

Abstract
The use of proteins for therapeutic applications requires the protein to maintain sufficient activity for the period of in vivo treatment. Many proteins exhibit a short half-life in vivo and, thus, require delivery systems for them to be applied as therapeutics. The relative biocompatibility and the ability to form functionalized bioconjugates via simple chemistry make gold nanoparticles excellent candidates as protein delivery systems. Herein, two protocols for coupling proteins to gold nanoparticles were compared. In the first, the strong biomolecular binding between biotin and streptavidin was used to couple catalase to the surface of gold nanoparticles. In the second protocol, the formation of an amide bond between carboxylic acid coated gold nanoparticles and free surface amines of catalase using carbodiimide chemistry was performed. The stability and kinetics of the different steps involved in these protocols were studied using UV-Visible spectroscopy, dynamic light scattering, and transmission electron microscopy. The addition of mercaptoundecanoic acid in conjugation with (N-(6-(biotinamido)hexyl)-3′-(2′-pyridyldithio)-propionamide increased the stability of biotinylated gold nanoparticles. Although the carbodiimide chemistry based bioconjugation approach exhibited a decrease in catalase activity, the carbodiimide chemistry based bioconjugation approach resulted in more active catalase per gold nanoparticle compared to that of mercaptoundecanoic acid stabilized biotinylated gold nanoparticles. Both coupling protocols resulted in gold nanoparticles loaded with active catalase. Thus, these gold nanoparticle systems and coupling protocols represent promising methods for the application of gold nanoparticles for protein delivery.
Keywords: Gold nanoparticles, carbodiimide chemistry, biotin-streptavidin binding, catalase
1. Introduction
Carrier systems of nanoscale dimensions such as liposomes, polymeric particles, and microemulsion droplets are being widely studied for the delivery of various biomolecules [1, 2]. In the case of proteins/enzymes, an important requirement for their immobilization to various carrier systems is that the protein should remain in its active form and be able to carry out necessary functions efficiently. Although physical adsorption of proteins via hydrophobic and electrostatic interactions is experimentally simple, the loss of the protein/enzyme from the carrier once in contact with the in vivo environment makes chemically mediated immobilization via covalent bonding attractive for increasing the half life of proteins for in vivo therapeutic applications. Additionally, irreversible covalent binding generally leads to high levels of surface coverage. The easy synthesis of inorganic nanoparticles, their high surface to volume ratio, and the ability to control their size prove vital for immobilizing proteins over their surfaces for therapeutic applications [3]. Added advantages of gold nanoparticles (GNPs) include biocompatibility, relative non-toxicity, and the ability to form functionalized bioconjugates via simple chemistry [4]. Some of the in vivo therapeutic applications of functionalized GNPs include tumor necrosis factor delivery [5], treatment of colon carcinomas with Paclitaxel grafted gold colloids [6], cellular drug delivery [7–9], gene therapy [10–14], thermoablative therapy [15, 16] and drug release [17–19]. With the advent of such successful applications, GNPs can also prove to be suitable carriers for therapeutic protein delivery.
Gold nanoparticles serve as excellent candidates for protein bioconjugation because they readily react with the amino and cysteine thiol groups of proteins. Unlike polymeric systems which prove effective in reducing the proteolytic degradation and deactivation of the therapeutic reagent [2, 20–22], proteins conjugated to GNPs are directly exposed to their environment and potential proteolysis. Although this exposure is a potential drawback, it overcomes substrate diffusion/accessibility limitations into encapsulated systems. One potential method to overcome proteolysis is to use polymeric brushes (e.g., poly(ethylene glycol); PEG-based) in conjunction with proteins, which can minimize the access of proteolytic enzymes to the surface of particles. Further, the inherent properties of their metal core make GNPs an effective drug carrier platform for combinatorial diagnosis through heavy metal imaging along with protein therapy [23]. Also, issues of rapid in vivo therapeutic clearance, poor inherent localization at targeted sites, and the simultaneous protection of untargeted sites from undesired reactions can be overcome through the functionalization of GNPs with polymeric brushes such as PEG [24], and biomolecular targeting agents [3, 25]. Several proteins such as insulin, pepsin, glucose oxidase, horse radish peroxidase, xanthine oxidase, fungal protease, etc have been directly conjugated to gold nanoparticles [2, 26–30]. In most cases, this strategy alters the conformational structure and active center of the protein similar to that of physical adsorption, thereby causing a reduction in activity [31]. Herein, we report and compare two simple procedures for coupling a model enzyme catalase to GNPs. Catalase catalyzes the conversion of hydrogen peroxide, a harmful oxidizing agent, to water and molecular oxygen. Its antioxidant properties have been studied extensively for potential effectiveness in antioxidative therapy. However, catalase undergoes rapid elimination from the blood stream and demonstrates poor intracellular delivery [32]. Figure 1 shows a schematic representation of the two approaches utilized in this study for the coupling of catalase to GNPs. The first involves the biotinylation of both the GNP and catalase and then coupling them together using a streptavidin crosslinker. The second method uses carbodiimide chemistry to form amide bonds between carboxylic acid coated GNPs and amino groups of catalase. The first method takes advantage of the high affinity between biotin and streptavidin to form a biotin-streptavidin complex, which is among the strongest interactions in biology. The advantage of using carbodiimide chemistry is that it employs mild reaction conditions, endowing considerable versatility of bioconjugation for a wide variety of protein and enzyme molecules. The common feature of both procedures is the exploitation of the reaction and attachment of thiol molecules onto the surface of GNPs in the form of thiol-gold chemistry. Characterization of the coupling kinetics, stability of the modified particle, and the activity of catalase bound GNPs were done using UV-Visible spectroscopy, dynamic light scattering, and transmission electron microscopy (TEM). With these simple methods of bioconjugation, protein coupled GNPs can be useful for the delivery of proteins for biomedical applications.
Figure 1.
