

Abstract
Hematopoietic cells, including lymphoid and myeloid cells, can develop into phenotypically distinct ‘subpopulations’ with different functions. However, evidence indicates that some of these subpopulations can manifest substantial plasticity (that is, undergo changes in their phenotype and function). Here we focus on the occurrence of phenotypically distinct subpopulations in three lineages of myeloid cells with important roles in innate and acquired immunity: macrophages, mast cells and neutrophils. Cytokine signals, epigenetic modifications and other microenvironmental factors can substantially and, in some cases, rapidly and reversibly alter the phenotype of these cells and influence their function. This suggests that regulation of the phenotype and function of differentiated hematopoietic cells by microenvironmental factors, including those generated during immune responses, represents a common mechanism for modulating innate or adaptive immunity.
In innate or adaptive immunity, differentiated hematopoietic cells must orchestrate complex functional programs to promote host defense while also limiting maladaptive collateral damage to the tissues in which such responses take place. There is mounting evidence that one of the mechanisms used to achieve this goal is the induction of alterations in the phenotype of various cells of the innate or adaptive immunity, which positions them to serve the appropriate functions in distinct contexts. However, the extent of such ‘functional plasticity’ of differentiated hematopoietic cells and the mechanisms that regulate such phenotypic changes remain to be fully understood.
In principle, substantial changes in cell phenotype and function can be achieved by exposure of susceptible differentiated cell populations to the appropriate mixtures of a relatively small number of signals. In the example of induced pluripotent stem cells, enforced expression of a limited set of transcription factors permits adult somatic cells to gain features of pluripotency, which in turn permits the directed differentiation of such induced pluripotent stem cells into various new cell lineages with highly specialized functions1. The direct reprogramming of adult somatic cells into those with features of another distinct cell type also has been achieved1.
Although the hematopoietic system has substantial developmental plasticity, as exemplified by the ability of hematopoietic stem cells to give rise to all hematopoietic lineages2,3, the ability of the main differentiated hematopoietic cell types to undergo substantial phenotypic changes is thought to be much more limited. However, some of these differentiated cell types (for example, T cells4, B cells5 and dendritic cells (DCs)6) consist of phenotypically distinct subpopulations that can serve different functions. In such settings, it has become customary to define and then name such subpopulations in part on the basis of their phenotypic characteristics and in part on the basis of their actions and then, when analyzing such cells in vitro or in vivo, to use such phenotypic features to infer function. This must be done cautiously, as some hematopoietic cells can have surface structures and other features of more than one ‘classical’ cell lineage, such as macrophages and DCs7 or DCs and B cells8.
Moreover, published work has called into question the stability of certain subpopulations of hematopoietic cells. For example, some T cell subpopulations can show considerable plasticity, at least under in vitro conditions9–11, including examples of situations in which T cell populations with features of one subset (for example, regulatory T cells) can acquire the phenotypic and functional characteristics of another (for example, interleukin 17 (IL-17)-producing helper T cells (TH17 cells))11. Given such findings, the following general questions might arise: to what extent is the phenotype and function of differentiated hematopoietic cell types intrinsically variable, either at the level of the single cell or the population; how is such variation regulated; and, equally important, what are the implications of such phenotypic plasticity for the roles of such cells in health or disease?
In this review we have addressed those questions in the context of three populations of myeloid cells that function mainly in the peripheral tissues and can have important roles in both innate and adaptive immune responses: macrophages, mast cells and neutrophils. We review the evidence that there are phenotypically and functionally distinct subpopulations or activation phenotypes of these three main lineages of myeloid cells and consider briefly some of what is known about the consequences of such plasticity in the context of the functions of these cell types during innate and adaptive immunity.
Basic biology of macrophages
A committed progenitor cell in the bone marrow is responsible for generating the mononuclear phagocyte system3. The two main populations in this lineage are the macrophages and DCs, which display many common cell surface receptors but have distinct functional activities. In contrast to neutrophils, which are short-lived, there is great complexity in the lifespan of macrophages and DCs, which varies from hours to possibly years depending on the nature of the immune response7. Tissue macrophages differentiate from circulating monocytes when they enter tissues and are distinguished from DCs by their expression of F4/80, CD11b and Fc receptors; also, in contrast to DCs, which serve as the main inducers of adaptive T cell responses, macrophages have proteolytic and catabolic activities and are more skilled at ingesting pathogens by phagocytosis, scavenging dead cells and cellular debris, and remodeling tissues after injury12–14 (Table 1).
Table 1.
Natural history and main functions of macrophages, mast cells and neutrophils
| Characteristic | Macrophages | Mast cells | Neutrophils |
|---|---|---|---|
| Difference in phenotype | F4/80+ (mouse) or EMR1+ (human), CD107b+ (Mac-3+), CD68+ | c-Kithi, FcεRIhi (including the αβγγ form of FcεRI; some macrophages and neutrophils can express the αγγ form of FcεRI); prominent cytoplasmic granules, some of which contain tryptase and/or contain heparin | Ly6G+, MPO+, polylobed nucleus |
| Origin of precursor cells | Bone marrow | Bone marrow | Bone marrow |
| Site of maturation | Almost all tissues (a few in the bone marrow) | Almost all tissues (a few in the bone marrow) | Bone marrow |
| Mature cells in the circulation | No (or very few) | No (except in mast cell disorders such a mastocytosis) | Yes |
| Mature cells recruited into tissues from circulation | No (immature monocytes migrate into tissues) | No (mast cell progenitors migrate into tissues) | Yes (during innate or acquired immune responses) |
| Mature cells normally reside in connective tissues | Yes | Yes | No (not detectable by microscopy) |
| Proliferative ability of mature cells | May vary by subpopulation (M2 macrophages can proliferate under certain circumstances) | Yes (under certain circumstances) | None reported |
| Lifespan | Weeks to months | Weeks to months (on the basis of studies of rodents) | Days (like other granulocytes) |
| Phenotypically distinct subpopulations in different tissues | Yes (Fig. 1) | Yes (Fig. 2) | Not reported |
| Phagocytosis | Yes | Reported, but biological importance not fully understood | Yes |
| Detect pathogens and danger signals and help to initiate inflammation | Yes (including near surfaces exposed to the environment) | Yes (including near surfaces exposed to the environment) | Yes (for example, in the blood or at sites of ongoing inflammation) |
| Enhance inflammation | Yes | Yes | Yes |
| Limit or suppress inflammation | Yes | Yes | Yes |
| Promote tissue repair | Yes | Yes | Yes |
| Antigen presentation | Yes | Reported, but biological importance is uncertain | Reported, but biological importance is uncertain |
| Degrade or detoxify components of animal venoms | Not reported | Yes | Not reported |
Macrophages and DCs can be further subcategorized into subpopulations on the basis of their anatomical location and functional phenotype15 (Fig. 1). Tissue-resident macrophages include osteoclasts (bone), microglia (brain), alveolar macrophages (lung), histiocytes (interstitial connective tissue) and Kupffer cells (liver). There are also many mononuclear phagocyte subpopulations in the circulation and in the spleen that can differentiate into macrophages12,16. Although their phenotypes and names vary on the basis of their anatomical location, they all act like macrophages and acquire similar functional abilities when stimulated appropriately17.
Figure 1.
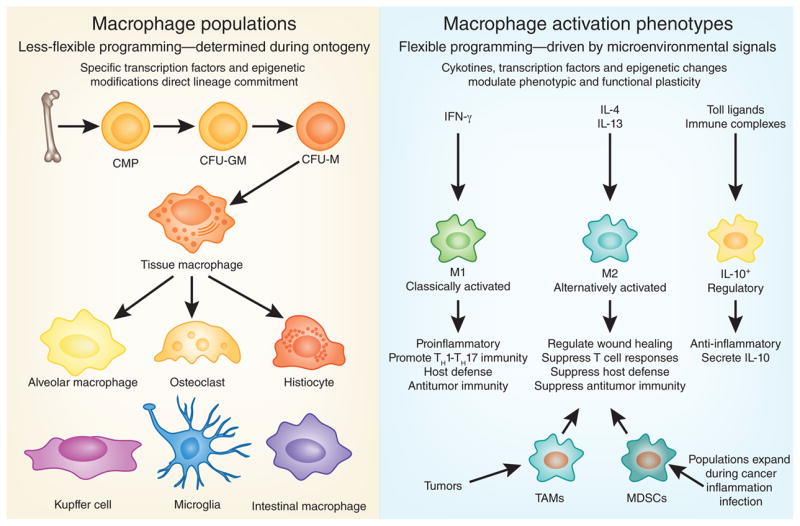
Macrophage populations and functional subsets. Macrophages can be subcategorized into specific populations on the basis of their anatomical location (left) and functional phenotype (right). Tissue-resident macrophages include alveolar macrophages (lungs), histiocytes (interstitial connective tissue), osteoclasts (bone), microglia (brain), intestinal macrophages, Kupffer cells (liver) and so on. Mononuclear phagocyte subpopulations in the circulation can also differentiate into tissue macrophages after entering different anatomical sites; when activated by the appropriate stimuli, these cells differentiate into various subsets with distinct phenotypic and functional characteristics.
