

Abstract
The MUC1 cytoplasmic tail (MUC1.CT) conducts signals from spatial and extracellular cues (growth factor and cytokine stimulation) to evoke a reprogramming of the cellular transcriptional profile. Specific phosphorylated forms of the MUC1.CT achieve this function by differentially associating with transcription factors and redirecting their transcriptional regulatory capabilities at specific gene regulatory elements. The specificity of interaction between MUC1.CT and several transcription factors is dictated by the phosphorylation pattern of the 18 potential phosphorylation motifs within the MUC1.CT. To better appreciate the scope of differential gene expression triggered by MUC1.CT activation, we performed microarray gene expression analysis and ChIP-chip promoter analysis and identified the genome-wide transcriptional targets of MUC1.CT signaling in pancreatic cancer. On a global scale, MUC1.CT preferentially targets genes relating to invasion, angiogenesis and metastasis, suggesting that MUC1.CT signaling contributes to establishing a reactive tumor microenvironment during tumor progression to metastatic disease. We examined in detail the molecular mechanisms of MUC1.CT signaling that induces expression of connective tissue growth factor (CTGF/CCN2), a potent mediator of ECM remodeling and angiogenesis. We demonstrate a robust induction of CTGF synthesis and secretion in response to serum factors that is enabled only when MUC1 is highly expressed. We demonstrate the requirement of phosphorylation at distinct tyrosine motifs within the MUC1.CT for MUC1-induced CTGF expression and demonstrate a phosphorylation-specific localization of MUC1.CT to the CTGF promoter. We found that MUC1 reorganizes transcription factor occupancy of genomic regions upstream of the CTGF gene, directing β-catenin and mutant p53 to CTGF gene regulatory elements to promote CTGF expression and destabilizing the interaction at these regions of the transcriptional repressor, c-Jun. With this example we illustrate the capacity of MUC1.CT to mediate transcription factor activity in a context-dependent manner to achieve widespread and robust changes in gene expression and facilitate creation of the reactive tumor microenvironment.
Keywords: MUC1, CTGF, Signal Transduction, Transcriptional Activation, Pancreatic Cancer
Introduction
Acquisition of the metastatic phenotype requires a variety of changes within a tumor cell, including activation of signaling pathways involved in motility and invasion. Successful primary tumor progression to metastatic disease is dependent on other nearby cell types and the extracellular composition of the niche in which the tumor cell resides, collectively referred to as the tumor microenvironment. A highly reactive tumor environment facilitates tumor cell invasion, angiogenesis, and metastasis in part by providing cues in the form of growth factors and cytokines that promote the dissemination of tumor cells to distal sites. The tumor itself participates in formation of this dynamic niche via synthesis and release of secreted factors that contribute to establishment of a reactive tumor microenvironment, by recruiting fibroblasts and endothelial cells, promoting neovascularization, and regulating the activity of protease capable of remodeling the extracellular matrix. Identifying the key players acting within tumor cells that orchestrate formation of the reactive tumor microenvironment identifies promising targets for the treatment of metastatic carcinoma.
Expression of the cell surface mucin MUC1 is elevated in most tumor types, including pancreatic ductal adenocarcinoma (PDAC) (Burdick et al. , 1997; Qu et al. , 2004). MUC1 promotes tumor pathogenesis by influencing cell proliferation (Tsutsumida et al. , 2006; Yuan et al. , 2008) and apoptosis (Agata et al. , 2008; Gao et al. , 2009; Raina et al. , 2008) and also contributes to the invasive and metastatic potential of pancreatic tumors (Kohlgraf et al. , 2003; Masaki et al. , 1999; Singh et al. , 2007; Tsutsumida et al. , 2006). MUC1 is a type I transmembrane protein comprised primarily of a large extracellular domain, containing a variable number of differentially O-glycosylated, tandemly repeated sequences (VNTR) rich in serine, threonine, and proline residues, and an autoproteolytic SEA (sperm protein-enterokinase-agrin) domain. Much of the oncogenic activity of MUC1 in malignant tissues can be attributed to robust reprogramming of the cellular transcriptional profile that is dictated by the small, 72 amino acid cytoplasmic tail, which functions as a molecular sensor and displays a unique capacity to convey information to the nucleus about the activation status and spatial orientation of receptor tyrosine kinases and other cell surface signaling molecules‥
The highly conserved cytoplasmic tail of MUC1 (MUC1.CT) receives post-translational modifications from growth factor receptor tyrosine kinases (RTKs) and engaging in specific interactions with a variety of transcription factors, directly impacting their transcriptional regulatory capacity (Carson, 2008; Singh and Hollingsworth, 2006). MUC1.CT contains 18 serine, threonine, and tyrosine sites of potential phosphorylation and the phosphorylation pattern of MUC1.CT directs its physical association with cell signaling mediators and transcription factors (Singh and Hollingsworth, 2006). Several intracellular kinases and receptor tyrosine kinases (RTKs) have been shown to phosphorylate MUC1.CTin vivo and in vitro (Li et al. , 1998; Li et al. , 2001a; Li et al. , 2001b; Singh et al. , 2007; Singh et al. , 2008). Activation of Met by HGF stimulation induces physical association of MUC1.CT and Met, as well as phosphorylation of MUC1.CT at the tyrosine residue within its YHPM motif. The specific phosphorylation of MUC1.CT at YHPM enables its interaction with p53 (Singh et al. , 2008). Similarly, phosphorylation of MUC1.CT at the tyrosines within the YVPP and YHPM motifs by PDGFR-β following PDGF-BB stimulation enhances colocalization with β-catenin (Singh et al. , 2007), demonstrating a specificity in the binding of MUC1.CT to transcription factors that is encoded by the phosphorylation pattern of MUC1.CT. The cytoplasmic tail of MUC1 has also been detected in the nucleus (Wen et al. , 2003), and MUC1.CT colocalizes with mutant p53 and β-catenin in cell nuclei following HGF and PDGF-BB stimulation, respectively (Singh et al. , 2007; Singh et al. , 2008). Though the precise mechanism of cleavage/internalization and subsequent nuclear translocation of MUC1.CT has not been fully elucidated, it is clear that MUC1.CT affects the activity of transcriptional regulators once in the nucleus. MUC1.CT has been shown to regulate the transcription of several genes implicated in tumor pathogenesis and has been detected by chromatin immunoprecipitation at gene regulatory elements in transcriptional complexes (Singh et al. , 2008; Wei et al. , 2005; Wei et al. , 2007). With no intrinsic DNA binding domain, MUC1.CT acts as a mediator that indirectly alters cellular transcription profiles by interacting with and redirecting the regulatory activity of transcription factors. Understanding the regulatory functions of MUC1.CT is crucial to appreciating its role in the onset and progression of pancreas tumors.
We illustrate here an example of the capability of MUC1.CT to conduct extracellular signals in a manner that demonstrates both specificity and complexity, redirecting transcription factor regulatory function to influence gene expression and dramatically alter the behavior of the cell. Signaling through the MUC1.CT directly regulates expression of connective tissue growth factor (CTGF/CCN2), a 36-38 kDa protein in the CCN (CTGF/Cyr61/Nov) family of secreted factors that have diverse biological activities, including roles in chemotaxis, motility, adhesion, apoptosis, and cell growth (Bennewith et al. , 2009; Brigstock, 2003). CTGF expression is observed in a variety of cell types, including endothelial cells and fibroblasts and is elevated in many tumor types and cell lines, including PDAC, suggesting a role in tumor progression and metastasis (Chu et al. , 2008; Wenger et al. , 1999). CTGF has been detected in tumor cells, stromal cells, as well as proliferating endothelial cells, implicating CTGF in the formation of a reactive tumor microenvironment. Studies using recombinant human CTGF in vitro have demonstrated a potency in inducing angiogenesis that is greater than the well-studied angiogenic factors bFGF and VEGF (Shimo et al. , 1999). Targeting CTGF in vivo using a humanized monoclonal antibody recognizing CTGF (FG-3019) significantly reduced tumor cell proliferation, tumor volume, and CD31 immunoreactivity of tumor sections in an orthotopic mouse model of PDAC (Aikawa et al. , 2006). We report CTGF is a novel target of MUC1.CT signaling and illustrate for the first time a molecular mechanism for the phosphorylation-dependent regulation of transcription factor activity and gene expression by MUC1.CT.