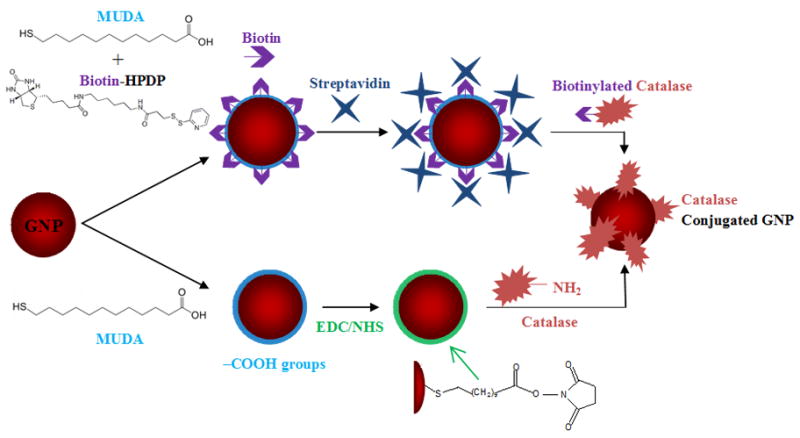
Schematic representation of the two approaches involved in the coupling of catalase to gold nanoparticles.
2. EXPERIMENTAL SECTION
2.1. Synthesis of biotinylated and carboxylic coated GNPs
Monodispersed gold nanoparticles were prepared using the Turkevich reduction of gold salts [33]. Briefly, a 1 mM aqueous solution of chloroauric acid (HAuCl4) was boiled under stirring. To this solution, 3 mM trisodium citrate in water was added to reduce HAuCl4 producing GNPs of sub-nanometer size. Tween 20 at 38 mg/ml was added to a 1 mg/ml final concentration to stabilize the particles for further functionalization. To incorporate biotin into the GNP a 4 mM stock solution of (N-(6-(biotinamido)hexyl)-3′-(2′-pyridyldithio)-propionamide (biotin-HPDP; Soltech Ventures) in dimethyl sulfoxide was added to the Tween stabilized GNPs so that the final concentration of biotin-HPDP was 0.5 mM, and the mixture incubated at room temperature for 4 hr. The biotinylated GNPs were then dialyzed using12 kDa molecular weight cutoff tubing against water to remove excess unreacted biotin-HPDP and stored for further use.
For the carbodiimide chemistry coupling method, carboxylic groups were introduced to the surface of GNPs by adding a 10 mM stock solution of mercaptoundecanoic acid in ethanol (MUDA; Asemblon) to Tween stabilized GNPs to a final concentration of 0.5 mM, followed by stirring the solution for 12 hrs at room temperature. The biotinylated GNPs and MUDA coated GNPs were dialyzed against water to remove free MUDA. Additionally GNPs containing varying concentrations of surface biotin were synthesized using MUDA as the stabilizing reagent. For this, the final concentration of MUDA was fixed at 0.5 mM, while biotin-HPDP was added at 50 μM for 1:10 biotin:MUDA GNPs and at 25 μM for 1:20 GNPs. Both biotin-HPDP and MUDA were added to the stabilized GNPs at the same time, and the reaction was allowed to proceed for approximately 12 hrs.
2.2. Biotinylation of Catalase
The biotinylation of catalase was done using succinimide chemistry. Briefly, to a 2 mg/ml catalase (Calbiochem) solution in 100 mM HEPES buffer, pH 8.0, was added biotin-N-hydroxysuccinimide (BNHS; Vector Labs) at 25 mg/ml in DMSO so that the final concentration of BNHS was 10 wt% of the enzyme to be biotinylated. The mixture was stirred occasionally for 3 hours, after which time 10 μl of 16.4 M ethanolamine was added to stop the reaction by reacting with any free biotin-NHS. The biotinylated catalase was dialyzed against 1 liter of 100 mM HEPES buffer, pH 8.0, with three buffer changes.
2.3. Coupling of catalase to the GNP surface via biotin-streptavidin binding
Purified biotinylated GNPs were dialyzed against HEPES prior to catalase coupling. Biotinylated GNPs in HEPES were treated with a threefold excess of streptavidin (Invitrogen; 100 μM) in 50 mM MES buffer, pH 5.5, for 3 hrs at room temperature. Excess streptavidin was used to avoid any crosslinking between biotinylated GNP particles. In order to remove any unbound streptavidin that may interact with biotinylated catalase, the streptavidin coated GNPs were subjected to three centrifugation and HEPES buffer wash cycles. To these particles biotinylated catalase (GNP:biotinylated catalase, 3:1 vol%) was added and permitted to bind for 3 hrs at room temperature. The resulting bioconjugated GNPs were centrifuged and washed with HEPES thrice and used as such.
2.4. Coupling of catalase to GNPs via carbodiimide chemistry
MUDA coated GNPs were dialyzed against 50 mM MES buffer, pH 5.5. A mixture of 17 μl of 100 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC; Thermo Scientific) and 63 μl of 50 mM N-hydroxysuccinimide (NHS; Thermo Scientific) in MES buffer was then added to 500 μl of MUDA coated GNPs. The reaction was allowed to proceed for 15–20 min, after which time 0.8 μl of β-mercaptoethanol was added to quench the unreacted excess EDC. The concentration of β-mercaptoethanol was then reduced to less than 5 mM by adding 1.75 ml of HEPES buffer, pH 7.4 containing varying concentrations of catalase. Conjugation via amide linkage was allowed to occur for 3 hrs at room temperature. Catalase conjugated GNPs were then centrifuged at 12,500 rpm for 10 min in an Accuspin centrifuge (Fisher Scientific) and washed thrice with 100 mM HEPES buffer, and used as such for further studies.
2.5. Characterization of nanoparticles
Biotinylation, streptavidin coating, binding of catalase, and the various steps of carbodiimide chemistry were followed by the change in the surface plasmon resonance peak using UV-Vis spectroscopy. Dynamic light scattering (DLS) was also used to observe changes in particle size with conjugation/functionalization. Transmission electron microscopy (TEM; Jeol 2010) was used to confirm the presence of enzyme over the surface of the GNPs. Staining of the particles was carried out using 2% phosphotungstic acid (Sigma), pH 7.4 (neutralized using 1 M potassium hydroxide). The presence of active catalase over the surface of GNPs was determined using the o-phenylene diamine assay (OPD assay; Sigma). Briefly, catalase conjugated GNPs were centrifuged, washed, and resuspended in PBS at pH 7.4. To the GNP-catalase, 3 ml of 2 mM H2O2 was added. Aliquots of 150 μl were removed at 1 min intervals over 5 min and the reactive oxygen product assayed using 100 μl of 40 μM OPD and 20 μl of 3 μM horseradish peroxidase (HRP; Calbiochem). The change in absorbance of o-phenylene diamine (OPD) at 490 nm, characteristic of the quantity of hydrogen peroxide remaining was measured and plotted as a function of time. The rate was calculated from the initial linear phase of the reaction.
