

Abstract
The high-yield expression and purification of Shewanella oneidensis cytochrome c nitrite reductase (ccNiR), and its characterization by a variety of methods, notably Laue crystallography, is reported. A key component of the expression system is an artificial ccNiR gene in which the N-terminal signal peptide from the highly expressed S. oneidensis protein “Small Tetra-heme c” replaces the wild-type signal peptide. This gene, inserted into the plasmid pHSG298 and expressed in S. oneidensis TSP-1 strain, generated ~20 mg crude ccNiR/L culture, compared with 0.5–1 mg/L for untransformed cells. Purified ccNiR has nitrite and hydroxylamine reductase activities comparable to those previously reported for E. coli ccNiR, and is stable for over two weeks in pH 7 solution at 4° C. UV/Vis spectropotentiometric titrations and protein film voltammetry identified 5 independent 1-electron reduction processes. Global analysis of the spectropotentiometric data also allowed determination of the extinction coefficient spectra for the 5 reduced ccNiR species. The characteristics of the individual extinction coefficient spectra suggest that, within each reduced species, the electrons are distributed amongst the various hemes, rather than being localized on specific heme centers. The purified ccNiR yielded good quality crystals, with which the 2.59 Å resolution structure was solved at room temperature using the Laue diffraction method. The structure is similar to that of E. coli ccNiR, except in the region where the enzyme interacts with its physiological electron donor (CymA in the case of S. oneidensis ccNiR, NrfB in the case of the E. coli protein).
Keywords: Cytochrome c nitrite reductase, NrfA, Laue crystallography, UV/Vis spectropotentiometry, Protein film voltammetry
Introduction
Ammonia-nitrite interconversion, an important part of the biological nitrogen cycle, is carried out by a variety of bacteria as part of their respiratory process. In one direction the ammonia oxidizing bacteria (AOB) such as Nitrosomonas europaea use ammonia as an electron donor in respiration, and oxidize it to nitrite [1]. A different class of bacteria uses nitrite as a terminal electron acceptor in the absence of oxygen, reducing it to ammonia in a process known as nitrite ammonification [2]. Hydroxylamine oxidoreductase (HAO), the AOB enzyme that catalyzes the 4-electron oxidation of hydroxylamine to nitrite, shows some structural similarities to the enzyme cytochrome c nitrite reductase (ccNiR, also called NrfA), which catalyzes the 6-electron reduction of nitrite to ammonia in ammonifying bacteria [3–10]. Moreover, formal catalytic cycles suggest that HAO and ccNiR probably go through many comparable reactive intermediates [11, 12]. Given the structural similarities between HAO and ccNiR, and the fact that they catalyze similar reactions in opposing directions, our research group is undertaking a long-term comparative study of HAO and ccNiR, aimed at determining how each enzyme is optimized to preferentially operate in one or the other direction. In two recent papers we described experiments in which HAO was forced to run in reverse (as a ccNiR) [12, 13]; herein we turn our attention towards ccNiR, and lay the groundwork for using the powerful evolving technique of Laue crystallography to study this enzyme in action.
CcNiR catalyzes the six-electron reduction of nitrite to ammonia (Scheme 1, Eq. 1), as well as the five-electron reduction of nitric oxide (Scheme 1, Eq. 2) and the two-electron reduction of hydroxylamine (Scheme 1, Eq. 3) to ammonia [11, 14]. The enzyme has also been shown to perform the six-electron reduction of sulfite to sulfide (Scheme 1, Eq. 4), albeit at much lower rates [15]. CcNiR is a soluble homodimeric protein that contains 5 hemes per monomer (Fig. 1). The molecular weight of a ccNiR monomer ranges from 52–65 kDa, depending on the bacterial species from which it was isolated. A number of crystal structures of the enzyme are available [3–9], that share many features in common. The ccNiR active site is a 5-coordinate heme with the unusual axial ligand lysine in the proximal position. During catalysis the substrate binds to the distal position and is reduced. The remaining four hemes in each subunit are all typical 6-coordinate c-type hemes with two His ligands each. These hemes are low-spin and closely spaced, so they can efficiently shuttle electrons to the active site. An interesting feature of the hemes used solely for electron transport is that in all known ccNiR structures they form two branches leading to the active site. Furthermore, this branched pattern is seen not just in the ccNiRs, but also in HAO [10]. From our HAO studies we have proposed that the branching hemes allow electrons to be added or removed from the active site in pairs, with each electron in a pair traveling along a different branch, in order to minimize Coulomb repulsion between the electrons (Fig. 1) [16].
Scheme 1.

Figure 1.

Arrangement of the 10 hemes within the ccNiR dimer. The yellow and green hemes are 6-coordinate and used for electron transport only, while the two orange hemes are the active sites. The red arrows show likely paths of electron flow. Electrons are believed to enter via the green hemes, but can move between subunits as shown (the dotted line separates the monomeric subunits).
A reaction mechanism for the catalytic reduction of nitrite by ccNiR was proposed in 2002 based on crystallographic and computational analysis [11], and this mechanism has recently been refined on the basis of a detailed density-functional study of the first reduction step [17]. The mechanistic scheme is buttressed by extensive literature precedent in other biological and heme model systems [18, 19], and serves as an excellent starting point for guiding future research. Moreover, a formally comparable catalytic cycle can be used as the starting point for guiding the investigation of HAO-catalyzed hydroxylamine oxidation [12, 13].
Laue crystallography is potentially a very powerful tool for investigating redox-active enzymes such as ccNiR. During data collection in conventional crystallography, the crystal is exposed to monochromatic X-rays for an extended period of time, in the range of several tens of minutes to hours for a bright source such as that available at a synchrotron. However, exposing a crystal to X-rays often changes the crystal on the time scale of the conventional experiment; in particular, metalloproteins such as ccNiR and HAO are extremely susceptible to photoreduction in an X-ray beam [20–22]. As a result, the structure one obtains from crystallography is often not that of the original molecule. Instead it reflects the average of the original structure and those of the protein after modification by the X-rays. In photography an analogy would be the photograph of a moving object taken at a slow shutter speed: such a photograph would be blurred because it would show the object in several positions at once. By contrast, using the Laue method is analogous to taking the same photograph with a fast shutter speed. A Laue diffraction pattern is obtained by exposing a crystal to polychromatic X-rays for only 100 ps, too short a time for radiation damage to appear. As a consequence, the Laue method is ideally suited to the investigation of proteins such as ccNiR and HAO that are susceptible to X-ray damage. Another major advantage of the Laue method is that it can be used to monitor reactions within crystals as a function of time [23–36], which is possible because Laue diffraction patterns are collected over very short time scales. If a reaction can be rapidly initiated within a crystal, for example by using a laser to activate a photochemical trigger, then a Laue diffraction pattern can provide the crystal structure at a defined time after reaction initiation. By piecing together Laue patterns obtained at different times after initiation, a time-resolved sequence can be obtained. One other feature aids in the logistics of applying the Laue technique. Since the polychromatic X-ray beam used in Laue crystallography is tightly focused, it typically strikes only a small part of the crystal in a given experiment. Consequently, the same crystal can be used for several experiments, simply by moving the crystal so that the beam strikes a different region each time.
One long-term goal in our investigation of ammonia-nitrite interconversion is to exploit Laue crystallography in order to obtain “snapshots” and “movies” of ccNiR and HAO in action. Such an ambitious goal first requires the availability of very large quantities of the enzymes in question. Furthermore, Laue crystallography typically requires crystals of higher quality than those required for conventional crystallography. In particular, low mosaicity is essential in order to interpret the more complex diffraction patterns obtained with polychromatic X-rays. Herein we introduce an expression and purification protocol for obtaining fully active ccNiR from Shewanella oneidensis in high yield and purity. We characterize the protein using a variety of techniques, and present the crystal structure obtained using the Laue method.
