

Abstract
A cofactor for HIV-1 (human immunodeficiency virus-type 1) fusion and entry was identified with the use of a novel functional complementary DNA (cDNA) cloning strategy. This protein, designated “fusin,” is a putative G protein–coupled receptor with seven transmembrane segments. Recombinant fusin enabled CD4-expressing nonhuman cell types to support HIV-1 Env-mediated cell fusion and HIV-1 infection. Antibodies to fusin blocked cell fusion and infection with normal CD4-positive human target cells. Fusin messenger RNA levels correlated with HIV-1 permissiveness in diverse human cell types. Fusin acted preferentially for T cell line–tropic isolates, in comparison to its activity with macrophage-tropic HIV-1 isolates.
The primary receptor for HIV-1, CD4, supports viral entry only when expressed on a human cell type (1–3). Experiments with CD4-expressing hybrids of nonhuman and human cells have revealed that the defect in nonhuman cells is due to the absence of a human-specific cofactor required for membrane fusion (4–10). This cofactor is essential both for entry of HIV-1 virions into CD4+ cell lines and for fusion between cells expressing the HIV-1 envelope glycoprotein (Env) and cells expressing CD4, Functional studies have suggested that the cofactor is present in a wide variety of human cell lines (1–3), though some exceptions have been noted (3,11). The identity of the fusion cofactor remains unresolved.
We previously reported a recombinant vaccinia virus–based transient expression and assay system in which fusion between Env-expressing and CD4-expressing cells leads to activation of a reporter gene (Escherichia coli lacZ) (12). We adapted this system for functional expression cloning of a fusion cofactor complementary DNA (cDNA) (13, 14) The approach made no assumptions about the mode of action of the cofactor, except that it can allow a CD4-expressing nonhuman cell type to undergo fusion (15)
NIH 3T3 cells expressing vaccinia-encoded T7 RNA polymerase and CD4 were transfected with a HeLa cDNA plasmid library (inserts linked to the T7 promoter). These cells were mixed with NIH 3T3 cells expressing vaccinia-encoded Env and containing the Escherichia coli lacZ gene linked to the T7 promoter. After incubation, the cultures were stained for (β-galactosidase (β-Gal) in situ. Consistently more (β-Gal–positive cells were observed with the CD4-expressing cells transfected with the entire library compared to control CD4-expressing cells transfected with a single random plasmid from the library. For example, in one experiment we detected an average of 76 cells per well with the library compared to 16 cells per well with the single plasmid. In an additional negative control, we observed only background numbers of stained cells with the library when the partner cells expressed a mutant uncleavable (Unc) Env rendered nonfusogenic by deletion of the gp120/gp41 cleavage cite. These results suggested that the library contained at least one cDNA encoding a fusion cofactor. After repeated subfractionation and screening, we isolated individual colonies on agar plates, and a single plasmid clone was identified that was capable of allowing the CD4-expressing NIH 3T3 cells to undergo fusion. The size of the cDNA insert was ~1.7 kb.
DNA sequence analysis was performed on both strands of the insert. The cDNA contained 1659 base pairs, and the longest open reading frame of the coding strand was 352 amino acids (16). Analysis of this sequence revealed that the protein is a member of the superfamily of G protein–coupled receptors with seven transmembrane segments. The nucleotide sequence of the open reading frame has been reported previously by several laboratories (17–21) investigating this receptor superfamily (22). Attempts by those researchers to identify a ligand have been unsuccessful, and the normal function of the putative receptor remains unknown. In particular, no relation to HIV has been suggested. Because of the role of this protein as an HIV-1 fusion cofactor, we propose that its name be “fusin.”
The cDNA insert was cloned (23) into plasmid pSC59 (24), which contains a strong synthetic vaccinia promoter supporting early and late transcription, flanked by sequences of the gene encoding vaccinia thymidine kinase. The resulting plasmid (pYF1-fusin) was used to generate a vaccinia recombinant (vCBYF1-fusin) from the parental WR strain by thymidine kinase selection (25). Expression of recombinant fusin was achieved either by transfection of pYF1-fusin into vaccinia-infected cells or by infection of cells with vCBYF1-fusin. Figure 1 shows a protein immunoblot analysis of cell extracts with rabbit polyclonal antisera raised against a synthetic peptide representing the predicted extracellular NH2-terminal region of fusin (26). With extracts from cells infected with vCBYF1-fusin, the immune serum detected a major protein species of ~46 kD, consistent with the deduced amino acid sequence (predicted relative molecular weight = 39,745) and the two potential N-linked glycosylation sites. This band was not observed in extracts from cells infected with control vaccinia virus WR or upon staining with pre-immune serum.
Fig. 1.