2.6. In vivo injection and characterization of catalase bound GNP activity
To test the feasibility of injecting catalase bound GNPs into a mouse, we first tested the stability of our particles in mouse blood ex vivo. Both carbodiimide and biotin-streptavidin protocol synthesized GNP-catalase in buffer was added to mouse blood at a 1:40 dilution to approximate the dilution that would be obtained when injecting 50 μl of particles into the ~2ml of blood found in a mouse. The GNP-catalase-blood mixture was incubated at 37°C. Blood samples were taken every 15 minutes for 90 minutes. For each time point, RBCs were collected by centrifugation at 10,000rpm for 10 minutes and catalase activity in the plasma was measured in the plasma. For in vivo injection, 50 μl of 1 mg/ml of biotin protocol based GNP-catalase in HEPES buffer was injected into the retro-orbital sinus of mice. At time points of 5, 10, 30, and 60 min following injection 50 μl of blood samples were collected from the opposite retro-orbital sinus into tubes containing 100 units/ml heparin. In order to assay catalase in blood samples, red blood cells (RBCs) were removed from the serum via centrifugation (10000 rpm, 10 min); conditions that are known not to remove GNP-catalase. In triplicate for each mouse and time point, 5 μl of supernatant plasma was exposed to 195 μl of 1.5 mM H2O2 for 2 minutes at room temperature. 110 μl of each H2O2/plasma solution was then added to an assay well on a 96 well plate containing 75 μl HRP and 15 μl OPD and incubated for 5 min at room temperature. Absorbance was then measured at 590 nm to quantify catalase activity. For these animal studies, we complied with institutional ethical use protocols.
3. Results and Discussion
3.1. Demonstration of biotinylation of GNPs
Monodispersed GNPs synthesized via the Turkevich method were characterized in terms of size and their surface plasmon resonance (SPR) peak using dynamic light scattering and UV-Vis spectrometry. The size of GNPs was determined to be 9–11 nm showing a SPR peak at 525 nm. Using these parameters, the concentration of GNPs was calculated using the number density relationship equation as described by Haiss et al [34] to be about 10 nM. Biotinylation of GNPs was followed by measuring pyridine-2-thione release at 353 nm. Although preliminary studies of GNP biotinylation with biotin-HPDP showed product formation (result not shown), agglomeration to micron sized particles was observed. This was indicated by a time dependent red shift and subsequent broadening of the GNP SPR peak from 525 nm to about 683 nm. Agglomeration of biotinylated GNPs has been shown to be caused by hydrogen bonding between the inter-particle biotin molecules [35].
In order to avoid agglomeration associated with inter-particle hydrogen bonding, MUDA was added in varying ratios. We synthesized GNPs containing biotin:MUDA ratios of 1:10 and 1:20 (molar ratio). The negative charge on the surface of GNPs caused by deprotonation of the carboxylic groups provides sufficient repulsion between nanoparticles. For example, figure 2 shows the UV-Vis profiles of a 1:20 biotinylated GNP system at increasing reaction times. As seen in figure 2, the absorbance of pyridine-2-thione at 353 nm increases over time indicating the coupling of biotin to the GNP. Additionally, the GNPs undergo a red shift from 525 nm to 552 nm during the reaction indicating particle size increase (see below) and some minor agglomeration. The biotinylated GNPs were then dialyzed to remove unreacted biotin-HPDP and used for further analyses.
Figure 2.
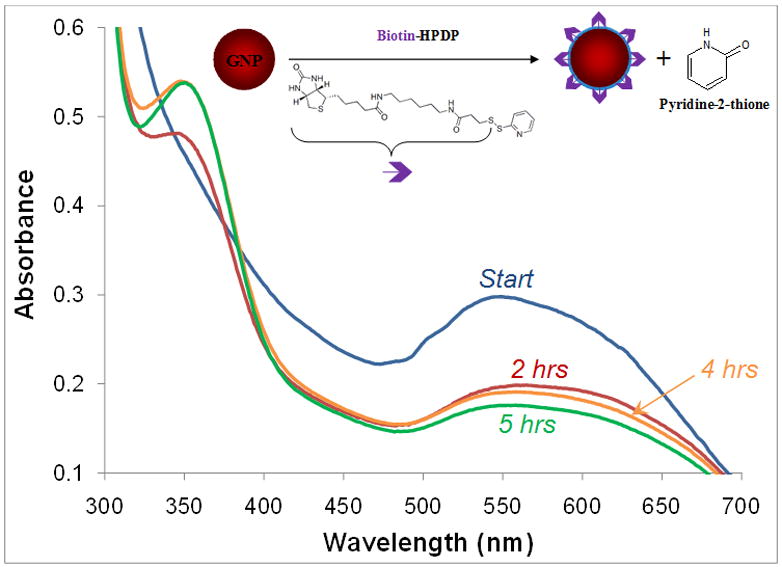
UV-Vis spectra showing biotinylation of GNPs as a function of time. The presence of the product pyridine-2-thione peak at 353 nm confirms biotinylation of GNPs.
The stability of biotinylated GNPs in the presence of tethered MUDA of differing ratios was analyzed to determine the working range of pH for coupling. Figure 3 shows the DLS size change of 100 % MUDA coated GNPs and 1:10, and 1:20 biotin:MUDA coated GNPs at different pHs. As observed from figure 3, the particle size/agglomeration of the GNPs decreases with increasing MUDA content for all pHs. The increase in MUDA content leads to increased negative charge of the GNPs at higher pH, thereby enabling the stabilization of the biotinylated GNPs. Therefore, the more MUDA over the surface, the more stable the biotinylated particles. Although an increase in pH should result in more deprotonation and subsequent stabilization of MUDA coated GNPs, Aslan et al. [36] reported a similar effect of agglomeration as shown in Figure 3. At longer time periods (>5 hrs), a fraction of larger sized peaks in the micron size range appeared due to continuous agglomeration, and these larger agglomerates precipitating out of solution. In order to avoid issues of particle aggregation at higher standing periods, we carried out streptavidin coating of the particle suspended buffer solutions immediately, which in turn stabilized the particles from aggregation. We chose 1:20 GNPs as the system for additional studies as it was stable enough for streptavidin binding and subsequent coupling of catalase.