Because phenotypic markers have not been particularly helpful in distinguishing the many subpopulations of macrophages13, a more useful approach has been to define macrophages on the basis of their specific functional activities18. Cells with a variety of functional phenotypes have been described, including classically activated macrophages (M1 macrophages; which mediate host defense and antitumor immunity), alternatively activated macrophages (M2 macrophages; which are suppressors and regulate wound healing), regulatory macrophages (which secrete IL-10), tumor-associated macrophages (which suppress tumor immunity) and the monocytic subset of myeloid-derived suppressor cells (MDSCs; which are functionally similar to tumor-associated macrophages), to name just a few19. Although there are some differences among the M2, regulatory, tumor-associated and MDSC subsets, each of these populations has mainly immunosuppressive activity17. Consequently, macrophages have one of two major phenotypes: they either induce host defense, antitumor immunity and inflammatory responses or suppress those functions. Therefore, macrophages, like T cells, can have active roles in both the induction and the resolution of immune responses.
Regulation of the phenotype and function of macrophages
Macrophages maintain tissue homeostasis by serving various housekeeping functions that apparently require no special activating stimuli or are activated during ontogeny. Macrophages are phagocytic cells that constitutively express a variety of scavenger receptors that facilitate the removal of aged red blood cells, necrotic tissues and toxic molecules from the circulation12. However, to keep up with greater demand, these homeostatic functions are increased by a variety of activating stimuli after tissue injury or during infection. Thus, like DCs, macrophages serve as sentinel cells for the immune response. They express pattern-recognition receptors that identify pathogen-associated or damage-associated molecular patterns expressed by microbial pathogens (for example, lipopolysaccharide) or during cellular stress (for example, nuclear and cytosolic proteins), respectively12. These receptors ‘instruct’ macrophages to produce a variety of mediators that recruit neutrophils and promote inflammation20.
Pathogen-associated and damage-associated molecular patterns also act in synergy with natural killer cell–derived interferon-γ to polarize macrophages toward the M1 phenotype, which is characterized by the production of reactive oxygen and nitrogen species that facilitate the killing of microbial pathogens19,21. M2 polarization, in contrast, is a programmed response facilitated by the signal transducer STAT6–activating cytokines IL-4 and IL-13 (ref. 12). Although CD4+ type 2 helper T cells (TH2 cells) are important inducers of M2 macrophages, a variety of innate IL-4- and IL-13-producing cells, such as basophils, nuocytes and natural helper cells, also may contribute to M2 polarization22–24. In addition to IL-4 and IL-13, IL-10, IL-21, the cell-signaling molecule GM-CSF, IL-33 and unique transcription factors regulate the differentiation of M2 cells25,26.
Epigenetic changes regulated by Jmjd3, a demethylase of histone 3 Lys27 (H3K27), have also been shown to control IL-4-induced M2 polarization27. IL-4 activates Jmjd3 expression, which results in less H3K27-dimethylation and H3K27-trimethylation marks in the promoters of several M2 marker genes, including Chi3l3, Retnla and Arg1, leading to their transcriptional activation28. It also induces expression of the transcription factor IRF4, which promotes M2 polarization while inhibiting the transcription of many M1-associated genes26. A role for CD4+CD25+Foxp3+ regulatory T cells in the polarization of human M2-like macrophages has also been described29. In contrast to the proinflammatory M1 cells, M2 macrophages suppress inflammation and antitumor immunity, facilitate wound repair, regulate glucose metabolism and mediate the expulsion of extracellular parasites from the gut18,30,31.
A third main class of macrophages called ‘regulatory macrophages’, similar to suppressive M2 cells, have also been described19. These macrophages are induced by Toll-like receptor (TLR) agonists in the presence of immunoglobulin G (IgG) immune complexes, apop-totic cells and prostaglandins and are defined by production of the immunosuppressive cytokines IL-10 and TGF-β1 (ref. 19). These cells are poor antigen-presenting cells and have a propensity to induce TH2 and regulatory T cell responses, which can further suppress chronic inflammatory and antitumor responses. Although distinct subpopulations of macrophages with unique functional abilities have been described, it is generally thought that macrophages represent a spectrum of activated phenotypes rather than discrete stable subpopulations19. Indeed, many studies have documented flexibility in macrophage programming, with macrophages readily switching from one functional phenotype to another in response to new microenvironmental signals32–35.
Macrophages in innate immunity
Macrophages are cells that function in both innate and adaptive immunity and can exert protective and pathogenic activity. They express a variety of pattern-recognition receptors, including TLRs, C-type lectin receptors, helicase RIG–like receptors and biosensor Nod–like receptors that recognize danger signals associated with invading pathogens, foreign substances (for example, silica and asbestos), and dead or dying cells7,12. These receptors all have important roles in the activation of the innate immune response. They ‘instruct’ macrophages to engulf and destroy foreign particles and bacteria through the generation of a respiratory burst, thus promoting an M1-like phenotype. They also activate signaling by the adaptor MyD88 and regulate inflammasome formation19,36, which can enhance the innate antimicrobial activity of M1 cells by stimulating the production of cytokines such as TNF and IL-1β.
In addition to their innate phagocytic activity and role in antimicrobial immunity, macrophages are also intimately involved in wound repair14. Macrophages are rapidly recruited to wounds after platelet degranulation, which may reflect in part the response of macrophages to platelet-derived growth factor37, and macrophages can secrete a wide variety of cytokines and chemokines, as well as matrix metalloproteinases and their tissue inhibitors, that regulate the recruitment of cells to and deposition of extracellular matrix components at sites of tissue injury38. Macrophages with a wound-healing M2-like phenotype also regulate important metabolic functions39. These macrophages are induced by signaling via the receptor PPAR-γ and maintain adipocyte function, insulin sensitivity and glucose tolerance, which prevents the development of diet-induced obesity40,41. It has been reported that IL-4-producing eosinophils are needed to maintain this particular macrophage population42. Those studies suggest that as obesity progresses, adipose-associated macrophages switch from an M2-like phenotype to a classically activated M1-like cell with potent proinflammatory activity39, with the NLRP3 inflammasome serving as the molecular switch by sensing obesity-associated danger signals43.
Macrophages in adaptive immunity
Although they are not as efficient as DCs, macrophages function as antigen-presenting cells and participate in the activation of the adaptive arm of the immune response12. Natural killer cells often provide the initial interferon-γ that ‘instructs’ macrophages to develop a classically activated M1 phenotype19. These innate inflammatory macrophages produce large amounts of TNF, IL-12 and IL-23 and therefore are important drivers of antigen-specific type 1 helper T cell (TH1 cell) and TH17 cell responses44. T cell–derived interferon-γ in turn feeds back, providing a positive amplification loop that expands the population of M1 cells while also increasing their microbicidal and tumoricidal activity19. M1 cells are generally believed to serve a protective role in tumorigenesis by antagonizing the suppressive activities of tumor-associated macrophages, MDSCs, alternatively activated macrophages and regulatory macrophages, which promote tumor growth, invasion and metastasis by suppressing adaptive antitumor immune responses45. Because M1 macrophages secrete large amounts of TNF and IL-1β and participate in the differentiation of TH17 cells, they are also believed to be important drivers of chronic inflammatory and autoimmune diseases, including rheumatoid arthritis, atherosclerosis, pulmonary fibrosis and Crohn’s disease46–48.
In contrast to M1 cells, M2 cells have mainly suppressive or immunoregulatory activity. They antagonize toxic M1 responses, dampen inflammation, suppress antitumor immunity and promote wound healing, tissue remodeling and angiogenesis12,49. Regulatory macrophages that secrete IL-10 have similar roles in adaptive immune responses, although they are particularly adept at suppressing antimicrobial immunity19. Regulatory macrophages also facilitate the maintenance of immune homeostasis in the gut by inducing the development of regulatory T cells50, whereas M2 cells mediate secondary immunity to gastrointestinal worms30. Although alternatively activated macrophages are induced by a variety of innate IL-4- and IL-13-producing cells, including basophils51, TH2 cells are thought to serve as the main inducers of M2 cells when the adaptive immune response is activated, as in many chronic inflammatory and fibrotic diseases14,18,31.
