

Abstract
The amyloid cascade hypothesis of Alzheimer's disease (AD) postulates that accumulation in the brain of amyloid β-peptide (Aβ) is the primary trigger for neuronal loss specific to this pathology. In healthy brain, Aβ levels are regulated by a dynamic equilibrium between Aβ release from the amyloid precursor protein (APP) and its removal by perivascular drainage or by amyloid-degrading enzymes (ADEs). During the last decade, the ADE family was fast growing, and currently it embraces more than 20 members. There are solid data supporting involvement of each of them in Aβ clearance but a zinc metallopeptidase neprilysin (NEP) is considered as a major ADE. NEP plays an important role in brain function due to its role in terminating neuropeptide signalling and its decrease during ageing or after such pathologies as hypoxia or ischemia contribute significantly to the development of AD pathology. The recently discovered mechanism of epigenetic regulation of NEP by the APP intracellular domain (AICD) opens new avenues for its therapeutic manipulation and raises hope for developing preventive strategies in AD. However, consideration needs to be given to the diverse physiological roles of NEP. This paper critically evaluates general biochemical and physiological functions of NEP and their therapeutic relevance.
1. Introduction
The amyloid cascade hypothesis of Alzheimer's disease (AD) was originally proposed 20 years ago [1, 2], and during this period it has significantly influenced development of AD-related research. Although it provided a huge amount of data confirming that the accumulation of the amyloid β-peptide (Aβ), especially Aβ 1–42, is directly linked to the development of neurodegeneration, it also to some extent detracted attention from understanding the normal physiological role both of Aβ and its precursor protein, APP. Recently, several attempts have been made to reevaluate the amyloid hypothesis and to suggest new directions in AD research [3–5]. Although our knowledge of the processes involved in Aβ production is rather extensive this has not resulted in any viable therapy despite several promising trials of inhibitors preventing Aβ formation [6]. Moreover, during the last two decades, Aβ toxicity was studied and reexamined in various animal and cellular models suggesting that the toxic Aβ species might be represented by oligomers rather than monomers, fibrils, or plaques [7, 8], and much research has been devoted to the search for pharmacological approaches to prevent Aβ oligomerization as a therapy in AD [9].
One of the important concepts developed from the amyloid cascade hypothesis is the realisation that amyloid metabolism is a dynamic process represented by production of Aβ (by β- and γ-secretases) and its removal from the brain (via perivascular or enzymatic mechanisms) rather than an irreversible pathway of its accumulation leading to cell death and cognitive impairment. As such the enzymes capable of degrading Aβ became a major research and therapeutic target [10–12]. Evaluation of the normal physiological role of Aβ suggests that complete elimination of Aβ from the brain would not be a target in AD therapy since it most likely has a normal physiological role as a regulatory peptide or even as a transcription factor [13–16]. However, by manipulating its levels through improved perivascular drainage or proteolytic degradation might help to prevent accumulation of harmful amyloid species causing cell death and AD pathology [12, 17]. One of the amyloid-degrading enzymes, neprilysin (NEP), has been the main target of our research over many years, and in this paper we will summarize current knowledge of this metallopeptidase and mechanisms to manipulate its activity in disease states.
2. General Properties of NEP
Neutral endopeptidase, or neprilysin (NEP), was first described as a neutral proteinase in rat kidney brush border membranes and then purified from rabbit kidney and characterised as a zinc metallopeptidase [18]. Although NEP is abundant in the kidney (about 4% of all membrane proteins), its content in other organs, including the brain, is much lower. NEP was later rediscovered as a brain enzyme responsible for inactivation of the enkephalin family of neuropeptides and given the name enkephalinase [19]. However, it was subsequently shown that NEP is not enkephalin-specific but that it can cleave a wide range of biologically relevant peptide substrates, for example, substance P, and as such it was given the common name, endopeptidase-24.11 [20]. In the literature, NEP is also known as the common acute lymphoblastic leukaemia antigen (CALLA or CD10) since it turned out to be identical with this leukocyte cell surface antigen [21], although to date the substrate(s) and functions of NEP in the immune system have not been identified. NEP was also reported to be identical with a recently described activity termed skin fibroblast elastase which plays a role in skin aging and UVA-induced skin damage [22].
NEP is an oligopeptidase which cleaves peptides containing up to 40–50 amino acids and the most efficiently hydrolyzed substrate is substance P [23]. NEP substrate specificity is rather wide but those for which NEP action has a physiological role in metabolism are rather limited. The principal substrates of NEP in vivo appear to be enkephalins, atrial natriuretic peptide, tachykinins, bradykinin, endothelins, adrenomedullin, members of the vasoactive intestinal peptide family, glucagon, thymopentin, and, most significantly in pathophysiological terms, the Alzheimer's disease Aβ peptide.
NEP is a type II integral membrane zinc metalloprotein and does not have a proenzyme form. It is an ectoenzyme with the bulk of its structure, including the active site, facing the extracellular space. Depending on tissue source the M r of NEP ranges from about 85, 000 to 110, 000 due to differences in its glycosylation [24]. The cDNA cloning of NEP revealed that rat and human enzymes consist of 742 amino acids [25]. The high similarity between human and rodent NEP proteins makes the rat a useful animal model for studying NEP functions and regulation. To date, there are only few characterised endogeneous tissue specific inhibitors of NEP. The first, isolated from bovine spinal cord, was a heptapeptide spinorphin which also inhibited dipeptidyl peptidases and angiotensin-converting enzyme [26]. A decade later, Rougeot and colleagues discovered sialorphin, an exocrine and endocrine signaling mediator, synthesized mostly in the submandibular gland and prostate of rats [27]. The first human NEP inhibitor isolated from saliva was opiorphin which had some pain-suppressive potency [28]. The most potent and widely used NEP inhibitors include phosphoramidon and thiorphan, and the 3D structure of the extracellular domain of NEP in a complex with phosphoramidon has been resolved allowing better understanding of the catalytic properties of the enzyme [29]. One particular feature of the NEP catalytic site is its restricted size which prevents access of large peptides and proteins but allows peptides containing up to 50 amino acid residues. This is consistent with Aβ as a preferred substrate of NEP. Another characteristic feature of NEP is its sensitivity to inhibition by phosphoramidon and thiorphan at nanomolar concentrations. Although a closely related NEP homologue endothelin-converting enzyme (ECE-1) is also inhibited by phosphoramidon, it is only sensitive to micromolar concentrations of the inhibitor and is not affected by thiorphan.
Despite being originally considered as a unique mammalian membrane endopeptidase, it was subsequently demonstrated that the human genome contains at least seven NEP-like enzymes. This metallopeptidase family is even more abundant in Drosophila melanogaster (24 predicted members) and Caenorhabditis elegans (22 members), and phosphoramidon-sensitive activities have been identified in these species [30, 31] which makes them useful models for studying functional properties of NEP. In the brain, NEP levels are much lower than in the kidney, and it appears to have mostly neuronal localisation [32] although it was recently reported to be expressed by activated astrocytes [33] and microglia [34]. In peripheral tissues NEP was also found to be transiently expressed on the surface of certain haematopoietic cells and increased NEP levels were found on mature lymphocytes in certain disease states (for review see [35]). It has also been implicated in the progression of a number of cancers, including prostate [36], renal [37], and lung [38] cancer. Another important role of NEP is related to inactivation of the natriuretic peptides in vivo and as such NEP inhibitors have been explored as potential cardiovascular and renal therapeutics.
The human NEP gene is located on chromosome 3 and exists in a single copy which spans more than 80 kb. It is composed of 24 exons and is highly conserved among mammalian species [39]. Expression of the NEP gene is controlled through two distinct promoters [40] whose role differs between cell types, although both promoters show similar characteristics and activity. Three distinct NEP mRNAs have been identified in human and rat which differ only in their 5′-noncoding regions [39, 40]. A gene knockout of NEP in mice has been reported in which the animals appeared developmentally normal but the NEP null mice were highly sensitive to endotoxic shock [41]. This observation may reflect a general role of NEP in the metabolism of proinflammatory peptides. NEP knockout mice also showed enhanced aggressive behaviour in the resident-intruder paradigm and altered locomotor activity as assessed in the photobeam system [42]. They also had an increased alcohol and food consumption [43].