Figure 3.

Effect of MUDA concentration on stabilizing biotinylated GNPs. DLS data showing the agglomeration of MUDA GNP, 1:10 GNP, and 1:20 GNP at pH 5.5, 6.5, and 7.5 respectively. As the amount of MUDA to biotin increased (MUDA GNP>1:20 GNP>1:10 GNP), the stability of the particles also increased. (N=3)
3.2. Demonstration of streptavidin binding to biotinylated GNPs
The reactive availability/affinity of biotinylated GNP to undergo biomolecular binding to streptavidin was analyzed using UV-Vis spectroscopy. Streptavidin has four binding sites and a hydrodynamic diameter of 5 nm. Based on the surface area availability from a 10 nm sized GNP, a maximum of ~16 streptavidin molecules can coat a biotinylated GNP. In other words, for a 10 nM concentration of GNP solution that is biotinylated, a 160 nM streptavidin concentration can be estimated as being needed for coating all GNPs. In order to confirm the binding between biotin and streptavidin, which is also an indirect way to confirm the presence of biotin over the surface of GNPs, a low concentration of 9 nM streptavidin in MES buffer was added to the 1:20 GNPs. This low concentration of available biotin binding sites (36 nM available biotin binding sites) was less than the total amount of biotinylated GNPs and should cause crosslinking between the biotinylated GNPs (with streptavidin being the crosslinker). As observed in Figure 4, at the initiation of binding, biotinylated GNPs exhibit their characteristic SPR peak value at 526 nm similar to that of the non-biotinylated GNP control sample (100% MUDA coated GNPs; right image of the inset). With time, biotin-streptavidin binding occurred and led to crosslinking between particles as indicated by the broadening of the spectra (0–2 hours). After 24 and 48 hours, all the streptavidin appeared bound as evidenced by no further crosslinking and the settling of agglomerated crosslinked particles leaving a clear solution on the top (left image of the inset). This confirmed the availability of biotin over the surface of GNPs for coupling with biotinylated catalase via intermediate streptavidin binding. However, the agglomeration under these low streptavidin concentrations precluded them for use for catalase coupling. Instead, using excess streptavidin (100 μM) provides an increased number of available biotin binding sites and results in a decreased probability of adjacent biotinylated particles crosslinking (black dotted absorbance profile). In other words, the streptavidin concentration was high enough to coat the entire biotinylated surface of GNPs, thereby avoiding interparticle biotin-streptavidin crosslinking. These streptavidin coated GNPs were then treated with biotinylated catalase, synthesized as described in methods. An assay of biotinylation (Quant Tag Biotin Kit, Vector Labs) gave a value of 128 nmoles of biotin/mg of catalase (~30 nmols biotin/nmol catalase). Similar to the binding between the biotin of the biotinylated GNP and streptavidin, binding between the binding sites of streptavidin and the biotin in biotinylated catalase was used for the coupling reaction.
Figure 4.
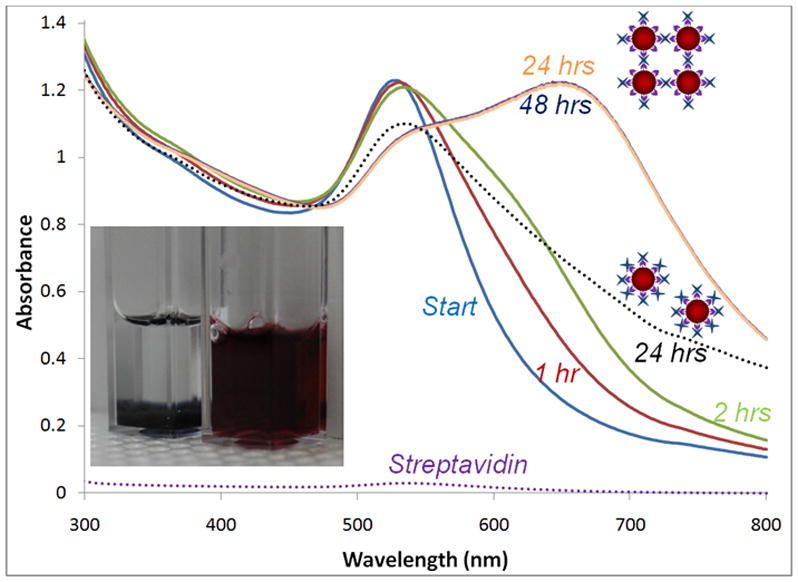
Kinetic study showing the crosslinking of biotinylated GNP in the presence of low streptavidin concentration (9 nM) confirming biotin-streptavidin interaction. Also shown as a dotted line is the non-crosslinked high concentration streptavidin (100 μM) coated GNP profile. Inset shows the initial non-crosslinked dispersed GNPs in the right and the final crosslinked settled GNPs in the left. Also shown in black dotted line is the streptavidin coated biotinylated GNPs (using high streptavidin concentration).
3.3. Confirmation of the presence of active catalase over biotin-streptavidin protocol GNPs
Streptavidin coupled catalase-GNPs were assayed for catalase activity by measuring the oxidation of OPD at 490 nm in the presence of H2O2 and horse radish peroxidase. One unit of catalase activity (U) is defined as the amount of enzyme cleaving 1 μmol of H2O2 in one minute. The activity of catalase was 44,350 U and 41,600 U before and after biotinylation, respectively, showing that biotinylation per se did not significantly affect catalase activity, which is similar to the result obtained by Muzykantov et al. [37]. Table 1 shows the activity for catalase bound to the surface of GNPs via the biotin-streptavidin interaction. Non-biotinylated catalase was used as the control. Very little non-specific adsorption of catalase to streptavidin coated GNPs is observed. As the amount of biotinylated catalase increased, an increase in the activity of catalase-bound GNPs was observed, which leveled off presumably due to the saturation of the GNP surface.
Table 1.
Increased activity of biotin-streptavidin bound catalase to GNPs with increasing amounts of biotinylated catalase added to streptavidin coated GNPs.