Basic biology of mast cells
Mast cells are derived from hematopoietic stem cells but, as is true for macrophages, the mature cells do not ordinarily circulate52 (Table 1). Instead, the precursor cells differentiate and mature locally after their migration into the vascularized tissues or (in rodents) serosal cavities in which mast cells will ultimately reside52. In vertebrates, mast cells are widely distributed throughout tissues, especially near surfaces exposed to the environment (for example, skin, airways and the gastrointestinal and genitourinary tracts), where pathogens, allergens and other environmental agents are frequently encountered52. This distribution permits them, along with DCs and tissue macrophages, to be among the first cells of the immune response to interact with environmental antigens and allergens, invading pathogens or environmentally derived toxins53. Like macrophages, mast cells can be long-lived, and at least some tissue mast cells can proliferate after the appropriate stimulation52,54. Although the extent to which various macrophage subpopulations have proliferative ability remains controversial, it has been reported that M2 macrophages can proliferate55. Mast cell populations also can expand via enhanced recruitment, survival and maturation of progenitors52,54. Many innate or acquired immune responses, including those associated with TH2-type responses, and many diseases processes are associated with changes in number of mast cells at the affected sites52,54.
Although mast cells share many structures and functions with macrophages (including many pathogen-associated and damage-associated molecular patterns) and have been reported to perform antigen presentation and phagocytosis56, mast cells are particularly specialized to serve functions that can amplify or suppress innate or acquired immune responses52,53,56–61. In large part, such functions reflect the ability of mast cells to secrete a wide spectrum of preformed or newly synthesized biologically active products, many of which can potentially mediate proinflammatory, anti-inflammatory and/or immunosuppressive functions52,54,56,58,59 (Fig. 2). Mast cells can participate in many cycles of activation for the release of mediators and can be activated to release distinct patterns of mediators or cytokines depending on the type and strength of the activating stimuli52,54,57,62 (Fig. 2). Moreover, the strength and nature of the response of mast cells to various activating stimuli, including aggregation of surface high-affinity IgE receptors (FcεRI receptors) by IgE and specific antigen (the main stimulus of mast-cell activation in allergic disorders and immune responses to parasites), may be influenced by genetic or microenvironmental factors that affect the expression pattern or functional properties of the surface receptors or signaling molecules that contribute to such responses52,54,57,62.
Figure 2.

Mast-cell populations and patterns of functional activation. Mast cells (MCs) in mice or humans can be subcategorized (left) into populations defined by anatomical location and/or mediator content (such as proteoglycans (heparin versus chondroitin sulfates) or proteases (tryptases, chymases or MC-CPA)). In IgE-associated immune responses to allergens or parasites (top right), the activation of mast cells via crosslinking of IgE bound to high-affinity receptors for IgE (FcεRI) on the cell surface by bi- or multivalent antigens results in rapid exocytosis of the cytoplasmic granules (degranulation) and the production of lipid mediators (such as leukotrienes and prostaglandins) and the more sustained secretion of many cytokines, chemokines and growth factors. Although many of these mediators have proinflammatory effects, others can have effects that suppress inflammation or promote tissue remodeling or repair. Signals not dependent on IgE (bottom right) can elicit different patterns of mediator release in mast-cell populations that express receptors appropriate for such ligands. Microenvironmental factors can influence the phenotype of mast cells that develop under basal conditions in different anatomic sites (left), including those phenotypic features that permit mast cells to respond to various ligands (such as the pattern of expression of receptors for those ligands) or to produce different mediators (right). TLRs are examples of the many pattern-recognition receptors expressed by various populations of mast cells. MCT, mast cell containing mainly tryptase; MCTC, mast cell containing both tryptase and chymase; C3a and C5a, anaphylatoxins of the complement system.
Regulation of the phenotype and function of mast cells
Environmental and genetic factors can finely control or ‘tune’ many key characteristics of mast-cell populations, such as their proliferation, survival and ability to store and/or produce various secreted products, and the magnitude and nature of their secretory responses to specific stimuli of activation generated during innate or acquired immune responses52. Stem-cell factor (SCF; also known as the ligand for the receptor c-Kit (CD117)) is the main survival and developmental factor for mast cells, but many growth factors, cytokines and chemokines can influence the number and mediator content of mast cells and other aspects of their phenotype, including IL-3, which is especially important in mice, TH2-associated cytokines (such as IL-4 and IL-9), and TGF-β1 (refs. 52, 54).
Important aspects of mast-cell phenotype can vary according to animal species, anatomical location, individual genetics or strain background, systemic or local changes in the amount of factors that can alter various properties of the cell, and whether the cell is analyzed in vivo or in vitro52,63 (Fig. 2). As the main functions of mast cells are thought to reflect the biological activities of their secreted products, it is widely thought that factors that influence either the spectrum of stimuli that can activate the release of mast-cell mediators (such as the types of immunoglobulins or pattern-recognition receptors expressed by the cells) or the ability of the cells to produce various mediators will in turn alter the functions of the cells in vivo52,54,56,58,59
Mast cells in innate immunity
Like macrophages, mast cells also can function in both innate and adaptive immune responses and can have protective and pathogenic activity. A chief role of mast cells in innate immunity is to enhance the local recruitment of neutrophils, a function that can either enhance host resistance or contribute to pathology56,64,65. Although there are many examples of how changes in mast-cell phenotype might influence the function of the cells of innate immunity, variation in the ability of mast cells to synthesize and store different proteases or proteoglycans is of particular interest. Stored serine proteases and proteoglycans represent a substantial fraction of the mass of a mast cell, and individual mast-cell subpopulations can store different mixtures of such proteases and proteoglycans (Fig. 2). In mice, there is evidence that the ability of mast cells to release large amounts of proteases (and perhaps heparin) permits these cells to enhance host resistance to the venoms of poisonous reptiles and arthropods and also to limit the toxicity of certain endogenous peptides, when exposure to such substances induces mast-cell degranulation and rapid release of the stored mediators66–70 (Fig. 2). Some mast-cell proteases (such as mMCP-6) contribute to the ability of the cells to recruit neutrophils to sites of bacterial infection64. Indeed, enhancing host resistance to toxins and inducing acute inflammation in response to pathogens may represent phylogenetically ancient innate functions of this cell type.
In humans, mast cells have been categorized on the basis of their protease content into those that contain predominantly tryptase or (more rarely) chymase or both major proteases71 (Fig. 2). As chymase and tryptase have distinct substrate specificities72,73, factors that can regulate the content of these two proteases in mast cells will in turn regulate the types of functions that the cells can serve after secretion of their stored mediators. Similarly, in mice, different mast-cell populations vary in their proteoglycan content, with some populations containing abundant heparin in their granules (connective tissue or serosal mast cells) or some having little or no heparin (mucosal mast cells)52 (Fig. 2), and this will obviously influence the types of proteoglycan-dependent functions of each mast-cell subset.
However, in vitro studies show that populations of human mast cells that contain tryptase but little or no chymase and are maintained in SCF-containing media can have more chymase after incubation with IL-4 (refs. 74–77), IL-6 or IL-1β77, TGF-β1 (ref. 78) or lipopolysaccharide77 and that mouse mast cells lacking heparin can be induced to synthesize and store heparin after contact with fibroblasts79 or exposure to SCF52. Although the extent to which such changes are reversible in individual cells is not clear, it has been shown that single mouse peritoneal mast cells that do contain heparin can give rise in vitro to clonal populations of mast cells that contain little or no heparin and that when such mast-cell populations that lack heparin are injected in vivo into mice genetically deficient in mast cells (WBB6F1-KitW/W–v mice), the cells become or give rise to cells that contain heparin80.
Mast cells in adaptive immunity
Mast cells in mice can store a substantially larger number of different proteases than can those in humans73,81, and the protease content of mast-cell populations in particular anatomical sites, such as the small intestine, can change during the course of infections associated with considerable increases in mast-cell numbers (such as infection with Trichinella spiralis)81,82. Because important mast-cell functions in such adaptive immune responses are thought to depend on the secretion of proteases from the cells, then factors that regulate this aspect of the cellular phenotype will in turn regulate mast-cell functions.