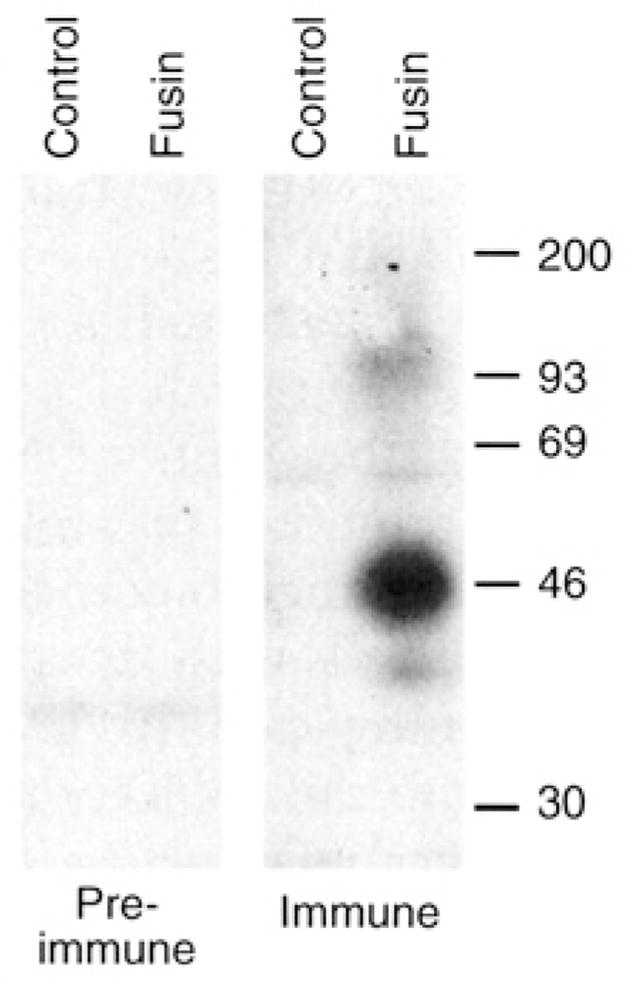
Protein immunoblot analysis of fusin produced by a recombinant vaccinia virus. BS-C-1 cells were infected with vCBYFI -fusin or with control virus WR (multiplicity of infection = 10). After overnight incubation at 37°C, the cells were washed twice with phosphate-buffered saline (PBS), pelleted, and lysed in buffer containing 1 % (v/v) Nonidet P-40,150 mM NaCl, and 10 mM tris (pH 7.4) plus protease inhibitors. The lysates were incubated 30 min at 4°C, then clarified by centrifugation at 10,000g for 5 min at 4°C. For protein immunoblot analysis, 10-μl aliquots of each lysate (representing ~5 × 104 cells) were mixed with 30 μl of 1 × reducing sample buffer supplemented with 8 M urea; the samples were incubated at 37°C overnight, then at 100°C for 3 min and subjected to SDS-polyacrylamide gel electrophoresis on 10% gels. Proteins were electrophoretically transferred to a nitrocellulose membrane, blocked in a solution containing 1% (w/v) bovine serum albumin and 0.15% (v/v) Tween-20, and incubated overnight with immune or pre-immune sera (1:200 dilution in blocking buffer). Bound antibodies were detected with [125I]Protein A (Amersham) and autoradiography. Size markers are shown on the right in kilodaltons.
Transient expression of recombinant fusin allowed nonhuman cells coexpressing recombinant CD4 to undergo fusion (Fig. 2). Murine NIH 3T3 cells expressing combinations of vaccinia-encoded proteins (CD4, fusin, or both) were mixed with HeLa cells expressing vaccinia-encoded HIV-1 Env (Lai, IIIB BH8). The NIH 3T3 cells also expressed vaccinia- encoded T7 RNA polymerase, and the Env-expressing cells contained the lacZ gene linked to the T7 promoter. NIH 3T3 cells expressing either CD4 alone or fusin alone did not permit fusion with cells expressing the wild-type (WT) Env, as judged by the absence of syncytia and the low background β-Gal levels. By contrast, cells coexpressing CD4 and fusin were highly permissive, as revealed by large syncytia and high β-Gal activity. Fusion was thus dependent on expression of both CD4 and fusin. The requirement for Env was demonstrated by the absence of fusion when the mutant Unc Env was used and by the strong fusion inhibition by monoclonal antibody D47, an antibody directed against the V3 loop of gp120 (27). The specificity of fusion conferred by fusin in the transient vaccinia system thus parallels that observed for HIV-1 infection.
Fig. 2.

Fusin permits nonhuman cells to undergo Env-CD4 –mediated cell fusion. Cell fusion mediated by vaccinia-encoded proteins was analyzed by syncytia formation and by the quantitative reporter gene activation assay (12). NIH 3T3 cells were co-infected with vTF7-3 (encoding T7 RNA polymerase) (49) plus the designated combinations of vCB-3 encoding CD4 (6) and vCBYFI-fusin; the multiplicity of infection (MOI) was 10 plaque-forming units per cell for each virus. Where appropriate, the cells were also co-infected with wild-type vaccinia virus WR to keep a constant total MOI. As the fusion partners, HeLa cells were co-infected with vCB21 R-LacZ (containing the lacZ gene linked to the T 7 promoter) (35) plus either vSC60 encoding wild-type HIV-1 Env (24) or vCB-16 encoding the mutant Unc Env (34) (both derived from the Lai, IIIB BH8 isolate). Cells were incubated in suspension overnight at 31°C to allow production of recombinant proteins. The cells were washed, and the indicated pairwise combinations were mixed in two, 96-well flat-bottom microtiter plates (1 × 105 cells per well for each cell type; total volume = 0.2 ml in culture medium plus 40 μg/ml of cytosine arabinoside). After incubation for 3.5 hours at 37°C, samples on one plate were fixed, stained with crystal violet, examined microscopically for syncytia, and photographed. Samples (duplicates) on the second plate were treated with Nonidet P-40 (0.5% final concentration), and 50-μl aliquots of each sample were mixed with 50 μl of 2× substrate solution (chlorphenol red- β-D-galactopyranoside) in 96-well plates. We monitored the rates of substrate hydrolysis at ambient temperature by measuring absorbance at 570 nm using a microplate absorbance reader (Molecular Dynamics). Values (insets) represent absorbance/min multiplied by 103 (mean of duplicate samples).