3. NEP and Neuronal Functions
In the brain, NEP is mainly located on neuronal cells, especially in the striatonigral pathway [44], although it is also present in the hippocampus, where it functions to inactivate somatostatin, and in cortical regions [45]. Pre- and postsynaptic localization of NEP in the nervous system further emphasizes its important role in neuronal function [46] and this is schematically reflected in Figure 1. The enzyme has also been found in Schwann cells in the peripheral nervous system [47]. The significant increase in the expression of NEP by Schwann cells after axonal damage suggests that this enzyme could play a role in axonal regeneration [48].
Figure 1.
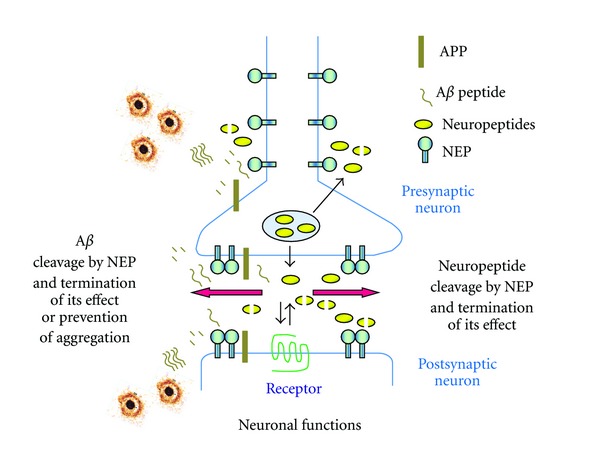
Schematic presentation of NEP localization and functional activity in the brain. NEP being localised pre- and postsynaptically in neuronal cells cleaves its neuropeptide substrates (including Aβ) terminating their properties and as such regulating cellular response to their action and neuronal functions. In the case of Aβ, NEP also prevents accumulation and aggregation of toxic amyloid oligomers. All symbols are explained in the figure.
The functional role of NEP in the brain is primarily determined by the physiological properties of its substrates and the roles they play in the nervous system (see Table 1). As such NEP was linked to such brain functions as LTP, synaptic plasticity, motor functions and locomotion, memory, anxiety, pain, hyperalgesia, circadian rhythms, sleep, fatigue, water homeostasis, blood-brain barrier integrity, and neuroinflammation. It plays a certain role in stroke pathology [49], pathophysiology of itch [50], attenuates central functions of baroreceptors [51], food intake, hormonal release, cardiovascular regulation, thermoregulation, stress [52], and anxiolytic response [53]. It also participates in dendrite elongation and the maturation of dendritic spines [54]. NEP was also suggested to play a major role in nociception activating the initial stage of nociceptin metabolism at the spinal cord level [55]. A role for NEP in memory has been confirmed in our experiments with i.c. injections of its inhibitors (phosphoramidon and thiorphan) to rats resulting in disruption of memory and neuronal plasticity [56–60]. In addition to these important neuronal functions of NEP, it is also now considered as a major amyloid-degrading enzyme and mechanisms of its regulation and reactivation have been extensively studied in the last decade [12]. Although the precise physiological properties of Aβ peptides are still far from being fully understood, the accumulating evidence suggests that they can act as modulators of neuronal function and synaptic plasticity [61] and the role of NEP in regulating concentrations of Aβ at functional levels can be important for normal brain activity.
Table 1.
Functional role of NEP and some of its substrates in the CNS.
| NEP substrates | Functions |
|---|---|
| Adrenomedullin | Vasodilator; tolerance to oxidative stress and hypoxia; inhibition of dendrite formation in the cerebral cortex [62], anxiety, pain [63] |
| Amyloid β-peptide | LTP, synaptic plasticity, memory, AD pathology [64] |
| Angiotensin I | Precursor to angiotensin II; enhances baroreceptor sensitivity [51] |
| Angiotensin II | Central cardiovascular regulation; attenuates baroreceptor sensitivity [51] |
| Bradykinin | Vasodilator; pain, hyperalgesia [65]; regulation of astrocyte calcium levels [66] |
| Cholecystokinin-8 | Feeding behaviour, satiety, anxiety, obesity [67] |
| Corticotropin | Sleep, fatigue [68] |
| Dynorphins | Learning and memory, emotional control, stress response, pain [69] |
| Endomorphin | Pain, analgesic effect [70] |
| Enkephalins | Pain perception, cognitive functions, affective behaviours, locomotion [71] |
| Endothelin-1 | Vasoconstriction, effects on water homeostasis and blood-brain barrier integrity, neuroinflammation, stroke [72] |
| Gastrin | Circadian rhythms [73], pathophysiology of itch [50] |
| Neuropeptide Y | Food intake, hormonal release, circadian rhythms, cardiovascular regulation, thermoregulation, stress response, anxiety and sleep [52] |
| Neurotensin | Modulation of dopamine signalling; dendrite elongation and the maturation of dendritic spines [54] |
| Oxytocin | Sexual arousal, bonding, stress, anxiolytic response [53] |
| Somatostatin | Motor activity, sleep, sensory processes, cognitive functions [74] |
| Substance P | Pain and inflammation [75], drug addiction [76], learning and memory [77], depression and anxiety [78, 79], itching [80] |
| VIP | Circadian rhythm [81] |
4. NEP and Amyloid Metabolism
The ability of neprilysin to catabolise β-amyloid peptide was first demonstrated in vitro by Howell and colleagues [82] and then confirmed in vivo [83, 84]. It was demonstrated that NEP knockout mice have increased levels of Aβ peptides in the brain and administration of the neprilysin inhibitor thiorphan to rats led to increased Aβ levels [83, 85]. On the contrary, NEP gene transfer to AD transgenic mice was able to reverse amyloid-like pathology and improve animal behaviour [86–88]. Importantly, it was shown that NEP is the most potent Aβ-degrading enzyme in the brain [89] and can degrade not only monomeric forms of Aβ but also its more toxic oligomers [90]. N-terminally truncated forms of Aβ (Aβ x-42) and pyroglutamyl modified Aβ 3–42 are also major contributors to the amyloid pathology of AD due to their abundance in AD brain and their cell toxicity [91]. Although the pyroglutamyl Aβ species have increased resistance to degradation by aminopeptidases [92], the comparative susceptibility of these peptides to NEP activity has not been adequately quantified to date.
Studies both in vivo and in vitro have now strongly linked NEP with the pathogenesis of AD and made it a viable therapeutic target. Further in vivo studies, including our own work, have indeed demonstrated that NEP mRNA, protein and activity levels decline with age in the cortex and hippocampus of rodents and humans [58–60, 93, 94] and also are reduced in the AD brain [95]. Decreased NEP levels and activity were also reported under such pathological conditions leading to AD, as ischemia or hypoxia [33, 93]. Our studies also demonstrated that prenatal hypoxia leads to reduced NEP protein and activity levels in the cortex and hippocampus of rats during their postnatal life [58–60].
Decreased NEP expression in the vasculature was also suggested to be responsible for the development of cerebral amyloid angiopathy found in AD patients [96]. However, along with the age-related and pathology-induced decrease of NEP expression seen in neuronal cells, it was reported that NEP is upregulated in reactive astrocytes surrounding amyloid plaques in AD transgenic mice which could contribute to some compensatory mechanisms [97]. On the contrary, Hickman and colleagues have reported an age-dependent decline of NEP and other amyloid-degrading enzyme expression in microglia resulting in decreased Aβ clearance [34]. Apart from the decline in NEP expression, age-related decrease of NEP capability to degrade Aβ might be due to enzyme oxidation [98] or conformational inactivation, for example, by amyloid peptide [99].
In addition to NEP, its homologue, neprilysin-2 (NEP2), was also characterised in the brain [100]. Although NEP2 is the closest NEP homologue, it has different properties, in particular, in cellular localization. NEP2 has two alternatively spliced forms, one of which is a soluble secreted form, also known as soluble, secreted endopeptidase (SEP) [101]. In the CNS, NEP2 is mainly localized in the cortex and hippocampus and is characteristic to specific neuronal populations [100, 102]. Despite the fact that NEP2 has a broad repertoire of substrates, its physiological role, apart from in male fertility, still is largely unknown. NEP2 was shown to degrade Aβ in vitro [89, 103] and recently Hafez and colleagues using gene knockout and transgenic animals have demonstrated that NEP2 contributes to Aβ degradation in vivo [104]. Recently it was demonstrated that NEP2 and NEP mRNA expression is altered in the AD-susceptible brain areas of patients with MCI compared to nonimpaired subjects. Moreover, NEP2 enzymatic activity in the mid-temporal and mid-frontal gyri of MCI and AD subjects was lower compared to controls and was associated with the level of cognitive decline [105]. However, at present, mechanisms of NEP2 cell specificity and regulation of its expression and activity have not been sufficiently addressed and further studies are required to estimate the role of this NEP homologue in pathogenesis of AD and to estimate its therapeutic value.