Results
MUC1 increases CTGF/CCN2 expression in pancreatic tumors cells
The MUC1.CT conducts signals from the cell surface by associating with and modifying the activity of transcriptional activators and co-activators (Singh et al. , 2007), which in turn affects cellular transcriptional profiles. We performed microarray analysis of mRNA expression of S2013 pancreatic tumor cells overexpressing MUC1 (S2013.MUC1) as compared to controls (S2013.Neo) to investigate the molecular mechanisms that enable MUC1-overexpressing pancreatic cancer cell lines to exhibit increased tumor growth, angiogenesis, and metastasis in vivo and identified several genes that were differentially expressed in MUC1 overexpressing cells, including a subset of genes implicated in tumor metastasis (Table 1). Among these, connective tissue growth factor (CTGF/CCN2) was identified as a target of MUC1 regulation. Real-time PCR analysis confirmed that CTGF mRNA levels are increased in S2013.MUC1 cells versus S2013.Neo (Figure 1a) and knockdown of MUC1 using siRNA in S2013 cells (S2013.MUC1KD) further reduced CTGF mRNA compared to S2013.Neo cells. An increase in CTGF mRNA was confirmed with overexpression of MUC1 in Capan1 cells (Capan1.MUC1). Further, HPAF2 adenocarcinoma cells, which naturally express high levels of MUC1, exhibit high levels of CTGF. Western blot analysis demonstrated correlative changes in intracellular dimeric CTGF (76 kDa) with MUC1 overexpression in the S2013, Capan1, and Panc1 pancreatic cancer cell lines (Figure 1b). Several secreted isoforms of CTGF have been reported, ranging in size from 30-38 kDa and differing primarily in their glycosylation (Masuko et al. , 2010; Yang et al. , 1998; Zarrinkalam et al. , 2003). Metabolic radiolabeling of S2013.Neo and S2013.MUC1 cells demonstrated an increase in secretion of CTGF isoforms of 36 and 38 kDa into culture media with MUC1 overexpression (Figure 1c).
Table 1. Differential expression of invasion and metastasis-related genes by MUC1.
| Genes more highly expressed (>2.5-fold) in S2013.MUC1 versus S2013.Neo | ||
|---|---|---|
| Accession ID | Gene Name | Fold Change |
| NM_000891 | potassium inwardly-rectifying channel, subfamily J, member 2 (KCNJ2) | 8.07 |
| NM_003246 | thrombospondin 1 (THBS1) | 6.46 |
| NM_001901 | connective tissue growth factor (CTGF) | 5.78 |
| X56692 | C-reactive protein | 3.85 |
| NM_004791 | integrin, beta-like 1 (with EGF-like repeat domains) (ITGBL1) | 2.87 |
| AF289028 | transmembrane protein B7-H2 ICOS ligand | 2.75 |
| NM_005069 | single-minded (Drosophila) homolog 2 (SIM2), transcript variant SIM2 | 2.70 |
| NM_001618 | ADP-ribosyltransferase (NAD+; poly (ADP-ribose) polymerase) (ADPRT) | 2.69 |
| AF118124 | myeloid cell leukemia sequence 1 (MCL1) | 2.69 |
| AF134894 | SWAP-70 homolog | 2.60 |
| NM_002317 | lysyl oxidase (LOX) | 2.57 |
| NM_019074 | delta (Drosophila)-like 4 (DLL4) | 2.50 |
| Genes more highly expressed (>2.5-fold) in S2013.Neo versus S2013.MUC1 | ||
| Accession ID | Gene Name | Fold Change |
| NM_002421 | matrix metalloproteinase 1 (MMP1) | 29.53 |
| NM_002961 | S100 calcium-binding protein A4 (S100A4) | 15.18 |
| NM_002165 | inhibitor of DNA binding 1 (ID1) | 7.75 |
| NM_002276 | keratin 19 (KRT19) | 6.83 |
| NM_000210 | integrin, alpha 6 (ITGA6) | 6.69 |
| NM_000582 | secreted phosphoprotein 1 (SPP1) | 5.77 |
| NM_004878 | prostaglandin E synthase (PTGES) | 5.63 |
| NM_014624 | S100 calcium-binding protein A6 (calcyclin) (S100A6) | 4.73 |
| M62403 | insulin-like growth factor binding protein 4 (IGFBP4) | 4.34 |
| NM_001657 | amphiregulin (schwannoma-derived growth factor) (AREG) | 3.83 |
| NM_001153 | annexin A4 (ANXA4) | 3.47 |
| NM_003810 | tumor necrosis factor (ligand) superfamily, member 10 (TNFSF10) | 3.35 |
| NM_001432 | epiregulin (EREG) | 3.33 |
| NM_004433 | E74-like factor 3 (ets domain transcription factor) (ELF3) | 3.26 |
| NM_000596 | insulin-like growth factor binding protein 1 (IGFBP1) | 3.19 |
| NM_001831 | clusterin (CLU) | 3.18 |
| NM_003236 | transforming growth factor, alpha (TGFA) | 3.09 |
| NM_004945 | dynamin 2 (DNM2) | 2.82 |
| NM_000612 | insulin-like growth factor 2 (IGF2) | 2.81 |
| NM_001066 | tumor necrosis factor receptor superfamily, member 1B (TNFRSF1B) | 2.77 |
| NM_000177 | gelsolin (amyloidosis, Finnish type) (GSN) | 2.69 |
| NM_003811 | tumor necrosis factor (ligand) superfamily, member 9 (TNFSF9) | 2.63 |
| NM_002429 | matrix metalloproteinase 19 (MMP19) | 2.60 |
Figure 1.
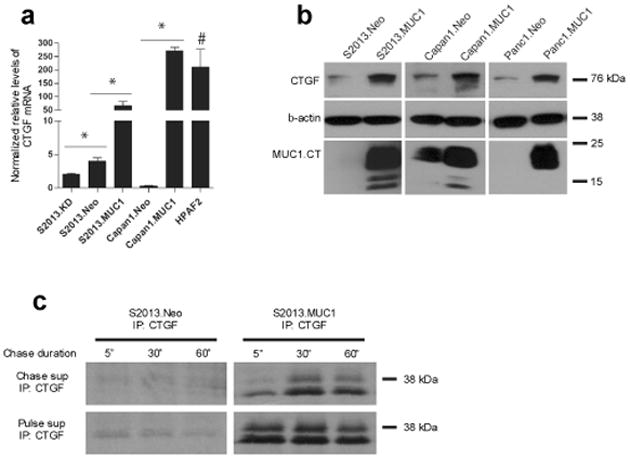
Overexpression of MUC1 increases CTGF levels in pancreatic cancer cell lines. (a) MUC1 overexpression increases CTGF mRNA transcripts in S2013 and Capan1 cells. Stable knockdown of MUC1 in S2013 cells further reduces CTGF transcripts when compared to S2013.Neo. Elevated relative levels of CTGF mRNA levels are observed in HPAF2 cells (#, P=0.094 when compared to S2013.Neo). All values have been normalized to levels of β-actin and are expressed as the average of three reactions +/− standard error mean (*, P<0.05). (b) Elevated levels of CTGF are observed at the protein level with MUC1 overexpression in pancreatic cancer cell lines S2013, Capan1, and Panc1. (c) Following metabolic radiolabeling using 35S-containing culture media for 1 hour (Pulse), media was collected (Pulse supe) and replaced with “cold” Cysteine and Methionine-containing media (Chase supe). Media was collected at indicated time points (5 minutes, 30 minutes, and 1 hour). Secreted CTGF was immunoprecipitated from Pulse supernatant to show equal secretion from each flask during the Pulse period, and from Chase supernatant to demonstrate increased secretion of CTGF over time from S2013.MUC1 cells when compared to S2013.Neo.