We extended this analysis to diverse cell types known to be nonpermissive for fusion even when expressing CD4 (Table 1). These include several nonhuman cell lines as well as unusual human cell lines known to not allow fusion when expressing CD4. In a positive control, we examined HeLa cells, which support fusion. Populations of each cell type were prepared expressing vaccinia-encoded CD4, without or with vaccinia-encoded fusin. These cells were mixed with cells expressing HIV-1 Env (WT or Unc), and fusion was scored by quantitation of β-Gal in detergent cell lysates. In the absence of recombinant fusin, all the CD4-expressing nonpermissive cell types failed to undergo fusion, consistent with the phenotypes reported when other methods were used (1 –3, 11). Coexpression of fusin along with CD4 enabled all the cell types to fuse with cells expressing WT Env; as expected, no fusion was observed with the control Unc Env. The fusion efficiencies of nonhuman cells expressing recombinant fusin were comparable to that observed with the highly permissive CD4-expressing HeLa cells that express fusin endogenously; recombinant fusin expression produced some fusion enhancement with HeLa cells as well.
Table 1.
Cell fusion–promoting activity of fusin-1 for different CD4-expressing cell types.
| Cell type | Species | Fusin | WT | Unc |
|---|---|---|---|---|
| NIH 3T3 | Murine | − | 1 | |
| + | 49 (0.5) | 1 | ||
| BS-C-1 | African green monkey | − | 1 | |
| + | 60 (0.5) | 1 | ||
| MV 1 Lu | Mink | − | 2 | |
| + | 286 (3.0) | 2 | ||
| U-87 MG | Human | − | 1 | |
| + | 67 (1.0) | 1 | ||
| SCL1 | Human | − | 0 | |
| + | 17 (0.1) | 0 | ||
| HeLa | Human | − | 124 (3.0) | |
| + | 186 (1.0) | 1 |
The cell lines examined were as follows: NIH 3T3, murine embryo; BS-C-1, African green monkey primary kidney; Mv 1 Lu, mink fetal lung; U-87 MG, human astroglioma; SCL1, human squamous cell carcinoma; and HeLa, human cervical carcinoma. Each cell type was infected with vCB-3 encoding CD4 and cotransfected with plasmid pG1NT7β-Gal (containing the lacZ gene linked to the T7 promoter) plus either pYF1-fusin (+) or control plasmid pSC59 (−). NIH 3T3 cells were co-infected with vP11gene1 (encoding T7 RNA polymerase), plus either vSC60 or vCB-16 encoding wild-type (WT) or mutant (Unc) Envs, respectively. Cell mixtures (in duplicate) were prepared in flat-bottom 96-well plates. Each well contained the indicated combinations of CD4-expressing and Env-expressing cells (1 × 105 cells of each type in 0.2 ml of culture medium plus 40 μg/ml of cytosine arabinoside). Plates were incubated 3 hours at 37°C, and β-Gal activity was measured as de scribed in Fig. 2. Values represent absorbance/ min multiplied by 103; numbers in parentheses indicate the sample standard deviations of duplicate samples.
As a second approach for testing the functional activity of recombinant fusin, we selected fusin transformants of a CD4-expressing nonhuman cell type and tested them for susceptibility to productive HIV-1 infection (28). We started with a neomycin-resistant CD4+ transformant of the mink Mv 1 Lu cell line, as this transformant is refractory to HIV-1 entry yet allows the post-entry steps in the replication cycle (3); furthermore, CD4-expressing Mv 1 Lu cells could be rendered competent for fusion by transient expression of fusin (Table 1).
We transfected the CD4+ Mv 1 Lu transformant with a plasmid vector containing the fusin cDNA linked to the SV40 promoter (as well as a gene conferring resistance to Zeocin); we transfected control cells with the same plasmid vector containing an irrelevant gene (lacZ) linked to the SV40 promoter. After selection in the presence of both G418 and Zeocin, individual colonies were tested for fusion with cells expressing Env by the vaccinia expression–assay system (using luciferase as the reporter); all colonies were positive (29). The three colonies with the best fusion activity were examined for susceptibility to HIV-1 infection (Fig. 3). All supported productive infection, whereas the control lacZ/CD4 transformant did not. Additional experiments indicated that p24 production in the fusin-CD4 cells was strongly inhibited by inclusion of 2.5 μM azidothymidine, which thereby verified that p24 production represented true productive infection (29). The fusin-CD4 mink cell transformants were sufficiently susceptible to HIV-1 infection to enable direct p24 measurement without the need for cocultivation with permissive human target cells. However, the infection efficiency for the mink cell transformants was considerably less than that for normal human CD4+ T cells, which is consistent with findings that fusin mRNA levels were much lower than in permissive human cells (29).
Fig. 3.
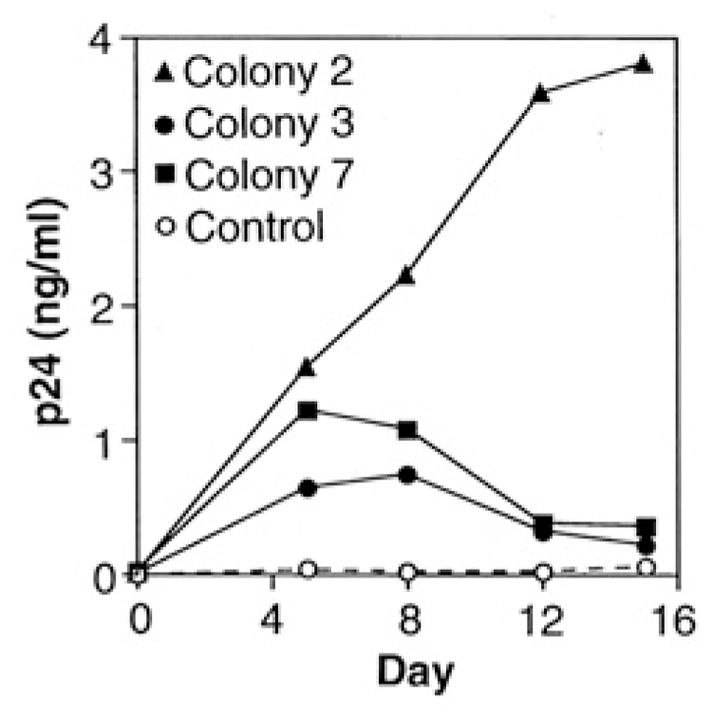
Fusin renders CD4+ nonhuman cells susceptible to productive HIV-1 infection. Transformants (filled symbols, fusin; open symbols, the control lacZ) of the CD4+ Mv 1 Lu cell line were seeded as monolayers in 24-well tissue culture plates (50,000 cells per well). To each well, 1000 median tissue culture infectious doses (TCID50) of HIV-1 (Lai. LAV isolate) were added, and the plates were incubated 4.5 hours at 37°C. The wells were washed four times with PBS, and 1.2 ml of culture medium supplemented with G418 was added. An aliquot (0.2 ml) was removed immediately as a zero time point; at the indicated times, aliquots (0.25 ml) were removed and replaced with the same volume of fresh media. Samples were assayed for p24 content with an enzyme-linked immunosorbent assay kit (DuPont).
