

Abstract
Antibodies against cholinergic and adrenergic receptors (adrenoceptors) are frequent in serum of patients with chronic heart failure. Their prevalence is associated with Chagas' disease, idiopathic dilated cardiomyopathy (DCM), and ischaemic heart disease. Among the epitopes targeted are first and second extracellular loops of the β-adrenergic (β-adrenoceptor) and M2 muscarinic receptor. β1-adrenoceptor autoantibodies affect radioligand binding and cardiomyocyte function similar to agonists. Corresponding rodent immunizations induce symptoms compatible with chronic heart failure that are reversible upon removal of the antibodies, transferable via the serum and abrogated by adrenergic antagonists. In DCM patients, prevalence and stimulatory efficacy of β1-adrenoceptor autoantibodies are correlated to the decline in cardiac function, ventricular arrhythmia and higher incidence of cardiac death. In conclusion, such autoantibodies seem to cause or promote chronic human left ventricular dysfunction by acting on their receptor targets in a drug-like fashion. However, the pharmacology of this interaction is poorly understood. It is unclear how the autoantibodies trigger changes in receptor activity and second messenger coupling and how that is related to the pathogenesis and severity of the associated diseases. Here, we summarize the available evidence regarding these issues and discuss these findings in the light of recent knowledge about the conformational activation of the human β2-adrenoceptor and the properties of bona fide cardiopathogenic autoantibodies derived from immune-adsorption therapy of DCM patients. These considerations might contribute to the conception of therapy regimen aimed at counteracting or neutralizing cardiopathogenic receptor autoantibodies.
Keywords: β1-adrenergic receptors, β2-adrenergic receptors, M2-acetylcholine receptors, autoantibodies, allosteric receptor regulation, dilated cardiomyopathy, Chagas' disease, Chagas' cardiomyopathy, atrial fibrillation, chronic heart failure
Humoral receptor autoimmunity and chronic heart disease
At least three human diseases are most certainly caused by autoantibodies that bind to the receptors of neuroendocrine transmitters and alter their function. Grave's disease (M. Basedow), a pathologically enhanced growth and endocrine function of the thyroid gland is caused by autoantibodies that stimulate the receptor of the thyroid-stimulating hormone (TSH-R). Myasthenia gravis, an intermittent weakness of skeletal muscles, is caused by autoantibodies blocking the nicotinic acetylcholine receptor at the neuromuscular endplate. Autoimmune autonomic ganglionopathy is an idiopathic acquired disorder of the autonomic nervous system associated with antibodies blocking the ganglionic nicotinic acetylcholine receptor found in sympathetic, parasympathetic and enteric ganglia.
Over the past two decades, various renal and cardiovascular pathologies have been added to this list, which are associated with humoral autoimmunity against G-protein coupled receptors involved in autonomous vegetative regulation. These encompass malignant (Fu et al., 1994), primary (Luther et al., 1997) or refractory hypertension (Wenzel et al., 2008) associated with humoral autoimmunity against α1-adrenergic receptors (α1-adrenoceptors), and preeclampsia (Wallukat et al., 1999) or renal allograft rejection (Dragun et al., 2005) associated with the occurrence of autoantibodies against angiotensin receptors. It has been demonstrated that immunization of rodents against these receptors leads to alterations in the regulation of blood pressure and kidney function, which may entail cardiac failure as a secondary complication (Dragun et al., 2009). Further experimental data also suggest a blood pressure-independent effect of these autoantibodies on cardiac remodelling (Zhou et al., 2005). Autoantibodies against β-adrenoceptors and muscarinic acetylcholine receptors are thought to be a primary cause of chronic heart failure and a causal factor in the pathogenesis of dilated cardiomyopathy (DCM). Although chronic heart failure can be a consequence of hypertension induced by autoantibodies against α1-adrenoceptors (Fu et al., 1994; Luther et al., 1997; Wenzel et al., 2008), primary forms such as DCM seem to be caused by humoral autoimmunity against heart-specific antigens. Our review will focus on the latter entity.
DCM denominates a disease characterized by a chronic decline in cardiac function and progressive ventricular dilation and dysfunction due to non-ischaemic myocardial damage. A DCM subgroup of about 30% is addressed as ‘idiopathic’ because its origin remains unclear despite efforts at reclassification (Maron et al., 2006). Data accumulated over the past three decades strongly suggest that at least a fraction of this DCM subgroup could represent a later stage of a heart-specific autoimmune disease triggered by viral (Yoshikawa et al., 2009) or protozoic (Cunha-Neto et al., 2006) infections or possibly induced by autoimmunization with heart-specific antigens in genetically predisposed individuals (Caforio et al., 2008b). Humoral autoimmunity seems to play a crucial role in DCM, as heart-reactive autoantibodies found in patients and relatives from familial and non-familial pedigrees predict disease development among healthy relatives (Caforio et al., 2008a). Some of these antibodies have functional effects on cardiac myocytes in vitro and in animal models (Caforio et al., 2005) and possibly in a DCM subset characterized by clinical responsiveness to extracorporeal antibody elimination (Felix et al., 2002). Moreover, chronic cardiac dysfunction can be induced in rodents by peritoneal injection of immune competent B-lymphocytes from DCM patients (Omerovic et al., 2000).
Among the cardiac autoantibodies that are pathognomonic and possibly pathogenic in DCM, those that modulate the function of receptors transducing the regulation of cardiac contraction frequency and force by the autonomous nervous system seem to play a particularly prominent role. Antibodies stimulating cholinergic and β-adrenoceptor signalling are frequently found in serum of patients with chronic heart failure. The epitopes most frequently targeted by such autoantibodies are the first and second extracellular loops of the β1- and β2-adrenoceptors (Magnusson et al., 1990) and the M2 muscarinic acetylcholine receptor (m2AChR) (Fu et al., 1993). In the case of the human β1-adrenoceptor the second extracellular loop is the only extracellular receptor domain capable of inducing antibody production with pharmacological effects on the receptor (Tate et al., 1994). This may be also true for the β2-adrenoceptor and m2AChR, given their structural similarity to the β1-adrenoceptor. Such autoantibodies are associated with cardiomyopathy evolving in the course of Chagas' disease (Sterin-Borda et al., 1976; Rosenbaum et al., 1994; Hernandez et al., 2003; Labovsky et al., 2007; Munoz-Saravia et al., 2010), DCM (Magnusson et al., 1990; 1994; Fu et al., 1993; Rosenbaum et al., 1994; Matsui et al., 1995; Wallukat et al., 1995; Jahns et al., 1999b; Staudt et al., 2001), congestive heart failure (Zhang et al., 2002), ischaemic heart disease (Jahns et al., 1999b), atrial tachyarrhythmia (Baba et al., 2004; Chiale and Ferrari, 2001; Del Corsso et al., 2004; Stavrakis et al., 2009; 2011; Yu et al., 2009), but not cardiomyopathies of other aetiology (Magnusson et al., 1996; Jahns et al., 1999a). In DCM patients, prevalence (Jahns et al., 1999b) and cAMP stimulatory efficacy (Nikolaev et al., 2007) of β1-adrenoceptor autoantibodies are correlated to reduced cardiac function (Jahns et al., 1999b), increased mortality (Stork et al., 2006), severe ventricular arrhythmia (Chiale et al., 2001), and higher incidence of sudden cardiac death (Iwata et al., 2001). Interestingly, atrial fibrillation in Graves' disease is also associated with the occurrence of autoantibodies stimulating cholinergic and β-adrenoceptors and these autoantibodies are distinct from the ones causing hyperthyroidism through stimulation of the TSH-R (Stavrakis et al., 2009). This observation suggests that syndromes associated with autoantibodies against β-adrenergic and cholinergic receptors can cross the borders of organ-specific aetiologies. On the other hand, low levels of autoantibodies against β1-adrenoceptor, β2-adrenoceptor and m2AChR are also present in the bloodstream of many healthy individuals (Liu et al., 1999) and are thought to be a part of the natural immunologic repertoire (Fraser and Venter, 1984; Rose, 2001; Jahns et al., 2006b). This raises the question of whether such autoantibodies can indeed cause heart failure, or are just an epiphenomenon.