CTGF expression is selectively induced in MUC1-overexpressing cells by EGF, PDGF-BB, and HGF and requires tyrosine phosphorylation of MUC1.CT
The MUC1.CT is phosphorylated by several RTKs, and has previously been investigated in cells stimulated with different growth factors, including EGF, PDGF-BB, and HGF (Li et al. , 2001b; Singh et al. , 2007; Singh et al. , 2008). We found that CTGF mRNA levels were reduced in S2013.MUC1 cells cultured in serum-free media to levels similar to those observed in S2013.Neo cells. Stimulation with 10% FBS-containing culture media restored CTGF mRNA levels in S2013.MUC1 cells, while no effect was observed in S2013.Neo cells (Figure 2a). We investigated the involvement of growth factors EGF, PDGF-BB, and HGF in induction of CTGF mRNA expression. Following overnight serum deprivation, stimulation with each of these factors individually led to an increase in CTGF mRNA in S2013.MUC1 cells, with no observed change in S2013.Neo cells (Figure 2b).
Figure 2.
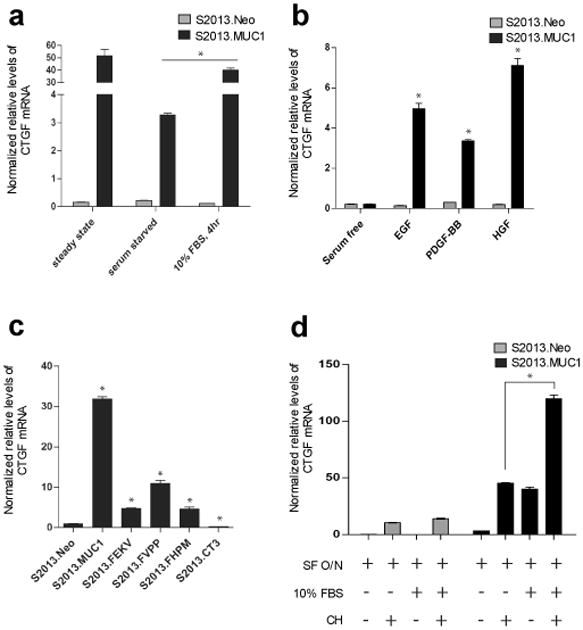
Induction of CTGF expression in MUC1-overexpressing cells. (a) Sustained CTGF expression in S2013.MUC1 cells is dependent upon factors present in fetal calf serum as overnight serum deprivation reduces CTGF mRNA levels. Four hour stimulation with 10% FBS-containing media rescues CTGF mRNA levels in S2013.MUC1 cells (*, P<0.05). No change in CTGF mRNA levels is observed with serum deprivation or serum stimulation in S2013.Neo cells. All values have been normalized to levels of β-actin and are expressed as the average of three reactions +/− SEM. (b) Induction of CTGF expression in MUC1 expressing cells occurs following stimulation with EGF, PDGF-BB, or HGF. S2013.Neo and S2013.MUC1 cells were serum deprived overnight and treated with 100 ng/ml recombinant human EGF, 100 ng/ml PDGF-BB, or 200 ng/ml HGF for 4 hours (*, P<0.05). No induction of CTGF expression was observed upon growth factor stimulation in S2013.Neo cells. (c) Expression of MUC1 harboring specific Y to F mutations in the cytoplasmic tail (S2013.FHPM, S2013.FVPP, and S2013.FEKV) and expression of a cytoplasmic tail-deleted MUC1 mutant (S2013.CT3) significantly reduces CTGF mRNA levels compared to overexpression of wild-type MUC1 (S2013.MUC1) (*, P<0.05). (d) Cycloheximide treatment was used to determine necessity of de novo protein synthesis to MUC1-induced CTGF expression. S2013.Neo and S2013.MUC1 cells were serum starved (SF) overnight and treated with 10% FBS-containing media, 2.0 ug/ml cycloheximide (CH), or both for 4 hours and CTGF mRNA levels were assayed. A general effect on CTGF mRNA was observed upon CH treatment in both S2013.Neo and S2013.MUC1 cells. The presence of CH does not inhibit induction of CTGF expression following stimulation with 10% FBS-containing media in S2013.MUC1 cells (*, P<0.05). No induction was observed with FBS stimulation in S2013.Neo cells.
We hypothesized that induction of CTGF expression by EGF, PDGF-BB, and HGF involved phosphorylation of MUC1.CT by the cognate RTKs of these growth factors: EGFR, PDGFR-β and Met, respectively. The levels of CTGF mRNA were significantly reduced in cells expressing MUC1.CT mutants harboring Tyrosine (Y) to Phenylalanine (F) substitutions at the reported EGFR, PDGFR-β, and Met motifs (YEKV to FEKV, YVPP to FVPP, and YHPM to FHPM, respectively) as compared to levels observed with overexpression of wild type MUC1 (Figure 2c). Expression levels of MUC1.CT in S2013.MUC1, S2013.FEKV, S2013.FHPM, and S2013.FVPP cell lines were similar (Supplementary Figure 1). Consistent with these findings, expression of cytoplasmic tail-deleted MUC1 in S2013 cells (S2013.CT3) significantly reduced CTGF mRNA levels (Figure 2c). These results confirm that induction of CTGF in MUC1-overexpressing cells is regulated by EGF, PDGF-BB, and HGF mediated phosphorylation of the MUC1.CT.
Analysis of S2013.MUC1 and S2013.Neo cells stimulated with FBS-containing media in the presence or absence of cycloheximide (CH), an inhibitor of protein synthesis, showed that CTGF transcripts were increased in S2013.MUC1 cells even in the presence of cycloheximide (Figure 2d). There is a general effect on CTGF mRNA levels with CH treatment in both S2013.Neo and S2013.MUC1 cells, yet a significant increase was still observed above this level with overexpression of MUC1 while no effect was observed in S2013.Neo. An induction of CTGF expression despite the presence of CH suggested that MUC1.CT influenced CTGF transcript levels by directly regulating transcription of the CTGF gene and led us to investigate this possibility.
MUC1.CT localizes to genomic regions within and surrounding the CTGF promoter and regulates transcription of the CTGF gene
MUC1.CT has been detected at gene regulatory elements in transcriptional complexes and regulates expression of the transcription factor target genes (Singh et al. , 2007; Singh et al. , 2008; Wei et al. , 2005; Wei et al. , 2007). We evaluated direct occupancy of genomic regions spanning the 10 kilobases (kb) upstream and 2 kb downstream of the CTGF gene transcription start site by complexes containing MUC1.CT by ChIP-chip using the MUC1.CT monoclonal antibody CT2 (Figure 3a). This analysis revealed that MUC1.CT localized to the proximal promoter of CTGF, to intragenic sequences, and to several regions upstream of the minimal CTGF promoter. Oligonucleotide primers were designed to amplify these regions and real-time PCR analysis confirmed their enrichment in anti-MUC1 immunoprecipitates versus IgG immunoprecipitates (Figure 3b).