We emphasize that fusin supports HIV-1 infection only in cells coexpressing CD4. This conclusion is based on studies with several human cell lines found to express fusin, such as HeLa (Fig. 4) (20), Raji, and Daudi (18). Each of these CD4-negative cell types does not allow HIV-1 infection, whereas the corresponding CD4+ transformants do (1, 3). Thus, as shown above in the cell fusion assay, fusin promotes HIV-1 infection by serving as a cofactor in CD4+ cells.
Fig. 4.
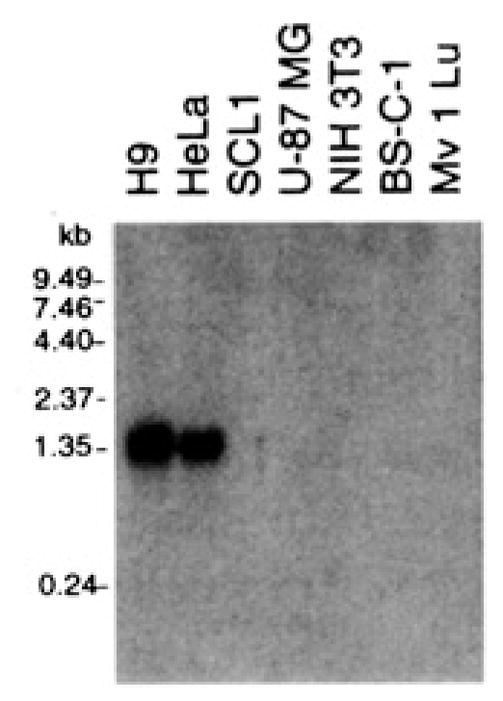
Northern (RNA) blot analysis of fusin mRNA in different cell types. Polyadenylated RNA was prepared from the indicated cell types with the Micro-Fast Track Kit (Invitrogen). Samples (1 μg of RNA) were electrophoresed through a 1 % argarose-formaldehyde gel and transferred to a nitrocellulose membrane. A [α-32P]guanosine triphosphate–labeled hybridization probe was prepared from the full-length fusin cDNA template by random primer synthesis. Hybridization was carried out with QuikHyb (Promega). The membrane was washed twice with 2% saline sodium citrate (SSC) and 0.1% SDS at room temperature for 30 min and once with 0.1 % SSC and 0.1% SDS at 65°C for 1 hour. Labeled bands were detected by autoradiography.
The ability of recombinant fusin to allow various nonhuman and human cell types to undergo fusion suggests that their fusion defects are due to the absence of functional fusin. The antibodies to fusin obtained thus far have not been suitable for detection of surface fusin by flow cytometry. Therefore, we examined various cell lines by Northern (RNA) blot analysis to test for a correlation between fusin mRNA levels and susceptibility to HIV-1 (Fig. 4). With permissive human cell types (HeLa and the H9 T cell line), a prominent RNA band at ~1.7 kb was detected; the size of this transcript is consistent with the length of the cDNA insert and is similar to that previously reported for various human cell types (18–21). By contrast, this transcript was not detected in the human cell types U-87 MG and SCL1, which do not permit fusion, nor was it observed in any of the nonhuman cell types. Other researchers have also detected this transcript in various HIV-1-permissive human cell types, including peripheral blood mononuclear cells (PBMCs) (20, 21) and CD4+ cell lines such as Jurkat T lymphocytes (20, 21), THP-1 (20) and U937 (21) promonocytes, and HL-60 promyelocytes (18, 20, 21).
Individual HIV-1 isolates vary greatly in their cytotropisms for infection of diverse CD4+ cell types. Some isolates (T cell line–tropic) efficiently infect CD4+ continuous cell lines (for example, T cell lines and HeLa-CD4 transformants), but only weakly infect primary macrophages; other isolates (macrophage-tropic) show the opposite selectivity. Both types of isolates readily infect PBMCs. These distinct cytotropisms, though not absolute (30), have major implications for HIV-1 pathogenesis and transmission between individuals (31–33). The viral determinants for this infection tropism have been mapped to the gp120 subunit of Env (31). Recently, we demonstrated a marked correlation between the infection cytotropisms of different HIV-1 isolates and the fusion specificities of the corresponding recombinant Envs (34); moreover, we obtained evidence that cytotropism is associated with distinct, cell type–specific fusion cofactors (35). We predict that fusin should function preferentially for Envs from T cell line–tropic isolates, because it was identified by screening a cDNA library from a human continuous cell line (HeLa) for the ability to allow a T cell line–tropic Env (Lai, IIIB) to undergo fusion. Indeed, when NIH 3T3 cells coexpressing recombinant CD4 and fusin were tested for fusion with cells expressing Envs from HIV-1 isolates with distinct cytotropisms, fusin showed preferential activity with Envs from T cell line–tropic isolates compared to its activity with macrophage-tropic isolates (Fig. 5).