Evidence that stimulatory β1-adrenoceptor autoantibodies cause heart failure
In Chagas' disease immune responses to the C-terminal end of the ribosomal P2β protein of T. cruzi give rise to antibodies cross-reacting with first and second extracellular loops of human β1-adrenoceptor, β2-adrenoceptor and m2AChR and trigger sustained humoral autoimmunity against these receptors (Lopez Bergami et al., 2005). What triggers receptor autoimmunization in DCM is unclear. Autoantibodies associated with DCM seem to be directed against a different portion of the second extracellular receptor loop than those associated with Chagas' disease (Magnusson et al., 1996). Various viral and microbial candidate proteins have been proposed that could trigger a bystander effect analogous to the one triggered by ribosomal P2β protein of T. cruzi in Chagas' disease (Levin and Hoebeke, 2008), but firm evidence of such a causative microbial immunogen is still lacking.
IgG autoantibodies from Chagas' patients increase cellular cAMP (Sterin-Borda et al., 1976; Rosenbaum et al., 1994) and impair L-type Ca2+ currents (Hernandez et al., 2003) in isolated cardiomyocytes, indicating that they can promote receptor coupling to stimulatory (Gs) as well as inhibitory (Gi) G-proteins, consistent with a varied spectrum of agonist-like actions on β1-adrenoceptor, β2-adrenoceptor and m2AChR. The clinical syndromes of chronic Chagas' disease developing with a latency of several decades after T. cruzi infection can be predicted from the cross-reactivity patterns of these receptor-stimulating autoantibodies: The development of cardiomyopathy is associated with the induction of autoantibodies against β1-adrenoceptor and m2AChR, whereas the development of mega-colon is associated with the induction of autoantibodies against β2-adrenoceptor and m2AChR (Wallukat et al., 2010). These observations also reported from animal models (Mijares et al., 1996b; Silvina Lo Presti et al., 2008) suggest that Chagas' cardiomyopathy could be caused or at least promoted by the continuous action of autoantibodies activating the β1-adrenoceptor, whereas vagal dysfunctions associated with the disease (Davila et al., 2005) seem to be linked to the occurrence of autoantibodies activating the m2AChR. The role of autoantibodies stimulating the β2-adrenoceptor that are also present in many of these patients remains unclear.
Direct immunization of rodents with peptides or fusion proteins representing sequences of the second extracellular loop of the β1-adrenoceptor have been demonstrated to induce left ventricular dilation and dysfunction (Jahns et al., 2004; Buvall et al., 2006) among other effects compatible with chronic cardiac dysfunction (Matsui et al., 1999; Omerovic et al., 2000; Fukuda et al., 2004; Jane-wit et al., 2007; Zuo et al., 2011). These effects were associated with the induction of cAMP-stimulatory β1-adrenoceptor autoantibodies and clinical signs consistent with chronic stimulation and desensitization of β1-adrenoceptor signalling in the heart (Jahns et al., 2004); they were reversible upon removal of the antibodies (Matsui et al., 2006b), transferable via serum transfusions (Jahns et al., 2004; Matsui et al., 2006a; Jane-wit et al., 2007; Liu et al., 2008) and at least partially abrogated by β1-adrenoceptor antagonists (Matsui et al., 2000). These findings support the assumption that autoantibodies stimulating Gs-coupling of the β1-adrenoceptor are a probable cause or decisive pathogenic cofactor of chronic heart failure. This assumption implies that there should be symptoms of increased heart rate or contractility during the early phase of immunization, which has not been observed. Moreover, there is evidence that receptor antibodies induced by the immunization of rodents act in a different fashion on receptor activity than human autoantibodies associated with DCM or Chagas' disease (Jahns et al., 2000).
Despite these inconsistencies, the paradigm of Chagas' cardiomyopathy in conjunction with the available results of experimental immunizations build a reasonably strong case for autoantibodies against the second extracellular loop domain of the human β1-adrenoceptor as a specific pathogen in chronic left ventricular dysfunction. These autoantibodies appear to somehow stimulate the receptor and thereby cause left ventricular failure in immunized animals. Currently, the pathogenic effect is blamed on inappropriate ino- and chronotropism leading to down-regulation and desentization of the cardiac β1-adrenoceptor (Jahns et al., 2004; 2006a). However, it has also been shown that such autoantibodies stimulate apoptosis (Staudt et al., 2003; Jane-wit et al., 2007) and stress responses of the endoplasmic reticulum (Liu et al., 2008) in isolated cardiomyocytes, suggesting that the pathogenic mechanism may also involve direct myocardial cytotoxicity. The high incidence of stimulatory β1-adrenoceptor autoantibodies in DCM (Magnusson et al., 1994; Jahns et al., 1999b; Staudt et al., 2001; Nikolaev et al., 2007) and Chagas' disease (Labovsky et al., 2007) and the predictive value of β1-adrenoceptor autoantibodies for the development of Chagas' cardiomyopathy (Wallukat et al., 2010) supports the hypothesis that these autoantibodies can trigger chronic left ventricular dysfunction not only in immunized rodents but also in human patients (Jahns et al., 2006b).
Role of cardiostimulatory versus -depressant autoantibodies
In contrast to β1-adrenoceptor autoantibodies, the role of β2-adrenoceptor or m2AChR autoantibodies in the pathogenesis of cardiomyopathy remains somewhat enigmatic. These autoantibodies are also frequently found in association with DCM (Fu et al., 1993; Magnusson et al., 1996) and Chagas' cardiomyopathy (Wallukat et al., 2010). However, they are expected to have the opposite effect to β1-adrenoceptor autoantibodies, that is, to act cardiodepressive via Gi-mediated inhibition of adenylate cyclase (Higgins et al., 1973) and impairment of L-type Ca2+ currents (He et al., 2005). Interestingly, the latter effect was found to dominate in some studies of IgG samples from Chagas' patients (Hernandez et al., 2003). Moreover, haemodynamic improvement following extracorporeal removal of bona fide cardiopathogenic IgG from DCM patients is poorly correlated to the extracorporeal removal of cardiostimulatory autoantibodies (Felix et al., 2002; Wallukat et al., 2002; Mobini et al., 2003; Dorffel et al., 2004; Kallwellis-Opara et al., 2007) or their subsequent reappearance (Felix et al., 2002). The parameter most closely related to the extent and time course of haemodynamic response to immune absorption therapy is the effect of eluated autoantibodies on L-type Ca2+ currents and cardiomyocyte contraction (Felix et al., 2002; Trimpert et al., 2010). However, these cardiodepressant effects may not solely be caused by stimulation of Gi-coupled cardiac receptors (e.g. m2AChR or β2-adrenoceptor). They may also be exerted through interactions of the Fc part of cardiac autoantibodies with Fc receptors that have been associated with the development of many human autoimmune diseases (Takai, 2002). Fcγ receptors IIa recently discovered on cardiomyocytes may be involved in the negative inotropic effects of cardiac antibodies obtained from DCM patients (Staudt et al., 2007). After autoantibody-binding to the respective myocardial antigen via the F(ab)2 part and cross-linking via the Fc part to Fcγ receptors IIa, these receptors then may induce an activating signal via the FC receptor's cytoplasmic domain, thereby possibly triggering a cardiodepressive effect (see Figure 1I). This novel mechanism is independent of the cardiac antigen specifically targeted by these antibodies. It seems particularly relevant for the response to immune-adsorption therapy, as patients with a polymorphism of the Fcγ receptor IIa that is associated with low affinity to the Fc fragment of antibodies exhibit significantly greater improvement in left ventricular function upon extracorporeal IgG elimination (Staudt et al., 2010).
Figure 1.