Mast-cell functions can also be altered via the types and amounts of receptors displayed by the cells, which renders the cells better able either to enhance or even (in some models) to suppress particular acquired immune responses. In TH2-associated allergic disorders or responses to parasite infection, high concentrations of circulating IgE result in high surface expression of FcεRI by tissue mast cells (and circulating basophils), which in turn can enhance the IgE-dependent effector functions of such cells83. In mice, IgE- and antigen-dependent mast-cell activation can be markedly enhanced by exposure of the cells to interferon-γ84, a cytokine that also enhances proinflammatory actions of macrophages (Fig. 1). In contrast, in certain models of severe contact-hypersensitivity responses, both in vitro and in vivo evidence suggests that the development of hapten-specific IgG1 antibodies during the response85, combined with a change in the cytokine milieu at the site of the pathology, permits mast cells that had been adoptively transferred to that site to limit the extent of inflammation and pathology observed in WBB6F1-KitW/W–v mice or C57BL/6-KitW–sh/W–sh mice genetically deficient in mast cells85,86, at least in part by enhancing the ability of the mast cells to secrete IL-10 via an FcγRIII-dependent mechanism85. The evidence that stimulation of skin mast cells via immune complexes of IgG1 may enhance their secretion of IL-10 is reminiscent of a similar effect of immune complexes on tissue macrophages (Fig. 1).
However, evidence from work with mice with mast cell–specific inactivation of Il10 has not confirmed a role for mast cell–derived IL-10 in the suppression of contact hypersensitivity87. Moreover, studies of mice rendered inducibly or constitutively deficient in mast cells independently of mutations affecting the gene encoding c-Kit have indicated, as did prior studies of IgE-deficient mice or mast cell–deficient WBB6F1-KitW/W–v mice88, that in the models of contact hypersensitivity tested, mast cells function mainly to enhance the response, including acting by means of effects on the sensitization phase87. Such findings suggest that the greater intensity of certain contact-hypersensitivity responses in WBB6F1-KitW/W–v or C57BL/6-KitW–sh/W–sh mice may reflect abnormalities other than simply the mast cell deficiency of these mice (even though adoptive transfer of mast cells to such mice can limit the extent of their contact-hypersensitivity responses)85,86.
In addition to producing IL-10, mast cells can produce an enormous spectrum of cytokines, chemokines and growth factors, as well as histamine and other autocoid mediators, many of which can participate in the transition from innate immunity to adaptive immunity52,54,57,59,61. Some of these mast cell–derived products may function in part to alter the phenotype and actions of other myeloid cells. For example, mast cells represent a potential source of TNF, which can influence the functions of macrophages and neutrophils, and of both IL-4 and IL-13, which can induce the M2 phenotype in macrophages (Fig. 1). Indeed, evidence from studies of C57BL/6-KitW–sh/W–sh mice indicates that mast cells are key drivers of an M2 response in a mouse model of chronic allergic inflammation of the airways, in which mast cells are required both for the much higher lung concentrations of IL-13 and for the greater production by the lungs of several factors associated with an M2 macrophage response84.
On the basis of evidence derived mainly from c-Kit-deficient mice, mast cell have also been linked to the promotion of peripheral tolerance to skin grafts89 and to modulating host stromal and immune responses to tumors (notably, depending on the model system, with consequences that can favor either the tumor or the host60,90). It will be important to reassess the roles of mast cells in such models with mice deficient in mast cells independently of mutations that affect c-Kit.
Basic biology of neutrophils
Neutrophils have shorter lives than do macrophages and mast cells, and unlike those cells, neutrophils are released into the blood as mature or nearly mature cells devoid of proliferative potential (Table 1). The estimate of the time that neutrophils spend in circulation has been extended more than tenfold from 5–10 hours to 5.4 days (ref. 91). Properly understanding how the lifespan and distribution of neutrophils are regulated is important for many reasons, including the obvious point that these features of neutrophil biology provide the temporal and spatial context in which neutrophil phenotypic and functional variation can occur. Neutrophils are estimated to be produced in the bone marrow at a rate of roughly 1 × 109 cells per kilogram body weight per day during the steady state92. However, maintaining a concentration of 3 × 106 neutrophils per ml blood would require that neutrophils disappear from the blood after 5 hours and not after 5 days (ref. 91). Thus, either the rate of production of neutrophils or the validity of the assumptions made to estimate the duration of neutrophils in the circulation must be reconsidered93. The hallmarks of neutrophils—cytoplasmic granules—are produced sequentially during maturation in the bone marrow, which results in a heterogeneous population of granules ranging from the azurophil granules produced at the promyelocyte stage to the gelatinase granules produced at the metamyelocyte and band-cell stage. The rate of granule production and the time allocated to maturation, and thus to the loading of cargo into granules, may be influenced by cytokines and growth factors in response to infection and inflammation30,94–99.
Regulation of the phenotype and function of neutrophils
Circulating neutrophils may be viewed as cells that have temporarily stopped their development, as they are not attached to any matrix and must remain functionally dormant so as not to obstruct the microcirculation100. Neutrophils become activated when caught by activated endothelial cells at sites of inflammation and/or infection and are activated further during their passage into tissues, where they can begin a new round of transcription of genes encoding modulators of the inflammatory response, such as IL-8, MIP-1α (CCL3) GRO-β (CXCL2), VEGF and IL-1β101, release their granules by exocytosis and mount a respiratory burst, which all contribute to the optimization of conditions for eradicating infecting microorganisms102 (Fig. 3). Type I interferons generated during microbial infection103 stimulate the recruitment of neutrophils and enhance phagocytosis via induction of the production of the chemokine CXCL10 (ref. 104).
Figure 3.
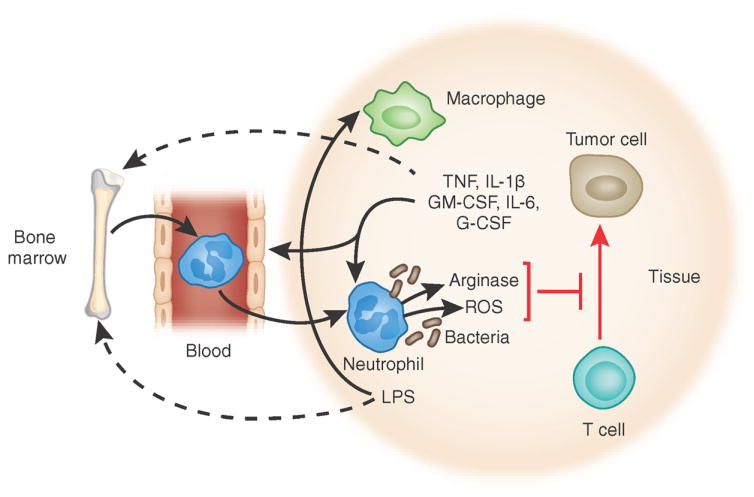
Features shared by ‘neutrophil MDSCs’ and neutrophils in mice with polymicrobial infection. Growth factors and cytokines generated by tumors and macrophages, as well as bacterial products, modulate the development and phenotype of neutrophils by acting both on developing neutrophils in the bone marrow and locally on neutrophils in tissues. Mast cells (not shown here) also can generate many cytokines and growth factors that can influence neutrophils, including TNF, IL-1β, GM-CSF and IL-6. Reactive oxygen species (ROS) and arginase secreted from activated neutrophils can inhibit T cell function and permit tumor growth. In this setting, such neutrophils constitute ‘neutrophil MDSCs’. LPS, lipopolysaccharide.
The oxygen tension in inflamed tissues is diminished by edema and the consumption of oxygen by the NADPH oxidase activity of the phagocytes that accumulate, as NADPH oxidase transfers electrons from intracellular NADPH to molecular oxygen to produce the reactive free radical superoxide. Superoxide, which can be produced in phagosomes or extracellularly, can spontaneously form hydrogen peroxide that undergoes further reactions to generate other reactive oxygen species. Hypoxia stabilizes hypoxia-induced factors, among which the transcription factor HIF-1α is prominent in phagocytes105. The transcription factor NF-κB is an important activator of HIF-1α and vice versa, and activation of NF-κB is a key activator of the proinflammatory activities of macrophages, neutrophils, mast cells and endothelial cells. In neutrophils, activation of NF-κB results in both the activation of inducible nitric oxide synthase and hence the production of nitric oxide and the synthesis and release of proinflammatory cytokines such as IL-8, TNF, IL-6 and IL-1β106. At sites of infection, NF-κB activation can be induced in neutrophils by activators of TLRs, TNF or IL-1β107. In such settings, transcription of the gene encoding HIF-1α will consequently be great and stabilization of HIF-1α mRNA by hypoxia will result in downstream effects of this transcription factor, including enhanced production of antimicrobial proteins and inducible nitric oxide synthase. The myeloid transcription factor KLF2 has been shown to be a key inhibitor of NF-κB-dependent transcription of the gene encoding HIF-1α and thus acts as a tonic repressor of myeloid-cell activation108.