(Chirra et al. Catalase coupled gold nanoparticles: Comparison between carbodiimide and biotin-streptavidin methods)
| Concn. of Biotinylated Catalase | Catalase Activity (μU)/GNP |
|---|---|
| 8 μM (non-biotinylated; control) | ~0.05 |
| 0.4 μM | 1.7 |
| 0.8 μM | 7 |
| 2 μM | 41 |
| 6 μM | 65 |
| 8 μM | 66 |
The enzymatic effect of coupling biotinylated catalase to streptavidin coated GNPs was also studied by binding 20 μl of biotinylated catalase (2 mg/ml; 41600 U) to streptavidin coated GNPs. The activity of the free catalase in the supernatant and bound catalase to the GNP pellet after centrifugation and washing was determined to be 15575 U and 25600 U (16 μU/GNP), respectively. Their combined activity adds to a total of 41175 U, which is within experimental error of the input biotinylated catalase activity. So, the binding of biotinylated catalase to GNPs via biotin-streptavidin binding did not appear to result in any significant loss of enzymatic activity.
3.4. Activation of MUDA coated GNPs to bind catalase via carbodiimide chemistry
In the case of coupling catalase to GNPs using carbodiimide chemistry, catalase was coupled to the GNPs through the reaction of its amino groups with EDC activated esters of MUDA carboxylic acids on the GNP surface. As shown in Figure 5, MUDA coated GNPs exhibited a characteristic surface plasmon resonance (SPR) peak value at 530 nm. With the addition of EDC and NHS, carboxyl groups were converted into thioesters that resulted in slight aggregation indicated by the red shift of the SPR peak value to 621 nm. The addition of catalase to the activated particles led to the formation of an amide bond between the amino groups of catalase and the activated carboxyl group of MUDA coated GNPs. The catalase coated particles showed a characteristic Soret iron (III) heme structure absorbance peak at 406 nm confirming the attachment of the enzyme [38]. Shoulder peaks of activated GNPs are also visible in the catalase spectra.
Figure 5.
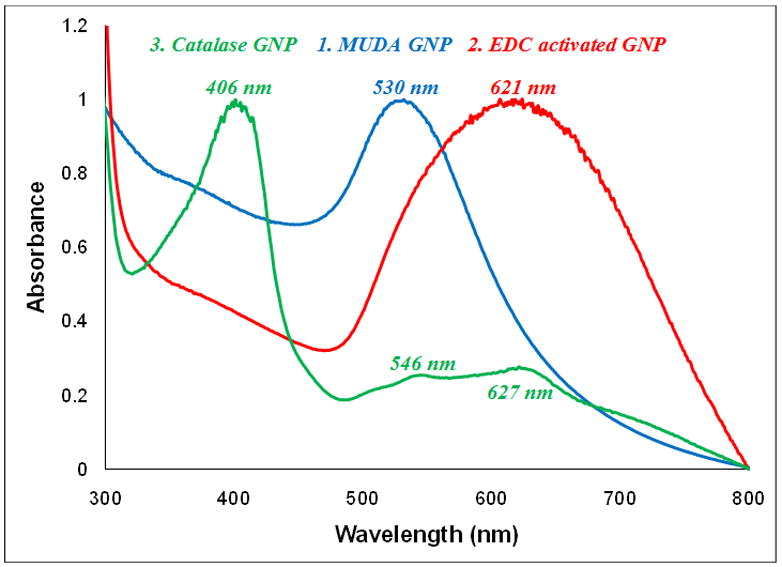
Normalized UV-Vis spectra of the carbodiimide chemistry protocol confirming the activation of MUDA coated GNPs and the subsequent attachment of enzyme catalase.
3.5. Confirmation of the presence of active carbodiimide coupled catalase on GNPs
The OPD assay was used to determine the amount of active catalase bound to GNPs through carbodiimide chemistry. Table 2 shows the activity for amide bound catalase to the surface of MUDA coated GNPs. Using the carbodiimide chemistry, we achieved catalase coated GNPs, similar to that of the biotinylated GNP protocol with minimal non-specific catalase adsorption onto the particle. As expected, as the concentration of catalase increased, the attachment of catalase via amide linkage to the GNP surface also increased. Saturation of catalase occurred as indicated by a constant activity with increasing added catalase. The enzymatic effect of covalently binding catalase to GNPs via an amide linkage was studied by binding 20 μl of catalase (2 mg/ml; 44,350 U) to MUDA coated GNPs. The activity of free unbound catalase in the supernatant and bound catalase to the GNP pellet was determined to be 8,150 U and 25,075 U (10 μU/GNP) respectively. The combined activity, which comes to a total of 33,225 U, is somewhat less than the activity of the input catalase. Assuming unbound catalase in the supernatant had no activity loss, it is estimated that the bound catalase exhibited minimally 70% activity.
Table 2.
Activity of the carbodiimide coupled catalase to GNPs increases with increasing concentrations of catalase until apparent saturation is reached.
(Chirra et al. Catalase coupled gold nanoparticles: Comparison between carbodiimide and biotin-streptavidin methods)
| Concn. of Catalase used | Catalase Activity (μU)/GNP |
|---|---|
| 10 μM (no EDC added inactive GNPs; control) | ~0.02 |
| 0.01 μM | 0.4 |
| 0.1 μM | 7 |
| 1 μM | 93 |
| 5 μM | 125 |
| 10 μM | 131 |
3.6. Size and stability analysis of the catalase bound GNPs
TEM analysis of the active particles was compared to that of the Turkevich reduced GNPs. Citrate reduced GNPs (Figure 6A) showed minimal presence of an amorphous layer over its surface indicative of the charge stabilization of the particle provided by citrate ions. In the case of the biotin-streptavidin affinity based active GNP an amorphous layer of thickness ~ 6.5 nm was observed (Figure 6B). Although the size of catalase and streptavidin together is > 6.5 nm (hydrodynamic diameter of catalase is around 9 nm [39], while for streptavidin it is 5 nm), the TEM sample of catalase bound via streptavidin to the particle surface is in a dehydrated form. This amorphous layer of the enzyme is absent in the case of the initial citrate stabilized GNP. A similar result is observed in the case of catalase bound to GNP via carbodiimide chemistry (Figure 6C), where the thickness of the amorphous layer is around 4 nm. DLS analysis of the agglomeration as a function of time with catalase bound GNPs was also done over a period of one week. While, the size of biotin-streptavidin protocol based GNP-catalase remained stable in the size range of 65–85 nm over a period of one week, GNP-catalase synthesized using carbodiimide chemistry showed agglomeration to about 400 nm. The agglomeration seen with GNPs containing catalase coupled via carbodiimide chemistry can be attributed to the fact that the surface MUDA groups are modified to amide bonds, thereby leading to charge neutralization and subsequent destabilization. With biotin-streptavidin GNP-catalase, there is free MUDA that can be deprotonated and thus stabilize the particles via charge repulsion.