Materials and Methods
Overexpression of S. oneidensis ccNiR
A DNA sequence encoding for ccNiR was synthesized (Genscript) using codon optimization parameters derived from E. coli. The wild-type N-terminal signal peptide was replaced with the signal peptide from the S. oneidensis protein “Small Tetra-heme c” (STC), which is constitutively expressed in high levels [37, 38]. PCR was performed using Phusion® Hot-Start High-Fidelity DNA polymerase (Finnzymes) and the primers ccNiRopt-F (5’-GACTGAATTCGGAGGATACAATTA-3’) and ccNiRopt-R (5’-GATCTCTAGATCACTTGTAGGTCG-3’). The resulting DNA fragment as well as the plasmid pHSG298 (Takara Bio) were digested with EcoRI and XbaI (New England Biolabs) and ligated together using T4 DNA Ligase (New England Biolabs). The plasmid construct was used to transform chemically-competent NEB5-α cells (New England Biolabs). Positive transformants were selected for by plating onto LB agar plates containing 50µg/mL kanamycin and screened using colony-PCR. Plasmid DNA was isolated from the E. coli cells and used to transform electro-competent S. oneidensis TSP-C cells (rifampicin-resistant) [39] via electroporation with the Gene-Pulser MXcell Electroporation System® (Bio-Rad). Positive transformants were screened for by plating onto LB agar plates containing 50µg/mL kanamycin, 30µg/mL rifampicin, and 20mM MgSO4. A single colony was used to inoculate a 5 mL overnight culture, which was then used to inoculate multiple 1 L cultures that were harvested after ~20 hours of growth. Cell pellets were re-suspended in 50 mM HEPES, pH 7.0, containing 1mM EDTA, 10 µM leupeptin, and 500 µM AEBSF, and then frozen at −80°C until needed.
Purification of S. oneidensis ccNiR from S. oneidensis TSP-C cells
CcNiR-containing cells were lysed via sonication using a Sonic Dismembrator Model 500 (Fisher Scientific), and centrifuged at 48,000 × g for 15 minutes. Ammonium sulfate was added to the supernatant to obtain 50% saturation at 0°C [40], and the sample was again centrifuged. The supernatant was applied to a 2.5 cm × 10 cm Octyl Sepharose column equilibrated with 20mM HEPES, 1mM EDTA, 3 M ammonium sulfate, pH 7.0 (Buffer A). The column was first washed with Buffer A, and then with 40% Buffer B (20mM HEPES, 1mM EDTA, pH 7.0). In each case the wash continued until the absorbance at 280 nm (A280) returned to baseline. Finally, ccNiR was eluted with 80% Buffer B. The ccNiR eluent was exchanged into Buffer B using Ultracell® 10K MWCO centrifugal filters (Millipore) via repeated concentration of the sample and dilution into Buffer B. The sample was then applied to a 1.3 cm × 10 cm Q-Sepharose column and the flow-through was collected and exchanged into Buffer C (20mM HEPES, 1mM EDTA, 2M ammonium sulfate, pH 7.0) using centrifugal filters as previously described. The sample, concentrated to a volume of ~5 mL, was applied to a 1 mL RESOURCE Isopropyl column (GE Healthcare), and a gradient of 0–40% Buffer B was run. CcNiR typically eluted at 20–30% Buffer B, exhibited an A410/A280 ratio of >3.8, and ran as a single band when analyzed by SDS-PAGE (Fig. 2a).
Figure 2.

(a) SDS-Page gel documenting the ccNiR purification procedure; (b) CcNiR activity recovered after each purification step; 1 enzyme unit is the amount required to reduce 1 µmol nitrite per minute under the assay conditons. HIC: hydrophobic interaction chromatography; IEx: ion exchange chromatography; IP: isopropyl.
Steady-state Kinetic Analysis
Steady-state kinetic parameters for the ccNiR-catalyzed reduction of nitrite or hydroxylamine by methyl viologen monocation radical (MVred) were obtained in a manner similar to that reported previously by Atkinson et al [41]. Briefly: MVred was generated via bulk electrolysis of methyl viologen (MV) at a potential of −550mV vs. the standard hydrogen electrode (SHE). An assay solution buffered at pH 7.0 with 50 mM HEPES, and containing a known concentration of ccNiR (typically 100 pM), ~75 µM MVred, 1 mM MV, and varying substrate concentration (2 – 500 µM for nitrite, 0.5 – 250 mM for hydroxylamine), was then prepared in a cuvette thermostated at 25°C. The final reagent added was MVred, and its addition initiated the reaction. The re-oxidation of MVred was monitored by tracking the absorbance decrease at 600 nm in the visible spectrum. Reactions were completed anaerobically in a nitrogen-filled glovebox (O2 < 2ppm).
Spectropotentiometry of ccNiR
UV/Vis spectropotentiometry experiments were performed using a BASi Epsilon EC potentiostat to set the potential, and a CARY Bio 50 UV/Vis spectrophotometer to obtain the spectra at each applied potential. The complete apparatus was housed in an anaerobic glove box. Controlled potentiometric electrolysis of the solution was performed in an optically transparent thin-layer electrode (OTTLE) cell, which was a modification of a previously reported design [42, 43], and is described in detail elsewhere [44]. A solution of ccNiR and mediators (Table 1) was prepared in a buffer containing 50mM HEPES, 200mM NaCl, pH 7.0. All mediators were used at concentrations of 25µM, except hexaammineruthenium(III) chloride which had a concentration of 100 µM. UV/Vis spectra in the range from 250nm – 800 nm were collected at 10mV intervals between +50mV and −600mV (vs. SHE). An Ag/AgCl electrode (BASi, Model RE-5B) was used as a reference. Cyclic voltammograms of MV were collected before and after collecting the datasets, and the calculated midpoint potentials were used to account for any drift in the reference electrode (the midpoint potential of MV was taken to be −0.449 V vs SHE [45]). Datasets were also collected with identical mediator solutions in the absence of ccNiR, and these were subtracted from the corresponding ccNiR data sets to account for any spectral changes from the mediators during the titration. The corrected datasets were analyzed using the commercially available software packages Origin V.6.0 (Microcal Software, Inc), and Mathcad 13 (Mathsoft Engineering and Education, Inc).
Table 1.
Mediators used for spectropotentiometry
| Mediator | εo (V vs SHE) |
|---|---|
| Hexaammineruthenium(III) chloride | −0.020 |
| Indigo carmine | −0.125 |
| Anthraquinone 1,5-disulfonic acid | −0.175 |
| Anthraquinone 2-sulfonic acid | −0.255 |
| Safranine O | −0.289 |
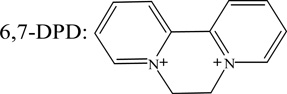
| 6,7-DPD1 | −0.390 |
| Methyl viologen | −0.449 |
Voltammetric analysis of ccNiR adsorbed on an electrode
Protein film voltammetry (PFV) experiments were performed by immobilizing ccNiR on a pyrolytic graphite edge (PGE) electrode. Prior to generation of a protein film, PGE electrodes were polished with an aqueous slurry of 1 µm alumina followed by a brief sonication. Films were generated by pipetting 3 µL concentrated ccNiR onto the electrode, waiting approximately 20 seconds, then pipetting off excess protein solution. The electrode was then immersed in a mixed buffer system (5 mM sodium acetate, MES, MOPS, TAPS, CHES, CAPS; 100 mM NaCl) in a three-electrode water jacketed electrochemical cell. A platinum wire counter-electrode and a saturated calomel reference electrode were used. PFV experiments were performed using a PGSTAT 30 AutoLab (Ecochemie) potentiostat equipped with FRA and ECD modules. PFV experiments were performed on the bench top with argon gas bubbling through the cell solution to remove oxygen. The electrochemical cell was housed in a Faraday cage to reduce noise. Data was collected using the GPES software package (Ecochemie). Cyclic voltammograms produced by PFV experiments were background subtracted to remove electrode capacitance using the SOAS software package [46]. The broad envelope of signal produced by a PFV experiment was deconvoluted by using a fit derived from the Nernst equation, assuming a one-electron contribution of equal surface coverage for each heme of ccNiR.
Crystallization of S. oneidensis MR-1 ccNiR
Initial crystallization screens were performed on ccNiR in the laboratory of Dr. J. Fu (Medical College of Wisconsin) using a robotic high throughput screening system, and the custom in-house screen MCW192. Crystals were obtained from solutions containing 50mM triethanolamine at pH 8.25 with 19% PEG4K. Further optimization of the crystallization conditions was then performed using the hanging drop method, where 2 µL drops were prepared by mixing 1 µL ccNiR stock with 1 µL precipitant solution, and suspended over a reservoir containing 500 µL precipitant. Optimal crystals were obtained when the ccNiR stock had a concentration of 10mg/mL, while the precipitant consisted of a solution containing 50mM triethanolamine and 14% PEG 4K, pH 8.25. Trays of hanging drop samples were incubated at 23° C. Crystals typically appeared overnight. The onset of crystal formation appears to be critically dependent on the temperature; no change in drop appearance was observed at temperatures above 23° C, whereas below this temperature liquid-liquid phase separation invariably occurred. Some batches of purified protein resisted crystallization even at 23° C. In such cases micro-seeding as follows proved useful. Previously grown crystals were pulverized in mother liquor using a 3 mL tissue grinder (Wheaton), and then mixed 1:1 with the ccNiR crystallization solution described above. This solution was incubated at 25° C for 6–18 hours, and then slowly cooled by transferring the sample to a 20° C room. Crystals grown in this manner typically appeared in 2–3 days.