5. Modulation of Neprilysin Expression
Reports on age- and AD-related NEP decline have induced an intensive search for means to upregulate NEP gene expression and enzyme activity. NEP gene delivery studies have suggested that not only intracerebral injections of NEP-bearing constructs can have an antiamyloid effect in AD animal models [87] but that intraperitoneal injections of a lentivirus vector expressing NEP fused with the ApoB transport domain could also reduce Aβ burden and increase synaptic density in the brain of AD transgenic mice [106]. This opened up the development of non-invasive therapeutic approaches for potential treatment in patients with AD. One such approach has utilised a novel system for injection of an NEP coding plasmid into skeletal muscle via a syringe electrode [107]. Injected in this way, hNEP was detected in the muscle, serum, and brain of treated mice even 30 days after injection with minimal damage at the site of electrotransfer. Another, ex vivo NEP gene delivery method, was also suggested by Selkoe and colleagues who implanted primary fibroblasts, expressing a secreted form of NEP, into the brain of APP transgenic mice which induced robust clearance of amyloid plaques at the site of engraftment [108]. An interesting approach based on the observation that brain and plasma Aβ are in equilibrium through transport mechanisms [109] was developed by Hersh and colleagues, who found that in AD transgenic mice overexpressing NEP in erythrocytes or leukocytes there was a reduced Aβ burden in the brain [110, 111]. An alternative strategy of expressing a secreted, soluble form of NEP in the plasma through an adenovirus construct was also effective in clearing brain Aβ yet did not affect the plasma levels of other peptide substrates of NEP such as bradykinin or substance P [112]. Expressing NEP in plasma in this way could also provide a simple but effective system to maintain and monitor long-term activity of this amyloid-β-degrading peptidase. Along with developing methods of NEP upregulation, the optimal timing of NEP overexpression has also been examined suggesting that earlier upregulation of NEP levels was more beneficial in alleviating symptoms in a mouse model of AD [113].
Apart from targeted gene delivery, strategies for pharmacological NEP regulation have also been intensively studied in the last ten years. Cell culture studies have demonstrated that NEP activity can be increased by, among other compounds, a component of green tea extract, EGCG [114] and other plant extracts and polyphenols (e.g., [115]). Saido and colleagues have suggested that elevated levels of NEP substrates could upregulate NEP by a feedback control mechanism [116]. However, after screening a wide range of NEP neuropeptide substrates, they have found that only somatostatin was capable of upregulating NEP activity in primary neuronal cells. They have also suggested a possible mechanism of NEP activation involving somatostatin receptor subtypes 2 or 4, but these studies have not resulted in any further development of somatostatin receptor agonists for therapeutic application in AD. A 24-residue peptide, humanin, originally isolated from the brain of an AD patient, which has neuroprotective properties and decreases brain Aβ levels in animal models, was shown to mediate its Aβ-lowering effects by increasing NEP expression levels and could also provide a strategy for enhancing amyloid clearance [117]. Another receptor-mediated mechanism for pharmacological upregulation of NEP is the peroxisome proliferator activated receptor-δ (PPARδ) whose selective agonist, GW742, was shown to activate the NEP promoter driving luciferase expression in transfected HEK293 cells [118].
A completely new direction of studies linking the amyloid cascade hypothesis and NEP to the pathogenesis of AD has emerged from studies of the role of the C-terminal APP intracellular domain (AICD), released by γ-secretase activity, in the regulation of NEP transcription [119]. AICD is an approximately 6 kDa peptide which is present as a number of species of which the major form is 50 amino acids long but AICD48 and 51 species are also detectable [120, 121]. It is still unclear whether all of the isoforms of AICD are equally competent in transcriptional regulation. Despite being controversial and disputed by some other authors (e.g., [122–124]), the role of AICD in regulation of NEP has been confirmed by demonstrating that AICD binds to the NEP promoter in neuronal cells expressing high levels of NEP while in low NEP expressing cells, the NEP promoter is repressed by histone deacetylases (HDACs) [125]. This AICD activating effect was shown to be cell specific and even cell age dependent which may explain some of the contradictions in the literature [126–128]. Moreover, it was established that formation of transcriptionally active AICD depends on the particular APP isoform expressed (specifically APP695) and requires the active β-secretase (amyloidogenic) pathway [126, 129]. Apart from NEP, AICD activates expression of several genes and their number is steadily increasing [130, 131]. An important functional link confirming the role of AICD and gene activation was reported by Xu and colleagues [132] who found that AICD binds the MED12 unit of the mediator RNA polymerase II complex. This finding confirms AICD transcriptional activity [133] and validates other AICD-dependent genes such as aquaporin-1, MICAL2, and fibronectin-1 [132].
The fact that NEP gene expression is repressed in neuronal cells via competitive binding of HDACs to its promoter [125] has prompted us to look at the HDAC inhibitors which might reactivate NEP gene expression. As we have found in human neuroblastoma SH-SY5Y cells, trichostatin was able to activate NEP expression at the mRNA and protein levels and also increase its activity. More important from the therapeutic point of view was our observation that a clinically available antiepileptic drug valproic acid (VA) was also able to activate the NEP gene not only in cellular but in animal models as well [59, 125]. Moreover, injections of VA to AD transgenic mice were shown to decrease amyloid-related toxicity and improve animal behaviour although the authors had not considered to analyse levels of NEP expression and activity in their paradigm [134]. Our own animal studies have further demonstrated that administration of VA to rats with reduced levels of NEP expression in the brain due to prenatal hypoxia resulted in increased NEP activity in the cortex and hippocampus and improvement of animal short-term memory [58] which can be linked with the role of NEP in dendritic spine formation and restoration of neuronal circuits [59, 60]. The role of histone modifications in downregulation of the NEP promoter under hypoxic conditions has also been demonstrated by Wang and colleagues in primary cortical neuronal cells who demonstrated that NEP mRNA levels could be restored by VA administration to cells prior to hypoxia [72]. These studies revise the role of such a widely used antiepileptic drug as VA in regulation of neuronal gene expression and its protective role in neurodegeneration [135]. However, they also underlie the necessity for design of more specific HDAC inhibitors for targeted activation of NEP or other neuronal and, specifically, AD-related genes. Indeed, a recent report specified that inhibitors of class 1 HDACs reverse contextual memory deficits in an AD-mouse model [136]. This opens an avenue for retrospective analysis of the effect of VA or other HDAC inhibitors on development of AD.
Another therapeutically approved compound which was shown to modulate NEP expression via AICD-dependent mechanisms is the tyrosine kinase inhibitor, Gleevec (imatinib, STI-571), which was shown to elevate AICD levels and increase NEP mRNA and protein levels [137]. Although other authors failed to support this observation [138], recent work by Bauer and colleagues clearly demonstrated that the imatinib- (Gleevec-) induced NEP increase is APP and AICD dependent [127].
Importantly, in prostate cancer, NEP expression is downregulated by extensive hypermethylation of the promoter region and reexpression of neprilysin by treating the animals with the demethylating agent 5-aza-2′-deoxycytidine was able to inhibit tumor formation in the prostates of athymic mice [139, 140]. According to our data, downregulation of NEP in neuronal cells is not due to hypermethylation of its promoter and cannot be reactivated by 5-aza-2′-deoxycytidine which confirms cell specificity of NEP gene regulation [125].
As mentioned above, green tea extracts EFLA85942 and EGCG increase NEP expression and activity in human neuroblastoma SH-SY5Y, SK-N-SH, and NB7 cells ([114] and our own unpublished data). Extending these studies to animal models, we have found that prolonged EGCG administration to rats via osmotic minipumps was able to increase NEP activity in hypoxic rats to the levels recorded in control age-matched animals. Moreover, administration of EGCG has also improved performance of animals in the radial maze and improvement of short-term and long-term memory in the novel object recognition test [57]. This further supports the role of NEP in memory and extends the list of biologically active compounds which might be beneficial for prevention of cognitive deficit characteristic to AD pathology (Figure 2).
Figure 2.