Figure 3.
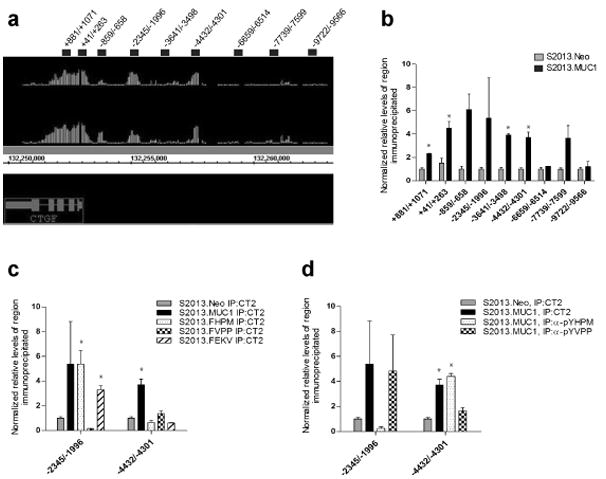
Occupancy of the CTGF promoter by MUC1.CT. (a) ChIP-chip revealed MUC1.CT occupies regions within the CTGF gene, the proximal promoter, and several regions upstream of the CTGF promoter. Image from Integrated Genome Browser (Affymetrix) displays two experimental MUC1.CT versus IgG ChIP-chip replicates where vertical lines represent individual oligonucleotide probes whose height represents degree of enrichment. Oligonucleotide primer pairs used to confirm MUC1.CT occupancy are represented by black bars and their sequences are reported in Supplementary Figure 2. (b) Occupancy of MUC1.CT at CTGF promoter regions was confirmed by chromatin immunoprecipitation (ChIP) using anti-MUC1.CT antibody (CT2) or IgG, followed by real-time PCR analysis. Enrichment of several CTGF promoter regions was detected in S2013.MUC1 cells while no enrichment was observed in S2013.Neo cells (*, P<0.05). (c) Chromatin immunoprecipitation of CTGF promoter regions using CT2 in S2013.FHPM, S2013.FVPP, or S2013.FEKV mutants revealed a phosphorylation-specific pattern of MUC1.CT localization at the −2345/−1996 and −4432/−4301 regions of the CTGF promoter (*, P<0.05 when compared to S2013.Neo) (d) ChIP using anti-pYHPM or anti-pYVPP rabbit antisera revealed occupancy of these MUC1 phosphorylated isoforms at the −2345/−1996 and −4432/−4301 regions of the CTGF promoter (*, P<0.05 when compared to S2013.Neo). For ChIP analysis, values for each region of interest were normalized to enrichment of a region within the β-glucuronidase gene. Values have been normalized to the enrichment detected using IgG and are expressed as the average of three reactions +/− standard error mean.
We sought to determine if there was a requirement for tyrosine phosphorylation of the YHPM, YVPP, and YEKV motifs to achieve elevated CTGF expression by performing chromatin immunoprecipitation with CT2 on S2013 cell lines that harbor mutations within these motifs. For S2013.FHPM cells, MUC1.CT occupancy remained unchanged at the −2345/-1996 promoter region while occupancy at the −4432/−4301 region was diminished (Figure 3c). This suggested that phosphorylation at YHPM is not required for MUC1.CT localization at −2345/−1996 yet is required for localization to −4432/−4301. This was confirmed by chromatin immunoprecipitation using an antibody recognizing MUC1.CT phosphorylated at YHPM (anti-pYHPM), which showed no enrichment of −2345/−1996 and enrichment of −4432/−4301 (Figure 3d). S2013.FVPP cells showed that occupancy at −2345/−1996 was diminished and occupancy at −4432/−4301 was also reduced. This suggested that phosphorylation at YVPP is required for localization of MUC1.CT to −2345/−1996 but only partially responsible for localization to −4432/−4301. ChIP using an anti-pYVPP antiserum revealed enrichment of −2345/−1996 to a degree similar to that observed using the CT2 antibody, but reduced enrichment of −4432/−4301. S2013.FEKV cells showed slightly reduced MUC1.CT localization to −2345/1996 and elimination of localization to −4432/−4301. ChIP analysis of the CTGF regulatory regions using the MUC1.CT Y to F mutants reveals a phosphorylation-specific pattern of MUC1.CT localization.
MUC1.CT influences binding of p53 and β-catenin to CTGF promoter regions
MUC1.CT has been reported to interact with p53 and β-catenin following HGF and PDGF-BB stimulation, respectively (Singh et al. , 2007; Singh et al. , 2008). We therefore investigated whether MUC1.CT influenced the occupancy of either of these transcription factors at the CTGF promoter. ChIP studies demonstrated increased occupancy of p53 at −2345/−1996 upon MUC1 overexpression in S2013 cells, which express the p53 mutant R273H. No change in occupancy of either the proximal promoter or a region approximately 1 kb upstream was detected (Figure 4a).
Figure 4.
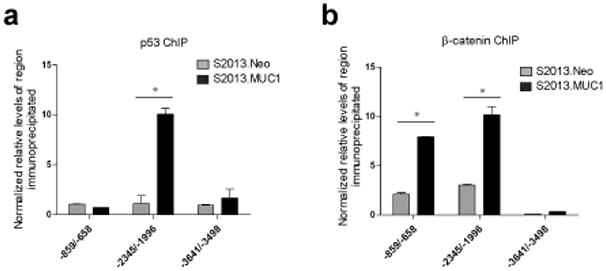
Effects of MUC1 overexpression on p53 and β–catenin occupancy of the CTGF promoter. (a) Chromatin immunoprecipitation reveals a significant increase in p53 occupancy of the –2345/−1996 region in cells overexpressing MUC1 (*, P<0.05 when compared to S2013.Neo), while occupancy of the proximal promoter (−859/−658) and an upstream region (−3641/−3498) remain unchanged. (b) MUC1 overexpression increased occupancy of β-catenin at the proximal promoter (*, P<0.05 when compared to S2013.Neo), while occupancy at an upstream region remains unchanged. For ChIP analysis, values for each region of interest were normalized to enrichment of a region within the β-glucuronidase gene. Values have been normalized to the enrichment detected using IgG and are expressed as the average of three reactions +/− standard error mean.
β-catenin has been reported to drive expression of the CTGF gene in esophageal squamous cell carcinoma cells from a TCF/Lef element within the proximal promoter of CTGF (Deng et al. , 2007). Localization of β-catenin to the CTGF promoter was investigated and we demonstrated increased occupancy of −859/−658 as well as −2345/−1996 upon MUC1 overexpression, while no change in enrichment of −3641/−3498 was detected (Figure 4b). These studies demonstrated an increase in localization of p53 and β-catenin to CTGF gene regulatory elements upon MUC1 overexpression, and support the hypothesis that interactions with phosphorylated MUC1.CT enable CTGF promoter binding by mutant p53 and β-catenin.
Gain-of-function studies of mutant p53 indicate a propensity of mutant p53 to abrogate AP−1 mediator complex interactions with basal transcriptional machinery at gene regulatory elements (Sun et al. , 2004; Singh et al. , 2008). Based on the robust induction of CTGF by HGF stimulation in MUC1 overexpressing cells, we hypothesized that MUC1.CT-p53 complexes in the CTGF promoter hinder c-Jun binding and investigated whether the presence of high levels of MUC1 influenced the occupancy of c-Jun at any regions near the CTGF promoter.