Crystallographic Data Collection
Crystals with dimensions of 200µm × 60µm × 60µm were grown as described above. For conventional crystallographic analysis crystals were cryoprotected by transferring them to a solution of mother liquor supplemented with 10% (w/v) PEG 20K, and then flash frozen. Weak monochromatic crystallographic data to a resolution of 3.2 Å were collected at BioCARS 14BM-C beamline at cryogenic temperatures (100K) using an ADSC Q315 detector. Data were collected in 0.5° steps for a total of 180°. Data were analyzed by Mosflm [47] and scaled and merged by the Collaborative Computational Project Number 4 (CCP4) program 'scala' [48]. Crystals appeared to be monoclinic, spacegroup P21, with cell parameters a=47.1 Å b=92.7 Å c=216.5 Å α=90° β=91.04° γ=90°. Data statistics are given in Table 2.
Table 2.
Data and refinement statistics
| Monochromatic | Laue | |
|---|---|---|
| Temperature | 100 K | 273 K |
| Crystal Size | 60 µm × 60 µm × 200 µm | 60 µm × 60 µm × 200 µm |
| Mosaicity [°] | 0.9† | < 0.1† |
| Resolution | 3.20 Å | 2.72 Å |
| cell parameters | a=47.1 Å b=92.7 Å c=216.5 Å α=90° β=91.04° γ=90° |
a=51.5 Å b=95.5 Å c=223.0 Å α=90° β=90° γ=90° |
| Vcell [Å3] | 945120 | 1096769 |
| Space group | P21 monoclinic | P212121 orthorhombic |
| Rsym | 21.9 % | 9.9% |
| I/sigI | 2.3 | 16.7 |
| Redundancy | 2.9 | 8.9 |
| Completeness (last shell) | 73.9 % (63.8 %, 3.37 Å - 3.20 Å) | 75.2 % (63.5 %, 2.86 Å – 2.72 Å) |
| Models for molecular replacement | E. coli ccNIR (2RDZ) subunits A and B | E.coli ccNIR oriented and refined against monochromatic data subunits A and B |
| asymmetric unit | tetramer | dimer |
| Rcryst/R-free % | ||
| molecular replacement | 47.7 to 3.5 Å | 31.1 (to 2.59 Å)‡ |
| rigid body | 41.1 (to 3.2 Å) | 25.6 |
| refinement (no water) | 32.9/37.8 | 21.6/26.7 |
| protein + water | 19.7/25.7 | |
As estimated by the data reduction program. Laue: estimated from shape of the reflection, mono + Laue: contain also contributions from beam geometry (crossfire) that might be slightly different at beamlines 14-IDB and 14-BMC.
Refinement includes all data to 2.59 Å
Laue crystallographic data were collected at the BioCARS 14ID-B Laue beamline using X-ray radiation from a double undulator (U23 and U27 [49]). Crystals were mounted in glass capillaries, and data were collected at room temperature. The synchrotron operated in hybrid mode, and radiation from a single electron bunch was selected. Each X-ray pulse contained about 3×1010 photons, and 7 pulses were used per Laue diffraction pattern. Waiting time between the pulses was 1 s. After each Laue pattern the crystal was translated along its long axis to expose a fresh volume of crystal to the X-ray beam. The X-ray beam size was 90µm × 60 µm. Laue data extended to a resolution of 2.59 Å with sufficient completeness to 2.72 Å. The complete dataset consisted of 90 Laue patterns covering 180°. Data were indexed, integrated, and scaled by Precognition/Epinorm (RenzResearch). The spacegroup was determined to be P212121, orthorhombic, with cell parameters a=51.5 Å b=95.5 Å c=223.0 Å α=90° β=90° γ=90°. Data statistics of the Laue data are also given in Table 2.
Results
Overexpression of ccNiR
Several approaches to overexpressing S. oneidensis ccNiR were explored in this project. In the ultimately successful approach an optimized ccNiR gene was incorporated into a pHSG298 expression vector, and then S. oneidensis (TSP-C strain) was transformed with this vector. PHSG298 is very similar to pUC plasmids, except that the latter use the ampicillin resistance gene as the selective agent for isolating transformed cells, whereas pHSG298 uses the kanamycin resistance gene instead. Attempts to obtain ampicillin-resistant colonies of S. oneidensis (containing ccNiR in a pUC18 plasmid) were unsuccessful. The S. oneidensis TSP-C strain was used, as it has natural rifampicin resistance and allows for double antibiotic selection of transformed cells.
A comparison between the wild-type and optimized ccNiR genes is provided in Supplementary Material. The most notable difference between the two is that in the optimized gene the ccNiR N-terminal signal sequence for periplasmic translocation has been replaced by the signal sequence for another S. oneidensis protein, STC, which is typically expressed in larger amounts than ccNiR [38]. S. oneidensis cells transformed with the pHSG298 expression vector containing the optimized gene produced 20–30 fold more ccNiR than untransformed cells. Typical yields were ~20 mg crude ccNiR/L culture for the transformed cells, compared with 0.5–1 mg/L for the untransformed cells. Significantly, S. oneidensis TSP-C cells transformed with pHSG298 plasmids containing the wild type ccNiR gene, complete with wild-type signal peptide, did not produce ccNiR in concentrations above the background seen from chromosomal expression.
Purification of overexpressed ccniR
CcNiR was obtained in high yield and purity using a 5-step purification procedure. Most impurities were removed after the third step (hydrophobic interaction chromatography [HIC] using octyl sepharose), as shown in Fig. 2a. One of the remaining impurities (~70 kDa) was removed by passing the protein mixture through a Q-sepharose anion exchange column, which doesn’t bind CcNiR; this step also removed other minor impurities. A final HIC step using a high-resolution column brought all remaining impurities down to acceptable background concentrations (Fig. 2a). The overall yield for the purification was typically between 30 – 40% (Fig. 2b) of the initial activity. This translates to 7 – 10 mg of pure ccNiR obtained per liter of cell culture, which is ample for protein-intensive experiments such as crystallography or spectropotentiometry.
Stability of the pure ccNiR was assessed using the steady-state assay described in the following section. Micromolar solutions of the protein are stable for over two weeks at pH 7 and 4° C. Very dilute (nanomolar) solutions kept in microcentrifuge tubes appeared to lose activity with half-lives of about 1.5 h. This apparent loss may be due to slight adsorption of the protein into the matrix of the microcentrifuge tube. Alternatively ccNiR in dilute solutions may slowly dissociate from an active dimeric to an inactive monomeric form. The stability of pure ccNiR did not depend on the presence of added Ca2+.
Steady-state Kinetic Analysis of ccNiR
The steady-state kinetics for ccNiR-catalyzed nitrite and hydroxylamine reduction by MVred were obtained for the purified protein. As expected initial rates showed first-order dependence on [ccNiR], zero-order dependence on [MVred] and hyperbolic dependence on [NO2−] and [NH2OH]. The Km and kcat values obtained from Michaelis-Menten plots of Vo vs. [NO2−] were 23±4 µM and 824±33 s−1, respectively, where the kcat value reflects the number of µmol of NO2− turned over per second. For the Vo vs. [NH2OH] plots the corresponding Km and kcat values were 8.3±2.4 mM and 2380±160 s−1, respectively. All of these values are similar to the Michaelis-Menten parameters obtained for ccNiR from E. coli (Km = 28 µM, kcat = 770 s−1 for NO2− and 30 mM, 2380 s−1 for NH2OH [6]).
Initially MV for use in the steady-state experiments was reduced using dithionite. However sulfite, a product of dithionite oxidation, was recently shown to be a substrate for the E. coli ccNiR, getting reduced to sulfide albeit at a very slow rate [15]. To date we haven’t conclusively shown that the S. oneidensis ccNiR can catalyze the reduction of sulfite by MVred; however, what is evident is that sulfite is a competitive inhibitor of the nitrite reduction by MVred. Thus, in experiments where MVred was generated by reduction with dithionite, the obtained Km values were consistently higher than those obtained when MVred was generated by bulk electrolysis. By contrast, the kcat values were unaffected by choice of protocol. The KI value for sulfite inhibition of nitrite reductase activity was subsequently determined to be approximately 600 µM, by monitoring the dependence of the observed Km on sulfite concentration in independent experiments (data not shown).
