.7, 70.1, 62.5, 57.7, 50.1, 47.4, 43.6, 37.1, 31.4, 31.1, 29.5, 25.4, 23.9, 22.7, 22.4, 14.0 ppm. MS(ESI) m/z (%) 427 (MH+). HRMS calcd for C26H35N2O4 (MH+), 427.2597; found, 427.2591. HPLC purity: 96.0%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-(dodecylamido)morphinan (35)
Compound 35 was synthesized according to the general procedure (II) described above using β-naloxamine, dodecanoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 202 mg (65%); mp 95–98 °C; [α] 20D −99.9 (c 0.1, CHCl3); 1H NMR (500 MHz, CDCl3): δ 6.71 (d, J = 8.2 Hz, 1H), 6.55 (d, J = 8.2 Hz, 1H,), 6.07 (d, J = 9.2 Hz, 1H,), 5.76 (m, 1H), 5.18 (d, J = 17.4 Hz, 1H), 5.14 (d, J = 10.1 Hz, 1H), 4.34 (d, J = 6.4 Hz, 1H), 3.91 (m, 1H), 3.10-0.86 (m, 38 H) ppm. 13C NMR (150 MHz, CDCl3) δ 173.1, 142.9, 139.7, 130.7, 119.2, 117.7, 93.7, 70.1, 62.5, 57.7, 53.4, 50.1, 47.4, 43.7, 37.1, 31.9, 29.6, 29.5, 29.3, 29.2, 25.7, 23.9, 22.7, 14.1 ppm. MS(ESI) m/z (%) 511 (MH+). HRMS calcd for C31H47N2O4 (MH+), 511.3536; found, 511.3550. HPLC purity: 95.3%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-(cyclohexylamido)morphinan (36)
Compound 36 was synthesized according to the general procedure (II) described above using β-naloxamine, cyclohexanoic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 174 mg (65%); mp 236–239 °C dec; [α] 20D −135.2 (c 0.1, CHCl3); 1H NMR (500 MHz, CDCl3): δ 6.71 (d, J = 8.1 Hz, 1H), 6.55 (d, J = 8.0 Hz, 1H), 6.14 (d, J = 9.1 Hz, 1H), 5.77 (m, 1H), 5.18 (d, J = 17.4 Hz, 1H), 5.14 (d, J = 10.0 Hz, 1H), 4.33 (d, J = 6.1 Hz, 1H), 3.93 (m, 1H), 3.15-0.80 (m, 26 H) ppm. 13C NMR (150 MHz, CDCl3) δ 176.0, 143.1, 139.5, 135.3, 130.8, 124.7, 119.1, 118.0, 117.6, 93.7, 70.1, 62.5, 57.7, 49.7, 47.3, 45.7, 43.6, 31.3, 29.7, 29.6, 29.3, 25.8, 25.7, 23.6, 22.7ppm. MS (ESI) m/z (%) 439 (MH+). HRMS calcd for C26H35N2O4 (MH+), 439.2597; found, 439.2602. HPLC purity: 93.2%.
17-Allyl-3,14β-dihydroxy-4,5α-epoxy-6β-[2-adamantylamido]morphinan (37)
Compound 37 was synthesized according to the general procedure (II) described above using β-naloxamine, 1-Adamantyl carboxylic acid, BOP and DIEA in DCM followed by base hydrolysis. A white solid was obtained. Yield: 227 mg (76%); mp 153–156 °C; [α]20D −129.0 (c 0.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 6.71 (d, J = 8.2 Hz, 1H,), 6.55 (d, J = 8.2 Hz, 1H), 6.22 (d, J = 9.5 Hz, 1H), 5.77 (m, 1H), 5.28 (s, 1H), 5.18 (d, J = 17.2 Hz, 1H), 5.14 (d, J = 10.2 Hz, 1H), 4.31 (d, J= 5.9 Hz, 1H), 3.97 (m, 1H), 3.15-0.76 (m, 29 H) ppm. 13C NMR (150 MHz, CDCl3) δ 178.1, 162.6, 143.2, 139.6, 135.3, 130.7, 124.4,119.0,118.0, 117.8, 93.0, 70.2, 62.4, 57.8, 49.6, 47.1, 43.6, 40.7, 39.1, 38.6, 36.5, 31.7, 31.5, 28.9, 28.1, 23.2, 22.6 ppm. MS (ESI) m/z (%) 491 (MH+). HRMS calcd for C30H39N2O4 (MH+), 491.2910; found, 491.2912. HPLC purity: 91.9%.