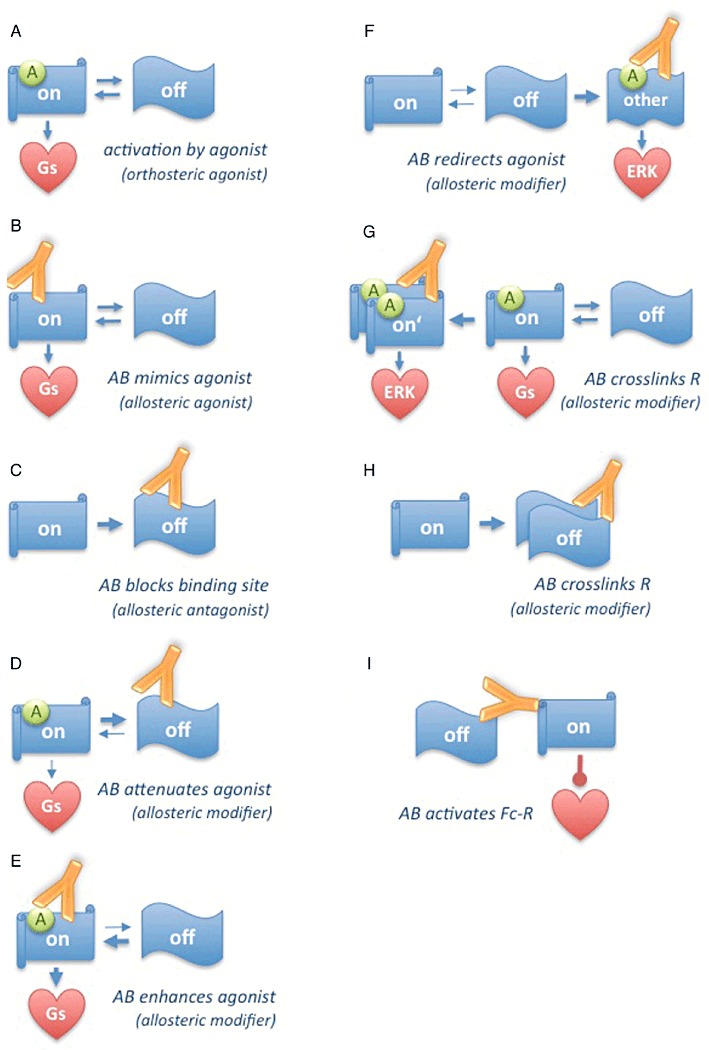
Modes of modulation of receptor conformation and function by autoantibodies. G-protein coupled receptors idle between various conformations with different abilities to couple to specific signalling pathways; orthosteric agonists act by stabilizing receptor conformations linked to one specific signalling pathway (A). Autoantibodies can act as direct allosteric agonists by mimicing agonists (B) or act as direct allosteric antagonists by blocking the agonist's binding site (C). Autoantibodies can act as allosteric modulators by promoting inactive (D) or active (E) conformations of the unliganded receptor thereby enhancing or attenuating subsequent agonist actions. Autoantibodies can act as allosteric modulators by inducing alternative receptor conformations that predispose for coupling to other signalling pathways (F). Autoantibodies can also modify agonist action by promoting receptor polymerization, which may enhance, redirect (G) or attenuate agonist action (H) depending on which receptor is targeted. Autoantibodies can activate as yet unknown cardiodepressive signalling pathways by cross-linking G-protein coupled receptors with Fc-receptors (I).
It is unclear whether cardiodepressant autoantibodies acting via stimulation of the m2AChR or possibly the β2-adrenoceptor also play a causal role in cardiomyopathy. Data of immunization experiments with the second extracellular loop of the β2-adrenoceptor are not available. Immunizations against the second extracellular loop of the m2AChR had inconclusive results, as symptoms consistent with cardiomyopathy were only inducible by combined immunization with peptides corresponding to the second loop of the β1-adrenoceptor (Matsui et al., 1999). However, m2AChR autoantibodies arising from such immunizations directly induce fibrillation in isolated atria (Hong et al., 2009), and this finding is consistent with the increased incidence of m2AChR autoantibodies in patients suffering from atrial fibrillation in conjunction with DCM (Baba et al., 2004) or Graves' hyper-thyreoidism (Stavrakis et al., 2009). In a recent study the combined impact of autoantibodies against β1-adrenoceptor, β2-adrenoceptor and m2AChR retrieved from patients with cardiomyopathy and/or atrial tachy-arrhythmias on isolated canine Purkinje fibre contractility was addressed in a systematic manner (Stavrakis et al., 2011). This study revealed that in most samples the positive inotropic effects of β1-adrenoceptor autoantibodies were negatively modulated by coincident β2-adrenoceptor and m2AChR autoantibodies, prompting the conclusion that β1-selective antagonists routinely used in these clinical conditions may place the patients at a disadvantage due to the unopposed muscarinic effect of m2AChR autoantibodies and the possible unmasking of Gi-signalling by β2-adrenoceptor autoantibodies. Both effects could blunt the contractile response of the failing heart mandating an adjustment of medication to the individual ‘mix’ of receptor autoantibodies present in a given patient. In principle, the same reasoning applies to the treatment of DCM with cyclic peptides specifically neutralizing β1-adrenoceptor autoantibodies (proposed by Jahns et al., 2006a; 2010), while it should not play a role in therapy regimen employing extracorporeal elimination of all IgG.
In summary, the available data are consistent with a model where stimulatory autoantibodies directed against the β1-adrenoceptor cause chronic heart failure through continuous inappropriate ino- and chronotropism and/or cytotoxic effects on cardiomyocytes. It seems conceivable that neutralization of these autoantibodies by peptides could prevent or postpone clinical manifestations of the disease as demonstrated in immunized rodents (Jahns et al., 2006a; 2010). However, the clinical phenotype of the full-blown disease seems to be influenced more significantly by the simultaneous occurrence of cardiodepressant autoantibodies. In this respect autoantibodies acting through the m2AChR and possibly also the β2-adrenoceptor seem to promote the incidence of atrial tachy-arrhythmia and may indicate the selection of appropriate receptor-directed medication, whereas cardiodepressant effects due to simultaneous interaction of IgG autoantibodies with specific myocardial antigens and Fcγ receptors IIa seem to be a relevant criterion for the prediction of the haemodynamic response to immune-adsorption therapy, irrespective of which myocardial antigen is specifically targeted.
What happens at the level of the receptor?
It is frequently proposed that stimulatory autoantibodies against second extracellular loops of adrenergic and cholinergic receptors are allosteric receptor agonists (Jahns et al., 2006b). This hypothesis is mainly based on the longstanding observation that human autoantibodies and antibodies raised in rodents against second extracellular loops of the human β1-adrenoceptor or m2AChR decrease affinity and maximal capacity of equilibrium radioligand binding to the receptor in a dose-dependent fashion (Magnusson et al., 1990; Fu et al., 1993; Jahns et al., 2000). This observation suggests a classical non-competitive type of interaction typical for allosteric receptor modulation (Kenakin, 2004; Kenakin, 2003). However, given the close position of the targeted epitope relative to the binding site (Cherezov et al., 2007), this observation could also be interpreted in terms of a steric hindrance of ligand access to the ligand binding pocket. The impact of human β1-adrenoceptor autoantibodies on ligand binding is frequently associated with moderately increased cAMP stimulation through the otherwise unliganded receptor (Magnusson et al., 1994; Jahns et al., 2000; Nikolaev et al., 2007). However, this weak agonist-like activity intrinsic to the autoantibodies is notably different from that of classical agonists: The autoantibodies have a weaker chrono- and ino-tropic potency and are less prone to induce receptor desensitization and down regulation of β-adrenergic signal transduction (Christ et al., 2006); they have a higher potency to trigger apoptosis (Staudt et al., 2003; Jane-wit et al., 2007); they induce stress responses of the endoplasmic reticulum (Liu et al., 2008); they stimulate the ERK1/2 pathway through a different intracellular signal cascade (Tutor et al., 2007). Moreover, maximal cAMP stimulation by a classical agonist can be potentiated or attenuated when a stimulatory autoantibody is bound at the same time (Jahns et al., 2000). Taken together, these observations suggest that the autoantibodies can have three distinct effects on the receptor: (i) they modulate the binding of true ligands; (ii) they activate per se various effector pathways downstream of the receptor; (iii) they modulate the receptor's disposition and response to simultaneous agonist binding in a varied manner.