Spectropotentiometry of ccNiR
For spectropotentiometric analysis the spectrum collected in the absence of an applied potential (that of the fully oxidized protein) was first subtracted from those collected under applied potentials, in order to generate difference spectra. The absorbance difference data collected between 379 nm and 454 nm, for all potentials between +34 mV and −516 mV vs NHE, were then subjected to a global analysis described in detail in Supplementary Material. Briefly, the data were first subjected to singular value decomposition (SVD) [50, 51], which revealed five components above the noise level. The SVD-processed data were next analyzed using a model in which each SVD component is assumed to correlate with the addition of 1 electron to each ccNiR
| 5a |
where
| 5b |
and
| 5c |
| 6 |
monomer (Scheme 2, Eqs. 5 and 6). Equation 5 is derived from the Nernst equation in exponential form, and correlates Cn with the applied potential (see Supplementary Material). In Eq. 5 εon is the midpoint potential associated with 1-electron reduction of the (n − 1)th reduced species, εapp is the applied potential, and CT is the total ccNiR concentration in solution. Equation 6 is a matrix version of Beer’s law such that: Cred is a matrix of concentrations in which each column corresponds to a unique reduced species and each row to a specific applied potential; Δε is an extinction coefficient difference matrix in which each column corresponds to a unique reduced species and each row to a wavelength; ΔA is the SVD-processed absorbance difference matrix in which each column contains a spectrum at a fixed applied potential, and each row shows how absorbance varies with potential at a fixed wavelength; and l is a scalar representing the path length of the OTTLE cell. The data were fitted to Eqs. 5 and 6 using a program created with Mathcad 13. This program first allows the user to manually enter trial values of the five midpoint potentials. For a given set of midpoint potential values the program then uses Eq. 5 to calculate the concentration of each reduced species at a given applied potential, and stores the concentrations in Cred (Eq. 6). Next the program calculates the extinction coefficient spectra Δε using Eq. 6 [52]. Finally, the program uses l and the Δε and Cred matrices to generate an absorbance matrix ΔAcalc, and the sum of squares for ΔA − ΔAcalc is computed. By varying the trial values of the five ccNiR midpoint potentials, this sum of squares can be minimized.
Scheme 2.

Fully oxidized ccNiR is referred to as Ox, while C1 – C5 refer to the 1 – 5 electron reduced species, respectively.
Figure 3 shows the results of the global analysis. Figure 3a compares the experimental and calculated spectra at selected applied potentials, while Fig. 3b shows the fitted ΔA vs. εapp slice at 426 nm. Figure 4a provides the calculated extinction coefficient difference spectra Δεn (the columns of matrix Δε) for each of the reduced ccNiR species, while Fig. 4b provides the absolute extinction coefficient spectra for the species, obtained by adding the extinction coefficient spectrum of the fully oxidized ccNiR to each of the extinction coefficient difference spectra. Finally, Fig. 4c shows the calculated concentrations of the reduced species Cn as a function of applied potential. Table 3 lists the five calculated midpoint potentials, comparing them with those obtained by direct voltammetric methods (next section), and with those previously reported for E. coli ccNiR. The uncertainties quoted for each of the εon values represent the changes that could be made to each trial εon without increasing the sum of squares by 0.001 units or more (~6% of the sum of squares value).
Figure 3.

(a) Spectra obtained at applied potentials of 0.034V, −0.106V, −0.196V, −0.256V, −0.316V and −0.506V vs SHE. Solid blue lines show the experimentally obtained data, while the dashed red lines show the fit obtained using the matrix Eq. 6 (b) The least-squares best fit of the data by Eq. 6 at λ=426 nm.
Figure 4.

(a) Extinction coefficient difference spectra corresponding to each of the reduced ccNiR species C1 – C5 (Scheme 2), as calculated by fitting the experimental spectropotentiometric titration data using Eq. 6. The vertical line shows the point at which a high-spin ferroheme should have an absorbance maximum. (b) Similar to (a), but here the calculated absolute extinction coefficient spectra of C1 – C5, together with the spectrum of Cox, are shown. (c) Concentrations of the various ccNiR species present in solution at a given applied potential (vs SHE), obtained by fitting the data to Eq. 6.
Table 3.
Midpoint potentials of ccNiR hemes (in V vs. SHE) obtained in this work for the S. oneidensis protein, and in previous investigations of ccNiR from E.coli.
| This Work | E. coli ccNiR | |||
|---|---|---|---|---|
| Spectropotentiometry | Film Voltammetry | MCD Spectropotentiometry1 | Film Voltammetry2 | EPR Spectropotentiometry3 |
| −0.062±0.007 | −0.036 | −0.020±0.015 (ls) | +0.022±0.015 | −0.037 (ls) |
| −0.140±0.005 | −0.105 | −0.108±0.009 (active site) | −0.056±0.015 | −0.107 (active site) |
| −0.25±0.01 | −0.166 | −0.153±0.011 (ls) | −0.117±0.015 | −0.107 (ls) |
| −0.283±0.007 | −0.230 | −0.206±0.019 (ls) | −0.189±0.015 | −0.323 (ls) |
| −0.342±0.004 | −0.295 | −0.292±0.034 (ls) | −0.275±0.015 | −0.323 (ls) |
The procedure described above was also used to investigate the spectral region from 500 nm – 650 nm, where the characteristic α, β bands are observed for reduced low-spin hemes. The complete analysis is provided as Supplementary Material.
Voltammetric analysis of ccNiR adsorbed on an electrode
PFV experiments performed on S. oneidensis ccNiR films in the absence of substrate produced a broad envelope of reversible signals that span approximately 450 mV (Fig. 5a). These signals correspond to the reduction and subsequent oxidation of the heme cofactors within the enzyme, and scan-rate dependencies of the maximal currents indicate that the redox couples are due to adsorbed species (data not shown). The non-turnover voltammograms for S. oneidensis ccNiR reported here are similar to data previously reported for E. coli ccNiR [53]. E. coli ccNiR voltammograms also appear as a broad envelope of signal spanning approximately the same region of potential; however E. coli ccNiR voltammograms are more featured and the range of potential covered is slightly larger than that of S. oneidensis ccNiR. The overall shape of S. oneidensis ccNiR voltammograms changes with pH, indicating the presence one or more pH-dependent redox centers. At high pH values the envelope appears as a single peak, while at pH values lower than 7 the envelope appears to be composed of two large overlapping peaks. At pH values less than 6 and at 0°C, the envelope of signal can be better resolved and more than two peaks can be observed. This resulting envelope of signal can be deconvoluted as the sum of five one-electron peaks, each corresponding to one of the five hemes in a ccNiR monomer (Fig. 5b). Voltammograms reported for E. coli ccNiR also fit to five one-electron centers [53]. Alternate fitting schemes, such as the combination of three one-electron features and one two-electron couple, could not account for the data shown in Fig. 5.
Figure 5.

Protein Film Voltammetry of S. oneidensis ccNiR. (a) Typical signal on a graphite electrode. Experiment was carried out at pH 5.15, 0°C, scan rate 45 mV/s. Both the full cyclic voltammogram and background-subtracted data are shown. (b) Baseline-subtracted non-turnover voltammogram recorded at 0°C, pH 6, 250 mV/s. Black trace is the baseline-subtracted voltammogram, red trace and dotted lines are from the resulting fit; equation for fitting derived from the Nernst equation. Sub-plots show the residuals for fits of the oxidative (Ox) and reductive (Red) scans. Calculated Em values are −0.295 V, −0.230 V, −0.166 V, −0.105 V, and −0.036 V vs. SHE.
Crystal Structure Model Building and Refinement
For an initial molecular replacement the monoclinic (P21) monochromatic data obtained at 100 K were used. The A and B subunits (one dimer) of the E. coli ccNiR without water or calcium ions were used as a template (E. coli ccNiR PDB-ID: 2RDZ). Molecular replacement was attempted to 3.5 Å using the CCP4 program “phaser” [54]. One solution comprising two dimers was identified. The molecular replacement solution was quickly refined using “refmac5” (CCP4 program collection [48]) by rigid body and by conventional refinement including non-crystallographic symmetry restraints. The Rcryst settled to 32.9%. No further refinement was attempted due to the poor data quality. However, one dimer consisting of subunits A and B of this initial model was then used for molecular replacement against the Laue data, collected at room temperature. A single, distinct molecular replacement solution was found in the orthorhombic crystal form. After rigid-body refinement of the individual subunits A and B, the resulting model was inspected at positions where differences in the sequence between E. coli and S. oneidensis ccNiR occur. Differences were immediately visible in the electron density, and the atomic model was modified according to the correct S. oneidensis ccNiR sequence. All model manipulations were done using “coot” [55]. The electron density map around residues 170 and 220 could be easily re-traced. Both regions accommodate loops which are different in E. coli and S. oneidensis ccNiR. Very large difference electron density within the protein indicated the presence of a strong scatterer. The electron density was modeled by a calcium ion in accordance to similar findings for the E. coli and other ccNiRs previously characterized crystallographically. Finally, water molecules were inserted. The final R-factor dropped to 19.7% (see Table 2).