The cytokine G-CSF is an important activator of neutrophils during bacterial infection, and one critical downstream target of G-CSF is CEACAM1 (CD66a), a transmembrane glycoprotein of specific granules and gelatinase granules of neutrophils109. Stimulation of neutrophils with G-CSF results in phosphorylation of the immunoreceptor tyrosine-based inhibitory motif of CEACAM1. Such phosphorylated CEACAM1 recruits the tyrosine phosphatase SHP-1, which acts as a bridge between CEACAM1 and the receptor for G-CSF and dephosphorylates that receptor, thus inhibiting its further signaling110. CEACAM-1-deficient mice not only have moderately more neutrophils but also a higher rate of death after challenge with Listeria monocytogenes due to the enhanced production of proinflammatory cytokines such as IL-1β and TNF by CEACAM-1-deficient neutrophils, which shows that enhanced stimulation of phagocytes can come at a price110.
Neutrophils in innate immunity
It is apparent that multiple signals that may be present at sites of inflammation or infection, including cytokines and growth factors, can influence individual phenotypic features of neutrophils in the tissues, and if such signals reach the bone marrow, they may influence the phenotype of neutrophils during their production at that site. Such factors can regulate the maturation of neutrophils, as well as the local responses of the cells to inflammation (Fig. 3). However, neutrophils are relatively short-lived cells that lack proliferative potential and are destined to die in the tissues to which they are recruited, and it is not yet clear to what extent locally induced phenotypic changes in neutrophils are stable or fully reversible. Accordingly, in contrast to the definition of subsets of T cells, macrophages and mast cells, there have been few attempts to define ‘subsets’ of neutrophils on the basis of their phenotype.
However, functionally important phenotypic features of neutrophils can vary stably from person to person. For example, human neutrophils can variably express CD177, a glycosylphosphatidylinositol-anchored protein located mainly on the membrane of specific granules and to the plasma membrane111. Its expression is influenced by polymorphisms of the gene encoding CD177 (ref. 112). CD177 is present in 0–100% of neutrophils, but the frequency of CD177+ neutrophils seems to be constant in each person113. CD177 interacts with CD31 (PECAM-1), which is present on platelets and endothelial cells and anchors the anti-neutrophil cytoplasmic autoantibody (ANCA) antigen PR3 to the surface of the CD177+ subset of neutrophils114. Only neutrophils that express CD177 are activated by ANCA to generate superoxide anions and to degranulate. The signal transduction depends on the association of CD177 with the integrin αMβ2 (Mac-1; CD11b) in lipid rafts and is mediated by Mac-1 (ref. 115). Therefore, it is possible that variation in the extent of neutrophil expression of CD177 among different people may influence the extent to which neutrophils can contribute to pathology in PR3-ANCA–associated small-vessel vasculitis115.
Neutrophils in adaptive immunity
Although the innate and adaptive immune systems operate very efficiently to prevent and combat infections, their ability to combat cancers is dismal. Many publications have indicated that a subset of neutrophil-like myeloid cells is the main suppressor of T cells in tumor-bearing mice and in patients with cancer and thus inhibit the elimination of tumors by T cells. These are the aforementioned MDSCs116. In the mouse, MDSCs were initially characterized as having a CD11b+Gr-1+ phenotype, which encompasses both neutrophils and monocytes117, but these can be distinguished by the expression of the marker Ly6G, which is present on neutrophils but not on monocytes, and by the expression of the marker Ly6C, which is high on monocytes but low on neutrophils118. It is now thought that there are two distinct MDSC subsets: granulocytic MDSCs have a CD11b+Ly6G+Ly6Clo phenotype, whereas MDSCs with monocytic morphology are CD11b+Ly6G–Ly6Chi. GM-CSF and IL-6 are particularly potent inducers of MDCSs that do so by activating the myeloid transcription factor C/EBPβ119. Evidence indicates that Ly6G+ granulocytic MDSCs have a major biological role in mouse models, and they can be detected in patients with cancer, but it is not clear that these cells constitute a particular developmentally regulated, stable subset of neutrophils. Instead, it seems more likely that such cells are neutrophils that are produced or phenotypically altered through the influence of cytokines and growth factors secreted by the tumors and/or by stromal cells reacting to the tumor cells. Such a hypothesis is consistent with the finding that cancers can induce a profile of myelopoiesis-stimulating growth factors and chemokines much the same as that seen during infection and inflammation120,121. Indeed, neutrophil MDSCs share many features with the neutrophils present in mice challenged by polymicrobial infection122,123. Tumors can produce many factors that can influence hematopoiesis, including IL-1β96,124, CCL2 (refs. 118,125), TGF-β126, G-CSF and GM-CSF127, which of course can also be produced during bacterial infection. In line with that, the transcription factor C/EBPβ, which is critical for emergency granulopoiesis in response to infection128, is also critical for the generation of MDSCs in both mice and humans119.
Neutrophil MDSCs contain large amounts of arginase I (a protein located in gelatinase granules of human neutrophils). Arginase I is probably one of the main suppressors of T cells, as deficiency in L-arginine inhibits T cell proliferation and function129 and arginase I converts L-arginine to L-ornithine, a precursor of polyamines and proline, which supports cell proliferation130 and collagen synthesis131 (Fig. 3). Neutrophil MDSCs also have relatively high NADPH oxidase activity; this results in generation of hydrogen peroxide, which again inhibits T cell function132. Nitric oxide synthase, another consumer of L-arginine, is induced in MDSCs and inhibits T cell function124,133; depletion of cysteine and cystine by MDSCs also has been demonstrated to inhibit T cell function134.
Concluding remarks
Having spent many years attempting to understand the biology and functions of the three myeloid cell types featured here, we are struck by how much there still is to learn. This is particularly true about the understanding of the regulation of macrophage, mast cell and neutrophil function in vivo; although many tools are available for investigating the functions of these cells in mice, the ability to assess the roles of these cells in humans is much more limited. Nevertheless, we have pointed out here that in both mice and humans, it is possible to identify ‘subpopulations’ of these cells that vary in phenotype and function. We therefore think that it is reasonable to conclude that regulating the development, stability and lifespan of such ‘subpopulations’ can represent a general strategy with which to ensure that macrophages, mast cells and neutrophils exert the appropriate functions during particular phases of innate and adaptive immune responses.
For both macrophages and mast cells, there is evidence (particularly from studies of mice) that cytokines and other microenvironmental factors present during an acquired immune response can alter the cells’ role from mainly promoting inflammation at the onset of the response to limiting the magnitude or duration of the response once it has developed31,85,86. Like macrophages and mast cells, neutrophils can also contribute to the resolution of inflammation via multiple mechanisms135,136. One such mechanism may be to undergo a change in phenotype that alters the production of lipid mediators by neutrophils from those that promote inflammation (such as prostaglandins and leukotrienes, produced early in the inflammatory response) to those that can have anti-inflammatory effects (such as lipoxins)137. In addition, the ingestion of apoptotic neutrophils by macrophages can induce the macrophages to produce derivatives of omega-3-polyunsaturated fatty acids (resolvins and protectins) that contribute to the blockade of further recruitment of neutrophils and that have other effects that actively promote resolution of the inflammation137. The last example is just one of many potentially important interactions among the three cell types featured here, some of which may enhance and others that may help resolve inflammatory responses.
Although each of the myeloid cells discussed here can have variation in phenotypic features that may influence cellular functions, there may be substantial differences among these cell types in the stage in their development at which such changes can occur. For cells that retain proliferative potential (mast cells and perhaps macrophages), phenotypic alterations may occur both in individual post-mitotic cells and in the population as a whole as it undergoes expansion. In contrast, neutrophils do not proliferate, so their phenotypic plasticity must be regulated in the context of the relatively short lifespan of this terminally differentiated cell type.