MUC1.CT binds the CTGF repressor c-Jun and influences its occupancy at CTGF promoter regions
Previous studies indicated a role of the transcription factor c-Jun in maintaining low levels of CTGF expression in differentiated cells, though the precise mechanism has not yet been reported (Leask et al. , 2003). We sought to confirm that c-Jun repressed CTGF expression in S2013.Neo cells. There was a significant increase in CTGF mRNA levels with increasing concentration of the Jun N-terminal Kinase inhibitor SP600125 while no effect was observed in S2013.MUC1 cells (Figure 5a), indicating that the low levels of CTGF in S2013 cells is due at least in part to c-Jun repressive activity. We sought to determine if MUC1 regulated c-Jun activity. Immunoprecipitation experiments show for the first time a direct interaction between MUC1.CT and c-Jun in nuclei of cells overexpressing MUC1 (Figure 5b). Little interaction was observed in S2013.Neo cells and no interaction was detected in IgG immunoprecipitates. We confirmed by chromatin IP that MUC1 overexpression reduced c-Jun binding at the −2345/−1996 region of the CTGF promoter, while its occupancy at the proximal promoter and an upstream control region remain unchanged (Figure 5c).
Figure 5.
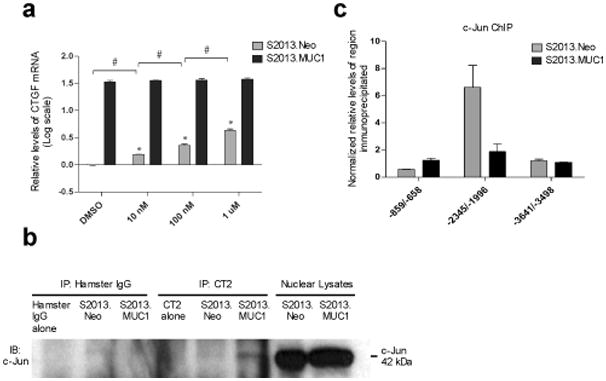
Effects of MUC1 overexpression on c-Jun activity. (a) Cells were cultured in the presence of 10 nM, 100 nM, or 1 uM SP600125, or DMSO only for 24 hours and total RNA was isolated. All volumes were equilibrated with DMSO. Treatment of cells with a Jun N-terminal Kinase inhibitor SP600125 results in increased expression of CTGF mRNA transcripts in S2013.Neo cells (*, P<0.05 when compared to DMSO treatment). The increase in CTGF mRNA levels in S2013.Neo cells is dose-dependent as increasing concentration of SP600125 significantly increases CTGF mRNA levels in S2013.Neo cells. (#, P<0.05 when comparing S2013.Neo cells with increasing inhibitor concentration). Treatment with SP600125 has no effect on CTGF expression in S2013.MUC1 cells. Real-time PCR values have been normalized to β-actin and CTGF mRNA levels are reported as the log fold increase over the levels of CTGF mRNA observed in DMSO treated S2013.Neo cells. All values are expressed as the average of three reactions +/− SEM. (b) An interaction between MUC1.CT and c-Jun was detected in the nuclei of cells overexpressing MUC1 while no interaction was observed in S2013.Neo cells or IgG immunoprecipitates. MUC1.CT or IgG was immunoprecipitated from 500 ug S2013.Neo or S2013.MUC1 nuclear lysate and 50 ug S2013.Neo or S2013.MUC1 nuclear lysate only was run to show mobility of c-Jun at 42kDa. (c) ChIP was used to determine that MUC1 overexpression decreases occupancy at the CTGF promoter by c-Jun at –2345/−1996 and has no effect on its occupancy at the proximal promoter or at an upstream region. All values have been normalized to the enrichment detected using IgG and are expressed as the average of three experiments +/− SEM.
Discussion
The results presented here confirm previous reports that phosphorylation of the MUC1.CT contributes significantly to oncogenic signaling (Singh et al. , 2007; Singh et al. , 2008). The cytoplasmic tail of MUC1 contains 18 potential phosphorylation sites that are modified by specific kinases (Singh and Hollingsworth, 2006). The receptor tyrosine kinases examined here, EGFR, PDGFR-β, and Met, are known to catalyze both common and distinct phosphorylation events on the MUC1.CT; however, all three catalyze phosphorylation of the YHPM site in the MUC1.CT. We propose that specific combinations of phosphorylation events enable the specific association of MUC1.CT with mediators of signal transduction and transcriptional regulation, which in turn modify expression of specific target genes. In the case examined here, mutation of specific tyrosine residues within the MUC1.CT diminished its occupancy at certain regions within the CTGF promoter, while occupancy at other regions remained unchanged. Chip experiments with phosphorylation specific antibodies confirmed that differential phosphorylation of the MUC1.CT led to changes in CTGF promoter occupancy.
The transcriptional regulatory functions of MUC1.CT results from interactions with specific transcription factors, as the MUC1.CT contains no known DNA-binding motif and no clear nuclear localization sequence. Differential phosphorylation of the MUC1.CT facilitates binding to several transcription factors, including p53, βcatenin and reported here for the first time, c-Jun, which in turn altered their chromatin-binding patterns. We show that localization of p53 and β-catenin to the CTGF promoter is enabled in the presence of MUC1. Additionally, the presence of MUC1.CT eliminated binding of c-Jun at the −2345/−1996 region of the CTGF promoter. Notably, this is the same region occupied by p53 upon MUC1 overexpression, suggesting that the MUC1.CT directly affects competition among transcription factors for occupancy at this site. The propensity of p53 to disrupt AP−1 transcriptional regulation has been described (Sun et al. , 2004) and this is the second report from our laboratory to demonstrate that MUC1.CT contributes to this activity.
The induction of CTGF expression by MUC1.CT signaling is significant as CTGF exhibits angiogenic properties comparable to basic FGF and VEGF (Shimo et al. , 1999). As such, CTGF is an attractive therapeutic target as are the signaling events that lead to its expression. Little is known of how CTGF is regulated in epithelial cell types and tumors of epithelial origin, and we provide evidence that its induction in PDAC cells occurs in response to several growth factors and is dependent on the presence of MUC1. Regulation of CTGF expression has been most well characterized in fibroblasts, where expression is induced by TGF-β(Leask et al. , 2003). However, CTGF appears to be regulated in a TGF-β independent manner in carcinoma cells and has been identified as an immediate/early target gene of EGF/TGFα(Deng et al. , 2007; Wenger et al. , 1999). We found that CTGF expression was not induced by TGF-β treatment of pancreatic adenocarcinoma cells irrespective of MUC1 expression (data not shown), further suggesting that the regulation of CTGF differs in a cell-type specific manner.
MUC1 has long been implicated in tumorigenesis, but only in the past decade have we begun to appreciate the role of its small, cytoplasmic domain in morphogenetic and oncogenic signaling (Hollingsworth and Swanson, 2004). The results presented here and in our previous reports (Singh et al. , 2007; Singh et al. , 2008) show clearly that the MUC1.CT functions by receiving signals in the form of phosphorylation events by cell surface associated receptor tyrosine kinases (and other kinases), that enable it to associate with and alter the function of transcription factors and transcriptional coactivators. In the case examined here, signals from different receptor tyrosine kinases (EGFR, PDGFR-β and Met) are transmitted through MUC1 to activate a novel signal transduction pathway involving p53 and c-Jun that resulted in activation of expression of CTGF, whereas cells lacking MUC1 did not activate expression of CTGF. Why would cells expressing MUC1 induce expression of CTGF upon growth factor stimulation? One possibility relates to a posited role for MUC1 in morphogenetic signaling (Hollingsworth and Swanson, 2004). MUC1 is expressed on normal well-differentiated epithelial cells at the apical surface, where it is separated spatially from many receptor tyrosine kinases such as EGFR and PDGFR. Cells that have lost polarity allow MUC1 to spatially contact receptor tyrosine kinases, which may send a signal in response to stimulation with growth and differentiation factors such as PDGF, EGF, or HGF. In the normal setting, the presence of growth or differentiation factors is presumably a consequence of and an indicator of damage, inflammation, or wound healing in the vicinity. Epithelial cells that have lost polarity are placed in a position of deciding whether to: live or die (apoptosis or anti-apoptosis); divide to fill a gap in the epithelial layer; or undertake a program of tissue remodeling to restore the normal architecture by recruiting fibroblasts, blood vessels, lymphatics, other stromal cells, or by remodeling the extracellular matrix. This hypothesis is consistent with our observation that MUC1.CT signaling in response to stimulation with EGF, PDGF, or HGF induced expression of CTGF, which promotes fibroblast proliferation and extracellular matrix deposition, upregulates TIMPS, and has been suggested as a master mediator of ECM turnover, angiogenesis, and wound healing (Chu et al. , 2008). Pancreatic adenocarcinomas that overexpress MUC1 have appropriated its protective and signaling functions to facilitate oncogenic progression, metastasis, and survival in different organ environments. The induction of expression of potent tissue morphogens such as CTGF in pancreatic tumors represents one facet of this process.