17-Allyl-4,5α-epoxy-14β-hydroxy-3β-methoxy-6β-[(3′-iodo)benzamido]morphinan (38)
Compound 38 was synthesized according to the general procedure (I) described above using 3-OMe-β-naloxamine, NHS ester of 3-iodobenzoic acid 10 and DIEA in DCM. A white solid was obtained. Yield: 296 mg (85%); mp 125–127 °C; [α]20D −133.7 (c 0.1, CHCl3); 1H-NMR δ: 8.19 (s, 1H), 7.8 (m, 1H), 7.42 (m, 1H), 7.16 (m, 1H), 6.75 (d, J=10 Hz, 1H), 6.66 (d, J=10 Hz, 1H), 5.85 (m, 1H), 5.18 (m, 2H), 4.61 (d, 1H), 4.08 (m, 1H), 3.85 (s, 2H), 3.15-0.1(m, 14H). 13C NMR (150 MHz, CDCl3) δ 165.2, 144.9, 144.3, 136.4, 130.8, 130.1, 127.3, 126.3, 124.4, 120.9, 119.4, 115.8, 94.2, 91.9, 72.7, 71.8, 71.7, 60.8, 57.0, 56.9, 50.0, 47.1, 30.9, 28.1, 26.7, 22.6 ppm. MS (ESI) m/z (%) 573 (MH+). HRMS calcd for C27H30N2O4I (MH+), 573.1250; found, 573.1252. HPLC purity: 96.6%.
Receptor-Binding Assays
Competition-binding assays in CHO cells stably expressing MOR-1 (mu), DOR-1 (delta) or KOR-1 (kappa) were performed at 25°C in potassium phosphate buffer (50 mM; pH 7.4), with the inclusion of MgSO4 (5 mM) in the MOR-1 assays. All competition assays were carried out using 125I-BNtxA (1) as described.39 Specific binding was defined as the difference between total binding and nonspecific binding, determined in the presence of levallorphan (8 μM). Protein concentrations were between 30–40 μg/mL and incubation times were 90 minutes. 6TM/E11 competition binding assays using 125I-BNtxA (1; 0.15 nM) were carried out in whole brain membrane homogenates (0.5 ml; 0.5 mg protein) at 25°C in potassium phosphate buffer (50 mM; pH 7.4) with magnesium sulfate (5 mM) for 90 minutes in the presence of CTAP, U50488H and DPDPE, all at 200nM, to block traditional opioid binding sites. Specific binding was defined as the difference between total binding and nonspecific binding, determined in the presence of levallorphan (8 μM). Protein concentration was determined as described by Lowry et al., 40 using bovine serum albumin as the standard.
Tail Flick Analgesia Assays
Male CD-1 mice (25–35 g; Charles River Breeding Laboratories, Wilmington, MA) were maintained on a 12-hr light/dark cycle with Purina rodent chow and water available ad libitum. Mice were housed in groups of five until testing. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of Memorial Sloan-Kettering Cancer Center. Analgesia was determined using the radiant heat tail-flick technique41 using a machine from (Ugo Basile; model number 37360). The intensity was set to achieve a baseline between 2–3 sec. The latency to withdraw the tail from a focused light stimulus was measured electronically using a photocell. Baseline latencies (2.0–3.0 sec) were determined before experimental treatments for all animals as the mean of two trials. Post-treatment tail-flick latencies were determined as indicated for each experiment, and a maximal latency of 10 sec for tail-flick was used to minimize tissue damage. Drugs were given subcutaneously and cumulative dose-response experiments carried out with two independent assays with each group (n=10). The combined results presented as the ED50 with 95% confidence limits (n=20) presented. Analgesia was defined quantally as a doubling, or greater, of the baseline latency. Similar results were obtained analyzing the data in a graded response manner. Analgesic ED50’s and confidence limits were determined using non-linear regression analysis Graph Pad Prism (Graphpad Software, La Jolla, CA).