One possible mechanism to explain these pleiotropic effects is that the antibodies induce or stabilize changes in receptor conformation that mimic or modulate the ones induced or stabilized by true agonistic ligands (see Figure 1B). At least for β1-adrenoceptor autoantibodies it is known that they bind to conformational epitopes (Jahns et al., 1999b; 2000) and therefore have the potential to alter receptor conformation and function. They exert these putative effects through established interactions with a receptor domain, which, based on the known structure of the β2-adrenoceptor (Cherezov et al., 2007), is not a part of the ligand binding pocket, but forms a separate, extracellular helix (Cherezov et al., 2007). This helix can reach down into the ligand-binding pocket and touch the ligand; disulfide bonds crucial for keeping the entire helix out of the binding pocket (Cherezov et al., 2007) and thus ensuring proper ligand binding (Dohlman et al., 1990; Bywater, 2005) are located within the very epitopes targeted by the antibodies (Magnusson et al., 1990). Thus, interference with this domain's conformation and relative position within the receptor molecule seems a plausible mechanism by which the autoantibodies could hinder the access of ligands to the binding pocked and/or induce distortions of the ligand binding pocket that alter its ligand binding properties and/or mimic the effects of orthosteric agonist binding (Cherezov et al., 2007). Along the same lines it is conceivable that the autoantibodies block the ligand-binding site or stabilize the inactive receptor conformation, thus acting as allosteric antagonists (Figure 1C) or attenuators (Figure 1D). However, up to now, it has not been demonstrated that autoantibodies against second extracellular loops of the β1-adrenoceptor, β2-adrenoceptor or m2AChR are indeed capable of triggering changes in the conformation of these receptors; consequently it is not known whether such putative conformation changes could have any resemblance to the ones known to be triggered by true agonistic ligands when they occupy the ligand binding pocket (Cherezov et al., 2007).
Another possible mechanism of autoantibody action is the stabilization of transient default states of the receptor. It has been suggested that receptors can exist in (or even idle between) distinct states with different abilities of G-protein interaction. Certain receptor states are selected by agonists to promote particular receptor/G-protein combinations with different abilities to stimulate particular effectors (Figure 1A). This mechanism for instance plays a role in the pleiotropic response of the β2-adrenoceptor to the various β-agonists used in the treatment of obstructive lung disease (Swift et al., 2007). Moreover, receptors can regulate various cellular functions by direct recruitment, activation and scaffolding of cytoplasmic signalling complexes via β-arrestins (Lefkowitz and Shenoy, 2005), a mechanism playing a role in the divergent effects of various agonistic and inverse agonistic β-adrenergic ligands on cAMP- versus MAP-kinase signalling (Azzi et al., 2003). There are some indications that β1-adrenoceptor autoantibodies could exert their effects by changing the spectrum of receptor states. There is evidence that they can influence the stability of transient conformational states of the β1-adrenoceptor induced by true agonists (Hoebeke, 1996; Mijares et al., 1996a; Jane-wit et al., 2007). Based on these observations it is conceivable that the autoantibodies act as enhancers or attenuators of normal agonist action by stabilizing active or inactive intermediates of receptor conformations involved in normal signalling (Figure 1D, E). They could also redirect agonist action to alternative downstream signalling pathways, as they promote or stabilize transient conformations of the unliganded receptor, which then possibly pre-dispose to its selection by an agonist for the promotion of a particular set of downstream events, while the receptor would be selected for another set of downstream events when no autoantibody is bound (Figure 1F). Such hypothetical mechanisms could explain how autoantibodies can, at the same time, increase basal and decrease maximal receptor activity (Jahns et al., 2000) and how they can possibly alter the receptor's balance of signalling into distinct downstream pathways (Tutor et al., 2007).
The third possible mechanism is that the autoantibodies have an impact on receptor di-/ oligomerization. It is known that heterodimerization between β1-adrenoceptor and β2-adrenoceptor plays a crucial role in the regulation of cardiac contractility (Zhu et al., 2005) as well as receptor internalization and the activation of β-arrestin-dependent effector systems such as ERK1/2 (Lavoie et al., 2002). The m2AChR on the other hand has been demonstrated to exist as a constitutive homotetramer (Pisterzi et al., 2010) but the relevance of m2AChR oligomerization for receptor function is unclear (reviewed in Milligan, 2008; Smith and Milligan, 2010). Given their divalent binding domain, IgG autoantibodies are ideally suited to induce or redirect receptor polymerization and thereby modulate downstream signalling (Figure 1G, H). On the other hand they can form bulky receptor adducts that at high titers (where IgG concentration is in excess to receptor density) could lead to inhibition of receptor oligomerization or hetero-dimerization. The autoantibodies could thereby exert some or even all of their functional effects without the need to interfere with the activation associated intra-molecular conformation switch of the receptor. However, smoking gun experiments demonstrating the impact of autoantibodies on receptor oligomerization (or the absence of such effects) are not available to date.
Diagnostic issues
It is not clear how autoantibodies against receptors of the autonomous nervous system can be reliably measured in a clinical setting. Measurements of the impact of isolated IgG on Ca2+-transients or the contraction amplitude and frequency of isolated mammalian cardiomyocytes (Wallukat and Nisson, 2001) or cardiac fibres (Stavrakis et al., 2011) seems to be the most valid and unbiased diagnostic approach, as it detects the net outcome of pleiotropic autoantibody actions in an analytical setting with a high resemblance of the situation in the patient. It has been demonstrated that such assays have a high power to predict the development of cardiomyopathy in asymptomatic Chagas' patients (Wallukat et al., 2010). However, these bio-assays are difficult to standardize and for a number of other technical reasons are unsuitable as a routine clinical diagnostic test. The pillars of autoantibody detection in clinical studies have been solid phase immune assays based on peptide analogues of the crucial epitopes targeted in the β1-adrenoceptor (Magnusson et al., 1990) and the m2AChR (Fu et al., 1993). However, a number of considerations caution against the use of peptide binding as the sole criterion of autoantibody detection. First of all, these assays cannot discriminate between autoantibodies that stimulate the receptor and those that merely bind or even block the receptor. There is increasing evidence that this distinction is crucial, as it seems to be not the mere presence of the antibodies but their impact on receptor function that is related to cardiopathogenesis (Stork et al., 2006; Nikolaev et al., 2007). Secondly, there are huge discrepancies between the abilities of human β1-adrenoceptor autoantibodies to bind to peptide analogues as compared with binding to native receptors presented on the cell surface (Jahns et al., 1999a; Labovsky et al., 2007). Moreover, the autoantibody epitope is lost upon denaturation of the receptor and recovered upon its re-naturation (Jahns et al., 1999a). These observations suggest that the autoantibodies target a labile conformational epitope that is poorly represented by synthetic receptor analogues and difficult to preserve outside the living cell. Consequently, various live cell-based assays have been developed (Labovsky et al., 2007; Nikolaev et al., 2007) that are currently evaluated in clinical studies (Deubner et al., 2010) but have as yet not been made available for routine clinical diagnostics.
Therapeutic issues
Currently, β1-adrenoceptor antagonists and angiotensin converting enzyme inhibitors or angiotensin receptor blockers are considered equally effective first line therapeutics of DCM. There is, however, some debate as to which of the two strategies should be employed first (Funck-Brentano et al., 2011). It has been proposed that the effectiveness of β1-adrenoceptor blockade is to some extent due to the disruption of receptor stimulation by autoantibodies (Magnusson et al., 1996) and there are some recent indications that therapy responses to β-blockers are indeed correlated with the presence of β1-adrenoceptor autoantibodies (Nagatomo et al., 2009). However, in many DCM patients, the positive inotropic effects of β1-adrenoceptor autoantibodies are negatively modulated by coincident β2-adrenoceptor and m2AChR autoantibodies so that β1-selective antagonists will unmask unfavourable muscarinic effects of m2AChR autoantibodies and/or Gi-signalling by β2-adrenoceptor autoantibodies (Stavrakis et al., 2011).
Similar concerns apply to therapy approaches aiming at the specific removal of β1-adrenoceptor autoantibodies by adsorption to synthetic β1-adrenoceptor analogues (Wallukat et al., 2002; Mobini et al., 2003) or their specific neutralization by the systemic application of such analogues that has been demonstrated in vitro (Haberland et al., 2011) or in immunized rodents (Jahns et al., 2010) but as yet has not been tested in patients. Given the imperfect representation of the autoantibody epitope by synthetic analogues it is to be expected that only a subgroup of antibodies will be targeted by such procedures. Moreover, selective removal or blockade of β1-adrenoceptor autoantibodies could elicit negative effects due to the unmasking of m2AChR- and/or β2-adrenoceptor autoantibodies. This concern is clearly supported by the observation that β1-adrenoceptor antagonists are not counteracting all the adverse effects of cardio depressant autoantibodies, as end-stage DCM patients subjected to total unselective IgG exchange benefit from positive haemodynamic effects that are additive to those of a preceding therapy with β1-adrenoceptor antagonists (Felix et al., 2002). However, it is unclear whether these additional beneficial effects are due to the removal of autoantibodies targeting autonomous transmitter receptors or of autoantibodies targeting other myocardial antigens. It is moreover unclear whether these effects – if related to the removal of receptor autoantibodies – rely on the disruption of receptor signalling by the autoantibodies or abolishment of cardiodepressant effects delivered through simultaneous interactions of the autoantibodies with Fc-receptors (Staudt et al., 2007).