Fig. 5.
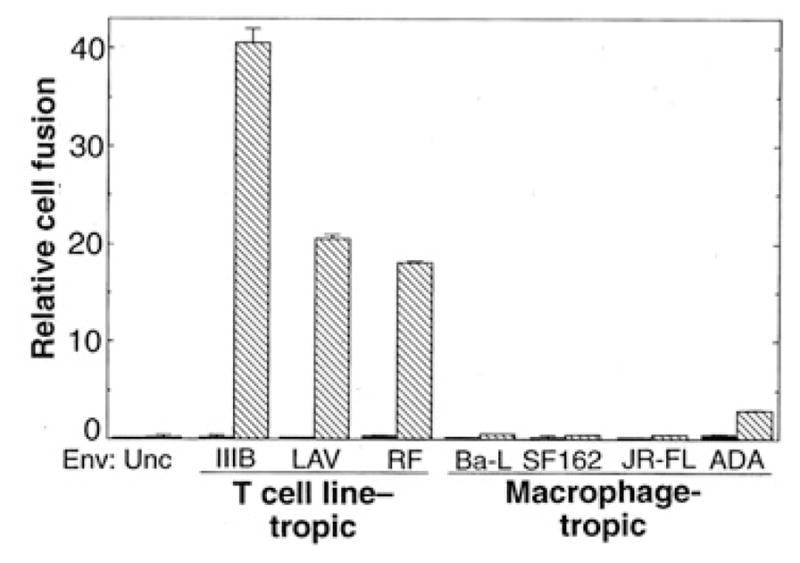
Fusin activity for Envs from T cell line–tropic versus macrophage-tropic HIV-1 isolates. One population of NIH 3T3 cells was cotransfected with pG1NT7β-Gal [containing the lacZgene linked to the T 7 promoter (50)] plus either pYF1 -fusin (cross-hatched bars) or control plasmid pSC59 (24) (filled bars); the cells were then infected with vCB-3 encoding CD4. Separate populations of NIH 3T3 cells were co-infected with vP11gene1 (encoding T7 RNA polymerase) (51) plus vaccinia recombinants encoding each of the indicated Envs. Previous results documented comparable expression of each vaccinia-encoded Env with the use of flow cytometry and radioimmunoprecipitation analyses (34). Cell mixtures were incubated at 37°C for 3 hours, and fusion was scored as described in Fig. 2. Each value represents absorbance/min multiplied by 103 (mean of duplicate samples; error bars indicate sample standard deviations).
Our finding that recombinant fusin allowed HIV-1 Env-CD4 –mediated fusion and infection prompted us to test whether fusin is required for HIV-1 infection of normal human CD4+ target cells. We therefore examined the effects of antibodies to fusin (anti-NH2-terminal peptide) on Env-mediated cell fusion and HIV-1 infection (Fig. 6). LAV and Ba-L served as prototypes for T cell line–tropic and macrophage-tropic isolates, respectively; PBMCs were used as the CD4+ cell type. Fusion mediated by the LAV Env was strongly inhibited by antibodies purified from the immune, but not the pre-immune, sera; by contrast, fusion mediated by the Ba-L Env was unaffected (Fig. 6A). Infection by LAV was strongly inhibited by antibodies from the immune, but not the pre-immune, serum, whereas infection by Ba-L was unaffected (Fig. 6B). The antibody resistance of Ba-L fusion and infection verifies that the inhibitory effects observed with LAV were not due to non-specific impairment of cell function. We conclude that for normal human CD4+ target cells distinct from those used to isolate fusin cDNA, fusin is involved in both Env-CD4– mediated cell fusion and HIV-1 infection. Moreover, our results provide strong additional evidence that fusin functions preferentially for T cell line–Tropic compared to macrophage-tropic isolates.
Fig. 6.

Inhibitory activities of antibodies to fusin on HIV-1 Env-mediated fusion and HIV-1 infection. Human PBMCs prepared from healthy donors were stimulated with phytohemagglutinin (M) for 3 to 4 days in RPMI 1640 medium supplemented with 10% fetal bovine serum. The effects of purified rabbit polyclonal antibodies to a synthetic peptide representing the NH2-terminus of fusin were examined (immune, filled squares; pre-immune, open circles). (A) Cell fusion, PBMCs were infected with vCB21R-LacZ (containing the lacZ gene linked to the T7 promoter). BS-C-1 cells were co-infected in the presence of 40 μg/ml of cytosine arabinoside with vTF7-3 (encoding T7 RNA polymerase) and vaccinia recombinants encoding the Env from either LAV (left) or Ba- L (right); the Unc Env was used as a background control. After overnight incubation at 31 °C, the cells were washed and resuspended. Duplicate aliquots of PBMCs in 96-well plates (1 × 105 cells per well in 70 μl of culture medium) were pre-incubated for 1 hour at room temperature with 10 μl of a dilution series of the indicated purified antibodies. Fusion reactions were initiated by addition of the Env expressing cells (1 × 105 cells per well in 20 μl of culture medium) The indicated antibody concentrations are the final concentrations used during the assay. The cultures were incubated at 37°C for 2.5 hours, and the β-Gal levels in detergent lysates were measured as described in Fig. 2. Results for each Env are expressed as the percent β-Gal activity observed at each antibody concentration compared to the control activity with no antibody (after subtraction of the low background level observed with the Unc Env). Error bars indicate sample standard deviations. (B) HIV-1 infection. PBMCs (1 × 106 cells/ml) were pre-incubated with the indicated concentrations of purified antibodies. HIV-1 LAV (left) or Ba-L (right) was added (70 TCID50 per milliliter) along with interleukin-2 (20 U/ml), and 100-μl aliquots of infected cells were dispensed into triplicate wells in 96-well plates. Aliquots of supernatant were assayed at day 7 for p24 content by enzyme- linked immunosorbent assay. Results for each isolate are expressed as the percentage p24 produced at each antibody concentration compared to the control value with no antibody (4.0 ng/ml for LAV; 3.6 ng/ml for Ba-L). Error bars indicate sample standard deviations.