Respiratory Depression assay
Respiratory rate was assessed in awake, freely-moving, adult male mice using the MouseOx Pulse Oximeter system (Starr Life Sciences, Pittsburgh, PA). Each animal was habituated to the device for 30 min and then tested. A five-second average breath rate was assessed at 5 minute intervals. A baseline for each animal was obtained over a 25 minute period prior to drug injection and testing began 15 minutes post-injection and continued for a period of 35 minutes. Groups of mice (n = 5) were treated subcutaneously with either morphine (20 mg/kg s.c.) or 15 (2.5 mg/kg s.c.) at doses approximately four times their analgesic ED50. Groups were compared with repeated measures ANOVA followed by Bonferroni as a post test.
Gastrointestinal motility assay
Gastrointestinal transit was determined as described.5 Animals received the indicated drug subcutaneously followed by a charcoal meal by gavage, were sacrificed 30 min later and the distance traveled measured.
Physical dependence studies
Tolerance was induced by twice daily injections for at least five days with either morphine (10 mg/kg s.c.) or 15 (1 mg/kg s.c.). Dependence was determined on Day 5 with naloxone or levallorphan (1 mg/kg, s.c.) to precipitate withdrawal and animals evaluated for signs of diarrhea and jumping.6, 42
Supplementary Material
Acknowledgments
This work was supported, in part, by research grants from the National Institute on Drug Abuse (DA06241, DA02165, and DA07242) to GWP, a Core Grant from the National Cancer Institute to MSKCC (CA08748) as well as the Technology Development Fund of Memorial Sloan-Kettering Cancer Center.
ABBREVIATIONS USED
- 6TM/E11
six transmembrane/exon 11
- MOR-1
mu opioid receptor
- KOR-1
kappa1 opioid receptor
- DOR-1
delta opioid receptor
- IBNtxA
3′-iodobenzoylnaltrexamide
- IBNalA
3′-iodobenzoylnaloxamide
- CPM
cyclopropylmethyl
- s.c
subcutaneous
- DIEA
diisopropyl ethyl amine
- K2CO3
potassium carbonate
- NHS
N-hydroxysuccinimide
- NH4OAc
ammonium acetate
- NaBH3CN
sodium cyanoborohydride
- ACN
acetonitrile
- NaOH
sodium hydroxide
- N2
nitrogen gas
- MeOH
methanol
- BOP
benzotriazol-1yl-oxy-tris(dimethylamino)phosphonium hexafluorophosphate
- CHO
chinese hamster ovary cells
- CPM
cyclopropylmethyl
- DPDPE
[DPen2,D-Pen5]Enkephalin
Footnotes
Supplementary data associated with this article i.e. HPLC’s of compounds 15–38. This material is available free of charge via the internet at http://pubs.acs.org
References
- 1.Serturner FWA. Trommsdorf’s J der Pharmazie. 1805;13:234. [Google Scholar]
- 2.Ling GSF, Spiegel K, Nishimura S, Pasternak GW. Dissociation of morphine’s analgesic and respiratory depressant actions. Eur J Pharmacol. 1983;86:487–488. doi: 10.1016/0014-2999(83)90203-0. [DOI] [PubMed] [Google Scholar]
- 3.Ling GSF, Spiegel K, Lockhart SH, Pasternak GW. Separation of opioid analgesia from respiratory depression: evidence for different receptor mechanisms. J Pharmacol Exp Ther. 1985;232:149–155. [PubMed] [Google Scholar]
- 4.Heyman JS, Koslo RJ, Mosberg HI, Tallarida RJ, Porreca F. Estimation of the affinity of Naloxone at supraspinal and spinal opioid receptors in vivo: Studies with receptor selective agonists. Life Sci. 1986;39:1795–1803. doi: 10.1016/0024-3205(86)90099-8. [DOI] [PubMed] [Google Scholar]