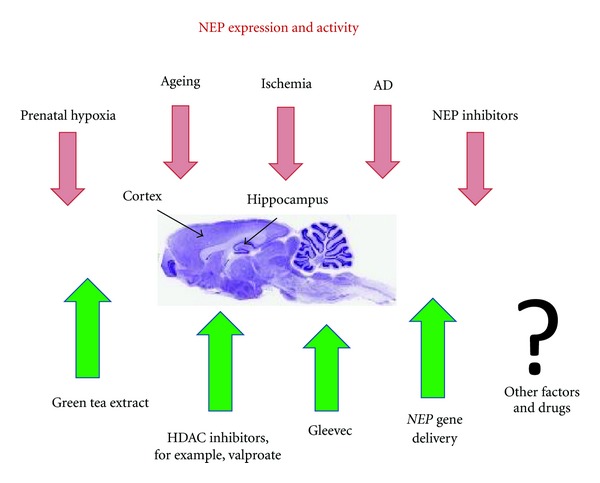
Effects of various experimental conditions on NEP activity in vivo. As explained in the text, NEP expression and activity in brain cortex and hippocampus (the structures which are characterised by accumulation of amyloid deposits) decreases with age and is also decreased after prenatal hypoxia, ischemia, or in the case of AD. In animal models, NEP activity can be modulated by its inhibitors affecting such brain functions as learning and memory. Mechanisms which can control and upregulate NEP expression and increase its activity include targeted NEP gene delivery, regulation of its promoter via inhibition of HDACs or pharmacologically by green tea extract (or EGCG) or Gleevec.
6. Concluding Remarks
Twenty years on from the formulation of the amyloid cascade hypothesis, there have been no successful clinical trials in AD. Several reasons for this can be suggested, for example, initiation of trials in patients in which neuronal loss and damage is already too far advanced, emphasizing the need for early diagnosis and good biomarkers. Also, late onset disease may well reflect defects in clearance mechanisms for Aβ rather than in the enhanced synthesis which occurs in early onset cases [141]. Hence, strategies to promote clearance, such as elevation of NEP expression and activity, may represent new opportunities for therapeutic intervention, either alone or in combination with other strategies. As follows from the detailed analysis of NEP properties and function, this enzyme plays an important role in brain function and disruption of its natural metabolic roles leads to various pathological conditions both centrally and in the periphery. Upregulation of NEP expression in such diseases as AD or prostate cancer has already been shown to be beneficial in animal models and various approaches have now been developed to activate this enzyme in cells and organisms. The discovery of epigenetic and pharmacological mechanisms for controlling NEP activity suggests a possibility for design of a preventive therapeutic strategy in AD and other age-related human diseases. Taking into account the wide substrate repertoire of NEP, there might be a cohort of functions which can be maintained by NEP modulators such as learning and memory, pain and inflammation, depression and anxiety, and further research of the precise molecular mechanisms involved in tissue and cell-specific regulation of this peptidase might give us a powerful tool to improve human health and wellbeing.
Acknowledgments
The authors thank the U.K. Medical Research Council, Alzheimer's Research UK, Russian Foundation for Basic Research (RFBR 10-04-01156), and the Programme of RAS “Fundamental Sciences for Medicine” for financial support for this work.
Abbreviations
- Aβ:
Amyloid β-peptide
- AD:
Alzheimer's disease
- ADE:
Amyloid-degrading enzyme
- AICD:
APP intracellular domain
- APP:
Amyloid precursor protein
- EGCG:
Epigallocatechin gallate
- HDAC:
Histone deacetylase
- IDE:
Insulin-degrading enzyme
- MED:
Mediator
- NEP:
Neprilysin
- PKC:
Protein kinase C
- PPAR:
Peroxisome proliferator-activated receptor
- SNP:
Single nucleotide polymorphism
- VA:
Valproic acid.
References
- 1.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6(4):487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. Journal of Neurochemistry. 2009;110(4):1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- 4.Castellani RJ, Smith MA. Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is ‘too big to fail’. Journal of Pathology. 2011;224(2):147–152. doi: 10.1002/path.2885. [DOI] [PubMed] [Google Scholar]
- 5.Goate A, Hardy J. Twenty years of Alzheimer’s disease-causing mutations. Journal of Neurochemistry. 2012;120(supplement 1):3–8. doi: 10.1111/j.1471-4159.2011.07575.x. [DOI] [PubMed] [Google Scholar]
- 6.Karran E, Mercken M, de Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nature Reviews Drug Discovery. 2011;10(9):698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 7.Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble a β oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. Journal of Neuroscience. 2011;31(18):6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larson and ME, Lesné SE. Soluble Aβ oligomer production and toxicity. Journal of Neurochemistry. 2012;120(supplement 1):125–139. doi: 10.1111/j.1471-4159.2011.07478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aisen PS. The development of anti-amyloid therapy for Alzheimer’s disease: from secretase modulators to polymerisation inhibitors. CNS Drugs. 2005;19(12):989–996. doi: 10.2165/00023210-200519120-00002. [DOI] [PubMed] [Google Scholar]
- 10.Miners JS, Barua N, Kehoe PG, et al. Aβ-degrading enzymes: potential for treatment of Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 2011;70(11):944–959. doi: 10.1097/NEN.0b013e3182345e46. [DOI] [PubMed] [Google Scholar]
- 11.Nalivaeva NN, Fisk LR, Belyaev ND, Turner AJ. Amyloid-degrading enzymes as therapeutic targets in Alzheimer’s disease. Current Alzheimer Research. 2008;5(2):212–224. doi: 10.2174/156720508783954785. [DOI] [PubMed] [Google Scholar]
- 12.Nalivaeva NN, Beckett C, Belyaev ND, Turner AJ. Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? Journal of Neurochemistry. 2012;120(supplement 1):167–185. doi: 10.1111/j.1471-4159.2011.07510.x. [DOI] [PubMed] [Google Scholar]
- 13.Pearson HA, Peers C. Physiological roles for amyloid β peptides. Journal of Physiology. 2006;575(1):5–10. doi: 10.1113/jphysiol.2006.111203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardy J. Does Aβ 42 have a function related to blood homeostasis? Neurochemical Research. 2007;32(4-5):833–835. doi: 10.1007/s11064-006-9221-9. [DOI] [PubMed] [Google Scholar]
- 15.Ohyagi Y, Asahara H, Chui DH, et al. Intracellular Aβ42 activates p53 promoter: a pathway to neurodegeneration in Alzheimer’s disease. The FASEB Journal. 2005;19(2):255–257. doi: 10.1096/fj.04-2637fje. [DOI] [PubMed] [Google Scholar]
- 16.Bailey JA, Maloney B, Ge YW, Lahiri DK. Functional activity of the novel Alzheimer’s amyloid β-peptide interacting domain (AβID) in the APP and BACE1 promoter sequences and implications in activating apoptotic genes and in amyloidogenesis. Gene. 2011;488(1-2):13–22. doi: 10.1016/j.gene.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hawkes CA, Härtig W, Kacza J, et al. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathologica. 2011;121(4):431–443. doi: 10.1007/s00401-011-0801-7. [DOI] [PubMed] [Google Scholar]
- 18.Kerr MA, Kenny AJ. The purification and specificity of a neutral endopeptidase from rabbit kidney brush border. Biochemical Journal. 1974;137(3):477–488. doi: 10.1042/bj1370477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malfroy B, Swerts JP, Guyon A, et al. High affinity enkephalin degrading peptidase in brain is increased after morphine. Nature. 1978;276(5687):523–526. doi: 10.1038/276523a0. [DOI] [PubMed] [Google Scholar]
- 20.Matsas R, Fulcher IS, Kenny AJ, Turner AJ. Substance P and [Leu]enkephalin are hydrolyzed by an enzyme in pig caudate synaptic membranes that is identical with the endopeptidase of kidney microvilli. Proceedings of the National Academy of Sciences of the United States of America. 1983;80(10 I):3111–3115. doi: 10.1073/pnas.80.10.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Letarte M, Vera S, Tran R, et al. Common acute lymphocytic leukemia antigen is identical to neutral endopeptidase. Journal of Experimental Medicine. 1988;168(4):1247–1253. doi: 10.1084/jem.168.4.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morisaki N, Moriwaki S, Sugiyama-Nakagiri Y, Haketa K, Takema Y, Imokawa G. Neprilysin is identical to skin fibroblast elastase: its role in skin aging and UV responses. The Journal of Biological Chemistry. 2010;285(51):39819–39827. doi: 10.1074/jbc.M110.161547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsas R, Kenny AJ, Turner AJ. The metabolism of neuropeptides. The hydrolysis of peptides, including enkephalins, tachykinins and their analogues, by endopeptidase-24.11. Biochemical Journal. 1984;223(2):433–440. doi: 10.1042/bj2230433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Relton JM, Gee NS, Matsas R, Turner AJ, Kenny AJ. Purification of endopeptidase-24.11 (“enkephalinase”) from pig brain by immunoadsorbent chromatography. Biochemical Journal. 1983;215(3):519–523. doi: 10.1042/bj2150519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malfroy B, Kuang WJ, Seeburg PH, Mason AJ, Schofield PR. Molecular cloning and amino acid sequence of human enkephalinase (neutral endopeptidase) The FEBS Letters. 1988;229(1):206–210. doi: 10.1016/0014-5793(88)80828-7. [DOI] [PubMed] [Google Scholar]