The identification of fusin as a fusion cofactor for T cell line–tropic HIV-1 isolates provides a new focus to elucidate the mechanism of HIV-1 entry into target cells. The simplest model is that fusin acts directly as a coreceptor in the fusion process. This could occur by interaction either with Env (perhaps via the V3 loop) or with CD4; multiple and possibly sequential interactions can be readily imagined. Alternatively, the role of fusin in entry might be indirect, possibly involving G protein signaling.
The preferential activity of fusin for T cell line–tropic HIV-1 isolates provides direct support for the model that T cell line versus macrophage trophism is due to distinct fusion cofactors required by each type of Env (34, 35). Identification of the fusion cofactor (or cofactors) for the macrophage-tropic isolates is thus critical. It is also important to emphasize that most primary HIV-1 isolates have some dual tropic character (30), perhaps reflective of their ability to function with more than one fusion cofactor. We also predict that distinct fusion cofactors exist for HIV-2 and simian immunodeficiency virus (SIV), as the entry requirements of these viruses for various CD4+ cells are clearly different from those of HIV-1 (3, 9, 36–38). It is thus reasonable to propose the existence of a family of fusin molecules, perhaps also members of the G protein–coupled receptor superfamily with seven transmembrane segments. Interestingly, other members of this superfamily have been exploited for target cell entry by diverse human pathogens. Examples include the Duffy antigen, which mediates erythrocyte invasion by the malarial parasite Plasmodium vivax (39), and the platelet-activating factor receptor, which mediates adhesion of Staphlococcus pneumoniae to endothelial and epithelial cells (40).
The identification of fusin suggests an immediate practical utility, namely in the production of a small animal model for the study of HIV-1 infection. Transgenic mice (41–43) and rabbits (44, 45) expressing human CD4 have been generated, but they support productive HIV-1 replication inefficiently at best. The block in mice will be difficult to overcome because the restrictions are multifaceted and not limited to viral entry; in particular, the accessory proteins tat and rev do not function well in murine cells (46). The rabbit model is more promising, because rabbit cells allow post-entry steps; specifically, they support the functions of tat, rev, and the HIV-1 long-terminal repeat (47). Of course, alternative species might be considered for transgenic small animal models.
The normal function and natural ligand for fusin are presently unknown. The inability to detect reactivity with diverse ligands (17–21) led to the description of the protein as an “orphan receptor” (20, 39). On the basis of the wide tissue and cell type distribution of the mRNA, it has been proposed that the natural ligand may have pleiotropic modulatory effects on cells of the immune and neuroendocrine systems (19–21). Particularly intriguing is the fact that among human proteins with known activities, the greatest homology (37% amino acid identity) is with a receptor for interleukin-8, a chemokine of the C-X-C class. The gene for fusin has been localized to chromosome 2 (17, 18), in the vicinity of the genes for other C-X-C chemokine receptors (39). These findings, coupled with other consensus structural features (39), have led to the suggestion that this protein is a member of the chemokine receptor subfamily within the G protein–coupled receptor superfamily (20, 21, 39). Chemokines might therefore be considered in experimental approaches for identifying the natural ligand for fusin, though the focus should not be restricted to this family of molecules.
Additional interest in this comes from the fact that the C-C chemokines RANTES, MIP-1-α, and MIP-1β suppress HIV-1 infection in vitro (48). Although the mechanisms for this are unknown, our results suggest the hypothesis that they act by binding to a receptor that also serves as a fusion cofactor. In contrast to antibodies to fusin, the C-C chemokines inhibited the macrophage-tropic Ba-L isolate much more than the T cell line–tropic Lai IIIB isolate (48). This may be a clue to the identity of the fusion cofactor used by the macrophage-tropic isolates. Resolution of these questions awaits identification of the corresponding surface molecules.
Finally, the interaction of HIV-1 with a putative G protein–coupled receptor raises interesting questions about pathogenic mechanisms. T cell line–tropic isolates tend to appear in infected individuals at the later stages of the infection, during the transition from the asymptomatic to the symptomatic state, coincident with the decline of CD4+ T lymphocytes (31, 32). Beyond the obvious importance for membrane fusion, Env interactions with CD4-fusin might trigger aberrant G protein–mediated signaling pathways that contribute to CD4+ T lymphocyte depletion, directly or indirectly.
Acknowledgments
Y.F. is a recipient of a National Research Council Research Associate fellowship, and C.C.B. is a recipient of an NIH intramural Research Training Award. Support was provided by the NIH Intramural AIDS Targeted Antiviral Program. We are grateful to P. Clapham and R. Weiss, Chester Beatty Laboratories, Institute for Cancer Research, London, for donation of the CD4+ transformant of Mv 1 Lu; to M. Garfield. J. Lukszo, and J. Coligan, Laboratory of Molecular Structure, NIAID, for synthetic peptide synthesis, purification, and coupling; and to J. Sisler. Laboratory of Viral Diseases, NIAID, for oligonucleotide synthesis and assistance with DNA sequencing. We thank P. Murphy, NIAID, and B. Moss, NIAID, for helpful discussions and comments on the manuscript.