- 5.Paul D, Pasternak GW. Differential blockade by naloxonazine of two μ opiate actions: analgesia and inhibition of gastrointestinal transit. Eur J Pharmacol. 1988;149:403–404. doi: 10.1016/0014-2999(88)90680-2. [DOI] [PubMed] [Google Scholar]
- 6.Ling GSF, MacLeod JM, Lee S, Lockhart SH, Pasternak GW. Separation of morphine analgesia from physical dependence. Science. 1984;226:462–464. doi: 10.1126/science.6541807. [DOI] [PubMed] [Google Scholar]
- 7.Payne R, Pasternak, Pain GW. In: Principles of drug therapy in neurology. Johnston MV, Macdonald RL, Young AB, editors. F. A. Davis; Philadelphia: 1992. pp. 268–301. [Google Scholar]
- 8.Chou R, Fanciullo GJ, Fine PG, Adler JA, Ballantyne JC, Davies P, Donovan MI, Fishbain DA, Foley KM, Fudin J, Gilson AM, Kelter A, Mauskop A, O’Connor PG, Passik SD, Pasternak GW, Portenoy RK, Rich BA, Roberts RG, Todd KH, Miaskowski C. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain. 2009;10:113–130. doi: 10.1016/j.jpain.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foley KM. Pain syndromes in patients with cancer. In: Portenoy RK, Kanner RM, editors. Pain management: theory and practice. F. A. Davis Company; Philadelphia: 1996. pp. 191–215. [Google Scholar]
- 10.Wolozin BL, Pasternak GW. Classification of multiple morphine and enkephalin binding sites in the central nervous system. Proc Natl Acad Sci USA. 1981;78:6181–6185. doi: 10.1073/pnas.78.10.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bare LA, Mansson E, Yang D. Expression of two variants of the human μ opioid receptor mRNA in SK-N-SH cells and human brain. FEBS Lett. 1994;354:213–216. doi: 10.1016/0014-5793(94)01129-x. [DOI] [PubMed] [Google Scholar]
- 12.Zimprich A, Bacher B, Hollt V. Cloning and expression of an isoform of the rmu-opioid receptor (rmuOR1B) Regul Pept. 1994;54:347–348. [Google Scholar]
- 13.Pan YX, Xu J, Rossi GC, Leventhal L, Wan BL, Zuckerman A, Pasternak GW. Cloning and expression of a novel splice variant of the mouse mu-opioid receptor (MOR-1) gene. Soc Neurosci. 1998;24:524. [Google Scholar]
- 14.Pan YX, Xu J, Bolan EA, Abbadie C, Chang A, Zuckerman A, Rossi GC, Pasternak GW. Identification and characterization of three new alternatively spliced mu opioid receptor isoforms. Mol Pharmacol. 1999;56:396–403. doi: 10.1124/mol.56.2.396. [DOI] [PubMed] [Google Scholar]
- 15.Pan YX, Xu J, Bolan E, Chang A, Mahurter L, Rossi G, Pasternak GW. Isolation and expression of a novel alternatively spliced mu opioid receptor isoform, MOR-1F. FEBS Lett. 2000;466:337–340. doi: 10.1016/s0014-5793(00)01095-4. [DOI] [PubMed] [Google Scholar]
- 16.Pan YX, Xu J, Mahurter L, Bolan EA, Xu MM, Pasternak GW. Generation of the mu opioid receptor (MOR-1) protein by three new splice variants of the Oprm gene. Proc Natl Acad Sci U S A. 2001;98:14084–14089. doi: 10.1073/pnas.241296098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan YX, Xu J, Mahurter L, Xu M, Gilbert AK, Pasternak GW. Identification and characterization of two new human mu opioid receptor splice variants, hMOR-1O and hMOR-1X. Biochem Biophys Res Commun. 2003;301:1057–1061. doi: 10.1016/s0006-291x(03)00089-5. [DOI] [PubMed] [Google Scholar]
- 18.Pasternak DA, Pan L, Xu J, Yu R, Xu MM, Pasternak GW, Pan YX. Identification of three new alternatively spliced variants of the rat mu opioid receptor gene: dissociation of affinity and efficacy. J Neurochem. 2004;91:881–890. doi: 10.1111/j.1471-4159.2004.02767.x. [DOI] [PubMed] [Google Scholar]