Rather than attempting to subcategorize these myeloid cells into phenotypically distinct ‘subpopulations’ with different names, we think that it is more useful to consider that macrophages, mast cells and neutrophils each consist of populations of individual cells with a broad spectrum of phenotypes and functions and that some key characteristics of these cells are subject to substantial microenvironmental modulation, particularly in the dynamic contexts of innate or adaptive immune responses (Box 1). The regulation of cellular functions may occur through the effects of factors that influence the phenotype and function of the mature cells and also by factors that alter the differentiation and/or maturation of the cells. For example, for mast cells analyzed in vitro, incubation with particular cytokines, such as SCF138, IL-4 (ref. 139) or TGF-β1 (ref. 78), that can induce evidence of more mast-cell maturation can also alter the types or amounts of mediators released from the cells after aggregation of FcεRI (the main mechanism for eliciting antigen-specific mast-cell function). This type of relatively rapid regulation, via transient increases in the amount of growth factors or cytokines, may be particularly important during innate or adaptive immune responses in which there is rapid population expansion of macrophages, mast cells and/or neutrophils. Regulation of function can also occur via alteration of the progenitor populations that give rise to the mature lineages. For example, evidence indicates that sepsis can result in epigenetic changes in hematopoietic progenitor populations that in turn may be reflected in altered (in this case, diminished) function of their differentiated progeny140. This kind of regulation may be especially important for neutrophils, given their short lifespan and lack of proliferative ability.
Box 1. Mechanisms that can contribute to changes in myeloid cell phenotype and function.
Epigenetic changes affecting progenitor cell populations (unless these also affect the hematopoietic stem cells, such changes may be transient as new progenitors are generated from hematopoietic stem cells)
Constitutive differences in anatomical microenvironments that induce local variation in the phenotype of the myeloid populations that develop at those sites; for example, the different types of macrophages, such as osteoclasts, Kuppfer cells, tissue macrophages, and so on (Fig. 1); connective tissue versus mucosal mast cells (in mice and rats); or mast cells containing tryptase versus mast cells containing tryptase and chymase (in humans; Fig. 2)
Factors that influence the maturation of the cells (for example, during population expansion in the context of inflammatory or immune responses) and factors that alter the cells’ functional repertoire (for example, the acquisition by mast cells, during their maturation, of the ability to synthesize and store heparin or particular serine proteases)
Factors that alter phenotypic features of the mature populations (for example, transition between M1 and M2 macrophage populations, acquisition by neutrophils or macrophages of a myeloid suppressor cell phenotype in a tumor-bearing host, development by mast cells of a phenotype that permits enhanced production of IL-10)
Together such findings suggest that in addition to genetic factors, the microenvironment can regulate the phenotype and function of differentiated myeloid cells at the level of their progenitors, during their lineage-specific differentiation and after they have matured into the fully differentiated cell types. Although we have focused on macrophages, mast cells and neutrophils here, it is likely that this general principle also applies to other myeloid cells and to lymphocytes.
Acknowledgments
We thank M. Tsai and J. Kalesnikoff for discussions; and J. Lilla for help with Figure 2. Supported by the US National Institutes of Health (AI70813, AI23990 and CA72074 to S.J.G.), the Danish National Research Council (N.B.) and the intramural research program of the National Institute of Allergy and Infectious Diseases, US National Institutes of Health (T.A.W.).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Cohen DE, Melton D. Turning straw into gold: directing cell fate for regenerative medicine. Nat Rev Genet. 2011;12:243–252. doi: 10.1038/nrg2938. [DOI] [PubMed] [Google Scholar]
- 2.Shizuru JA, Negrin RS, Weissman IL. Hematopoietic stem and progenitor cells: clinical and preclinical regeneration of the hematolymphoid system. Annu Rev Med. 2005;56:509–538. doi: 10.1146/annurev.med.54.101601.152334. [DOI] [PubMed] [Google Scholar]
- 3.Doulatov S, et al. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol. 2010;11:585–593. doi: 10.1038/ni.1889. [DOI] [PubMed] [Google Scholar]
- 4.Zhu J, Paul WE. Heterogeneity and plasticity of T helper cells. Cell Res. 2010;20:4–12. doi: 10.1038/cr.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allman D, Pillai S. Peripheral B cell subsets. Curr Opin Immunol. 2008;20:149–157. doi: 10.1016/j.coi.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palucka K, Banchereau J, Mellman I. Designing vaccines based on biology of human dendritic cell subsets. Immunity. 2010;33:464–478. doi: 10.1016/j.immuni.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geissmann F, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson BA, III, et al. B-lymphoid cells with attributes of dendritic cells regulate T cells via indoleamine 2,3-dioxygenase. Proc Natl Acad Sci USA. 2010;107:10644–10648. doi: 10.1073/pnas.0914347107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirahara K, et al. Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev. 2010;21:425–434. doi: 10.1016/j.cytogfr.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukasa R, et al. Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity. 2010;32:616–627. doi: 10.1016/j.immuni.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidl C, et al. Epigenetic reprogramming of the RORC locus during in vitro expansion is a distinctive feature of human memory but not naive Treg. Eur J Immunol. 2011;41:1491–1498. doi: 10.1002/eji.201041067. [DOI] [PubMed] [Google Scholar]
- 12.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 13.Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol. 2010;10:453–460. doi: 10.1038/nri2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. 2007;7:543–555. doi: 10.1038/nri2103. [DOI] [PubMed] [Google Scholar]
- 16.Krutzik SR, et al. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat Med. 2005;11:653–660. doi: 10.1038/nm1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol. 2011;89:557–563. doi: 10.1189/jlb.0710409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 19.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cros J, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33:375–386. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sindrilaru A, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. 2011;121:985–997. doi: 10.1172/JCI44490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuroda E, et al. SHIP represses the generation of IL-3-induced M2 macrophages by inhibiting IL-4 production from basophils. J Immunol. 2009;183:3652–3660. doi: 10.4049/jimmunol.0900864. [DOI] [PubMed] [Google Scholar]
- 23.Moro K, et al. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+Sca-1+ lymphoid cells. Nature. 2010;463:540–544. doi: 10.1038/nature08636. [DOI] [PubMed] [Google Scholar]
- 24.Neill DR, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pesce J, et al. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J Clin Invest. 2006;116:2044–2055. doi: 10.1172/JCI27727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satoh T, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11:936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 27.De Santa F, et al. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 28.Ishii M, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. 2009;114:3244–3254. doi: 10.1182/blood-2009-04-217620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tiemessen MM, et al. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci USA. 2007;104:19446–19451. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anthony RM, et al. Memory TH2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat Med. 2006;12:955–960. doi: 10.1038/nm1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pesce JT, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009;5:e1000371. doi: 10.1371/journal.ppat.1000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rutschman R, et al. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol. 2001;166:2173–2177. doi: 10.4049/jimmunol.166.4.2173. [DOI] [PubMed] [Google Scholar]
- 33.Hagemann T, et al. “Re-educating” tumor-associated macrophages by targeting NF-κB. J Exp Med. 2008;205:1261–1268. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mylonas KJ, Nair MG, Prieto-Lafuente L, Paape D, Allen JE. Alternatively activated macrophages elicited by helminth infection can be reprogrammed to enable microbial killing. J Immunol. 2009;182:3084–3094. doi: 10.4049/jimmunol.0803463. [DOI] [PubMed] [Google Scholar]
- 35.Kawanishi N, Yano H, Yokogawa Y, Suzuki K. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc Immunol Rev. 2010;16:105–118. [PubMed] [Google Scholar]
- 36.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 37.Uutela M, et al. PDGF-D induces macrophage recruitment, increased interstitial pressure, and blood vessel maturation during angiogenesis. Blood. 2004;104:3198–3204. doi: 10.1182/blood-2004-04-1485. [DOI] [PubMed] [Google Scholar]