Attempts to solve the ccNiR structure using monochromatic crystallographic data collected at cryogenic temperatures (100 K) were considerably less successful than those using the Laue method on data collected at room temperature. A variety of cryoprotectants were tested, including glycerol, MPD, PEG 400, PEG 8K, PEG 20K, mineral oil, and various mixtures of these. The best results were obtained using 10% PEG 20K as the cyoprotectant; however, even with this protectant very weak monochromatic data were collected (see I/sigI and Rmerge in Table 2). Multiple attempts to collect better monochromatic data failed. At room temperature the diffraction patterns fade away after a few degrees. When comparing the room temperature Laue data to the cryogenic monochromatic data, differences as large as 6.5 Å in cell parameters can be observed. In addition, freezing increases the mosaicity. It is difficult to estimate the mosaicity from the size of the Laue reflections, since factors such as crystal size and crossfire also contribute. However, the software (Precognition) estimated the mosaicity to be around 0.1°, which usually yields good Laue patterns. Freezing then increases the mosaicity to around 1°.
The ccNIR crystallizes as dimers, which form the building blocks of both the orthorhombic and monoclinic crystals. Two dimers occupy similar places in both space groups; however, numerous contacts are established between subunits of individual dimers, whereas contacts between two distinct dimers are few. In P212121 individual dimers are related by crystallographic symmetry, whereas in P21 two dimers reside in the same asymmetric unit. The difference is that in P21 the second dimer is slightly rotated relative to the corresponding dimer position in the orthorhombic form (Supplementary Material). Hence, freezing generates strain in the crystals that breaks the symmetry and increases the mosaicity, although the molecular packing in both crystals remains almost the same. These factors contribute to disorder in the crystals at cryogenic temperatures, increase (decrease) the temperature factor (the Debye-Waller factor), and hence contribute to the limited resolution and poor monochromatic data quality.
Discussion
Overexpression of ccNiR
Numerous strategies for the overexpression of multi-heme c-type cytochromes have been proposed [38, 39, 56, 57]. Initially, E. coli would appear to be a good organism to use for overexpressing c-heme proteins such as ccNiR, and indeed it has been used with some success [57–60]. A major disadvantage of E. coli though, is that it expresses the eight cytochrome c maturation (CCM) chaperones (ccmA-H) needed to incorporate c-type hemes post-translationally into apoproteins only under anaerobic conditions. Londer et al. overcame this problem by using an E. coli strain that contains the requisite chaperones within the pEC86 vector; however, ccNiR requires an additional three chaperones (nrfEFG) in order to correctly ligate lysine to the active-site heme, and E. coli only expresses these anaerobically [61].
Shewanella oneidensis has also been used to overexpress c hemes [38, 39, 62–64], and is in many ways a better host for this purpose. Crucially, unlike E. coli, this organism expresses both the ccmA-H and the nrfEFG operons even under aerobic conditions. In a suitably overexpressing system, this makes S. oneidensis a potential source of large quantities of c-hemes, since bacteria typically grow much more rapidly and to higher density under aerobic conditions.
Takayama and Akutsu recently used S. oneidensis to overexpress human cytochrome c by first substituting the human N-terminal signal peptide with that of the S. oneidensis protein STC [38]. They proposed this strategy as a general one for the heterologous expression of c-hemes within S. oneidensis. Our results suggest that in some cases it may be beneficial to change the N-terminal signal peptide prior to expression, even when the desired protein is from S. oneidensis and already has a signal peptide recognized by the bacterial CCM system. When we initially expressed the wild-type ccNiR gene, with its corresponding wild–type signal peptide, in S. oneidensis, the expression was no higher than the background seen from chromosomal expression. When the optimized gene bearing the STC signal peptide was used, expression was 20–30 fold above background. It is possible that the overexpression is due to better codon usage in the optimized gene, but for ccNiR the wild-type gene doesn’t really show any obvious poor usage sections (Supplementary Material). Instead, we suspect that the STC signal peptide leads to more efficient post-translational modification of the associated protein than the ccNiR signal peptide. For example, the protein responsible for translocating apoproteins across the periplasmic membrane might have more affinity for the STC signal peptide than for the ccNiR signal peptide. One can even see how such affinity variations might evolve: for proteins that are not needed by the bacteria in large quantities, such as the highly efficient ccNiR enzyme, there may be no evolutionary pressure to develop a signal peptide with high affinity for the translocation machinery.
The hypothesis that the rate of c-heme protein maturation can be increased by judicious choice of signal peptide is worthy of more systematic investigation in the future. Indeed it is possible that some other signal peptide, such as that for the highly expressed S. oneidensis protein Periplasmic Fumarate Reductase (SO2727), could in general lead to even higher overexpression than the STC peptide.
Electrochemical analysis of ccNiR
As shown in Figs. 3 and 5, the spectropotentiometric and film voltammetric data sets are very well modeled by Scheme 2, which assumes 5 uncoupled reduction events. Attempts to fit either data set with models that incorporated coupled reductions invariably produced poorer fits. Table 3 summarizes the midpoint potentials of S. oneidenis ccNiR obtained by spectropotentiometry and film voltammetry, and compares these values to ones previously reported for ccNiR from E. coli [6, 65]. For the S. oneidensis protein the midpoint potentials obtained by film voltammetry are consistently more positive than those obtained by spectropotentiometry, by anywhere from 30 to 80 mV. Attempts to fit the spectropotentiometric data set using the midpoint potentials obtained by film voltammetry yielded very poor fits. The voltammetric data were collected at pH 6 whereas the potentiometric data were obtained at pH 7, and this may partially explain the observed differences. However, we note that the various sets of midpoint potentials obtained for E. coli show similar inter-set variability (Table 3), even though all of those experiments were carried out at pH 7. Indeed, there is more variation between the midpoint potential values obtained from the three E. coli ccNiR datasets, or between the values obtained from the two S. oneidensis ccNiR datasets, than there is when comparing the S. oneidensis midpoint potentials obtained by film voltammetry with those obtained by MCD spectropotentiometry for the E. coli ccNiR. We therefore conclude that the ccNiRs from S. oneidensis and E. coli probably have very similar heme midpoint potentials at both pH 6 and 7, and that the observed differences are primarily due to experimental biases introduced by the various measuring methods. Given the complexity of the systems being investigated, these differences are comparatively minor.
We have found one surprising difference between the UV/Vis spectral characteristics of the S. oneidensis ccNiR and those reported for E. coli ccNiR. In the E. coli enzyme, reduction of the active site heme was reported to produce a distinct signal at 435.5 nm [65], consistent with its EPR identification as a high-spin species [6]. We see no such distinct signal in the S. oneidensis ccNiR UV/Vis as the protein is reduced; the five reduced species all have calculated extinction coefficient spectra with maxima below 430 nm, where low-spin ferrous hemes are expected to absorb (Fig. 4). The spectrum of the final reduced species C5 does shift to significantly longer wavelength, but remains below 430 nm. Moreover, by analogy with the E. coli ccNiR, whose reduction potentials have been assigned to specific hemes primarily on the basis of EPR spectropotentiometry [6], we would expect that the active site heme 1 is the second heme reduced (εo2 = −0.140 V). The extinction coefficient difference spectrum of the second reduced species, C2, has a maximum at 423 nm (Fig 4). However, we do note what could be an important pattern at 435 nm in the calculated extinction coefficient difference spectra (vertical line in Fig. 4a). Reduction of Ox to C1 results in a small increase in Δε435, which is followed by a significantly bigger increase as C1 is reduced to C2. Further reduction of C2 first to C3 and then to C4 results in virtually no change in Δε435, after which there is another sizeable increase as C4 is reduced to C5. This final increase in Δε435 can be explained by the shift in λmax as C4 is reduced to C5; however, the increase associated with the reduction of C1 to C2 arises from the fact that the C2 extinction coefficient difference spectrum is broader that the other spectra, with an unusually long tail stretching to longer wavelength.