Summary, conclusion and outlook
Over the last decade, humoral autoimmunity against β-adrenergic and cholinergic receptors has developed from a curious coincidence to a probable cause of chronic heart failure. Various therapeutic concepts of targeting this pathogenic process in a causal manner show promising results in the treatment of end-stage DCM. However, our knowledge about how the autoantibodies alter receptor function and how these effects possibly contribute to the pathogenesis and the clinical phenotype of chronic heart failure is still very limited. As a consequence, it is not known, which features of the autoantibodies should be assessed to indicate and control antibody-directed therapy. Peptide-directed binding assays have an insufficient sensitivity and specificity, because the autoantibodies target a conformational epitope that is only exposed when the receptor has its native conformation and is properly embedded in the cell membrane. On the other hand, the assessment of selected autoantibody effects on receptor function seems an insufficient criterion, given the pleotropic effects of β1-adrenoceptor autoantibodies on various functions of various receptors with opposing biological functions. Moreover, it is not clear to what extent the phenotypes of chronic heart failure are determined by the individual contributions of cardiostimulating autoantibodies promoting Gs-coupling of β1-adrenoceptor and cardiodepressive autoantibodies actin through Fcγ receptors IIa or promoting Gi-coupling of β2-adrenoceptor and m2AChR. Currently, the measurement of the impact of isolated IgG on Ca2+-transients or the contraction of isolated mammalian cardiomyocytes seems the most valid and unbiased diagnostic approach. More practical diagnostic assays could be designed, if more information were available regarding the molecular action of the autoantibodies at the level of the receptor. The current lack of such specific and practical diagnostic criteria and tools is particularly unfortunate, as these autoantibodies seem to constitute a part of the natural immunologic repertoire and therefore are also present in the bloodstream of many healthy individuals, which may or may not develop an autoimmune cardiomyopathy later on.
Acknowledgments
The authors acknowledge financial support by the Deutsche Forschungsgemeinschaft (DFG), collaborative research centres SFB TR 19 (Stephan B. Felix and Lars R. Herda), SFB 612, SFB 728, and research training group GK1089 (Fritz Boege) and the Allianz Industrie und Forschung (AIF) of the German Ministry of Economy and Technology (BMWi).
Glossary
- β-adrenoceptor
β-adrenergic receptor
- β1-adrenoceptor
β-adrenergic receptor type 1
- β2-adrenoceptor
β-adrenergic receptor type 2
- Fc
invariable domain of IgG
- G-protein
heterotrimeric GTPase protein
- Gi
G-protein inhibiting adenylate cyclase
- Gs
G-protein stimulating adenylate cyclase
- m2AChR
muscarininic actylcholine receptor type 2
- TSH-R
receptor of the thyroid-stimulating hormone
Conflict of interest
We state that there are no conflicts of interest to disclose by any of the authors.
References
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, et al. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci U S A. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba A, Yoshikawa T, Fukuda Y, Sugiyama T, Shimada M, Akaishi M, et al. Autoantibodies against M2-muscarinic acetylcholine receptors: new upstream targets in atrial fibrillation in patients with dilated cardiomyopathy. Eur Heart J. 2004;25:1108–1115. doi: 10.1016/j.ehj.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Buvall L, Bollano E, Chen J, Shultze W, Fu M. Phenotype of early cardiomyopathic changes induced by active immunization of rats with a synthetic peptide corresponding to the second extracellular loop of the human beta-adrenergic receptor. Clin Exp Immunol. 2006;143:209–215. doi: 10.1111/j.1365-2249.2005.02986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bywater RP. Location and nature of the residues important for ligand recognition in G-protein coupled receptors. J Mol Recognit. 2005;18:60–72. doi: 10.1002/jmr.685. [DOI] [PubMed] [Google Scholar]
- Caforio AL, Daliento L, Angelini A, Bottaro S, Vinci A, Dequal G, et al. Autoimmune myocarditis and dilated cardiomyopathy: focus on cardiac autoantibodies. Lupus. 2005;14:652–655. doi: 10.1191/0961203305lu2193oa. [DOI] [PubMed] [Google Scholar]
- Caforio AL, Tona F, Bottaro S, Vinci A, Dequal G, Daliento L, et al. Clinical implications of anti-heart autoantibodies in myocarditis and dilated cardiomyopathy. Autoimmunity. 2008a;41:35–45. doi: 10.1080/08916930701619235. [DOI] [PubMed] [Google Scholar]
- Caforio AL, Vinci A, Iliceto S. Anti-heart autoantibodies in familial dilated cardiomyopathy. Autoimmunity. 2008b;41:462–469. doi: 10.1080/08916930802031546. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiale PA, Ferrari I. Autoantibodies in Chagas' cardiomyopathy and arrhythmias. Autoimmunity. 2001;34:205–210. doi: 10.3109/08916930109007386. [DOI] [PubMed] [Google Scholar]
- Chiale PA, Ferrari I, Mahler E, Vallazza MA, Elizari MV, Rosenbaum MB, et al. Differential profile and biochemical effects of antiautonomic membrane receptor antibodies in ventricular arrhythmias and sinus node dysfunction. Circulation. 2001;103:1765–1771. doi: 10.1161/01.cir.103.13.1765. [DOI] [PubMed] [Google Scholar]
- Christ T, Schindelhauer S, Wettwer E, Wallukat G, Ravens U. Interaction between autoantibodies against the beta1-adrenoceptor and isoprenaline in enhancing L-type Ca2+ current in rat ventricular myocytes. J Mol Cell Cardiol. 2006;41:716–723. doi: 10.1016/j.yjmcc.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Cunha-Neto E, Bilate AM, Hyland KV, Fonseca SG, Kalil J, Engman DM. Induction of cardiac autoimmunity in Chagas heart disease: a case for molecular mimicry. Autoimmunity. 2006;39:41–54. doi: 10.1080/08916930500485002. [DOI] [PubMed] [Google Scholar]
- Davila DF, Santiago JJ, Odreman WA. Vagal dysfunction and the pathogenesis of chronic Chagas disease. Int J Cardiol. 2005;103:227–229. doi: 10.1016/j.ijcard.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Del Corsso C, de Carvalho AC, Martino HF, Varanda WA. Sera from patients with idiopathic dilated cardiomyopathy decrease ICa in cardiomyocytes isolated from rabbits. Am J Physiol Heart Circ Physiol. 2004;287:H1928–H1936. doi: 10.1152/ajpheart.00044.2004. [DOI] [PubMed] [Google Scholar]
- Deubner N, Berliner D, Schlipp A, Gelbrich G, Caforio AL, Felix SB, et al. Cardiac beta1-adrenoceptor autoantibodies in human heart disease: rationale and design of the Etiology, Titre-Course, and Survival (ETiCS) Study. Eur J Heart Fail. 2010;12:753–762. doi: 10.1093/eurjhf/hfq072. [DOI] [PubMed] [Google Scholar]
- Dohlman HG, Caron MG, DeBlasi A, Frielle T, Lefkowitz RJ. Role of extracellular disulfide-bonded cysteines in the ligand binding function of the beta 2-adrenergic receptor. Biochemistry. 1990;29:2335–2342. doi: 10.1021/bi00461a018. [DOI] [PubMed] [Google Scholar]