Our results show that MUC1, when expressed at high levels similar to those observed in most cases of pancreatic cancer, promotes expression of the CTGF gene by receiving signals in the form of phosphorylation events on the MUC1.CT from growth factor receptor tyrosine kinases at the cell surface. These signals are transmitted to transcriptional complexes in the nucleus where phosphorylated MUC1.CT isoforms associate with and modify the activity of regulatory complexes at promoters such as CTGF. There are several lines of evidence supporting this conclusion. Firstly, MUC1 overexpression significantly increased CTGF mRNA and protein levels in pancreatic cancer cells cultured in serum-containing media. Second, MUC1 expression in S2013 cells enabled induction of CTGF in response to serum and the growth factors EGF, PDGF-BB, and HGF, whereas control S2013.Neo cells exhibited no change in CTGF mRNA levels following stimulation with these growth factors or serum. In addition, preventing RTK phosphorylation of the MUC1.CT by expressing non-phosphorylatable MUC1 mutants reduced levels of CTGF. Third, transcription of the CTGF gene was correlated directly to MUC1.CT localization in complexes within and surrounding the CTGF minimal promoter. MUC1.CT was detected at the CTGF promoter only in cells in which MUC1 was overexpressed and the specific location of the MUC1.CT was dependent on its phosphorylation status. Finally, occupancy of several key transcription factors at the CTGF promoter was disrupted upon MUC1 overexpression. Localization of mutant p53 and β-catenin to the CTGF promoter was increased in S2013.MUC1 cells whereas c-Jun localization was decreased. This was likely due to functional interactions between these molecules and MUC1.CT as MUC1.CT-p53 and MUC1.CT-β-catenin interactions have previously been demonstrated. We also report for the first time an interaction between MUC1.CT and the CTGF repressor, c-Jun.
There is no doubt that signaling through MUC1.CT influences expression of many other genes that are likely involved in different programs of cellular responses. Though it is only implied from the microarray expression results, our unpublished ChIP-chip data identified MUC1.CT binding to over 300 promoters in the pancreatic cancer cells here examined. Thus, further elucidation of the network of signaling mediated by the MUC1.CT will provide insight into malignant progression of tumors and novel targets for potential therapeutic intervention.
Materials and Methods
Cell Culture
Panc1 and Capan1 pancreatic cancer cell lines were obtained from ATCC and S2013 cells have been described previously (Iwamura et al. , 1992). Stable expression of flag-epitope tagged MUC1 in Panc1 (Panc1.MUC1) and S2013 (S2013.MUC1) cells has been described previously (Wen et al. , 2003) and Capan1.MUC1 cells were similarly generated. S2013 transfectants containing cytoplasmic tail-deleted MUC1 (S2013.CT3) and stable knockdown of MUC1 (S2013.MUC1KD) were described previously (Kohlgraf et al. , 2003; McDermott et al. , 2001; Pemberton et al. , 1996). S2013.FHPM and S2013.FVPP cells expressing MUC1 mutants harboring tyrosine (Y) to phenylalanine (F) point mutations at Tyrosine of the YHPM and YVPP motif, respectively, of the cytoplasmic tail are described elsewhere (Singh et al. , 2007). S2013.FEKV cells expressing MUC1 harboring a Y to F point mutant at Tyrosine of the YEKV motif were similarly transduced. All S2013, Capan1, and Panc1 transfectants were maintained in Dulbecco's modified Eagle's medium plus 10% HI fetal calf serum, ciprofloxacin, and 200 ug/ml G418. HPAF2 cells were cultured in DMEM plus 10% HI fetal calf serum and cipro. All cells were maintained at 37°C in a humidified atmosphere with 5% CO2.
Growth Factor and Inhibitor Treatment
For all growth factor stimulations, cells were cultured in media lacking FBS for 16 hours prior to stimulation. Stimulations included 10% FBS-containing media, 100 ng/ml human recombinant EGF or PDGF-BB or 200 ng/ml HGF in DMEM, for 4 hours at 37°C (Peprotech, Rocky Hill, New Jersey, USA). Cells were cultured in the presence of SP600125 (Calbiochem, San Diego, California, USA) for 24 hours.
Immunoblotting
Cells were lysed in an NP−40 based buffer with commercial protease inhibitors (Complete mini inhibitor cocktail, Roche, Mannheim, Germany), phosphatase inhibitors (Phosphatase Inhibitor Cocktail II, Sigma-Aldrich, St. Louis, Missouri, USA) and 1 mM PMSF. Samples were run on 4−20% Tris-glycine denaturing polyacrylamide gels (Invitrogen, Carlsbad, California, USA). Gel electrophoresis and immunoblotting were performed as described previously (Singh et al. , 2008). CT2, an Armenian hamster monoclonal antibody recognizing the cytoplasmic tail of MUC1 was generously provided by Dr. Sandra J. Gendler (Mayo Clinic, Scottsdale, Arizona, USA). CTGF (R&D Systems, Minneapolis, Minnesota, USA), p53, c-Jun, (Santa Cruz Biotechnology, Santa Cruz, California, USA), and (catenin (BD Transduction Laboratories, San Jose, California, USA) antibodies were used at 1:1000 for western blotting.
Metabolic Radiolabeling and Immunoprecipitation
Metabolic radiolabeling using 250 uCi/ml 35S and immunoprecipitation of CTGF was performed as previously described (Takagi et al. , 2002). Briefly, media was removed from three T25 flasks of either S2013.Neo or S2013.MUC1 cells and was replaced with media lacking Cysteine and Methionine supplemented with 10% FBS for 1 hour. Media was removed and replaced with Cys-/Met- media containing 250 uCi/ml NEG-072 EXPRE35S35S 35S-protein labeling mix (Perkin Elmer, Boston, Massachusetts, USA) for 1 hour (pulse). 35S-containing media was removed (and saved as “Pulse supe”) and “cold” media containing cysteine and methionine was added. Media was collected from each flask at either5 minutes, 30 minutes, or 1 hour and immunoprecipitation was performed as described.
Coimmunoprecipitations
Nuclear fractions were obtained using a hypotonic cell lysis buffer (10mM Hepes pH 7.9, 10 mM KCl, 0.1 mM EDTA) and lysed using a hypertonic lysis buffer (20mM Hepes pH 7.9, 0.4M NaCl, 1mM EDTA) supplemented with protease and phosphatase inhibitors and PMSF. Coimmunoprecipitations were performed as described previously (Singh et al. , 2008) using isotype matched IgG as a control.
RNA isolation and Microarray analysis
Total RNA was isolated and microarray analysis was performed as described (Yi et al. , 2007).