- 26.Nishimura K, Ueki M, Kaneto H, Hazato T. Study of a new endogenous inhibitor of enkephalin-degrading enzymes; pharmacological function and metabolism of spinorphin. Japanese Journal of Anesthesiology. 1993;42(11):1663–1670. [PubMed] [Google Scholar]
- 27.Rougeot C, Messaoudi M, Hermitte V, et al. Sialorphin, a natural inhibitor of rat membrane-bound neutral endopeptidase that displays analgesic activity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8549–8554. doi: 10.1073/pnas.1431850100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wisner A, Dufour E, Messaoudi M, et al. Human Opiorphin, a natural antinociceptive modulator of opioid-dependent pathways. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(47):17979–17984. doi: 10.1073/pnas.0605865103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oefner C, D’Arcy A, Hennig M, Winkler FK, Dale GE. Structure of human neutral endopeptidase (neprilysin) complexed with phosphoramidon. Journal of Molecular Biology. 2000;296(2):341–349. doi: 10.1006/jmbi.1999.3492. [DOI] [PubMed] [Google Scholar]
- 30.Turner AJ, Isaac RE, Coates D. The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioassay. 2001;23(3):261–269. doi: 10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 31.Bland ND, Pinney JW, Thomas JE, Turner AJ, Isaac RE. Bioinformatic analysis of the neprilysin (M13) family of peptidases reveals complex evolutionary and functional relationships. BMC Evolutionary Biology. 2008;8(1, article 16) doi: 10.1186/1471-2148-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barnes K, Turner AJ, Kenny AJ. An immunoelectron microscopic study of pig substantia nigra shows co-localization of endopeptidase-24.11 with substance P. Neuroscience. 1993;53(4):1073–1082. doi: 10.1016/0306-4522(93)90490-7. [DOI] [PubMed] [Google Scholar]
- 33.Fisk L, Nalivaeva NN, Boyle JP, Peers CS, Turner AJ. Effects of hypoxia and oxidative stress on expression of neprilysin in human neuroblastoma cells and rat cortical neurones and astrocytes. Neurochemical Research. 2007;32(10):1741–1748. doi: 10.1007/s11064-007-9349-2. [DOI] [PubMed] [Google Scholar]
- 34.Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective β-amyloid clearance pathways in aging alzheimer’s disease mice. Journal of Neuroscience. 2008;28(33):8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LeBien TW, McCormack RT. The common acute lymphoblastic leukemia antigen (CD10). Emancipation from a functional enigma. Blood. 1989;73(3):625–635. [PubMed] [Google Scholar]
- 36.Papandreou CN, Usmani B, Geng YP, et al. Neutral endopeptidase 24.11 loss in metastatic human prostate cancer contributes to androgen-independent progression. Nature Medicine. 1998;4(1):50–57. doi: 10.1038/nm0198-050. [DOI] [PubMed] [Google Scholar]
- 37.Göhring B, Holzhausen HJ, Meye A, et al. Endopeptidase 24.11/CD10 is down-regulated in renal cell cancer. International Journal of Molecular Medicine. 1998;2(4):409–414. doi: 10.3892/ijmm.2.4.409. [DOI] [PubMed] [Google Scholar]
- 38.Cohen AJ, Bunn PA, Franklin W, et al. Neutral endopeptidase: variable expression in human lung, inactivation in lung cancer, and modulation of peptide-induced calcium flux. Cancer Research. 1996;56(4):831–839. [PubMed] [Google Scholar]
- 39.D’Adamio L, Shipp MA, Masteller EL, Reinherz EL. Organization of the gene encoding common acute lymphoblastic leukemia antigen (neutral endopeptidase 24.11): multiple miniexons and separate 5′ untranslated regions. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(18):7103–7107. doi: 10.1073/pnas.86.18.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li C, Booze RM, Hersh LB. Tissue-specific expression of rat neutral endopeptidase (neprilysin) mRNAs. The Journal of Biological Chemistry. 1995;270(11):5723–5728. doi: 10.1074/jbc.270.11.5723. [DOI] [PubMed] [Google Scholar]
- 41.Lu B, Gerard NP, Kolakowski LF, et al. Neutral endopeptidase modulation of septic shock. Journal of Experimental Medicine. 1995;181(6):2271–2275. doi: 10.1084/jem.181.6.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fischer HS, Zernig G, Schuligoi R, et al. Alterations within the endogenous opioid system in mice with targeted deletion of the neutral endopeptidase (“enkephalinase”) gene. Regulatory Peptides. 2000;96(1-2):53–58. doi: 10.1016/s0167-0115(00)00200-7. [DOI] [PubMed] [Google Scholar]
- 43.Siems WE, Maul B, Krause W, et al. Neutral endopeptidase and alcohol consumption, experiments in neutral endopeptidase-deficient mice. European Journal of Pharmacology. 2000;397(2-3):327–334. doi: 10.1016/s0014-2999(00)00222-3. [DOI] [PubMed] [Google Scholar]
- 44.Barnes K, Matsas R, Hooper NM, Turner AJ, Kenny AJ. Endopeptidase-24.11 is striosomally ordered in pig brain and, in contrast to aminopeptidase N and peptidyl dipeptidase A (“angiotensin converting enzyme”), is a marker for a set of striatal efferent fibres. Neuroscience. 1988;27(3):799–817. doi: 10.1016/0306-4522(88)90184-4. [DOI] [PubMed] [Google Scholar]
- 45.Barnes K, Doherty S, Turner AJ. Endopeptidase-24.11 is the integral membrane peptidase initiating degradation of somatostatin in the hippocampus. Journal of Neurochemistry. 1995;64(4):1826–1832. doi: 10.1046/j.1471-4159.1995.64041826.x. [DOI] [PubMed] [Google Scholar]
- 46.Barnes K, Turner AJ, Kenny AJ. Membrane localization of endopeptidase-24.11 and peptidyl dipeptidase A (angiotensin converting enzyme) in the pig brain: a study using subcellular fractionation and electron microscopic immunocytochemistry. Journal of Neurochemistry. 1992;58(6):2088–2096. doi: 10.1111/j.1471-4159.1992.tb10950.x. [DOI] [PubMed] [Google Scholar]
- 47.Kioussi C, Matsas R. Endopeptidase-24.11, a cell-surface peptidase of central nervous system neurons, is expressed by Schwann cells in the pig peripheral nervous system. Journal of Neurochemistry. 1991;57(2):431–440. doi: 10.1111/j.1471-4159.1991.tb03770.x. [DOI] [PubMed] [Google Scholar]
- 48.Kioussi C, Mamalaki A, Jessen K, Mirsky R, Hersh LB, Matsas R. Expression of endopeptidase-24.11 (common acute lymphoblastic leukaemia antigen CD10) in the sciatic nerve of the adult rat after lesion and during regeneration. European Journal of Neuroscience. 1995;7(5):951–961. doi: 10.1111/j.1460-9568.1995.tb01083.x. [DOI] [PubMed] [Google Scholar]
- 49.Wang Z, Yang D, Zhang X, et al. Hypoxia-induced down-regulation of neprilysin by histone modification in mouse primary cortical and Hippocampal neurons. PLoS ONE. 2011;6(4) doi: 10.1371/journal.pone.0019229.e19229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun YG, Chen ZF. A gastrin-releasing peptide receptor mediates the itch sensation in the spinal cord. Nature. 2007;448(7154):700–703. doi: 10.1038/nature06029. [DOI] [PubMed] [Google Scholar]
- 51.Diz DI, Garcia-Espinosa MA, Gallagher PE, Ganten D, Ferrario CM, Averill DB. Angiotensin-(1–7) and baroreflex function in nucleus tractus solitarii of (mRen2)27 transgenic rats. Journal of Cardiovascular Pharmacology. 2008;51(6):542–548. doi: 10.1097/FJC.0b013e3181734a54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Silva AP, Cavadas C, Grouzmann E. Neuropeptide Y and its receptors as potential therapeutic drug targets. Clinica Chimica Acta. 2002;326(1-2):3–25. doi: 10.1016/s0009-8981(02)00301-7. [DOI] [PubMed] [Google Scholar]