- Dorffel WV, Wallukat G, Dorffel Y, Felix SB, Baumann G. Immunoadsorption in idiopathic dilated cardiomyopathy, a 3-year follow-up. Int J Cardiol. 2004;97:529–534. doi: 10.1016/j.ijcard.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Dragun D, Muller DN, Brasen JH, Fritsche L, Nieminen-Kelha M, Dechend R, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med. 2005;352:558–569. doi: 10.1056/NEJMoa035717. [DOI] [PubMed] [Google Scholar]
- Dragun D, Philippe A, Catar R, Hegner B. Autoimmune mediated G-protein receptor activation in cardiovascular and renal pathologies. Thromb Haemost. 2009;101:643–648. [PubMed] [Google Scholar]
- Felix SB, Staudt A, Landsberger M, Grosse Y, Stangl V, Spielhagen T, et al. Removal of cardiodepressant antibodies in dilated cardiomyopathy by immunoadsorption. J Am Coll Cardiol. 2002;39:646–652. doi: 10.1016/s0735-1097(01)01794-6. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Venter JC. Anti-receptor antibodies in human disease. J Allergy Clin Immunol. 1984;74:661–673. doi: 10.1016/0091-6749(84)90227-6. [DOI] [PubMed] [Google Scholar]
- Fu LX, Magnusson Y, Bergh CH, Liljeqvist JA, Waagstein F, Hjalmarson A, et al. Localization of a functional autoimmune epitope on the muscarinic acetylcholine receptor-2 in patients with idiopathic dilated cardiomyopathy. J Clin Invest. 1993;91:1964–1968. doi: 10.1172/JCI116416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu ML, Herlitz H, Wallukat G, Hilme E, Hedner T, Hoebeke J, et al. Functional autoimmune epitope on alpha 1-adrenergic receptors in patients with malignant hypertension. Lancet. 1994;344:1660–1663. doi: 10.1016/s0140-6736(94)90456-1. [DOI] [PubMed] [Google Scholar]
- Fukuda Y, Miyoshi S, Tanimoto K, Oota K, Fujikura K, Iwata M, et al. Autoimmunity against the second extracellular loop of beta(1)-adrenergic receptors induces early afterdepolarization and decreases in K-channel density in rabbits. J Am Coll Cardiol. 2004;43:1090–1100. doi: 10.1016/j.jacc.2003.09.057. [DOI] [PubMed] [Google Scholar]
- Funck-Brentano C, van Veldhuisen DJ, van de Ven LL, Follath F, Goulder M, Willenheimer R. Influence of order and type of drug (bisoprolol vs. enalapril) on outcome and adverse events in patients with chronic heart failure: a post hoc analysis of the CIBIS-III trial. Eur J Heart Fail. 2011;13:765–772. doi: 10.1093/eurjhf/hfr051. [DOI] [PubMed] [Google Scholar]
- Haberland A, Wallukat G, Dahmen C, Kage A, Schimke I. Aptamer neutralization of beta1-adrenoceptor autoantibodies isolated from patients with cardiomyopathies. Circ Res. 2011;109:986–992. doi: 10.1161/CIRCRESAHA.111.253849. [DOI] [PubMed] [Google Scholar]
- He JQ, Balijepalli RC, Haworth RA, Kamp TJ. Crosstalk of beta-adrenergic receptor subtypes through Gi blunts beta-adrenergic stimulation of L-type Ca2+ channels in canine heart failure. Circ Res. 2005;97:566–573. doi: 10.1161/01.RES.0000181160.31851.05. [DOI] [PubMed] [Google Scholar]
- Hernandez CCQ, Barcellos LC, Gimenez LED, Cabarcas RAB, Garcia S, Pedrosa RC, et al. Human chagasic IgGs bind to cardiac muscarinic receptors and impair L-type Ca2+ currents. Cardiovasc Res. 2003;58:55–65. doi: 10.1016/s0008-6363(02)00811-8. [DOI] [PubMed] [Google Scholar]
- Higgins CB, Vatner SF, Braunwald E. Parasympathetic control of the heart. Pharmacol Rev. 1973;25:119–155. [PubMed] [Google Scholar]
- Hoebeke J. Structural basis of autoimmunity against G protein coupled membrane receptors. Int J Cardiol. 1996;54:103–111. doi: 10.1016/0167-5273(96)02586-7. [DOI] [PubMed] [Google Scholar]
- Hong CM, Zheng QS, Liu XT, Shang FJ, Wang HT, Jiang WR. Effects of autoantibodies against M2 muscarinic acetylcholine receptors on rabbit atria in vivo. Cardiology. 2009;112:180–187. doi: 10.1159/000149152. [DOI] [PubMed] [Google Scholar]
- Iwata M, Yoshikawa T, Baba A, Anzai T, Mitamura H, Ogawa S. Autoantibodies against the second extracellular loop of beta(1)-adrenergic receptors predict ventricular tachycardia and sudden death in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2001;37:418–424. doi: 10.1016/s0735-1097(00)01109-8. [DOI] [PubMed] [Google Scholar]
- Jahns R, Boivin V, Siegmund C, Boege F, Lohse MJ, Inselmann G. Activating beta-1-adrenoceptor antibodies are not associated with cardiomyopathies secondary to valvular or hypertensive heart disease. J Am Coll Cardiol. 1999a;34:1545–1551. doi: 10.1016/s0735-1097(99)00381-2. [DOI] [PubMed] [Google Scholar]
- Jahns R, Boivin V, Siegmund C, Inselmann G, Lohse MJ, Boege F. Autoantibodies activating human beta1-adrenergic receptors are associated with reduced cardiac function in chronic heart failure. Circulation. 1999b;99:649–654. doi: 10.1161/01.cir.99.5.649. [DOI] [PubMed] [Google Scholar]
- Jahns R, Boivin V, Krapf T, Wallukat G, Boege F, Lohse MJ. Modulation of beta(1)-adrenoceptor activity by domain-specific antibodies and-heart failure-associated autoantibodies. J Am Coll Cardiol. 2000;36:1280–1287. doi: 10.1016/s0735-1097(00)00881-0. [DOI] [PubMed] [Google Scholar]
- Jahns R, Boivin V, Hein L, Triebel S, Angermann CE, Ertl G, et al. Direct evidence for a beta(1)-adrenergic receptor-directed autoimmune attack as a cause of idiopathic dilated cardiomyopathy. J Clin Invest. 2004;113:1419–1429. doi: 10.1172/JCI20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahns R, Boivin V, Lohse MJ. Beta 1-adrenergic receptor-directed autoimmunity as a cause of dilated cardiomyopathy in rats. Int J Cardiol. 2006a;112:7–14. doi: 10.1016/j.ijcard.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Jahns R, Boivin V, Lohse MJ. beta(1)-adrenergic receptor function, autoimmunity, and pathogenesis of dilated cardiomyopathy. Trends Cardiovasc Med. 2006b;16:20–24. doi: 10.1016/j.tcm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Jahns R, Schlipp A, Boivin V, Lohse MJ. Targeting receptor antibodies in immune cardiomyopathy. Semin Thromb Hemost. 2010;36:212–218. doi: 10.1055/s-0030-1251506. [DOI] [PubMed] [Google Scholar]
- Jane-wit D, Altuntas CZ, Johnson JM, Yong S, Wickley PJ, Clark P, et al. Beta 1-adrenergic receptor autoantibodies mediate dilated cardiomyopathy by agonistically inducing cardiomyocyte apoptosis. Circulation. 2007;116:399–410. doi: 10.1161/CIRCULATIONAHA.106.683193. [DOI] [PubMed] [Google Scholar]
- Kallwellis-Opara A, Staudt A, Trimpert C, Noutsias M, Kuhl U, Pauschinger M, et al. Immunoadsorption and subsequent immunoglobulin substitution decreases myocardial gene expression of desmin in dilated cardiomyopathy. J Mol Med. 2007;85:1429–1435. doi: 10.1007/s00109-007-0263-5. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Ligand-selective receptor conformations revisited: the promise and the problem. Trends Pharmacol Sci. 2003;24:346–354. doi: 10.1016/S0165-6147(03)00167-6. [DOI] [PubMed] [Google Scholar]
- Kenakin T. G-protein coupled receptors as allosteric machines. Receptors Channels. 2004;10:51–60. doi: 10.1080/10606820490464316. [DOI] [PubMed] [Google Scholar]