Real-time PCR assays and Primers
cDNA was generated using Superscript III First-strand Synthesis Kit (Invitrogen). Reactions containing 2.0 ul cDNA for gene expression analysis or 3.0 ul purified chromatin for ChIP analysis were prepared in Sybr green master mix (Applied Biosystems, Foster City, California) and subjected to quantitative real-time PCR analysis using ABI 7500 thermocycler. Each reaction was repeated in triplicate and the experiments were repeated at least twice to confirm reproducibility. Values were obtained for the threshold cycle (Ct) for each gene or genomic region and data were analyzed using the standard curve method. For RT-PCR analysis, values were normalized to the expression of β-actin and average expression +/− SEM was reported. For ChIP PCR analysis, values were normalized to a genomic region located within the β-glucuronidase (GUSB) gene and expressed as a fold increase over enrichment detected using IgG. The average expression +/− SEM was reported. Primer sequences for β-actin (Gatto et al. , 2008) and CTGF (Husson et al. , 2002) are published. GUSB validated primer pair was purchased from SA Biosciences (Frederick, Maryland, USA). CTGF promoter primer sequences are reported in Supplementary Figure 2.
Chromatin Immunoprecipitations and ChIP-chip
Chromatin immunoprecipitations were performed according to the Affymetrix ChIP-chip protocol, with the following modifications: 5.0×107 cells were subjected to additional crosslinking using 5mM dimethyl 3,3′-dithiobispropioimidate (DTBP, Pierce Biotechnology, Rockford, Illinois, USA) for 30 minutes at 4°C. Protease and phosphatase inhibitors were added to all buffers used. Complexes were decrosslinked using 100mM DTT and 1 ug/ul Proteinase K at 65°C overnight. Decrosslinked chromatin was purified using Qiaquick PCR Purification Kit (Qiagen, Valencia, California, USA). Anti-pYHPM and anti-pYVPP rabbit antisera have been previously described (Singh et al. , 2007; Singh et al. , 2008). For ChIP-chip, 20 ng of purified chromatin was amplified using the Whole Genome Complete Amplification kit (Sigma) and digested using Dnase I (Invitrogen). For ChIP-chip, 20 ng of purified chromatin Amplification kit (Sigma) and digested using Dnase I (Invitrogen). 7.5 ug digested DNA was labeled and hybridized to Human 1.0R Promoter arrays (Affymetrix, Santa Clara, California, USA) as per the Affymetrix protocol. MUC1.CT targets were identified using the ChIP-on-chip workflow in Partek Genomics Suite (Partek Incorporated, Saint Louis, Missouri, USA). Enrichment of genomic regions within and surrounding the CTGF promoter was visualized in Integrated Genome Browser (Affymetrix) and confirmed using quantitative real-time PCR.
Statistical Analysis
The analysis of variance (ANOVA) method with Tukey-Kramer multiple comparison test was used to compare the mean value of normalized relative levels of CTGF mRNA among different pancreatic cancer cell lines or treaments, or the normalized relative levels of region immunoprecipitated at different CTGF promoter regions. The Levene's (absolute value and squared residuals) test is used to test for homogeneity of variances across the groups of comparison. For the data showing unequal variances among groups in comparison, the ANOVA method for unequal variance was used. A p-value of less than 0.05 was considered to be significant. All calculations used SAS version 9.2 (Cary, North Carolina, USA).
Supplementary Material
Footnotes
Confilct of interest: The authors declare no conflict of interest
Supplementary information is available at Oncogene's website
References
- Agata N, Ahmad R, Kawano T, Raina D, Kharbanda S, Kufe D. MUC1 oncoprotein blocks death receptor-mediated apoptosis by inhibiting recruitment of caspase-8. Cancer Res. 2008;68:6136–6144. doi: 10.1158/0008-5472.CAN-08-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aikawa T, Gunn J, Spong SM, Klaus SJ, Korc M. Connective tissue growth factor-specific antibody attenuates tumor growth, metastasis, and angiogenesis in an orthotopic mouse model of pancreatic cancer. Mol Cancer Ther. 2006;5:1108–1116. doi: 10.1158/1535-7163.MCT-05-0516. [DOI] [PubMed] [Google Scholar]
- Bennewith KL, Huang X, Ham CM, Graves EE, Erler JT, Kambham N, et al. The role of tumor cell-derived connective tissue growth factor (CTGF/CCN2) in pancreatic tumor growth. Cancer Res. 2009;69:775–784. doi: 10.1158/0008-5472.CAN-08-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigstock DR. The CCN family: A new stimulus package. J Endocrinol. 2003;178:169–175. doi: 10.1677/joe.0.1780169. [DOI] [PubMed] [Google Scholar]
- Burdick MD, Harris A, Reid CJ, Iwamura T, Hollingsworth MA. Oligosaccharides expressed on MUC1 produced by pancreatic and colon tumor cell lines. J Biol Chem. 1997;272:24198–24202. doi: 10.1074/jbc.272.39.24198. [DOI] [PubMed] [Google Scholar]
- Carson DD. The cytoplasmic tail of MUC1: A very busy place. Sci Signal. 2008;1:35. doi: 10.1126/scisignal.127pe35. [DOI] [PubMed] [Google Scholar]
- Chu CY, Chang CC, Prakash E, Kuo ML. Connective tissue growth factor (CTGF) and cancer progression. J Biomed Sci. 2008;15:675–685. doi: 10.1007/s11373-008-9264-9. [DOI] [PubMed] [Google Scholar]
- Deng YZ, Chen PP, Wang Y, Yin D, Koeffler HP, Li B, et al. Connective tissue growth factor is overexpressed in esophageal squamous cell carcinoma and promotes tumorigenicity through beta-catenin-T-cell factor/Lef signaling. J Biol Chem. 2007;282:36571–36581. doi: 10.1074/jbc.M704141200. [DOI] [PubMed] [Google Scholar]
- Gao J, McConnell MJ, Yu B, Li J, Balko JM, Black EP, et al. MUC1 is a downstream target of STAT3 and regulates lung cancer cell survival and invasion. Int J Oncol. 2009;35:337–345. [PMC free article] [PubMed] [Google Scholar]
- Gatto M, Drudi-Metalli V, Torrice A, Alpini G, Cantafora A, Blotta I, et al. Insulin-like growth factor−1 isoforms in rat hepatocytes and cholangiocytes and their involvement in protection against cholestatic injury. Lab Invest. 2008;88:986–994. doi: 10.1038/labinvest.2008.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth MA, Swanson BJ. Mucins in cancer: Protection and control of the cell surface. Nat Rev Cancer. 2004;4:45–60. doi: 10.1038/nrc1251. [DOI] [PubMed] [Google Scholar]
- Husson H, Carideo EG, Neuberg D, Schultze J, Munoz O, Marks PW, et al. Gene expression profiling of follicular lymphoma and normal germinal center B cells using cDNA arrays. Blood. 2002;99:282–289. doi: 10.1182/blood.v99.1.282. [DOI] [PubMed] [Google Scholar]
- Iwamura T, Taniguchi S, Kitamura N, Yamanari H, Kojima A, Hidaka K, et al. Correlation between CA19-9 production in vitro and histological grades of differentiation in vivo in clones isolated from a human pancreatic cancer cell line (SUIT−2) J Gastroenterol Hepatol. 1992;7:512–519. doi: 10.1111/j.1440-1746.1992.tb01030.x. [DOI] [PubMed] [Google Scholar]