All of our observations are more easily explained if we don’t try to associate each ccNiR reduction step with reduction of a single individual heme. Instead we propose that, for example, in 2-electron reduced ccNiR (species C2) the two electrons are delocalized among several of the hemes, and that the same may be true for the other stages of reduction. This would explain why the UV/Vis spectral changes associated with reduction of C1 to C2 appear to exhibit features characteristic of both low-spin and high-spin ferrohemes. The interpretation is also consistent with the EPR results reported by Bamford et al [6]. For example, these researchers showed that a signal at g = 10.8 observable in parallel mode decreased rapidly to about 20% of its initial intensity at applied potentials of ~ −0.05 – −0.2 V, but then decreased at a much slower rate thereafter. This signal was assigned to the high-spin ferric active site weakly exchange coupled to the low-spin ferric heme 3. Our interpretation would assign the rapid decrease in the g = 10.8 signal to production of the 2-electron reduced ccNiR (C2), but would allow a fraction of the C2 population to retain the original ferric heme 1. In this fraction another heme would be reduced instead. The appearance of such a distribution of reduced hemes within a multi-heme complex has been predicted in a theoretical investigation of HAO redox equilibria [16], and is likely to be a common phenomenon.
Structure of S. oneidensis ccNiR
Structure determination of the S. oneidensis ccNIR reveals what to our knowledge may be the first case in which room temperature polychromatic Laue X-ray data turned out to be better than monochromatic data collected at cryogenic temperatures. Without Laue crystallography we would have been limited to relatively low quality data with a resolution limit lower than 3 Å. The biggest challenge in cryo-crystallography is to find a good cryo-protectant. If this is difficult, and the overall crystal quality is poor in the first place, most data sets are of low quality and must be discarded. In addition, very often crystals even from the same batch do not scatter equally well. Room temperature monochromatic data collection with crystals held in capillaries, or freely mounted using a humidifier [66] offer one possible way out. Since crystals are not frozen, there is little strain that might generate disorder and increased mosaicity, and consequently data quality can be exquisite. However, we never succeeded in collecting complete monochromatic room-temperature data on this ccNIR, because after a few exposures the diffraction patterns fade away. Laue crystallography offers an alternative when monochromatic data collection at ambient or cryotemperatures obstinately fails. Laue crystallography tends to minimize damage to the crystal caused by the ionizing X-rays. Several publications report the effect of X-ray dose and dose rate on protein crystals [35, 67, 68]. The dose tolerance depends on the rate at which the X-ray dose is administered. The higher the rate the higher the tolerance[67], up to a certain limit where temperature increase and, presumably, hydrogen formation destroys the crystal rapidly [68]. Single-pulse Laue crystallography exhibits the highest peak dose rates at synchrotrons (in the order of 1013 Gy/s, [35]), but there is enough waiting time between the X-ray pulses to allow for the temperature to equilibrate and for potential hydrogen to diffuse out of the crystal. The combination of ultra-short brilliant X-ray pulses with about a second waiting time in between, and the displacement of the crystal along its axis to expose a fresh crystal volume, proved to be very advantageous for a successful collection of high-resolution data on this ccNIR.
Crystal structures of ccNiR were previously reported from 3 different branches of the bacterial phylogenetic tree [3–9]. Sulfurospirillum deleyianum and Wollinella succinogenes are ε-proteobacteria, Desulfovibrio desulfuricans and D. vulgaris are δ-proteobacteria, and E. coli and S. oneidensis are γ-proteobacteria [4, 7]. All structures reported so far, including the S. oneidensis form described herein, appear to be functional dimers, and exhibit highly conserved heme arrangements. Figure 6 shows an overlay of the S. oneidensis hemes within one monomer with the corresponding E. coli hemes. The arrangements of the histidine imidazole planes are of note because they often permit correlations to be made between a heme protein’s structure and its EPR spectroscopic features [6, 69]. The heme 2 imidazole planes are very nearly parallel in all of the ccNiRs reported to date, whereas the heme 4 and 5 imidazole planes are closer to perpendicular. Each of these arrangements gives rise to a distinct type of EPR spectrum. The imidazole arrangement of heme 2 is more variable than the others, being the most parallel in the S. oneidensis protein and less so in other ccNiRs. This heme is typically magnetically coupled to the high-spin active site, and thus exhibits an anomalous EPR signature [6].
Figure 6.

Comparison between the heme arrangement within a monomer of S. oneidensis ccNiR (lighter shade) and that within a monomer of E. coli (darker shade). Subunit A is shown. Irons are shown in yellow, while the heme color scheme matches that of Fig. 1.
In addition to sharing a common heme arrangement, all ccNiRs reported to date contain a Ca2+ ion that lies close to the active site in a highly conserved region of the protein [4]. Figure 7 shows the environment of the Ca2+ in S. oneidensis ccNiR. The ion is coordinated in bidentate fashion by Glu 205, and in monodentate fashion by the Tyr 206 and Lys 254 backbone carbonyls, and the Gln 256 side-chain carbonyl. In other ccNiRs two remaining (cis-oriented) coordination sites are occupied by waters; however in the S. oneidensis structure we only assigned one water to the Ca2+ in subunit B. In subunit A the difference electron density that represents this water is very close to the noise level, and it is difficult to identify even one water there. The carbonyl sidechain of Asp 242 and the hydroxyl of Tyr 235 come near to the open calcium coordination sites, but are not within bonding distance. Instead they interact with the water that is weakly coordinated to the Ca2+ (see Fig. 7). The ccNiR calcium ions appear to play a vital role in organizing the active site [4], and at least in the case of the E. coli protein, mutations to the Ca2+ binding amino acids have been shown to affect the active site heme midpoint potential [70]. In the case of ccNiR from S. deleyianum, enzyme activity increased in the presence of added calcium, and decreased when chelators such as EDTA were added to the buffers [71]. However the S. oneidensis enzyme showed no activity dependence on added calcium, or on the presence of added EDTA. It is not clear from structural comparisons why the Ca2+ should be labile in one case and not the other, as the calcium environment is so similar in both cases. In addition to the binding site conserved within all ccNiRs, the ccNiRs from D. desulfuricans and D. vulgaris exhibit a second, well-defined Ca2+ binding site in the vicinity of hemes 3 and 4 [7, 8]. The E. coli enzyme crystal structure exhibits a second Ca2+ in the same relative position, but the binding site in this case is not as clearly defined, and it is unclear if the calcium is physiologically relevant [6]. We did not detect a second Ca2+ ion in the S. oneidensis ccNiR, perhaps simply because Ca2+ was not included in the crystallization buffer.
Figure 7.

Coordination environment of the conserved Ca2+ site within the S. oneidensis ccNiR. Subunit B is shown here; in subunit A the difference electron density feature that corresponds to the Ca-bound water is only 3.5 sigma. This is extremely close to the noise level, and consequently the water was left out of the structure. The E. coli structure revealed two waters bound to the Ca2+.
Overall, the S. oneidensis ccNiR structure is very similar to that of E. coli; the structures are compared in Fig. 8. When corresponding subunits of E.coli and S. oneidensis ccNIRs are compared, the root mean square deviation between the cα-atoms is 0.7 Å with a 64% sequence identity [72]. A small rectangle on the lower right hand side of the figure highlights the region where the greatest sequence divergence is found. This region of the protein, which borders heme 2, was long suspected to be the entry point for electrons into ccNiR [4]; more recently a crystal structure of D. vulgaris ccNiR bound to its physiological partner has all but confirmed this hypothesis [8]. CcNiRs from the different phylogenetic branches use different physiological electron acceptors, which probably explains why the region around heme 2 generally shows significant inter-species sequence variability. CcNiRs from δ and ε proteobacteria use NrfH, a membrane-bound quinol dehydrogenase as the electron donor, while γ-proteobacteria normally use a soluble electron transport protein known as NrfB to shuttle electrons from the membrane-bound quinol dehydrogenase to ccNiR [4]. S. oneidensis and E. coli are both γ-proteobacteria; however the S. oneidensis ccNiR is unusual in that its electron donor is a membrane-bound tetraheme protein called CymA, rather than the more common NrfB [73]. Shewanella species appear to have evolved to use a large number of physiological electron donors interchangeably. Accordingly, CymA appears to be a general-purpose protein that can donate electrons equally well to a variety of proteins, such as ccNiR, and nitrate, fumarate and DMSO reductases among others [73].
Figure 8.

Overlay comparing the protein structures of S. oneidensis and E. coli ccNiRs. The rectangle in the lower right-hand side highlights the region near Hemes 2 that shows the greatest sequence divergence.