Figure 6.

TEM images showing (A) GN stabilized by citrate ions with the inset showing negligible amorphous layer, (B) biotin-streptavidin affinity protocol based catalase conjugated GNP (amorphous layer ~ 6.5 nm), and (C) carbodiimide chemistry protocol based catalase conjugated GNP (amorphous layer ~ 4 nm). (Scale bar = 5 nm)
3.7. Clearance of GNP-catalase from blood in vivo
Before injecting GNP-linked catalase into a mouse it was first necessary to determine the activity of the GNP-linked enzyme in mouse blood. This is necessary to determine the stability of the GNP-catalase in blood as well as the inactivation, if any, of the catalase itself from factors within the blood. Furthermore, blood removed from the animal could also be used to determine any basal level of catalase activity in mouse blood. Our results show that GNP-catalase was stable for at least 90 minutes in blood plasma at 37°C; however, a 20% reduction in activity was seen within the first time point in the plasma compared to a PBS control (data not shown). Following this initial drop in activity there was no additional loss of catalase activity over the 90 minute test period indicating the GNP-Catalase is stable in blood.
Knowing the GNP-that catalase activity on GNPs was stable in blood, we injected mice with 50 μl of GNP-catalase for in vivo half life studies. We chose the biotin-streptavidin protocol GNP-catalase particles for this purpose, since their size was smaller and more stable than that of the carbodiimide protocol derived GNP-catalase as noted above. Figure 7 shows the activity of GNP-catalase in blood at various times after injection. The activity/μl plasma at the initial injection condition is the theoretical rate measured by in vitro conditions assuming the overall volume of blood in mice is 2 ml. On average, mice have about 2 mL of blood; however, it can vary between 1.5mL and 2.5 mL, indicated by the dotted lines in Figure 7 [40]. The half life in blood was estimated to be around 6 min, likely reflecting reticuloendothelial system uptake of the GNPs mostly by liver, spleen, and lung [41]. As previously noted [42], longer blood circulation of GNPs with minimum uptake by the RES is possible by coating the particle surface with hydrophilic polymers, such as poly(ethylene glycol) (PEG) derivatives. A detailed study on the effects of PEGylation of enzyme or GNP on in vivo half life of GNP bound enzyme/protein will be analyzed, but was beyond the scope of this study.
Figure 7.
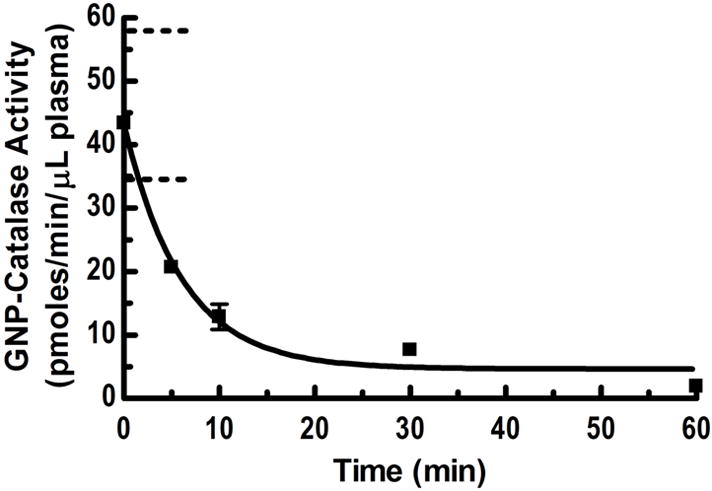
Clearance of GNP-catalase from the plasma of mice showing the half life of GNP-catalase is around 6 minutes (N=3). The zero time value is calculated theoretically at in vitro conditions based on a total blood volume of 2 ml in mice. The dotted lines represent the initial activity range for a 1.5 ml to 2.5 ml total plasma volume range in mice
3.8. Comparison between the biotin-streptavidin and carbodiimide methods
It is clear that both the biotin-streptavidin binding and carbodiimide chemistry methods readily attach catalase to the GNP surface. The carbodiimide chemistry protocol produces a larger amount of active catalase/GNP at saturation than that of the biotin-streptavidin protocol, which could prove useful for those cases of conjugation where higher loading capacity is required. In the case of the carbodiimide chemistry protocol, the entire surface area of MUDA coated GNP (~315 nm2) is available for binding with catalase. Based on a theoretical approximation, the surface area of 1:20 GNPs occupied by biotin is low (~15 nm2), and only this area is available for streptavidin and subsequent catalase binding. By increasing the ratio of biotin-HPDP to MUDA, the amount of assembled biotin or available surface for coupling catalase can be increased at the expense of biotinylated particle stability. The carbodiimide chemistry derived GNP-catalase showed increased aggregation with time as compared to biotin-streptavidin derived GNP-catalase which is likely due to absence of free MUDA. Thus, only the streptavidin derived GNP-catalase was utilized for in vivo studies. The aggregation could be reduced or eliminated by using sterically hindering carboxylic acid thiols such as PEGylated MUDA, thereby making them useful for in vivo applications. Also, the biotin-streptavidin protocol requires a two step synthesis procedure (synthesis of biotinylated catalase and then conjugation), while the carbodiimide chemistry protocol is carried out in a single step of activation and conjugation. Generalizing, the ideal procedure needed to bind a protein to GNP surface depends on several factors such as the effects of biotinylation on activity of the protein to be bound, size of the protein on binding to the GNP or streptavidin coated GNP, loss of activity with either of the protocols, and the stability of the final protein bound GNP.