- 53.Hernández J, Segarra AB, Ramírez M, et al. Stress influences brain enkephalinase, oxytocinase and angiotensinase activities: a new hypothesis. Neuropsychobiology. 2009;59(3):184–189. doi: 10.1159/000219306. [DOI] [PubMed] [Google Scholar]
- 54.Gandou C, Ohtani A, Senzaki K, Shiga T. Neurotensin promotes the dendrite elongation and the dendritic spine maturation of the cerebral cortex in vitro. Neuroscience Research. 2010;66(3):246–255. doi: 10.1016/j.neures.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 55.Sakurada C, Sakurada S, Orito T, Tan-No K, Sakurada T. Degradation of nociceptin (orphanin FQ) by mouse spinal cord synaptic membranes is triggered by endopeptidase-24.11: an in vitro and in vivo study. Biochemical Pharmacology. 2002;64(8):1293–1303. doi: 10.1016/s0006-2952(02)01295-9. [DOI] [PubMed] [Google Scholar]
- 56.Dubrovskaya NM, Nalivaeva NN, Plesneva SA, Feponova AA, Turner AJ, Zhuravin IA. Changes in the activity of amyloid-degrading metallopeptidases leads to disruption of memory in rats. Neuroscience and Behavioral Physiology. 2010;40(9):975–980. [PubMed] [Google Scholar]
- 57.Dubrovskaya NM, Nalivaeva NN, Vasilev DS, et al. Short-Term Memory: New Research. New York, NY, USA: Nova Science; 2012. Mechanisms of short-term working memory deficit. [Google Scholar]
- 58.Nalivaeva NN, Belyaev ND, Lewis DI, et al. Effect of sodium valproate administration on brain neprilysin expression and memory in rats. Journal of Molecular Neuroscience. 2012;46(3):569–577. doi: 10.1007/s12031-011-9644-x. [DOI] [PubMed] [Google Scholar]
- 59.Nalivaeva NN, Dubrovskaya NM, Vasiliev DS, et al. Changes in the activity of amyloid-degrading enzymes affect cognitive functions in rats via alteration of the synaptopodin-positive dendritic network. In: Babusikova E, Dobrota D, Lehotsky J, Martin, Slovakia, editors. New Frontiers in Molecular Mechanisms in Neurological and Psychiatric Disorders. Vol. 1. 2011. pp. 277–285. [Google Scholar]
- 60.Zhuravin IA, Dubrovskaya NM, Vasilev DS, Tumanova NL, Nalivaeva NN. Epigenetic and pharmacological regulation of the amyloid-degrading enzyme neprilysin results in modulation of cognitive functions in mammals. Doklady Biological Sciences. 2011;438(1):145–148. doi: 10.1134/S001249661103015X. [DOI] [PubMed] [Google Scholar]
- 61.Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses. Nature Neuroscience. 2009;12(12):1567–1576. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- 62.Harigai Y, Natsume M, Li F, Ohtani A, Senzaki K, Shiga T. Differential roles of calcitonin family peptides in the dendrite formation and spinogenesis of the cerebral cortex in vitro. Neuropeptides. 2011;45(4):263–272. doi: 10.1016/j.npep.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 63.Fernández AP, Serrano J, Tessarollo L, Cuttitta F, Martínez A. Lack of adrenomedullin in the mouse brain results in behavioral changes, anxiety, and lower survival under stress conditions. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(34):12581–12586. doi: 10.1073/pnas.0803174105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma T, Klann E. Amyloid β: linking synaptic plasticity failure to memory disruption in Alzheimer’s disease. Journal of Neurochemistry. 2011;120(supplement 1):140–148. doi: 10.1111/j.1471-4159.2011.07506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fischer HS, Zernig G, Hauser KF, Gerard C, Hersh LB, Saria A. Neutral endopeptidase knockout induces hyperalgesia in a model of visceral pain, an effect related to bradykinin and nitric oxide. Journal of Molecular Neuroscience. 2002;18(1-2):129–134. doi: 10.1385/JMN:18:1-2:129. [DOI] [PubMed] [Google Scholar]
- 66.Wang YB, Peng C, Liu YH. Low dose of bradykinin selectively increases intracellular calcium in glioma cells. Journal of the Neurological Sciences. 2007;258(1-2):44–51. doi: 10.1016/j.jns.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 67.Arora S, Anubhuti Role of neuropeptides in appetite regulation and obesity—a review. Neuropeptides. 2006;40(6):375–401. doi: 10.1016/j.npep.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 68.Ogawa T, Kiryu-Seo S, Tanaka M, et al. Altered expression of neprilysin family members in the pituitary gland of sleep-disturbed rats, an animal model of severe fatigue. Journal of Neurochemistry. 2005;95(4):1156–1166. doi: 10.1111/j.1471-4159.2005.03436.x. [DOI] [PubMed] [Google Scholar]
- 69.Schwarzer C. 30 years of dynorphins—new insights on their functions in neuropsychiatric diseases. Pharmacology and Therapeutics. 2009;123(3):353–370. doi: 10.1016/j.pharmthera.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Song B, Marvizón JCG. Peptidases prevent μ-opioid receptor internalization in dorsal horn neurons by endogenously released opioids. Journal of Neuroscience. 2003;23(5):1847–1858. doi: 10.1523/JNEUROSCI.23-05-01847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roques BP, Noble F, Daugé V, Fournie-Zaluski MC, Beaumont A. Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacological Reviews. 1993;45(1):87–146. [PubMed] [Google Scholar]
- 72.Wang HH, Hsieh HL, Yang CM. Nitric oxide production by endothelin-1 enhances astrocytic migration via the tyrosine nitration of matrix metalloproteinase-9. Journal of Cellular Physiology. 2011;226(9):2244–2256. doi: 10.1002/jcp.22560. [DOI] [PubMed] [Google Scholar]
- 73.Gamble KL, Kudo T, Colwell CS, McMahon DG. Gastrin-releasing peptide modulates fast delayed rectifier potassium current in Per1-expressing SCN neurons. Journal of Biological Rhythms. 2011;26(2):99–106. doi: 10.1177/0748730410396678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Viollet C, Lepousez G, Loudes C, Videau C, Simon A, Epelbaum J. Somatostatinergic systems in brain: networks and functions. Molecular and Cellular Endocrinology. 2008;286(1-2):75–87. doi: 10.1016/j.mce.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 75.Krämer HH, He L, Lu B, Birklein F, Sommer C. Increased pain and neurogenic inflammation in mice deficient of neutral endopeptidase. Neurobiology of Disease. 2009;35(2):177–183. doi: 10.1016/j.nbd.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 76.Loonam TM, Noailles PAH, Yu J, Zhu JPQ, Angulo JA. Substance P and cholecystokinin regulate neurochemical responses to cocaine and methamphetamine in the striatum. Life Sciences. 2003;73(6):727–739. doi: 10.1016/s0024-3205(03)00393-x. [DOI] [PubMed] [Google Scholar]
- 77.Huston JP, Hasenöhrl RU, Boix F, Gerhardt P, Schwarting RKW. Sequence-specific effects of neurokinin substance P on memory, reinforcement, and brain dopamine activity. Psychopharmacology. 1993;112(2-3):147–162. doi: 10.1007/BF02244906. [DOI] [PubMed] [Google Scholar]
- 78.Hasenöhrl RU, Souza-Silva MAD, Nikolaus S, et al. Substance P and its role in neural mechanisms governing learning, anxiety and functional recovery. Neuropeptides. 2000;34(5):272–280. doi: 10.1054/npep.2000.0824. [DOI] [PubMed] [Google Scholar]
- 79.McLean S. Do substance P and the NK1 receptor have a role in depression and anxiety? Current Pharmaceutical Design. 2005;11(12):1529–1547. doi: 10.2174/1381612053764779. [DOI] [PubMed] [Google Scholar]
- 80.Yamaoka J, Kawana S. Rapid changes in substance P signaling and neutral endopeptidase induced by skin-scratching stimulation in mice. Journal of Dermatological Science. 2007;48(2):123–132. doi: 10.1016/j.jdermsci.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 81.Hughes ATL, Guilding C, Piggins HD. Neuropeptide signaling differentially affects phase maintenance and rhythm generation in SCN and extra-SCN circadian oscillators. PLoS ONE. 2011;6(4) doi: 10.1371/journal.pone.0018926.e18926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Howell S, Nalbantoglu J, Crine P. Neutral endopeptidase can hydrolyze β-amyloid(1–40) but shows no effect on β-amyloid precursor protein metabolism. Peptides. 1995;16(4):647–652. doi: 10.1016/0196-9781(95)00021-b. [DOI] [PubMed] [Google Scholar]