- 19.Pan L, Xu J, Yu R, Xu MM, Pan YX, Pasternak GW. Identification and characterization of six new alternatively spliced variants of the human mu opioid receptor gene, Oprm. Neuroscience. 2005;133:209–220. doi: 10.1016/j.neuroscience.2004.12.033. [DOI] [PubMed] [Google Scholar]
- 20.Pan YX, Xu J, Bolan E, Moskowitz HS, Xu M, Pasternak GW. Identification of four novel exon 5 splice variants of the mouse mu-opioid receptor gene: functional consequences of C-terminal splicing. Mol Pharmacol. 2005;68:866–875. doi: 10.1124/mol.105.011858. [DOI] [PubMed] [Google Scholar]
- 21.Xu J, Xu M, Rossi GC, Pasternak GW, Pan YX. Identification and characterization of seven new exon 11-associated splice variants of the rat mu opioid receptor gene, OPRM1. Mol Pain. 2011;7:9. doi: 10.1186/1744-8069-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu J, Xu M, Hurd YL, Pasternak GW, Pan YX. Isolation and characterization of new exon 11-associated N-terminal splice variants of the human mu opioid receptor gene. J Neurochem. 2009;108:962–972. doi: 10.1111/j.1471-4159.2008.05833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gris P, Gauthier J, Cheng P, Gibson DG, Gris D, Laur O, Pierson J, Wentworth S, Nackley AG, Maixner W, Diatchenko L. A Novel alternatively spliced isoform of the mu-opioid receptor: functional antagonism. Mol Pain. 2010;6:33. doi: 10.1186/1744-8069-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cadet P, Mantione KJ, Stefano GB. Molecular identification and functional expression of mu 3, a novel alternatively spliced variant of the human mu opiate receptor gene. J Immunol. 2003;170:5118–5123. doi: 10.4049/jimmunol.170.10.5118. [DOI] [PubMed] [Google Scholar]
- 25.Majumdar S, Burgman M, Haselton N, Grinnell S, Ocampo J, Pasternak AR, Pasternak GW. Generation of novel radiolabeled opiates through site-selective iodination. Bioorg Med Chem Lett. 2011;21:4001–4004. doi: 10.1016/j.bmcl.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Majumdar S, Grinnell S, Le RV, Burgman M, Polikar L, Ansonoff M, Pintar J, Pan YX, Pasternak GW. Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proc Natl Acad Sci U S A. 2011;108:19776–19783. doi: 10.1073/pnas.1115231108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang JB, Hanson RN, Portoghese PS. Stereochemical studies on medicinal agents. 23.1 Synthesis and biological evaluation of 6-amino derivatives of naloxone and naltrexone. J Med Chem. 1977;20:1100–1102. doi: 10.1021/jm00218a023. [DOI] [PubMed] [Google Scholar]
- 28.Iltzsch MH, Uber SS, Tankersley KO, el Kouni MH. Structure-activity relationship for the binding of nucleoside ligands to adenosine kinase from Toxoplasma gondii. Biochem Pharmacol. 1995;49:1501–1512. doi: 10.1016/0006-2952(95)00029-y. [DOI] [PubMed] [Google Scholar]
- 29.Himmel DM, Das K, Clark AD, Jr, Hughes SH, Benjahad A, Oumouch S, Guillemont J, Coupa S, Poncelet A, Csoka I, Meyer C, Andries K, Nguyen CH, Grierson DS, Arnold E. Crystal structures for HIV-1 reverse transcriptase in complexes with three pyridinone derivatives: a new class of non-nucleoside inhibitors effective against a broad range of drug-resistant strains. J Med Chem. 2005;48:7582–7591. doi: 10.1021/jm0500323. [DOI] [PubMed] [Google Scholar]