4. Conclusion
Herein, we used the strong biomolecular interaction between biotin and streptavidin or covalent carbodiimide chemistry to couple catalase to gold nanoparticles. The biotinylation of GNPs was accomplished using biotin-HPDP. Dynamic light scattering showed that the interparticle aggregation between biotinylated GNPs decreased with increasing concentrations of the stabilizing reagent MUDA. The stability of the particles decreased with increasing pH. The OPD assay confirmed the presence of streptavidin bound active biotinylated catalase over GNPs. The different surface plasmon resonance peak values obtained via UV-Vis analysis confirmed the individual steps involved in the carbodiimide dependant coupling of catalase to MUDA coated GNPs. The OPD assay confirmed the presence of bound catalase to GNPs in high yields. The saturation values of activity showed that there was more active catalase on carbodiimide chemistry coupled GNPs than on biotin-streptavidin bound GNPs. The enzyme bound particles were stable enough to be used for studies in mice and exhibited a half life of around 6 min, which likely is a result of the uptake by the liver, spleen, and lung. Both protocols resulted in the attachment of active catalase to GNPs, and these methods are critical for the development of GNPs as potential carriers for protein delivery.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yokoyama M, Okano T. Targetable drug carriers: present status and a future perspective. Adv Drug Delivery Rev. 1996;21:77–80. [Google Scholar]
- 2.Dziubla TD, Muzykantov VR. Synthetic carriers for vascular delivery of protein therapeutics. Biotechnol Genet Eng Rev. 2006;22:267–298. doi: 10.1080/02648725.2006.10648074. [DOI] [PubMed] [Google Scholar]
- 3.Moghimi SM, Hunter AC, Murray JC. Nanomedicine: Current status and future prospects. FASEB J. 2005;19:311–330. doi: 10.1096/fj.04-2747rev. [DOI] [PubMed] [Google Scholar]
- 4.Chirra HD, Biswal D, Hilt JZ. Gold Nanoparticles and Surfaces: Nanodevices for Diagnostics and Therapeutics. In: Pathak Y, Thassu D, editors. Nanoparticulate Drug Delivery Systems (NPDDS) II: Formulation and Characterization. Informa Healthcare USA Inc; 2009. pp. 90–114. [Google Scholar]
- 5.Chen YH, Tsai CY, Huang PY, Chang MY, Cheng PC, Chou CH, Chen DH, Wang CR, Shiau AL, Wu CL. Methotrexate conjugated to gold nanoparticles inhibit tumor growth in a synergestic lung tumor model. Mol Pharmaceutics. 2007;4:713–722. doi: 10.1021/mp060132k. [DOI] [PubMed] [Google Scholar]
- 6.Gibson JD, Khanal BP, Zubarev ER. Paclitaxel functionalized gold nanoparticles. J Am Chem Soc. 2007;129:11653–11661. doi: 10.1021/ja075181k. [DOI] [PubMed] [Google Scholar]
- 7.Hone DC, Walker PI, Evans-Gowing R, FitzGerald S, Beeby A, Chambrier I, Cook MJ, Russell DA. Generation of cytotoxic singlet oxygen via phthalocyanine stabilized gold nanoparticles: a potential delivery vehicle for photodynamic therapy. Langmuir. 2002;18:2985–2987. [Google Scholar]
- 8.Saito G, Swanson JA, Lee KD. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv Drug Delivery Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 9.Sapsford KE, Berti L, Mendintz IL. Materials for fluorescence energy transfer analysis: beyond traditional donor acceptor combinations. Angew Chem Int Ed. 2006;45:4562–4589. doi: 10.1002/anie.200503873. [DOI] [PubMed] [Google Scholar]
- 10.McIntosh CM, Eposito EA, Boal AK, Simard JM, Martin CT, Rotello VM. Inhibition of DNA transcription using cationic mixed monolayer protected gold clusters. J Am Chem Soc. 2001;123:7626–7629. doi: 10.1021/ja015556g. [DOI] [PubMed] [Google Scholar]
- 11.Han G, Martin CT, Rotello VM. Stability of gold nanoparticle bound DNA towards biological, physical, and chemical agents. Chem Biol Drug Des. 2006;67:78–82. doi: 10.1111/j.1747-0285.2005.00324.x. [DOI] [PubMed] [Google Scholar]
- 12.Sandhu KK, McIntosh CM, Simard JM, Smith SW, Rotello VM. Gold nanoparticle mediated transfection of mammalian cells. Bioconjugate Chem. 2002;13:3–6. doi: 10.1021/bc015545c. [DOI] [PubMed] [Google Scholar]
- 13.Thomas M, Klibanov AM. Conjugation to gold nanoparticles enhances polyethyleneimines transfer of plasmid DNA into mammalian cells. Proc Natl Acad Sci U S A. 2003;100:9138–9143. doi: 10.1073/pnas.1233634100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AKR, Han MS, Mirkin CA. Oligonucleotide modified gold nanoparticles for intracellular gene regulation. Science. 2006;312:1027–1030. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- 15.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Nanoshell mediated near infrared thermal therapy of tumors under magnetic resonance guidance. Proc Natl Acad Sci U S A. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirsch LR, Jackson JB, Lee A, Halas NJ, West JL. A whole blood immunoassay using gold nanoshells. Anal Chem. 2003;75:2377–2381. doi: 10.1021/ac0262210. [DOI] [PubMed] [Google Scholar]
- 17.Radt B, Smith A, Caruso F. Optically addressable nanostructured capsules. Adv Mater. 2004;16:2184–2189. [Google Scholar]
- 18.Angelatos AS, Radt B, Caruso F. Light responsive polyelectrolyte/gold nanoparticle microcapsules. J Phys Chem B. 2005;109:3071–3076. doi: 10.1021/jp045070x. [DOI] [PubMed] [Google Scholar]
- 19.West JL, Halas NJ. Applications of nanotechnology to biotechnology. Curr Opin Biotechnol. 2000;11:215–217. doi: 10.1016/s0958-1669(00)00082-3. [DOI] [PubMed] [Google Scholar]