- 83.Iwata N, Tsubuki S, Takaki Y, et al. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nature Medicine. 2000;6(2):143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 84.Takaki Y, Iwata N, Tsubuki S, et al. Biochemical identification of the neutral endopeptidase family member responsible for the catabolism of amyloid β peptide in the brain. Journal of Biochemistry. 2000;128(6):897–902. doi: 10.1093/oxfordjournals.jbchem.a022839. [DOI] [PubMed] [Google Scholar]
- 85.Iwata N, Tsubuki S, Takaki Y, et al. Metabolic regulation of brain Aβ by neprilysin. Science. 2001;292(5521):1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 86.Hama E, Shirotani K, Masumoto H, Sekine-Aizawa Y, Aizawa H, Saido TC. Clearance of extracellular and cell-associated amyloid β peptide through viral expression of neprilysin in primary neurons. Journal of Biochemistry. 2001;130(6):721–726. doi: 10.1093/oxfordjournals.jbchem.a003040. [DOI] [PubMed] [Google Scholar]
- 87.Marr RA, Rockenstein E, Mukherjee A, et al. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. Journal of Neuroscience. 2003;23(6):1992–1996. doi: 10.1523/JNEUROSCI.23-06-01992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Spencer B, Marr RA, Rockenstein E, et al. Long-term neprilysin gene transfer is associated with reduced levels of intracellular Aβ and behavioral improvement in APP transgenic mice. BMC Neuroscience. 2008;9, article 109 doi: 10.1186/1471-2202-9-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shirotani K, Tsubuki S, Iwata N, et al. Neprilysin degrades both amyloid β peptides 1–40 and 1–42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. The Journal of Biological Chemistry. 2001;276(24):21895–21901. doi: 10.1074/jbc.M008511200. [DOI] [PubMed] [Google Scholar]
- 90.Kanemitsu H, Tomiyama T, Mori H. Human neprilysin is capable of degrading amyloid β peptide not only in the monomeric form but also the pathological oligomeric form. Neuroscience Letters. 2003;350(2):113–116. doi: 10.1016/s0304-3940(03)00898-x. [DOI] [PubMed] [Google Scholar]
- 91.Jawhar S, Wirths O, Bayer TA. Pyroglutamate amyloid-β (Aβ): a hatchet man in Alzheimer disease. The Journal of Biological Chemistry. 2011;286(45):38825–38832. doi: 10.1074/jbc.R111.288308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Drew SC, Masters CL, Barnham KJ. Alzheimer’s Aβ peptides with disease-associated NTerminal modifications: Influence of isomerisation, truncation and mutation on Cu2+ coordination. PLoS ONE. 2010;5(12) doi: 10.1371/journal.pone.0015875.e15875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nalivaeva NN, Fisk L, Kochkina EG, et al. Effect of hypoxia/ischemia and hypoxic preconditioning/reperfusion on expression of some amyloid-degrading enzymes. Annals of the New York Academy of Sciences. 2004;1035:21–33. doi: 10.1196/annals.1332.002. [DOI] [PubMed] [Google Scholar]
- 94.Caccamo A, Oddo S, Sugarman MC, Akbari Y, LaFerla FM. Age- and region-dependent alterations in Aβ-degrading enzymes: implications for Aβ-induced disorders. Neurobiology of Aging. 2005;26(5):645–654. doi: 10.1016/j.neurobiolaging.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 95.Wang DS, Lipton RB, Katz MJ, et al. Decreased neprilysin immunoreactivity in Alzheimer disease, but not in pathological aging. Journal of Neuropathology and Experimental Neurology. 2005;64(5):378–385. doi: 10.1093/jnen/64.5.378. [DOI] [PubMed] [Google Scholar]
- 96.Carpentier M, Robitaille Y, DesGroseillers L, Boileau G, Marcinkiewicz M. Declining expression of neprilysin in Alzheimer disease vasculature: possible involvement in cerebral amyloid angiopathy. Journal of Neuropathology and Experimental Neurology. 2002;61(10):849–856. doi: 10.1093/jnen/61.10.849. [DOI] [PubMed] [Google Scholar]
- 97.Apelt J, Ach K, Schliebs R. Aging-related down-regulation of neprilysin, a putative β-amyloid-degrading enzyme, in transgenic Tg2576 Alzheimer-like mouse brain is accompanied by an astroglial upregulation in the vicinity of β-amyloid plaques. Neuroscience Letters. 2003;339(3):183–186. doi: 10.1016/s0304-3940(03)00030-2. [DOI] [PubMed] [Google Scholar]
- 98.Wang DS, Iwata N, Hama E, Saido TC, Dickson DW. Oxidized neprilysin in aging and Alzheimer’s disease brains. Biochemical and Biophysical Research Communications. 2003;310(1):236–241. doi: 10.1016/j.bbrc.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 99.Dorfman VB, Pasquini L, Riudavets M, et al. Differential cerebral deposition of IDE and NEP in sporadic and familial Alzheimer’s disease. Neurobiology of Aging. 2010;31(10):1743–1757. doi: 10.1016/j.neurobiolaging.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tanja O, Facchinetti P, Rose C, Bonhomme MC, Gros C, Schwartz JC. Neprilysin II: a putative novel metalloprotease and its isoforms in CNS and testis. Biochemical and Biophysical Research Communications. 2000;271(3):565–570. doi: 10.1006/bbrc.2000.2664. [DOI] [PubMed] [Google Scholar]
- 101.Ikeda K, Emoto N, Raharjo SB, et al. Molecular identification and characterization of novel membrane-bound metalloprotease, the soluble secreted form of which hydrolyzes a variety of vasoactive peptides. The Journal of Biological Chemistry. 1999;274(45):32469–32477. doi: 10.1074/jbc.274.45.32469. [DOI] [PubMed] [Google Scholar]
- 102.Facchinetti P, Rose C, Schwartz JC, Ouimet T. Ontogeny, regional and cellular distribution of the novel metalloprotease neprilysin 2 in the rat: a comparison with neprilysin and endothelin-converting enzyme-1. Neuroscience. 2003;118(3):627–639. doi: 10.1016/s0306-4522(02)01002-3. [DOI] [PubMed] [Google Scholar]
- 103.Huang JY, Bruno AM, Patel CA, et al. Human membrane metallo-endopeptidase-like protein degrades both β-amyloid 42 and β-amyloid 40. Neuroscience. 2008;155(1):258–262. doi: 10.1016/j.neuroscience.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hafez D, Huang JY, Huynh AM, et al. Neprilysin-2 is an important β-amyloid degrading enzyme. American Journal of Pathology. 2011;178(1):306–312. doi: 10.1016/j.ajpath.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huang JY, Hafez DM, James BD, et al. Altered NEP2 expression and activity in mild cognitive impairment and Alzheimer’s disease. Journal of Alzheimer’s Disease. 2012;28(2):433–441. doi: 10.3233/JAD-2011-111307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Spencer B, Marr RA, Gindi R, et al. Peripheral delivery of a CNS targeted, metalo-protease reduces Aβ toxicity in a mouse model of Alzheimer’s disease. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0016575.e16575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li Y, Wang J, Liu J, Liu F. A novel system for in vivo neprilysin gene delivery using a syringe electrode. Journal of Neuroscience Methods. 2010;193(2):226–231. doi: 10.1016/j.jneumeth.2010.08.029. [DOI] [PubMed] [Google Scholar]