- 30.Benjahad A, Guillemont J, Andries K, Nguyen CH, Grierson DS. 3-iodo-4-phenoxypyridinones (IOPY’s), a new family of highly potent non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg Med Chem Lett. 2003;13:4309–4312. doi: 10.1016/j.bmcl.2003.09.045. [DOI] [PubMed] [Google Scholar]
- 31.Xu Z, Liu Z, Chen T, Chen T, Wang Z, Tian G, Shi J, Wang X, Lu Y, Yan X, Wang G, Jiang H, Chen K, Wang S, Xu Y, Shen J, Zhu W. Utilization of halogen bond in lead optimization: a case study of rational design of potent phosphodiesterase type 5 (PDE5) inhibitors. J Med Chem. 2011;54:5607–5611. doi: 10.1021/jm200644r. [DOI] [PubMed] [Google Scholar]
- 32.Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghirmai S, Azar MR, Polgar WE, Berzetei-Gurske I, Cashman JR. Synthesis and biological evaluation of alpha- and beta-6-amido derivatives of 17-cyclopropylmethyl-3, 14beta-dihydroxy-4, 5alpha-epoxymorphinan: potential alcohol-cessation agents. J Med Chem. 2008;51:1913–1924. doi: 10.1021/jm701060e. [DOI] [PubMed] [Google Scholar]
- 34.Ghirmai S, Azar MR, Cashman JR. Synthesis and pharmacological evaluation of 6-naltrexamine analogs for alcohol cessation. Bioorg Med Chem. 2009;17:6671–6681. doi: 10.1016/j.bmc.2009.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li G, Aschenbach LC, Chen J, Cassidy MP, Stevens DL, Gabra BH, Selley DE, Dewey WL, Westkaemper RB, Zhang Y. Design, synthesis, and biological evaluation of 6alpha- and 6beta-N-heterocyclic substituted naltrexamine derivatives as mu opioid receptor selective antagonists. J Med Chem. 2009;52:1416–1427. doi: 10.1021/jm801272c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan Y, Elbegdorj O, Chen J, Akubathini SK, Beletskaya IO, Selley DE, Zhang Y. Structure selectivity relationship studies of 17-cyclopropylmethyl-3,14beta-dihydroxy-4,5alpha-epoxy-6beta-[(4′-pyridyl) carboxamido]morphinan derivatives toward the development of the mu opioid receptor antagonists. Bioorg Med Chem Lett. 2011;21:5625–5629. doi: 10.1016/j.bmcl.2011.06.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacDougall JM, Zhang XD, Polgar WE, Khroyan TV, Toll L, Cashman JR. Synthesis and biological evaluation of some 6-arylamidomorphines as analogues of morphine-6-glucuronide. Bioorg Med Chem. 2004;12:5983–5990. doi: 10.1016/j.bmc.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 38.Yekkirala AS, Lunzer MM, McCurdy CR, Powers MD, Kalyuzhny AE, Roerig SC, Portoghese PS. N-naphthoyl-beta-naltrexamine (NNTA), a highly selective and potent activator of mu/kappa-opioid heteromers. Proc Natl Acad Sci U S A. 2011;108:5098–5103. doi: 10.1073/pnas.1016277108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Majumdar S, Burgman M, Haselton N, Grinnell S, Ocampo J, Pasternak AR, Pasternak GW. Generation of novel radiolabeled opiates through site-selective iodination. Bioorg Med Chem Lett. 2011;21:4001–4004. doi: 10.1016/j.bmcl.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 41.D’Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- 42.Gistrak MA, Paul D, Hahn EF, Pasternak GW. Pharmacological actions of a novel mixed opiate agonist/antagonist: naloxone benzoylhydrazone. J Pharmacol Exp Ther. 1989;251:469–476. [PubMed] [Google Scholar]
- 43.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibiton (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.