- 38.Wynn TA. Fibrotic disease and the TH1/TH2 paradigm. Nat Rev Immunol. 2004;4:583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. 2011;6:275–297. doi: 10.1146/annurev-pathol-011110-130138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Odegaard JI, et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu D, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332:243–247. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandanmagsar B, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krausgruber T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12:231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 45.Nardin A, Abastado JP. Macrophages and cancer. Front Biosci. 2008;13:3494–3505. doi: 10.2741/2944. [DOI] [PubMed] [Google Scholar]
- 46.Smith AM, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med. 2009;206:1883–1897. doi: 10.1084/jem.20091233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson MS, et al. Bleomycin and IL-1β-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med. 2010;207:535–552. doi: 10.1084/jem.20092121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol. 2010;7:77–86. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.London A, et al. Neuroprotection and progenitor cell renewal in the injured adult murine retina requires healing monocyte-derived macrophages. J Exp Med. 2011;208:23–39. doi: 10.1084/jem.20101202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hadis U, et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity. 2011;34:237–246. doi: 10.1016/j.immuni.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 51.Maizels RM, Pearce EJ, Artis D, Yazdanbakhsh M, Wynn TA. Regulation of pathogenesis and immunity in helminth infections. J Exp Med. 2009;206:2059–2066. doi: 10.1084/jem.20091903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galli SJ, et al. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol. 2005;23:749–786. doi: 10.1146/annurev.immunol.21.120601.141025. [DOI] [PubMed] [Google Scholar]
- 53.Galli SJ, Tsai M. Mast cells in allergy and infection: versatile effector and regulatory cells in innate and adaptive immunity. Eur J Immunol. 2010;40:1843–1851. doi: 10.1002/eji.201040559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryan JJ, et al. Mast cell homeostasis: a fundamental aspect of allergic disease. Crit Rev Immunol. 2007;27:15–32. doi: 10.1615/critrevimmunol.v27.i1.20. [DOI] [PubMed] [Google Scholar]
- 55.Jenkins SJ, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–1288. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol. 2010;10:440–452. doi: 10.1038/nri2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat Immunol. 2005;6:135–142. doi: 10.1038/ni1158. [DOI] [PubMed] [Google Scholar]
- 58.Boyce JA. Mast cells and eicosanoid mediators: a system of reciprocal paracrine and autocrine regulation. Immunol Rev. 2007;217:168–185. doi: 10.1111/j.1600-065X.2007.00512.x. [DOI] [PubMed] [Google Scholar]
- 59.Galli SJ, Grimbaldeston M, Tsai M. Immunomodulatory mast cells: negative, as well as positive, regulators of immunity. Nat Rev Immunol. 2008;8:478–486. doi: 10.1038/nri2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalesnikoff J, Galli SJ. New developments in mast cell biology. Nat Immunol. 2008;9:1215–1223. doi: 10.1038/ni.f.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sayed BA, Christy A, Quirion MR, Brown MA. The master switch: the role of mast cells in autoimmunity and tolerance. Annu Rev Immunol. 2008;26:705–739. doi: 10.1146/annurev.immunol.26.021607.090320. [DOI] [PubMed] [Google Scholar]
- 62.Blank U, Rivera J. The ins and outs of IgE-dependent mast-cell exocytosis. Trends Immunol. 2004;25:266–273. doi: 10.1016/j.it.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 63.Bischoff SC. Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat Rev Immunol. 2007;7:93–104. doi: 10.1038/nri2018. [DOI] [PubMed] [Google Scholar]
- 64.Thakurdas SM, et al. The mast cell-restricted tryptase mMCP-6 has a critical immunoprotective role in bacterial infections. J Biol Chem. 2007;282:20809–20815. doi: 10.1074/jbc.M611842200. [DOI] [PubMed] [Google Scholar]
- 65.Piliponsky AM, et al. Mast cell-derived TNF can exacerbate mortality during severe bacterial infections in C57BL/6-KitW-sh/W-sh mice. Am J Pathol. 2010;176:926–938. doi: 10.2353/ajpath.2010.090342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maurer M, et al. Mast cells promote homeostasis by limiting endothelin-1-induced toxicity. Nature. 2004;432:512–516. doi: 10.1038/nature03085. [DOI] [PubMed] [Google Scholar]
- 67.Metz M, et al. Mast cells can enhance resistance to snake and honeybee venoms. Science. 2006;313:526–530. doi: 10.1126/science.1128877. [DOI] [PubMed] [Google Scholar]
- 68.Schneider LA, Schlenner SM, Feyerabend TB, Wunderlin M, Rodewald HR. Molecular mechanism of mast cell mediated innate defense against endothelin and snake venom sarafotoxin. J Exp Med. 2007;204:2629–2639. doi: 10.1084/jem.20071262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Piliponsky AM, et al. Neurotensin increases mortality and mast cells reduce neurotensin levels in a mouse model of sepsis. Nat Med. 2008;14:392–398. doi: 10.1038/nm1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Akahoshi M, et al. Mast cell chymase reduces the toxicity of Gila monster venom, scorpion venoms, and vasoactive intestinal polypeptide in mice. J Clin Invest. 2011;121:4180–4191. doi: 10.1172/JCI46139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Craig SS, Schwartz LB. Tryptase and chymase, markers of distinct types of human mast cells. Immunol Res. 1989;8:130–148. doi: 10.1007/BF02919075. [DOI] [PubMed] [Google Scholar]
- 72.Caughey GH. Mast cell tryptases and chymases in inflammation and host defense. Immunol Rev. 2007;217:141–154. doi: 10.1111/j.1600-065X.2007.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pejler G, Abrink M, Ringvall M, Wernersson S. Mast cell proteases. Adv Immunol. 2007;95:167–255. doi: 10.1016/S0065-2776(07)95006-3. [DOI] [PubMed] [Google Scholar]
- 74.Xia HZ, et al. Effect of recombinant human IL-4 on tryptase, chymase, and Fcε receptor type I expression in recombinant human stem cell factor-dependent fetal liver-derived human mast cells. J Immunol. 1997;159:2911–2921. [PubMed] [Google Scholar]
- 75.Toru H, et al. Interleukin-4 promotes the development of tryptase and chymase double-positive human mast cells accompanied by cell maturation. Blood. 1998;91:187–195. [PubMed] [Google Scholar]
- 76.Ahn K, et al. Regulation of chymase production in human mast cell progenitors. J Allergy Clin Immunol. 2000;106:321–328. doi: 10.1067/mai.2000.108107. [DOI] [PubMed] [Google Scholar]
- 77.Kirshenbaum AS, Swindle E, Kulka M, Wu Y, Metcalfe DD. Effect of lipopolysaccharide (LPS) and peptidoglycan (PGN) on human mast cell numbers, cytokine production, and protease composition. BMC Immunol. 2008;9:45. doi: 10.1186/1471-2172-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gebhardt T, et al. Growth, phenotype, and function of human intestinal mast cells are tightly regulated by transforming growth factor β1. Gut. 2005;54:928–934. doi: 10.1136/gut.2004.054650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Levi-Schaffer F, Austen KF, Gravallese PM, Stevens RL. Coculture of interleukin 3-dependent mouse mast cells with fibroblasts results in a phenotypic change of the mast cells. Proc Natl Acad Sci USA. 1986;83:6485–6488. doi: 10.1073/pnas.83.17.6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kanakura Y, et al. Multiple bidirectional alterations of phenotype and changes in proliferative potential during the in vitro and in vivo passage of clonal mast cell populations derived from mouse peritoneal mast cells. Blood. 1988;72:877–885. [PubMed] [Google Scholar]
- 81.Stevens RL, Adachi R. Protease-proteoglycan complexes of mouse and human mast cells and importance of their β-tryptase-heparin complexes in inflammation and innate immunity. Immunol Rev. 2007;217:155–167. doi: 10.1111/j.1600-065X.2007.00525.x. [DOI] [PubMed] [Google Scholar]
- 82.Friend DS, et al. Mast cells that reside at different locations in the jejunum of mice infected with Trichinella spiralis exhibit sequential changes in their granule ultrastructure and chymase phenotype. J Cell Biol. 1996;135:279–290. doi: 10.1083/jcb.135.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kawakami T, Galli SJ. Regulation of mast-cell and basophil function and survival by IgE. Nat Rev Immunol. 2002;2:773–786. doi: 10.1038/nri914. [DOI] [PubMed] [Google Scholar]
- 84.Yu M, et al. Identification of an IFN-γ/mast cell axis in a mouse model of chronic asthma. J Clin Invest. 2011;121:3133–3143. doi: 10.1172/JCI43598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grimbaldeston MA, Nakae S, Kalesnikoff J, Tsai M, Galli SJ. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol. 2007;8:1095–1104. doi: 10.1038/ni1503. [DOI] [PubMed] [Google Scholar]
- 86.Norman MU, et al. Mast cells regulate the magnitude and the cytokine microenvironment of the contact hypersensitivity response. Am J Pathol. 2008;172:1638–1649. doi: 10.2353/ajpath.2008.070559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dudeck A, et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity. 2011;34:973–984. doi: 10.1016/j.immuni.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 88.Bryce PJ, et al. Immune sensitization in the skin is enhanced by antigen-independent effects of IgE. Immunity. 2004;20:381–392. doi: 10.1016/s1074-7613(04)00080-9. [DOI] [PubMed] [Google Scholar]