- 108.Hemming ML, Patterson M, Reske-Nielsen C, Lin L, Isacson O, Selkoe DJ. Reducing amyloid plaque burden via ex vivo gene delivery of an Aβ-degrading protease: a novel therapeutic approach to Alzheimer disease. PLoS Medicine. 2007;4(8, article e262) doi: 10.1371/journal.pmed.0040262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Deane R, Wu Z, Zlokovic BV. RAGE (Yin) versus LRP (Yang) balance regulates Alzheimer amyloid β-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35(11):2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- 110.Liu Y, Guan H, Beckett TL, et al. In vitro and in vivo degradation of Aβ peptide by peptidases coupled to erythrocytes. Peptides. 2007;28(12):2348–2355. doi: 10.1016/j.peptides.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Guan H, Liu Y, Daily A, et al. Peripherally expressed neprilysin reduces brain amyloid burden: a novel approach for treating Alzheimer’s disease. Journal of Neuroscience Research. 2009;87(6):1462–1473. doi: 10.1002/jnr.21944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu Y, Studzinski C, Beckett T, Murphy MP, Klein RL, Hersh LB. Circulating neprilysin clears brain amyloid. Molecular and Cellular Neuroscience. 2010;45(2):101–107. doi: 10.1016/j.mcn.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.El-Amouri SS, Zhu H, Yu J, Marr R, Verma IM, Kindy MS. Neprilysin: an enzyme candidate to slow the progression of Alzheimer’s disease. American Journal of Pathology. 2008;172(5):1342–1354. doi: 10.2353/ajpath.2008.070620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Melzig MF, Janka M. Enhancement of neutral endopeptidase activity in SK-N-SH cells by green tea extract. Phytomedicine. 2003;10(6-7):494–498. doi: 10.1078/094471103322331449. [DOI] [PubMed] [Google Scholar]
- 115.Kiss A, Kowalski J, Melzig MF. Effect of Epilobium angustifolium L. extracts and polyphenols on cell proliferation and neutral endopeptidase activity in selected cell lines. Pharmazie. 2006;61(1):66–69. [PubMed] [Google Scholar]
- 116.Sãito T, Iwata N, Tsubuki S, et al. Somatostatin regulates brain amyloid β peptide Aβ42 through modulation of proteolytic degradation. Nature Medicine. 2005;11(4):434–439. doi: 10.1038/nm1206. [DOI] [PubMed] [Google Scholar]
- 117.Niikura T, Sidahmed E, Hirata-Fukae C, Aisen PS, Matsuoka Y. A humanin derivative reduces amyloid beta accumulation and ameliorates memory deficit in triple transgenic mice. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0016259.e16259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kalinin S, Richardson JC, Feinstein DL. A PPARdelta agonist reduces amyloid burden and brain inflammation in a transgenic mouse model of Alzheimer’s disease. Current Alzheimer Research. 2009;6(5):431–437. doi: 10.2174/156720509789207949. [DOI] [PubMed] [Google Scholar]
- 119.Pardossi-Piquard R, Petit A, Kawarai T, et al. Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by intracellular domains of βAPP and APLP. Neuron. 2005;46(4):541–554. doi: 10.1016/j.neuron.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 120.Yu C, Kim SH, Ikeuchi T, et al. Characterization of a presenilin-mediated amyloid precursor protein carboxyl-terminal fragment γ: evidence for distinct mechanisms involved in γ-secretase processing of the APP and Notch1 transmembrane domains. The Journal of Biological Chemistry. 2001;276(47):43756–43760. doi: 10.1074/jbc.C100410200. [DOI] [PubMed] [Google Scholar]
- 121.Sato T, Dohmae N, Qi Y, et al. Potential link between amyloid β-protein 42 and C-terminal fragment γ 49–99 of β-amyloid precursor protein. The Journal of Biological Chemistry. 2003;278(27):24294–20301. doi: 10.1074/jbc.M211161200. [DOI] [PubMed] [Google Scholar]
- 122.Chen AC, Selkoe DJ. Response to: Pardossi-Piquard etal., “Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by intracellular domains of βAPP and APLP.” Neuron 46, 541–554. Neuron. 2007;53(4):479–483. doi: 10.1016/j.neuron.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 123.Hébert SS, Serneels L, Tolia A, et al. Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression of putative target genes. EMBO Reports. 2006;7(7):739–745. doi: 10.1038/sj.embor.7400704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Waldron E, Isbert S, Kern A, et al. Increased AICD generation does not result in increased nuclear translocation or activation of target gene transcription. Experimental Cell Research. 2008;314(13):2419–2433. doi: 10.1016/j.yexcr.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 125.Belyaev ND, Nalivaeva NN, Makova NZ, Turner AJ. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Reports. 2009;10(1):94–100. doi: 10.1038/embor.2008.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Belyaev ND, Kellett KAB, Beckett C, et al. The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a β-secretase-dependent pathway. The Journal of Biological Chemistry. 2010;285(53):41443–41454. doi: 10.1074/jbc.M110.141390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bauer C, Pardossi-Piquard R, Dunys J, et al. β-secretase-mediated regulation of neprilysin: influence of cell density and aging and modulation by Imatinib. Journal of Alzheimer’s Disease. 2011;27(3):511–520. doi: 10.3233/JAD-2011-110746. [DOI] [PubMed] [Google Scholar]
- 128.Hong Y, Beckett C, Belyaev ND, Turner AJ. The impact of amyloid precursor protein signalling and histone deacetylase inhibition on neprilysin expression in human prostate cells. International Journal of Cancer. 2012;130(4):775–786. doi: 10.1002/ijc.26028. [DOI] [PubMed] [Google Scholar]
- 129.Goodger ZV, Rajendran L, Trutzel A, Kohli BM, Nitsch RM, Konietzko U. Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. Journal of Cell Science. 2009;122(20):3703–3714. doi: 10.1242/jcs.048090. [DOI] [PubMed] [Google Scholar]
- 130.Aydin D, Filippov MA, Tschäpe JA, et al. Comparative transcriptome profiling of amyloid precursor protein family members in the adult cortex. BMC Genomics. 2011;12, article 160 doi: 10.1186/1471-2164-12-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pardossi-Piquard R, Checler F. The physiology of the β-amyloid precursor protein Intracellular domain AICD. Journal of Neurochemistry. 2012;120(supplement 1):109–124. doi: 10.1111/j.1471-4159.2011.07475.x. [DOI] [PubMed] [Google Scholar]
- 132.Xu X, Zhou H, Boyer TG. Mediator is a transducer of amyloid-precursor-protein-dependent nuclear signalling. EMBO Reports. 2011;12(3):216–222. doi: 10.1038/embor.2010.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Turner AJ, Belyaev ND, Nalivaeva NN. Mediator: the missing link in amyloid precursor protein nuclear signalling. EMBO Reports. 2011;12(3):180–181. doi: 10.1038/embor.2011.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Qing H, He G, Ly PTT, et al. Valproic acid inhibits Aβ production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. Journal of Experimental Medicine. 2008;205(12):2781–2789. doi: 10.1084/jem.20081588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nalivaeva NN, Belyaev ND, Turner AJ. Sodium valproate: an old drug with new roles. Trends in Pharmacological Sciences. 2009;30(10):509–514. doi: 10.1016/j.tips.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 136.Kilgore M, Miller CA, Fass DM, et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 2010;35(4):870–880. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Eisele YS, Baumann M, Klebl B, Nordhammer C, Jucker M, Kilger E. Gleevec increases levels of the amyloid precursor protein intracellular domain and of the amyloid-β degrading enzyme neprilysin. Molecular Biology of the Cell. 2007;18(9):3591–3600. doi: 10.1091/mbc.E07-01-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vázquez MC, Vargas LM, Inestrosa NC, Alvarez AR. C-Abl modulates AICD dependent cellular responses: transcriptional induction and apoptosis. Journal of Cellular Physiology. 2009;220(1):136–143. doi: 10.1002/jcp.21743. [DOI] [PubMed] [Google Scholar]
- 139.Usmani BA, Shen R, Janeczko M, et al. Methylation of the neutral endopeptidase gene promoter in human prostate cancers. Clinical Cancer Research. 2000;6(5):1664–1670. [PubMed] [Google Scholar]
- 140.Dai J, Shen R, Sumitomo M, et al. Tumor-suppressive effects of neutral endopeptidase in androgen-independent prostate cancer cells. Clinical Cancer Research. 2001;7(5):1370–1377. [PubMed] [Google Scholar]
- 141.Hama E, Saido TC. Etiology of sporadic Alzheimer’s disease: somatostatin, neprilysin, and amyloid β peptide. Medical Hypotheses. 2005;65(3):498–500. doi: 10.1016/j.mehy.2005.02.045. [DOI] [PubMed] [Google Scholar]