- Labovsky V, Smulski CR, Gomez K, Levy G, Levin MJ. Anti-beta1-adrenergic receptor autoantibodies in patients with chronic Chagas heart disease. Clin Exp Immunol. 2007;148:440–449. doi: 10.1111/j.1365-2249.2007.03381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie C, Mercier JF, Salahpour A, Umapathy D, Breit A, Villeneuve LR, et al. Beta 1/beta 2-adrenergic receptor heterodimerization regulates beta 2-adrenergic receptor internalization and ERK signaling efficacy. J Biol Chem. 2002;277:35402–35410. doi: 10.1074/jbc.M204163200. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Levin MJ, Hoebeke J. Cross-talk between anti-beta1-adrenoceptor antibodies in dilated cardiomyopathy and Chagas' heart disease. Autoimmunity. 2008;41:429–433. doi: 10.1080/08916930802031702. [DOI] [PubMed] [Google Scholar]
- Liu HR, Zhao RR, Zhi JM, Wu BW, Fu ML. Screening of serum autoantibodies to cardiac beta1-adrenoceptors and M2-muscarinic acetylcholine receptors in 408 healthy subjects of varying ages. Autoimmunity. 1999;29:43–51. doi: 10.3109/08916939908995971. [DOI] [PubMed] [Google Scholar]
- Liu J, Mao W, Iwai C, Fukuoka S, Vulapalli R, Huang H, et al. Adoptive passive transfer of rabbit beta1-adrenoceptor peptide immune cardiomyopathy into the Rag2-/- mouse: participation of the ER stress. J Mol Cell Cardiol. 2008;44:304–314. doi: 10.1016/j.yjmcc.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Bergami P, Gomez KA, Levy GV, Grippo V, Baldi A, Levin MJ. The beta1 adrenergic effects of antibodies against the C-terminal end of the ribosomal P2beta protein of Trypanosoma cruzi associate with a specific pattern of epitope recognition. Clin Exp Immunol. 2005;142:140–147. doi: 10.1111/j.1365-2249.2005.02885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther HP, Homuth V, Wallukat G. Alpha 1-adrenergic receptor antibodies in patients with primary hypertension. Hypertension. 1997;29:678–682. doi: 10.1161/01.hyp.29.2.678. [DOI] [PubMed] [Google Scholar]
- Magnusson Y, Marullo S, Hoyer S, Waagstein F, Andersson B, Vahlne A, et al. Mapping of a functional autoimmune epitope on the beta 1-adrenergic receptor in patients with idiopathic dilated cardiomyopathy. J Clin Invest. 1990;86:1658–1663. doi: 10.1172/JCI114888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson Y, Wallukat G, Waagstein F, Hjalmarson A, Hoebeke J. Autoimmunity in idiopathic dilated cardiomyopathy. Characterization of antibodies against the beta 1-adrenoceptor with positive chronotropic effect. Circulation. 1994;89:2760–2767. doi: 10.1161/01.cir.89.6.2760. [DOI] [PubMed] [Google Scholar]
- Magnusson Y, Hjalmarson A, Hoebeke J. Beta 1-adrenoceptor autoimmunity in cardiomyopathy. Int J Cardiol. 1996;54:137–141. doi: 10.1016/0167-5273(96)02590-9. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- Matsui S, Fu ML, Shimizu M, Fukuoka T, Teraoka K, Takekoshi N, et al. Dilated cardiomyopathy defines serum autoantibodies against G-protein-coupled cardiovascular receptors. Autoimmunity. 1995;21:85–88. doi: 10.3109/08916939508993354. [DOI] [PubMed] [Google Scholar]
- Matsui S, Fu ML, Hayase M, Katsuda S, Yamaguchi N, Teraoka K, et al. Active immunization of combined beta1-adrenoceptor and M2-muscarinic receptor peptides induces cardiac hypertrophy in rabbits. J Card Fail. 1999;5:246–254. doi: 10.1016/s1071-9164(99)90009-x. [DOI] [PubMed] [Google Scholar]
- Matsui S, Persson M, Fu HM, Hayase M, Katsuda S, Teraoka K, et al. Protective effect of bisoprolol on beta-1 adrenoceptor peptide-induced autoimmune myocardial damage in rabbits. Herz. 2000;25:267–270. doi: 10.1007/s000590050018. [DOI] [PubMed] [Google Scholar]
- Matsui S, Fu M, Hayase M, Katsuda S, Yamaguchi N, Teraoka K, et al. Transfer of immune components from rabbit autoimmune cardiomyopathy into severe combined immunodeficiency (SCID) mice induces cardiomyopathic changes. Autoimmunity. 2006a;39:121–128. doi: 10.1080/08916930500314855. [DOI] [PubMed] [Google Scholar]
- Matsui S, Larsson L, Hayase M, Katsuda S, Teraoka K, Kurihara T, et al. Specific removal of beta 1-adrenoceptor autoantibodies by immunoabsorption in rabbits with autoimmune cardiomyopathy improved cardiac structure and function. J Mol Cell Cardiol. 2006b;41:78–85. doi: 10.1016/j.yjmcc.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Mijares A, Lebesgue D, Argibay J, Hoebeke J. Anti-peptide antibodies sensitive to the ‘active’ state of the beta2-adrenergic receptor. FEBS Lett. 1996a;399:188–191. doi: 10.1016/s0014-5793(96)01321-x. [DOI] [PubMed] [Google Scholar]
- Mijares A, Verdot L, Peineau N, Vray B, Hoebeke J, Argibay J. Antibodies from Trypanosoma cruzi infected mice recognize the second extracellular loop of the beta 1-adrenergic and M2-muscarinic receptors and regulate calcium channels in isolated cardiomyocytes. Mol Cell Biochem. 1996b;163–164:107–112. doi: 10.1007/BF00408646. [DOI] [PubMed] [Google Scholar]
- Milligan G. A day in the life of a G protein-coupled receptor: the contribution to function of G protein-coupled receptor dimerization. Br J Pharmacol. 2008;153(Suppl 1):S216–S229. doi: 10.1038/sj.bjp.0707490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobini R, Staudt A, Felix SB, Baumann G, Wallukat G, Deinum J, et al. Hemodynamic improvement and removal of autoantibodies against beta1-adrenergic receptor by immunoadsorption therapy in dilated cardiomyopathy. J Autoimm. 2003;20:345–350. doi: 10.1016/s0896-8411(03)00042-8. [DOI] [PubMed] [Google Scholar]
- Munoz-Saravia SG, Haberland A, Wallukat G, Schimke I. Chronic Chagas' heart disease: a disease on its way to becoming a worldwide health problem: epidemiology, etiopathology, treatment, pathogenesis and laboratory medicine. Heart Fail Rev. 2010;17:45–64. doi: 10.1007/s10741-010-9211-5. [DOI] [PubMed] [Google Scholar]
- Nagatomo Y, Yoshikawa T, Kohno T, Yoshizawa A, Baba A, Anzai T, et al. A pilot study on the role of autoantibody targeting the beta1-adrenergic receptor in the response to beta-blocker therapy for congestive heart failure. J Card Fail. 2009;15:224–232. doi: 10.1016/j.cardfail.2008.10.027. [DOI] [PubMed] [Google Scholar]
- Nikolaev VO, Boivin V, Stork S, Angermann CE, Ertl G, Lohse MJ, et al. A novel fluorescence method for the rapid detection of functional beta1-adrenergic receptor autoantibodies in heart failure. J Am Coll Cardiol. 2007;50:423–431. doi: 10.1016/j.jacc.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Omerovic E, Bollano E, Andersson B, Kujacic V, Schulze W, Hjalmarson A, et al. Induction of cardiomyopathy in severe combined immunodeficiency mice by transfer of lymphocytes from patients with idiopathic dilated cardiomyopathy. Autoimmunity. 2000;32:271–280. doi: 10.3109/08916930008994101. [DOI] [PubMed] [Google Scholar]
- Pisterzi LF, Jansma DB, Georgiou J, Woodside MJ, Chou JT, Angers S, et al. Oligomeric size of the m2 muscarinic receptor in live cells as determined by quantitative fluorescence resonance energy transfer. J Biol Chem. 2010;285:16723–16738. doi: 10.1074/jbc.M109.069443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose NR. Infection, mimics, and autoimmune disease. J Clin Invest. 2001;107:943–944. doi: 10.1172/JCI12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum MB, Chiale PA, Schejtman D, Levin M, Elizari MV. Antibodies to beta-adrenergic receptors disclosing agonist-like properties in idiopathic dilated cardiomyopathy and Chagas' heart disease. J Cardiovasc Electrophysiol. 1994;5:367–375. doi: 10.1111/j.1540-8167.1994.tb01174.x. [DOI] [PubMed] [Google Scholar]