Summary
We have designed an expression and purification method for obtaining S. oneidensis ccNiR from S. oneidensis TSP-C strain, which yields 7 – 10 mg of purified protein per liter of culture, enough to sustain protein-intensive research projects such as X-ray crystallography or spectropotentiometry. The purified ccNiR has nitrite and hydroxylamine reductase activities comparable to those previously reported for E. coli ccNiR. Spectropotentiometric titration and film voltammetric data are readily fitted assuming 5 independent 1 – electron reduction processes. Global analysis of the spectropotentiometric data suggest that within each reduced species the electrons are distributed amongst the various hemes, rather than being localized on specific heme centers. The purified ccNiR yielded good quality crystals, which allowed us to solve the structure to 2.59 Å using the Laue diffraction method. The structure is similar to that of E. coli ccNiR, except in the region where the enzyme interacts with its physiological electron donor. This is expected as different electron donors are used by the two organisms. Efforts are now under way to improve the resolution of the ccNiR Laue structure, obtain structures of the protein in the presence of nitrite, nitric oxide and hydroxylamine, and to develop a method for photoinitiating ccNiR reduction within the crystals, in order to enable time-resolved crystallographic studies. UV/Vis and EPR- spectropotentiometric titrations at various pHs are also under way.
Supplementary Material
Acknowledgments
MS and AAP are supported by grants NSF-0843459, NSF-1121770 and UWM RGI 101X157. MS is supported by NSF CAREER grant 0952643. ETJ was supported by an NIH-NIGMS National Research Service Award (NRSA) fellowship (1F31GM099416-01). BS and TG gratefully acknowledge support from UW-Milwaukee’s Office of Undergraduate Research. Use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources, under grant number RR007707. The time-resolved set-up at Sector 14 was funded in part through a collaboration with Philip Anfinrud (NIH/NIDDK). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. The authors gratefully acknowledge UWM Profs. Nick Silvaggi, Chuck Wimpee, Graham Moran and Daad Safarini for helpful discussions, Mr. Victor Trussell of Rufus King High School for help in stability studies of ccNiR, Nick Silvaggi for the use of his high performance chromatography apparatus, and the Jianhua Fu research group (Medical College of Wisconsin) for the use of their automated crystallization machine in the preliminary crystallization screens.
Abbreviations used
- ccNiR
cytochrome c nitrite reductase
- HAO
hydroxylamine oxidoreductase
- AOB
ammonia oxidizing bacteria
- MV
methyl viologen
- MVred
methyl viologen monocation radical
- SHE
standard hydrogen electrode
- CCP4
collaborative computational project, number 4
- SVD
singular value decomposition
- PFV
protein film voltammetry
- PGE
pyrolytic graphite edge
- AEBSF
4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride
- CCM
cytochrome c maturation
- STC
small tetraheme c
References
- 1.Schmidt I, Steenbakkers PJM, op den Camp HJM, Schmidt K, Jetten MSM. J. Bacteriol. 2004;186:2781–2788. doi: 10.1128/JB.186.9.2781-2788.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ehrlich HL. Geomicrobiology. New York, NY: Marcel Dekker, Inc; 2002. [Google Scholar]
- 3.Einsle O, Messerschmidt A, Stach P, Bourenkov GP, Bartunik HD, Huber R, Kroneck PMH. Nature. 1999;400:476–480. doi: 10.1038/22802. [DOI] [PubMed] [Google Scholar]
- 4.Einsle O, Stach P, Messerschmidt A, Simon J, Kroger A, Huber R, Kroneck PMH. J. Biol. Chem. 2000;275:39608–39616. doi: 10.1074/jbc.M006188200. [DOI] [PubMed] [Google Scholar]
- 5.Pereira I, LeGall J, Xavier A, Teixeira M. Biochim. Biophys. Acta. 2000;1481:119–130. doi: 10.1016/s0167-4838(00)00111-4. [DOI] [PubMed] [Google Scholar]
- 6.Bamford VA, Angove HC, Seward HE, Thomson AJ, Cole JA, Butt JN, Hemmings AM, Richardson DJ. Biochemistry. 2002;41:2921–2931. doi: 10.1021/bi015765d. [DOI] [PubMed] [Google Scholar]
- 7.Cunha CA, Macieira S, Dias JM, Almeida G, Goncalves LL, Costa C, Lampreia J, Huber R, Moura JJG, Moura I, Romao MJ. J. Biol. Chem. 2003;278:17455–17465. doi: 10.1074/jbc.M211777200. [DOI] [PubMed] [Google Scholar]
- 8.Rodrigues M, Oliveira T, Pereira I, Archer M. EMBO Journal. 2006;25:5951–5960. doi: 10.1038/sj.emboj.7601439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodrigues ML, Oliveira T, Matias PM, Martins C, Valente FM, Pereira IA, Archer M. Acta Cryst. F. 2006;62:565–568. doi: 10.1107/S1744309106016629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Igarashi N, Moriyama H, Fujiwara T, Fukumori Y, Tanaka N. Nat. Struct. Biol. 1997;4:276–284. doi: 10.1038/nsb0497-276. [DOI] [PubMed] [Google Scholar]
- 11.Einsle O, Messerschmidt A, Huber R, Kroneck PMH, Neese F. J. Am. Chem. Soc. 2002;124:11737–11745. doi: 10.1021/ja0206487. [DOI] [PubMed] [Google Scholar]
- 12.Kostera J, Youngblut MD, Slosarczyk JM, Pacheco AA. J. Biol. Inorg. Chem. 2008;13:1073–1083. doi: 10.1007/s00775-008-0393-4. [DOI] [PubMed] [Google Scholar]
- 13.Kostera J, McGarry JM, Pacheco AA. Biochemistry. 2010;49:8546–8553. doi: 10.1021/bi1006783. [DOI] [PubMed] [Google Scholar]
- 14.Poock S, Leach E, Moir J, Cole J, Richardson D. J. Biol. Chem. 2002;277:23664–23669. doi: 10.1074/jbc.M200731200. [DOI] [PubMed] [Google Scholar]
- 15.Lukat P, Rudolf M, Stach P, Messerschmidt A, Kroneck P, Simon J, Einsle O. Biochemistry. 2008;47:2080–2086. doi: 10.1021/bi7021415. [DOI] [PubMed] [Google Scholar]
- 16.Kurnikov IV, Ratner MA, Pacheco AA. Biochemistry. 2005;44:1856–1863. doi: 10.1021/bi048060v. [DOI] [PubMed] [Google Scholar]
- 17.Bykov D, Neese F. J. Biol. Inorg. Chem. 2011;16:417–430. doi: 10.1007/s00775-010-0739-6. [DOI] [PubMed] [Google Scholar]
- 18.Ford PC, Lorkovic IM. Chem. Rev. 2002;102:993–1017. doi: 10.1021/cr0000271. [DOI] [PubMed] [Google Scholar]
- 19.Wolak M, van Eldik R. Coord. Chem. Rev. 2002;230:263–282. [Google Scholar]
- 20.Ravelli RBG, McSweeney SM. Structure. 2000;8:315–328. doi: 10.1016/s0969-2126(00)00109-x. [DOI] [PubMed] [Google Scholar]
- 21.Pearson AR, Pahl R, Kovaleva EG, Davidson VL, Wilmot CM. J. Synchrotron Rad. 2007;14:92–98. doi: 10.1107/S0909049506051259. [DOI] [PubMed] [Google Scholar]
- 22.Beitlich T, Kuhnel K, Schulze-Briese C, Shoeman RL, Schlichting I. J. Synchrotron Rad. 2007;14:11–23. doi: 10.1107/S0909049506049806. [DOI] [PubMed] [Google Scholar]
- 23.Moffat K, Szebenyi D, Bilderback D. Science. 1984;223:1423–1425. doi: 10.1126/science.223.4643.1423. [DOI] [PubMed] [Google Scholar]
- 24.Srajer V, Teng T, Ursby T, Pradervand C, Ren Z, Adachi S, Schildkamp W, Bourgeois D, Wulff M, Moffat K. Science. 1996;274:1726–1729. doi: 10.1126/science.274.5293.1726. [DOI] [PubMed] [Google Scholar]
- 25.Ren Z, Bourgeois D, Helliwell JR, Moffat K, Srajer V, Stoddard BL. J. Synchrotron Rad. 1999;6:891–917. [Google Scholar]