- 20.Dzuibla TD, Shuvaev VV, Hong NK, Hawkins BJ, Madesh M, Takano H, Simone E, Nakada MT, Fisher A, Albelda SM, Muzykantov VR. Endothelial targeting of semi-permeable polymer nanocarriers for enzyme therapies. Biomaterials. 2008;29:215–227. doi: 10.1016/j.biomaterials.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong J, Luxenhofer R, Yi X, Jordan R, Kabanov AV. Protein modification with amphiphilic block copoly(2-oxazoline)s as a new platform for enhanced drug delivery. Mol Pharm. 2010;7:984–992. doi: 10.1021/mp100102p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simone EA, Dziubla TD, Arguiri E, Vardon V, Shuvaev VV, Christofidou-Solomidou M, Muzykantov VR. Loading PEG-catalase into filamentous and spherical polymer nanocarriers. Pharm Res. 2009;26:250–260. doi: 10.1007/s11095-008-9744-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duncan B, Kim C, Rotello VM. Gold nanoparticle platforms as drug and biomacromolecule delivery systems. J Controlled Release. 2010;148:122–127. doi: 10.1016/j.jconrel.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eghtedari M, Liopo AV, Copland JA, Oraevsky AA, Motamedi M. Engineering of hetero-functional gold nanorods for the in vivo molecular targeting of breast cancer cells. Nano Letters. 2009;9:287–291. doi: 10.1021/nl802915q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dixit V, Bossche J, Sherman DM, Thompson DH, Andres RP. Synthesis and grafting of thioctic acid-PEG-folate conjugates onto Au nanoparticles for selective targeting of cancer cells. Bioconjugate Chem. 2006;17:603–609. doi: 10.1021/bc050335b. [DOI] [PubMed] [Google Scholar]
- 26.Bhumkar DR, Joshi HM, Sastry M, Pokharkar VB. Chitosan reduced gold nanoparticles as novel carriers for transmucosal delivery of insulin. Pharm Res. 2007;24:1415–1426. doi: 10.1007/s11095-007-9257-9. [DOI] [PubMed] [Google Scholar]
- 27.Phadtare S, Kumar A, Vinod VP, Dash C, Palaskar DV, Rao M, Shukla PG, Sivaram S, Sastry M. Direct assembly of gold nanoparticle shells on polyurethane microsphere cores and their application as enzyme immobilization templates. Chem Mater. 2003;15:1944–1949. [Google Scholar]
- 28.Xiao Y, Patolsky F, Katz E, Hainfeld JF, Willner I. Plugging into enzymes: nanowiring of redox enzymes by a gold nanoparticle. Science. 2003;299:1877–1881. doi: 10.1126/science.1080664. [DOI] [PubMed] [Google Scholar]
- 29.Stoneheurner JG, Zhao J, O’Daly JP, Crumbliss AL, Henkens RW. Comparison of colloidal gold electrode fabrication methods: the preparation of a horse radish peroxidase enzyme electrode. Biosens Bioelectron. 1992;7:421–428. doi: 10.1016/0956-5663(92)85041-8. [DOI] [PubMed] [Google Scholar]
- 30.Zhao J, O’Daly JP, Henkens RW, Stoneheurner JG, Crumbliss AL. A xanthine oxidase/colloidal gold enzyme electrode for amperometric biosensor applications. Biosens Bioelectron. 1996;11:493–502. [Google Scholar]
- 31.Patel N, Davies MC, Hartshorne M, Heaton RJ, Roberts CJ, Tendler SJB, Williams PM. Immobilization of protein molecules onto homogenous and mixed carboxylate-terminated self assembled monolayers. Langmuir. 1997;13:6485–6490. [Google Scholar]
- 32.Kozower BD, Christopher-Solomidou M, Sweitzer TD, Muro S, Buerk DG, Solomides CC, Albelda SM, Patterson GA, Muzykantov VR. Immunotargeting of catalase to the pulmonary endothelium alleviates oxidative stress and reduce acute lung transplantation injury. Nat Biotechnol. 2003;21:392–398. doi: 10.1038/nbt806. [DOI] [PubMed] [Google Scholar]
- 33.Turkevich J, Stevenson PC, Hillier J. A study of nucleation and growth process in the synthesis of gold colloids. Faraday Discuss. 1951;11:55–75. [Google Scholar]
- 34.Haiss W, Thanh NTK, Aveyard J, Fernig DG. Determination of size and concentration of gold nanoparticles from UV-Vis spectra. Anal Chem. 2007;79:4215–4221. doi: 10.1021/ac0702084. [DOI] [PubMed] [Google Scholar]
- 35.Strzelczyk AA, Dobrowolski JC, Mazurek AP. On the confirmation of biotin molecule. J Molec Struc (Theochem) 2001;541:283–290. [Google Scholar]
- 36.Aslan K, Perez-Luna PH. Quenched emission of fluorescence by ligand functionalizaed gold nanoparticles. Langmuir. 2002;18:6059–6065. doi: 10.1023/b:jofl.0000031821.74706.ea. [DOI] [PubMed] [Google Scholar]
- 37.Muzykantov VR, Atochina EN, Ischiropolous H, Danilov VM, Fisher AB. Immunotargeting of antioxidant enzyme to the pulmonary endothelium. Proc Natl Acad Sci U S A. 1996;93:5213–5218. doi: 10.1073/pnas.93.11.5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen X, Xie H, Kong J, Deng J. Characterization for didodecyldimethylammonium bromide liquid crystal film entrapping catalase with enhanced direct electron transfer rate. Biosens Bioelectron. 2001;16:115–120. doi: 10.1016/s0956-5663(00)00138-x. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Chi Q, Zhang B, Dong S, Wang E. Molecular characterization of beef liver catalase by scanning tunneling microscopy. Electroanalysis. 1998;10:738–746. [Google Scholar]
- 40.Riches AC, Sharp JG, Thomas DB, Smith SV. Blood volume determination in the mouse. J Physiology. 1973;228:279–284. doi: 10.1113/jphysiol.1973.sp010086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sonavane G, Tomoda K, Maknio K. Biodistribution of colloidal gold nanoparticles after intravenous administration: effect of particle size. Colloids Surf B. 2008;66:274–280. doi: 10.1016/j.colsurfb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Bergen JM, Recum HAV, Goodman TT, Massey AP, Pun SH. Gold nanoparticles as a versatile platform for optimizing physicochemical parameters for targeted drug delivery. Macromol Biosci. 2006;6:506–516. doi: 10.1002/mabi.200600075. [DOI] [PubMed] [Google Scholar]