REFERENCES AND NOTES
- 1.Maddon PJ, et al. Cell. 1986;47:333. doi: 10.1016/0092-8674(86)90590-8. [DOI] [PubMed] [Google Scholar]
- 2.Ashom PA, Berger EA, Moss B. J Virol. 1990;64:2149. doi: 10.1128/jvi.64.5.2149-2156.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clapham PR, Blanc D, Weiss RA. Virology. 1991;181:703. doi: 10.1016/0042-6822(91)90904-P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiner DB, Huebner K, Williams WV, Greene MI. Pathobiology. 1991;59:361. doi: 10.1159/000163679. [DOI] [PubMed] [Google Scholar]
- 5.Dragic T, Charneau P, Clavel F, Alizon M. J Virol. 1992;66:4794. doi: 10.1128/jvi.66.8.4794-4802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broder CC, Dimitrov DS, Blumenthal R, Berger EA. Virology. 1993;193:483. doi: 10.1006/viro.1993.1151. [DOI] [PubMed] [Google Scholar]
- 7.Harrington RD, Geballe AP. J Virol. 1993;67:5939. doi: 10.1128/jvi.67.10.5939-5947.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramarli D, et al. AIDS Res Hum Retroviruses. 1993;9:1269. doi: 10.1089/aid.1993.9.1269. [DOI] [PubMed] [Google Scholar]
- 9.Dragic T, Alizon M. J Virol. 1993;67:2355. doi: 10.1128/jvi.67.4.2355-2359.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dragic T, Picard L, Alizon M. ibid. 1995;69:1013. doi: 10.1128/jvi.69.2.1013-1018.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chesebro B, Buller R, Portis J, Wehrly K. ibid. 1990;64:215. doi: 10.1128/jvi.64.1.215-221.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nussbaum O, Broder CC, Berger EA. ibid. 1994;68:5411. doi: 10.1128/jvi.68.9.5411-5422.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger EA, Broder CC, Alkhatib G, Feng Y. Keystone Symposium. Cell Biology of Virus Entry, Replication and Pathogenesis. 1996;8:abstract 002. [Google Scholar]
- 14.Feng Y, Broder CC, Kennedy PE, Berger EA. Keystone Symposium: Immunopathogenesis of HIV Infection. 1996;21:abstract 116. [Google Scholar]
- 15.A HeLa cell cDNA library cloned unidirectionally into the plasmid vector pcDNA3 with the cDNAs linked to the T7 promoter was purchased from invitrogen; the library was derived from 4 × 106 clones. For functional expression cloning, an NIH 3T3 cell monolayer (2 × 106 cells) was cotransfected overnight with plasmid pTF7-3 (which contains the T7 RNA polymerase gene linked to a vaccinia promoter) (49) plus from the library cDNA (10 μg of each DNA); as a negative control, a parallel monolayer was cotransfected with pTF7-3 plus a random single plasmid obtained from the library. The transfected cells were then infected with vCB-3 encoding CD4 (6). The idea was that if the library contained a cDNA encoding the cofactor, a small number of the CD4-expressing murine cells would be allowed to undergo fusion; this would not occur with cells transfected with the single plasmid instead of the library. A separate population of NIH 3T3 cells was co-infected with vCB21R-LacZ, which contains the lacZ gene linked to the T 7 promoter (35) plus vSC60 encoding wild type HIV-1 Env (IIIB BH8) (24); as an additional negative control, cells were infected with vCB-16 encoding the mutant Unc Env (34). At 1,5 hours after infection, cells were detached by trypsinization and incubated in suspension overnight at 31°C to allow production of recombinant proteins (including those encoded by the library). In 96-well flat-bottom plates, replicate mixtures were prepared containing CD4-expressing and Env-expressing cells (1 × 105 of each cell type per well) and incubated 3 hours at 37°C. β-Gal was detected by in situ staining with X-Gal. Because more stained cells were consistently observed with the total library compared to the single plasmid control, the library was subdivided into 10G0 fractions (~4000 plasmids each), and pools of the fractions were screened in a similar fashion until a single positive fraction was identified. This fraction was further subdivided into 1000 subtractions (~4 plasmids each) and screened in the same way. A positive subtraction was identified and plated onto agar, and individual colonies were picked and assayed. A single positive clone was identified (designated pcDNA3-fusin).
- 16.There are five potential open reading frames in the cDNA sequence; the longest one encoding fusin starts with the first ATG, which has a surrounding sequence consistent with efficient translation initiation in vertebrate cells. To rule out the possibility that the functional activities might be encoded by the other open reading frames. we subcloned into pcDNA3 a cDNA fragment containing only the remaining four downstream open reading frames. Unlike the full length cDNA, the truncated fragment did not allow CD4-expressing NIH 3T3 cells to undergo fusion.
- 17.Herzog H, Hort YJ, Shine J, Selbie LA. DNA Cell Biol. 1993;12:465. doi: 10.1089/dna.1993.12.465. [DOI] [PubMed] [Google Scholar]
- 18.Federsppiel B, et al. Genomics. 1993;16:707. doi: 10.1006/geno.1993.1251. [DOI] [PubMed] [Google Scholar]
- 19.Jazin EE, et al. Regul Pept. 1993;47:247. doi: 10.1016/0167-0115(93)90392-l. [DOI] [PubMed] [Google Scholar]
- 20.Nomura H, Nielsen BW, Matsushima K. Int Immunol. 1993;5:1239. doi: 10.1093/intimm/5.10.1239. [DOI] [PubMed] [Google Scholar]
- 21.Loetscherer M, et al. J Biol Chem. 1994;269:232. [Google Scholar]
- 22.Compared to the nucleotide sequence reported for the cDNA (21), there is one difference in the 3′ untranslated region: whereas that sequence has eight consecutive T residues beginning at nucleotide 1076, the fusin sequence has seven.
- 23.Plasmid pcDNA3-fusin was digested with Xho I and blunt ended with the Klenow fragment of E. coli DNA polymerase. The fusin cDNA insert was excised by digestion with Eco RI and ligated into Stu I-Eco RI-digested pSC59 (24). The resulting plasmid, designated pYF1-fusin, was used to generate vaccinia recombinant vCBYF1-fusin.