- 26.Schmidt M, Rajagopal S, Ren Z, Moffat K. Biophys. J. 2003;84:2112–2129. doi: 10.1016/S0006-3495(03)75018-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt M, Pahl R, Srajer V, Anderson S, Ren Z, Ihee H, Rajagopal S, Moffat K. Proc. Natl. Acad. Sci. U S A. 2004;101:4799–4804. doi: 10.1073/pnas.0305983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajagopal S, Schmidt M, Anderson S, Moffat K. Acta Cryst. D. 2004;60:860–871. doi: 10.1107/S0907444904004160. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt M, Pahl R, Ihee H, Srajer V. In: Protein-ligand interactions: Methods and Applications. Nienhaus GU, editor. Totowa, NJ: Humana Press; 2005. [Google Scholar]
- 30.Rajagopal S, Anderson S, Srajer V, Schmidt M, Pahl R, Moffat K. Structure. 2005;13:55–63. doi: 10.1016/j.str.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 31.Ihee H, Rajagopal S, Srajer V, Pahl R, Schmidt M, Schotte F, Anfinrud PA, Wulff M, Moffat K. Proc. Natl. Acad. Sci, USA. 2005;102:7145–7150. doi: 10.1073/pnas.0409035102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Key J, Srajer V, Pahl R, Moffat K. Biochemistry. 2007;46:4706–4715. doi: 10.1021/bi700043c. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt M. In: Ultrashort Laser Pulses in Medicine and Biology. Zinth W, Braun M, Gilch P, editors. Springer Verlag; 2008. [Google Scholar]
- 34.Raicu V, Schmidt M, Stoneman M. Jakovlev V. Non-Linear Phenomena. 2008. pp. 1–32. [Google Scholar]
- 35.Schmidt M, Šrajer V, Purwar N, Tripathi S. J. Synchrotron Rad. 2012;19 doi: 10.1107/S090904951105549X. Available online, Jan 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tripathi S, Srajer V, Purwar N, Henning RW, Schmidt M. Biophys. J. 2012;102:325–332. doi: 10.1016/j.bpj.2011.11.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsapin AI, Nealson KH, Meyers T, Cusanovich MA, van Beuumen J, Crosby LD, Feinberg BA, Zhang C. J. Bacteriol. 1996;178:6386–6388. doi: 10.1128/jb.178.21.6386-6388.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takayama Y, Akutsu H. Protein Expr. Purif. 2007;56:80–84. doi: 10.1016/j.pep.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 39.Ozawa K, Yasukawa F, Fujiwara Y, Akutsu H. Biosci. Biotechnol. Biochem. 2001;65:185–189. doi: 10.1271/bbb.65.185. [DOI] [PubMed] [Google Scholar]
- 40.Dawson RMC, Elliot DC, Jones KM. London: Oxford University Press; 1969. [Google Scholar]
- 41.Atkinson SJ, Mowat CG, Reid GA, Chapman SK. FEBS Lett. 2007;581:3805–3808. doi: 10.1016/j.febslet.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 42.Heineman WR, Norris BJ, Goelz JF. Anal. Chem. 1975;47:79–84. doi: 10.1021/ac60351a001. [DOI] [PubMed] [Google Scholar]
- 43.Pilkington MBG, Coles BA, Compton RG. Anal. Chem. 1989;61:1787–1789. [Google Scholar]
- 44.Bodemer GJ. Ph. D dissertation. Milwaukee, WI: University of Wisconsin-Milwaukee; 2008. [Google Scholar]
- 45.Watanabe T, Honda K. J. Phys. Chem. 1982;86:2617–2619. [Google Scholar]
- 46.Fourmond V, Hoke K, Heering HA, Baffert C, Leroux F, Bertrand P, Leger C. Bioelectrochemistry. 2009;76:141–147. doi: 10.1016/j.bioelechem.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 47.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. Acta Cryst. D. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collaborative Computational Project Number 4. Acta Cryst. D. 1994;50:760–763. [Google Scholar]
- 49.Graber T, Anderson S, Brewer H, Chen YS, Cho HS, Dashdorj N, Henning RW, Kosheleva I, Macha G, Meron M, Pahl R, Ren Z, Ruan S, Schotte F, Rajer VS, Viccaro PJ, Westferro F, Anfinrud P, Moffat K. J. Synchrotron Rad. 2011;18:658–670. doi: 10.1107/S0909049511009423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Press WH, Teukolsky SA, Vetterling WT, Flannery BP. New York, NY: Cambridge University Press; 2007. pp. 65–75. [Google Scholar]
- 51.Henry ER, Hofrichter J. In: Meth. Enzymol. Brand L, Johnson ML, editors. San Diego: Academic Press; 1992. pp. 129–192. [Google Scholar]
- 52.Press WH, Teukolsky SA, Vetterling WT, Flannery BP. New York, NY: Cambridge University Press; 2007. pp. 773–839. [Google Scholar]
- 53.Gates AJ, Kemp GL, To CY, Mann J, Marritt SJ, Mayes AG, Richardson DJ, Butt JN. Phys. Chem. Chem. Phys. 2011;13:7720–7731. doi: 10.1039/c0cp02887h. [DOI] [PubMed] [Google Scholar]
- 54.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J. Appl. Cryst. 2002;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Cryst. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thony-Meyer L, Fischer F, Kunzler P, Ritz D, Hennecke H. J. Bacteriol. 1995;177:4321–4326. doi: 10.1128/jb.177.15.4321-4326.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Londer Y, Giuliani SE, Peppler T, Collart FR. Prot. Expr. Purif. 2008;62:128–137. doi: 10.1016/j.pep.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 58.Londer YY, Pokkuluri PR, Tiede DM, Schiffer M. Biochim. Biophys. acta. 2002;1554:202–211. doi: 10.1016/s0005-2728(02)00244-x. [DOI] [PubMed] [Google Scholar]
- 59.Londer YY, Pokkuluri PR, Erickson J, Orshonsky V, Schiffer M. Protein Expr. Purif. 2005;39:254–260. doi: 10.1016/j.pep.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 60.Londer YY, Pokkuluri PR, Orshonsky V, Orshonsky L, Schiffer M. Protein Expr. Purif. 2006;47:241–248. doi: 10.1016/j.pep.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 61.Eaves DJ, Grove J, Staudenmann W, James P, Poole RK, White SA, Griffiths L, Cole JA. Mol. Microbiol. 1998;28:205–216. doi: 10.1046/j.1365-2958.1998.00792.x. [DOI] [PubMed] [Google Scholar]
- 62.Ozawa K, Tsapin AI, Nealson KH, Cusanovich MA, Akutsu H. Appl. Environ. Microbiol. 2000;66:4168–4171. doi: 10.1128/aem.66.9.4168-4171.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shi L, Lin JT, Markillie LM, Squier TC, Hooker BS. BioTechniques. 2005;38:297–299. doi: 10.2144/05382PT01. [DOI] [PubMed] [Google Scholar]
- 64.Shi L, Chen B, Wang Z, Elias DA, Mayer MU, Gorby YA, Ni S, Lower BH, Kennedy DW, Wunschel DS, Mottaz HM, Marshall MJ, Hill EA, Beliaev AS, Zachara JM, Fredrickson JK, Squier TC. J. Bacteriol. 2006;188:4705–4714. doi: 10.1128/JB.01966-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marritt SJ, Kemp GL, Xiaoe L, Durrant JR, Cheesman MR, Butt JN. J. Am. Chem. Soc. 2008;130:8588–8589. doi: 10.1021/ja802641a. [DOI] [PubMed] [Google Scholar]
- 66.Kiefersauer R, Than ME, Dobbek H, Gremer L, Melero M, Strobl S, Dias JM, Soulimane T, Huber R. J. Appl. Cryst. 2000;33:1223–1230. [Google Scholar]
- 67.Southworth-Davies RJ, Medina MA, Carmichael I, Garman EF. Structure. 2007;15:1531–1541. doi: 10.1016/j.str.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 68.Rajendran C, Dworkowski FS, Wang M, Schulze-Briese C. J. Synchrotron Rad. 2011;18:318–328. doi: 10.1107/S090904951100968X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Walker FA. Coord. Chem. Rev. 1999;186:471–534. [Google Scholar]
- 70.Clarke TA, Kemp GL, Van Wonderen JH, Doyle RAS, Cole JA, Tovell N, Cheesman MR, Butt JN, Richardson DJ, Hemmings AM. Biochemistry. 2008;47:3789–3799. doi: 10.1021/bi702175w. [DOI] [PubMed] [Google Scholar]
- 71.Stach P, Einsle O, Schumacher W, Kurun E, Kroneck PMH. J. Inorg. Biochem. 2000;79:381–385. doi: 10.1016/s0162-0134(99)00248-2. [DOI] [PubMed] [Google Scholar]
- 72.Holm L, Park J. Bioinformatics. 2000;16:566–567. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- 73.Gao H, Yang ZK, Barua S, Reed SB, Romine MF, Nealson KH, Fredrickson JK, Tiedje JM, Zhou J. ISME Journal. 2009;3:966–976. doi: 10.1038/ismej.2009.40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.