- 89.Lu LF, et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. 2006;442:997–1002. doi: 10.1038/nature05010. [DOI] [PubMed] [Google Scholar]
- 90.Wasiuk A, de Vries VC, Hartmann K, Roers A, Noelle RJ. Mast cells as regulators of adaptive immunity to tumours. Clin Exp Immunol. 2009;155:140–146. doi: 10.1111/j.1365-2249.2008.03840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pillay J, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood. 2010;116:625–627. doi: 10.1182/blood-2010-01-259028. [DOI] [PubMed] [Google Scholar]
- 92.Mary JY. Normal human granulopoiesis revisited. II Bone marrow data. Biomed Pharmacother. 1985;39:66–77. [PubMed] [Google Scholar]
- 93.Summers C, et al. Neutrophil kinetics in health and disease. Trends Immunol. 2010;31:318–324. doi: 10.1016/j.it.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med. 2005;201:1771–1780. doi: 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine. 2008;42:277–288. doi: 10.1016/j.cyto.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ueda Y, Cain DW, Kuraoka M, Kondo M, Kelsoe G. IL-1R type I-dependent hemopoietic stem cell proliferation is necessary for inflammatory granulopoiesis and reactive neutrophilia. J Immunol. 2009;182:6477–6484. doi: 10.4049/jimmunol.0803961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Scumpia PO, et al. Cutting edge: bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. J Immunol. 2010;184:2247–2251. doi: 10.4049/jimmunol.0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang H, et al. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood. 2010;116:2462–2471. doi: 10.1182/blood-2009-12-259630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen CW, Sowden M, Zhao Q, Wiedmer T, Sims PJ. Nuclear phospholipid scramblase 1 prolongs the mitotic expansion of granulocyte precursors during G-CSF-induced granulopoiesis. J Leukoc Biol. 2011;90:221–223. doi: 10.1189/jlb.0111006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Theilgaard-Mönch K, Knudsen S, Follin P, Borregaard N. The transcriptional activation program of human neutrophils in skin lesions supports their important role in wound healing. J Immunol. 2004;172:7684–7693. doi: 10.4049/jimmunol.172.12.7684. [DOI] [PubMed] [Google Scholar]
- 102.Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657–670. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 103.Mancuso G, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. 2007;178:3126–3133. doi: 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- 104.Kelly-Scumpia KM, et al. Type I interferon signaling in hematopoietic cells is required for survival in mouse polymicrobial sepsis by regulating CXCL10. J Exp Med. 2010;207:319–326. doi: 10.1084/jem.20091959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Imtiyaz HZ, Simon MC. Hypoxia-inducible factors as essential regulators of inflammation. Curr Top Microbiol Immunol. 2010;345:105–120. doi: 10.1007/82_2010_74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 107.Verstrepen L, et al. TLR-4, IL-1R and TNF-R signaling to NF-kappaB: variations on a common theme. Cell Mol Life Sci. 2008;65:2964–2978. doi: 10.1007/s00018-008-8064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mahabeleshwar GH, et al. The myeloid transcription factor KLF2 regulates the host response to polymicrobial infection and endotoxic shock. Immunity. 2011;34:715–728. doi: 10.1016/j.immuni.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Feuk-Lagerstedt E, Jordan ET, Leffler H, Dahlgren C, Karlsson A. Identification of CD66a and CD66b as the major galectin-3 receptor candidates in human neutrophils. J Immunol. 1999;163:5592–5598. [PubMed] [Google Scholar]
- 110.Pan H, Shively JE. Carcinoembryonic antigen-related cell adhesion molecule-1 regulates granulopoiesis by inhibition of granulocyte colony-stimulating factor receptor. Immunity. 2010;33:620–631. doi: 10.1016/j.immuni.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Goldschmeding R, et al. Further characterization of the NB 1 antigen as a variably expressed 56–62 kD GPI-linked glycoprotein of plasma membranes and specific granules of neutrophils. Br J Haematol. 1992;81:336–345. doi: 10.1111/j.1365-2141.1992.tb08237.x. [DOI] [PubMed] [Google Scholar]
- 112.Wolff J, et al. Lack of NB1 GP (CD177/HNA-2a) gene transcription in NB1 GP- neutrophils from NB1 GP-expressing individuals and association of low expression with NB1 gene polymorphisms. Blood. 2003;102:731–733. doi: 10.1182/blood-2002-09-2831. [DOI] [PubMed] [Google Scholar]
- 113.Moritz E, et al. Human neutrophil alloantigens systems. An Acad Bras Cienc. 2009;81:559–569. doi: 10.1590/s0001-37652009000300019. [DOI] [PubMed] [Google Scholar]
- 114.von Vietinghoff S, et al. NB1 mediates surface expression of the ANCA antigen proteinase 3 on human neutrophils. Blood. 2007;109:4487–4493. doi: 10.1182/blood-2006-10-055327. [DOI] [PubMed] [Google Scholar]
- 115.Jerke U, et al. Complement receptor Mac-1 is an adaptor for NB1 (CD177)-mediated PR3-ANCA neutrophil activation. J Biol Chem. 2011;286:7070–7081. doi: 10.1074/jbc.M110.171256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gabrilovich DI, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425. doi: 10.1158/0008-5472.CAN-06-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kusmartsev S, Nagaraj S, Gabrilovich DI. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J Immunol. 2005;175:4583–4592. doi: 10.4049/jimmunol.175.7.4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sawanobori Y, et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008;111:5457–5466. doi: 10.1182/blood-2008-01-136895. [DOI] [PubMed] [Google Scholar]
- 119.Marigo I, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPβ transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 120.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 121.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Delano MJ, et al. MyD88-dependent expansion of an immature GR-1+CD11b+ population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204:1463–1474. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cuenca AG, et al. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med. 2011;17:281–292. doi: 10.2119/molmed.2010.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Elkabets M, et al. IL-1beta regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol. 2010;40:3347–3357. doi: 10.1002/eji.201041037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fridlender ZG, et al. CCL2 blockade augments cancer immunotherapy. Cancer Res. 2010;70:109–118. doi: 10.1158/0008-5472.CAN-09-2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fridlender ZG, et al. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ninck S, et al. Expression profiles of angiogenic growth factors in squamous cell carcinomas of the head and neck. Int J Cancer. 2003;106:34–44. doi: 10.1002/ijc.11188. [DOI] [PubMed] [Google Scholar]
- 128.Hirai H, et al. C/EBPβ is required for ‘emergency’ granulopoiesis. Nat Immunol. 2006;7:732–739. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 129.Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–191. doi: 10.1111/j.1600-065X.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Landau G, Bercovich Z, Park MH, Kahana C. The role of polyamines in supporting growth of mammalian cells is mediated through their requirement for translation initiation and elongation. J Biol Chem. 2010;285:12474–12481. doi: 10.1074/jbc.M110.106419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Curran JN, Winter DC, Bouchier-Hayes D. Biological fate and clinical implications of arginine metabolism in tissue healing. Wound Repair Regen. 2006;14:376–386. doi: 10.1111/j.1743-6109.2006.00151.x. [DOI] [PubMed] [Google Scholar]
- 132.Corzo CA, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182:5693–5701. doi: 10.4049/jimmunol.0900092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Movahedi K, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6Chigh monocytes. Cancer Res. 2010;70:5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 134.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70:68–77. doi: 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 136.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 137.Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators. Annu Rev Pathol. 2008;3:279–312. doi: 10.1146/annurev.pathmechdis.3.121806.151409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Murakami M, Matsumoto R, Urade Y, Austen KF, Arm JP. c-kit ligand mediates increased expression of cytosolic phospholipase A2, prostaglandin endoperoxide synthase-1, and hematopoietic prostaglandin D2 synthase and increased IgE-dependent prostaglandin D2 generation in immature mouse mast cells. J Biol Chem. 1995;270:3239–3246. doi: 10.1074/jbc.270.7.3239. [DOI] [PubMed] [Google Scholar]
- 139.Yamaguchi M, et al. IgE enhances Fcε receptor I expression and IgE-dependent release of histamine and lipid mediators from human umbilical cord blood-derived mast cells: synergistic effect of IL-4 and IgE on human mast cell Fc epsilon receptor I expression and mediator release. J Immunol. 1999;162:5455–5465. [PubMed] [Google Scholar]
- 140.Carson WF, Cavassani KA, Dou Y, Kunkel SL. Epigenetic regulation of immune cell functions during post-septic immunosuppression. Epigenetics. 2011;6:273–283. doi: 10.4161/epi.6.3.14017. [DOI] [PMC free article] [PubMed] [Google Scholar]