- 24.Chakrabarti S, Moss B. personal communication. National Institute of Allergy and Infectious Diseases; [Google Scholar]
- 25.Earl PL, Cooper N, Moss B. In: Current Protocols in Molecular Biology. suppl 15. Ausubel FM, et al., editors. Vol. 2. Wiley; New York: 1991. p. 16.15.1. [Google Scholar]
- 26.A synthetic peptide corresponding to the predicted extracellular NH2 terminus of fusin (MEGISIYTSDNYTEEMGSGDYDSMKEPCFREENANFNK; abbreviations for the amino acid residues are as follows; A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S. Ser; T, Thr; V. Val; W. Trp; and Y. Tyr) was synthesized by standard FMOC chemistry, purified by reversed-phase high-performance liquid chromatography (HPLC), and analyzed by analytical HPLC, mass spectrometry, and amino acid sequencing. The purified peptide was conjugated to keyhole limpet hemocyanin by means of the Cys residue with the use of m-maleimidobenzoyl-N-hydroxysuccinimide ester (108 peptides per carrier molecule). New Zealand White rabbits were immunized with 200 μg of conjugate emulsified in RiBi adjuvant (RiBi, Hamilton, MT) three times at 28-day intervals. For preparation of purified immunoglobulin G, the serum was subjected to affinity chromatography on protein G sepharose.
- 27.Richardson TM, et al. J Virol. 1996;70:753. doi: 10.1128/jvi.70.2.753-762.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The fusin cDNA insert was excised from pCDNA3-fusin with Hind III and Xho I and ligated to Hind III-Xho I-digested pZeoSV (Invitrogen). The resulting plasmid (pZeoSV-fusin) contained the fusin cDNA linked to the SV40 promoter, as well as the Zeocin resistance gene. A monolayer of the CD4+ transformant of Mv 1 Lu (also containing the neomycin resistance gene) (3) was transfected with pZeoSV-fusin (with the use of DOTAP; Boeringer Mannheim); as a negative control, a monolayer of the same cells was transfected with pZeoSV-LacZ (Invitrogen) containing the lacZ gene linked to the SV40 promoter plus the Zeocin resistance gene. After 24 hours, the cells were washed and cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum plus antibiotics (during the first 2 days with 1 mg/ml of G418 alone; subsequently, with G418 plus 1 mg/ml of zeocin). Colonies resistant to both antibiotics were picked, amplified, and screened for Env-dependent fusion permissiveness in the vaccinia assay system (with the use of luciferase as the reporter).
- 29.Feng Y, et al. data not shown. [Google Scholar]
- 30.Valentin A, Albert J, Fenyö EM, Asjö B. J Virol. 1994;68:6684. doi: 10.1128/jvi.68.10.6684-6689.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Planelles V, Li Q-X, Chen ISY. In: Human Retroviruses. Cullen BR, editor. Oxford Univ. Press; New York: 1993. p. 17. [Google Scholar]
- 32.Miedema F, et al. Immunol, Rev. 1994;140:35. doi: 10.1111/j.1600-065x.1994.tb00864.x. [DOI] [PubMed] [Google Scholar]
- 33.Connor RI, Ho DD. AIDS Res Hum Retroviruses. 1994;10:321. doi: 10.1089/aid.1994.10.321. [DOI] [PubMed] [Google Scholar]
- 34.Broder CC, Berger EA. Proc Natl Acad Sci USA. 1995;92:9004. doi: 10.1073/pnas.92.19.9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alkhatib G, Broder CC, Berger EA. J Virol. in press. [Google Scholar]
- 36.Koenig S, et al. Proc Natl Acad- Sci USA. 1989;86:2443. doi: 10.1073/pnas.86.7.2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoxie JA, et al. J Virol. 1988;62:2557. doi: 10.1128/jvi.62.8.2557-2568.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKnight A, Clapham PR, Weiss RA. Virology. 1994;201:8. doi: 10.1006/viro.1994.1260. [DOI] [PubMed] [Google Scholar]
- 39.Murphy PM. Annu Rev Immunol. 1994;12:593. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 40.Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. Nature. 1995;377:435. doi: 10.1038/377435a0. [DOI] [PubMed] [Google Scholar]
- 41.Gillespie FP, et al. Mol Cell Biol. 1993;13:2952. doi: 10.1128/mcb.13.5.2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Killeen N, Sawada S, Littman DR. EMBO J. 1993;12:1547. doi: 10.1002/j.1460-2075.1993.tb05798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanna Z, Simard C, Laperriere A, Jolicoeur P. Mol Cell Biol. 1994;14:1084. doi: 10.1128/mcb.14.2.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snyder BW, et al. Mol Reprod Dev. 1995;40:419. doi: 10.1002/mrd.1080400405. [DOI] [PubMed] [Google Scholar]
- 45.Dunn CS, et al. J Gen Virol. 1995;76:1327. doi: 10.1099/0022-1317-76-6-1327. [DOI] [PubMed] [Google Scholar]
- 46.Trono D. Clin Lab Med. 1994;14:203. [PubMed] [Google Scholar]
- 47.Cho S, Kindt TJ, Zhao TM, Sawasdikosol S, Hague BF. AIDS Res Ilum Retroviruses. 1995;11:1487. doi: 10.1089/aid.1995.11.1487. [DOI] [PubMed] [Google Scholar]
- 48.Cocchi F, et al. Science. 1995;270:1811. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 49.Fuerst TR, Niles EG, Studier FW, Moss B. Proc Natl Acad Sci USA. 1986;83:8122. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morgan RA. personal communication. National Center for Human Genome Research; [Google Scholar]
- 51.Alexander WA, Moss B, Fuerst TR. J Virol. 1992;66:2934. doi: 10.1128/jvi.66.5.2934-2942.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]