- Kohlgraf KG, Gawron AJ, Higashi M, Meza JL, Burdick MD, Kitajima S, et al. Contribution of the MUC1 tandem repeat and cytoplasmic tail to invasive and metastatic properties of a pancreatic cancer cell line. Cancer Res. 2003;63:5011–5020. [PubMed] [Google Scholar]
- Leask A, Holmes A, Black CM, Abraham DJ. Connective tissue growth factor gene regulation. requirements for its induction by transforming growth factor-beta 2 in fibroblasts. J Biol Chem. 2003;278:13008–13015. doi: 10.1074/jbc.M210366200. [DOI] [PubMed] [Google Scholar]
- Li Y, Bharti A, Chen D, Gong J, Kufe D. Interaction of glycogen synthase kinase 3beta with the DF3/MUC1 carcinoma-associated antigen and beta-catenin. Mol Cell Biol. 1998;18:7216–7224. doi: 10.1128/mcb.18.12.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kuwahara H, Ren J, Wen G, Kufe D. The c-src tyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3 beta and beta-catenin. J Biol Chem. 2001a;276:6061–6064. doi: 10.1074/jbc.C000754200. [DOI] [PubMed] [Google Scholar]
- Li Y, Ren J, Yu W, Li Q, Kuwahara H, Yin L, et al. The epidermal growth factor receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-src and beta-catenin. J Biol Chem. 2001b;276:35239–35242. doi: 10.1074/jbc.C100359200. [DOI] [PubMed] [Google Scholar]
- Masaki Y, Oka M, Ogura Y, Ueno T, Nishihara K, Tangoku A, et al. Sialylated MUC1 mucin expression in normal pancreas, benign pancreatic lesions, and pancreatic ductal adenocarcinoma. Hepatogastroenterology. 1999;46:2240–2245. [PubMed] [Google Scholar]
- Masuko K, Murata M, Yudoh K, Shimizu H, Beppu M, Nakamura H, et al. Prostaglandin E2 regulates the expression of connective tissue growth factor (CTGF/CCN2) in human osteoarthritic chondrocytes via the EP4 receptor. BMC Res Notes. 2010;3:5. doi: 10.1186/1756-0500-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott KM, Crocker PR, Harris A, Burdick MD, Hinoda Y, Hayashi T, et al. Overexpression of MUC1 reconfigures the binding properties of tumor cells. Int J Cancer. 2001;94:783–791. doi: 10.1002/ijc.1554. [DOI] [PubMed] [Google Scholar]
- Pemberton LF, Rughetti A, Taylor-Papadimitriou J, Gendler SJ. The epithelial mucin MUC1 contains at least two discrete signals specifying membrane localization in cells. J Biol Chem. 1996;271:2332–2340. doi: 10.1074/jbc.271.4.2332. [DOI] [PubMed] [Google Scholar]
- Qu CF, Li Y, Song YJ, Rizvi SM, Raja C, Zhang D, et al. MUC1 expression in primary and metastatic pancreatic cancer cells for in vitro treatment by (213)bi-C595 radioimmunoconjugate. Br J Cancer. 2004;91:2086–2093. doi: 10.1038/sj.bjc.6602232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina D, Ahmad R, Chen D, Kumar S, Kharbanda S, Kufe D. MUC1 oncoprotein suppresses activation of the ARF-MDM2-p53 pathway. Cancer Biol Ther. 2008;7:1959–1967. doi: 10.4161/cbt.7.12.6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimo T, Nakanishi T, Nishida T, Asano M, Kanyama M, Kuboki T, et al. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J Biochem. 1999;126:137–145. doi: 10.1093/oxfordjournals.jbchem.a022414. [DOI] [PubMed] [Google Scholar]
- Singh PK, Behrens ME, Eggers JP, Cerny RL, Bailey JM, Shanmugam K, et al. Phosphorylation of MUC1 by met modulates interaction with p53 and MMP1 expression. J Biol Chem. 2008;283:26985–26995. doi: 10.1074/jbc.M805036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PK, Hollingsworth MA. Cell surface-associated mucins in signal transduction. Trends Cell Biol. 2006;16:467–476. doi: 10.1016/j.tcb.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Singh PK, Wen Y, Swanson BJ, Shanmugam K, Kazlauskas A, Cerny RL, et al. Platelet-derived growth factor receptor beta-mediated phosphorylation of MUC1 enhances invasiveness in pancreatic adenocarcinoma cells. Cancer Res. 2007;67:5201–5210. doi: 10.1158/0008-5472.CAN-06-4647. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zeng XR, Wenger L, Firestein GS, Cheung HS. P53 down-regulates matrix metalloproteinase−1 by targeting the communications between AP−1 and the basal transcription complex. J Cell Biochem. 2004;92:258–269. doi: 10.1002/jcb.20044. [DOI] [PubMed] [Google Scholar]
- Takagi J, DeBottis DP, Erickson HP, Springer TA. The role of the specificity-determining loop of the integrin beta subunit I-like domain in autonomous expression, association with the alpha subunit, and ligand binding. Biochemistry. 2002;41:4339–4347. doi: 10.1021/bi016047u. [DOI] [PubMed] [Google Scholar]
- Tsutsumida H, Swanson BJ, Singh PK, Caffrey TC, Kitajima S, Goto M, et al. RNA interference suppression of MUC1 reduces the growth rate and metastatic phenotype of human pancreatic cancer cells. Clin Cancer Res. 2006;12:2976–2987. doi: 10.1158/1078-0432.CCR-05-1197. [DOI] [PubMed] [Google Scholar]
- Wei X, Xu H, Kufe D. Human mucin 1 oncoprotein represses transcription of the p53 tumor suppressor gene. Cancer Res. 2007;67:1853–1858. doi: 10.1158/0008-5472.CAN-06-3063. [DOI] [PubMed] [Google Scholar]
- Wei X, Xu H, Kufe D. Human MUC1 oncoprotein regulates p53-responsive gene transcription in the genotoxic stress response. Cancer Cell. 2005;7:167–178. doi: 10.1016/j.ccr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Wen Y, Caffrey TC, Wheelock MJ, Johnson KR, Hollingsworth MA. Nuclear association of the cytoplasmic tail of MUC1 and beta-catenin. J Biol Chem. 2003;278:38029–38039. doi: 10.1074/jbc.M304333200. [DOI] [PubMed] [Google Scholar]
- Wenger C, Ellenrieder V, Alber B, Lacher U, Menke A, Hameister H, et al. Expression and differential regulation of connective tissue growth factor in pancreatic cancer cells. Oncogene. 1999;18:1073–1080. doi: 10.1038/sj.onc.1202395. [DOI] [PubMed] [Google Scholar]
- Yang DH, Kim HS, Wilson EM, Rosenfeld RG, Oh Y. Identification of glycosylated 38-kDa connective tissue growth factor (IGFBP-related protein 2) and proteolytic fragments in human biological fluids, and up-regulation of IGFBP-rP2 expression by TGF-beta in Hs578T human breast cancer cells. J Clin Endocrinol Metab. 1998;83:2593–2596. doi: 10.1210/jcem.83.7.5097. [DOI] [PubMed] [Google Scholar]
- Yi CH, Smith DJ, West WW, Hollingsworth MA. Loss of fibulin−2 expression is associated with breast cancer progression. Am J Pathol. 2007;170:1535–1545. doi: 10.2353/ajpath.2007.060478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Liu X, Wong S, Machan JT, Chung MA. MUC1 knockdown with RNA interference inhibits pancreatic cancer growth. J Surg Res. 2008 doi: 10.1016/j.jss.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Zarrinkalam KH, Stanley JM, Gray J, Oliver N, Faull RJ. Connective tissue growth factor and its regulation in the peritoneal cavity of peritoneal dialysis patients. Kidney Int. 2003;64:331–338. doi: 10.1046/j.1523-1755.2003.00069.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.