- Silvina Lo Presti M, Walter Rivarola H, Bustamante JM, Fernandez AR, Enders JE, Levin G, et al. Some components of the cardiac beta-adrenergic system are altered in the chronic indeterminate form of experimental Trypanosoma cruzi infection. Int J Parasitol. 2008;38:1481–1492. doi: 10.1016/j.ijpara.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Smith NJ, Milligan G. Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol Rev. 2010;62:701–725. doi: 10.1124/pr.110.002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt A, Mobini R, Fu M, Grosse Y, Stangl V, Stangl K, et al. beta(1)-adrenoceptor antibodies induce positive inotropic response in isolated cardiomyocytes. Eur J Pharmacol. 2001;423:115–119. doi: 10.1016/s0014-2999(01)01113-x. [DOI] [PubMed] [Google Scholar]
- Staudt A, Eichler P, Trimpert C, Felix SB, Greinacher A. Fc(gamma) receptors IIa on cardiomyocytes and their potential functional relevance in dilated cardiomyopathy. J Am Coll Cardiol. 2007;49:1684–1692. doi: 10.1016/j.jacc.2006.11.051. [DOI] [PubMed] [Google Scholar]
- Staudt A, Herda LR, Trimpert C, Lubenow L, Landsberger M, Dorr M, et al. Fcgamma-receptor IIa polymorphism and the role of immunoadsorption in cardiac dysfunction in patients with dilated cardiomyopathy. Clin Pharmacol Ther. 2010;87:452–458. doi: 10.1038/clpt.2009.246. [DOI] [PubMed] [Google Scholar]
- Staudt Y, Mobini R, Fu M, Felix SB, Kuhn JP, Staudt A. beta(1)-Adrenoceptor antibodies induce apoptosis in adult isolated cardiomyocytes. Eur J Pharmacol. 2003;466:1–6. doi: 10.1016/s0014-2999(03)01431-6. [DOI] [PubMed] [Google Scholar]
- Stavrakis S, Yu X, Patterson E, Huang S, Hamlett SR, Chalmers L, et al. Activating autoantibodies to the beta-1 adrenergic and m2 muscarinic receptors facilitate atrial fibrillation in patients with Graves' hyperthyroidism. J Am Coll Cardiol. 2009;54:1309–1316. doi: 10.1016/j.jacc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrakis S, Kem DC, Patterson E, Lozano P, Huang S, Szabo B, et al. Opposing cardiac effects of autoantibody activation of beta-adrenergic and M2 muscarinic receptors in cardiac-related diseases. Int J Cardiol. 2011;148:331–336. doi: 10.1016/j.ijcard.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterin-Borda L, Cossio PM, Gimeno MF, Gimeno AL, Diez C, Laguens RP, et al. Effect of chagasic sera on the rat isolated atrial preparation: immunological, morphological and function aspects. Cardiovasc Res. 1976;10:613–622. doi: 10.1093/cvr/10.6.613. [DOI] [PubMed] [Google Scholar]
- Stork S, Boivin V, Horf R, Hein L, Lohse MJ, Angermann CE, et al. Stimulating autoantibodies directed against the cardiac beta1-adrenergic receptor predict increased mortality in idiopathic cardiomyopathy. Am Heart J. 2006;152:697–704. doi: 10.1016/j.ahj.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Swift SM, Schwarb MR, Mihlbachler KA, Liggett SB. Pleiotropic beta-agonist-promoted receptor conformations and signals independent of intrinsic activity. Am J Respir Cell Mol Biol. 2007;36:236–243. doi: 10.1165/rcmb.2006-0257OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol. 2002;2:580–592. doi: 10.1038/nri856. [DOI] [PubMed] [Google Scholar]
- Tate K, Magnusson Y, Viguier M, Lengagne R, Hjalmarson A, Guillet JG, et al. Epitope analysis of T- and B-cell response against the human beta 1-adrenoceptor. Biochimie. 1994;76:159–164. doi: 10.1016/0300-9084(94)90008-6. [DOI] [PubMed] [Google Scholar]
- Trimpert C, Herda LR, Eckerle LG, Pohle S, Muller C, Landsberger M, et al. Immunoadsorption in dilated cardiomyopathy: long-term reduction of cardiodepressant antibodies. Eur J Clin Invest. 2010;40:685–691. doi: 10.1111/j.1365-2362.2010.02314.x. [DOI] [PubMed] [Google Scholar]
- Tutor AS, Penela P, Mayor F., Jr Anti-beta1-adrenergic receptor autoantibodies are potent stimulators of the ERK1/2 pathway in cardiac cells. Cardiovasc Res. 2007;76:51–60. doi: 10.1016/j.cardiores.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Wallukat G, Nisson E. Anti beta(1)-adrenoceptor autoantibodies analyzed in spontaneously beating neonatal rat heart myocyte cultures – Comparison of methods. Cell Dev Biol. 2001;37:175–176. doi: 10.1290/1071-2690(2001)037<0175:aaaais>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Wallukat G, Wollenberger A, Morwinski R, Pitschner HF. Anti-beta 1-adrenoceptor autoantibodies with chronotropic activity from the serum of patients with dilated cardiomyopathy: mapping of epitopes in the first and second extracellular loops. J Mol Cell Cardiol. 1995;27:397–406. doi: 10.1016/s0022-2828(08)80036-3. [DOI] [PubMed] [Google Scholar]
- Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103:945–952. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallukat G, Muller J, Hetzer R. Specific removal of beta1-adrenergic autoantibodies from patients with idiopathic dilated cardiomyopathy. N Engl J Med. 2002;347:1806. doi: 10.1056/NEJM200211283472220. [DOI] [PubMed] [Google Scholar]
- Wallukat G, Munoz Saravia SG, Haberland A, Bartel S, Araujo R, Valda G, et al. Distinct patterns of autoantibodies against G-protein-coupled receptors in Chagas' cardiomyopathy and megacolon. Their potential impact for early risk assessment in asymptomatic Chagas' patients. J Am Coll Cardiol. 2010;55:463–468. doi: 10.1016/j.jacc.2009.06.064. [DOI] [PubMed] [Google Scholar]
- Wenzel K, Haase H, Wallukat G, Derer W, Bartel S, Homuth V, et al. Potential relevance of alpha(1)-adrenergic receptor autoantibodies in refractory hypertension. PLoS ONE. 2008;3:e3742. doi: 10.1371/journal.pone.0003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa T, Baba A, Nagatomo Y. Autoimmune mechanisms underlying dilated cardiomyopathy. Circ J. 2009;73:602–607. doi: 10.1253/circj.cj-08-1151. [DOI] [PubMed] [Google Scholar]
- Yu X, Patterson E, Stavrakis S, Huang S, De Aos I, Hamlett S, et al. Development of cardiomyopathy and atrial tachyarrhythmias associated with activating autoantibodies to beta-adrenergic and muscarinic receptors. J Am Soc Hypertens. 2009;3:133–140. doi: 10.1016/j.jash.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Hu D, Li J, Wu Y, Liu X, Yang X. Autoantibodies against the myocardial beta1-adrenergic and M2-muscarinic receptors in patients with congestive heart failure. Chin Med J (Engl) 2002;115:1127–1131. [PubMed] [Google Scholar]
- Zhou Z, Liao YH, Wei Y, Wei F, Wang B, Li L, et al. Cardiac remodeling after long-term stimulation by antibodies against the alpha1-adrenergic receptor in rats. Clin Immunol. 2005;114:164–173. doi: 10.1016/j.clim.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Zhu WZ, Chakir K, Zhang S, Yang D, Lavoie C, Bouvier M, et al. Heterodimerization of beta1- and beta2-adrenergic receptor subtypes optimizes beta-adrenergic modulation of cardiac contractility. Circ Res. 2005;97:244–251. doi: 10.1161/01.RES.0000176764.38934.86. [DOI] [PubMed] [Google Scholar]
- Zuo L, Bao H, Tian J, Wang X, Zhang S, He Z, et al. Long-term active immunization with a synthetic peptide corresponding to the second extracellular loop of beta(1)-adrenoceptor induces both morphological and functional cardiomyopathic changes in rats. Int J Cardiol. 2011;149:89–94. doi: 10.1016/j.ijcard.2009.12.023. [DOI] [PubMed] [Google Scholar]