

Abstract
In Huntington's disease (HD), the mutant huntingtin (mhtt) protein is associated with striatal dysfunction and degeneration. Excitotoxicity and early synaptic defects are attributed, in part, to altered NMDA receptor (NMDAR) trafficking and function. Deleterious extrasynaptic NMDAR localization and signalling are increased early in yeast artificial chromosome mice expressing full-length mhtt with 128 polyglutamine repeats (YAC128 mice). NMDAR trafficking at the plasma membrane is regulated by dephosphorylation of the NMDAR subunit GluN2B tyrosine 1472 (Y1472) residue by STriatal-Enriched protein tyrosine Phosphatase (STEP). NMDAR function is also regulated by calpain cleavage of the GluN2B C-terminus. Activation of both STEP and calpain is calcium-dependent, and disruption of calcium homeostasis occurs early in the HD striatum. Here, we show increased calpain cleavage of GluN2B at both synaptic and extrasynaptic sites, and elevated extrasynaptic total GluN2B expression in the YAC128 striatum. Calpain inhibition significantly reduced extrasynaptic GluN2B expression in the YAC128 but not wild-type striatum. Furthermore, calpain inhibition reduced whole-cell NMDAR current and the surface/internal GluN2B ratio in co-cultured striatal neurons, without affecting synaptic GluN2B localization. Synaptic STEP activity was also significantly higher in the YAC128 striatum, correlating with decreased GluN2B Y1472 phosphorylation. A substrate-trapping STEP protein (TAT-STEP C-S) significantly increased VGLUT1-GluN2B colocalization, as well as increasing synaptic GluN2B expression and Y1472 phosphorylation. Moreover, combined calpain inhibition and STEP inactivation reduced extrasynaptic, while increasing synaptic GluN2B expression in the YAC128 striatum. These results indicate that increased STEP and calpain activation contribute to altered NMDAR localization in an HD mouse model, suggesting new therapeutic targets for HD.
INTRODUCTION
NMDA receptors (NMDARs) play an important role in learning and memory (1). However, NMDAR dysregulation contributes to pathogenesis in some neurological disorders, including Huntington's disease (HD) (2–4). HD is characterized by striatal dysfunction and neurodegeneration (5), caused by a polyglutamine expansion in the protein huntingtin (mutant huntingtin, mhtt) (6). NMDAR current, surface expression and excitotoxicity are enhanced early in HD transgenic mice (7–14), and the function and trafficking of NMDARs that contain the GluN2B subunit are altered (8,15–18).
Mhtt induces faster NMDAR trafficking to the surface membranes in striatal neurons (8), where receptors accumulate at extrasynaptic sites in young yeast artificial chromosome mice expressing full-length human huntingtin with 128 polyglutamine repeats (YAC128) (17). Synaptic and extrasynaptic NMDARs (Ex-NMDARs) are linked to survival and apoptotic signalling cascades, respectively (19,20). At low concentrations, the selective Ex-NMDAR antagonist memantine restores YAC128 cyclic AMP-response element-binding (CREB) protein pro-survival signalling deficits, and improves motor learning, behaviour and neuropathology (17,21). However, the mechanisms by which mhtt alters NMDAR surface distribution are unknown.
The NMDAR C-terminus contains multiple protein-interaction domains that regulate receptor trafficking and downstream signalling (22–24). Posttranslational modifications such as proteolytic cleavage and phosphorylation modulate NMDAR membrane trafficking, as well as lateral diffusion between synaptic and extrasynaptic sites (3). Several studies have reported that the Ca2+-dependent protease calpain cleaves the GluN2B C-terminus (25,26) and thereby may alter surface NMDAR distribution. In human postmortem brains and HD mouse models, calpain activation is elevated, and the GluN2B subunit is one of several cleaved substrates (27,28).
The membrane-associated STriatal-Enriched protein tyrosine Phosphatase 61 (STEP61) downregulates synaptic NMDAR expression by dephosphorylating a regulatory tyrosine (Y1472) residue on GluN2B (29–31). STEP61 activity is downregulated in excitotoxin-resistant (late stage) HD mouse models (32); however, in early stage HD, disruption of Ca2+ homeostasis might result in enhanced STEP activity via calcineurin activation (33,34). Whether STEP activity is altered in synaptic or extrasynaptic compartments in an excitotoxin-sensitive (early stage) model such as young YAC128 mice is unknown.
Here, we compare the effects of two Ca2+-dependent enzymes, calpain and STEP, on NMDAR subcellular localization in YAC128 striatal neurons, prior to onset of the motor phenotype. We find that increased activation of both enzymes contributes to enhanced extrasynaptic GluN2B expression and activity, at a stage when these mechanisms could contribute to early cognitive deficits.
RESULTS
Enhanced calpain cleavage of NMDARs contributes to increased non-synaptic GluN2B in YAC128 striatal tissue
Previous studies report that calpain cleavage of NMDARs as well as the expression of extrasynaptic full-length GluN2B is elevated in presymptomatic YAC128 mice (17,27). To determine whether calpain cleavage of NMDARs occurs at both synaptic and extrasynaptic sites, striatal subcellular fractionation was performed from 1- to 2-month-old wild-type (WT) and YAC128 mice. The purity of the fractionation was indicated by enrichment of postsynaptic density protein-95 (PSD-95) and synaptophysin in postsynaptic density (PSD) and non-PSD fractions, respectively (data not shown). An N-terminal GluN2B antibody was used that recognizes both full-length (180 kDa) and cleaved (115 kDa) GluN2B. Full-length GluN2B was abundantly expressed in the PSD fraction, whereas cleaved GluN2B was enriched in the non-PSD (extrasynaptic-containing) fraction from WT and YAC128 mice (Fig. 1A). There was a significant increase in full-length, cleaved and total (full-length plus cleaved) GluN2B expression in the YAC128 non-PSD fraction compared with the WT (Fig. 1A and B). Cleaved GluN2B expression was also significantly increased in the YAC128 versus WT PSD fraction. However, increased levels of cleaved GluN2B were not associated with reduced full-length GluN2B expression in either fraction from YAC128 striata, suggesting the involvement of a secondary upregulatory mechanism (Fig. 1A and B).
Figure 1.

Synaptic and extrasynaptic full-length and cleaved GluN2B expression and effect of calpain inhibition. (A) Representative immunoblots of WT and YAC128 striatal non-PSD and PSD fractions probed for GluN2B (using an N-terminal antibody) and β-actin. (B) Quantification of non-PSD and PSD full-length, calpain-cleaved and total (full-length plus cleaved) GluN2B in YAC128 mice relative to WT mice. Synaptic (PSD) cleaved GluN2B and non-PSD full-length, cleaved and total GluN2B are elevated in YAC128 mice relative to the WT. Significance tested using one-sample t-tests (n = 8, *P< 0.05, **P< 0.01). (C) Representative immunoblots of striatal non-PSD and PSD fractions from WT or YAC128 parasagittal slices treated with either vehicle (Veh; DMSO) or calpeptin (Cal), and probed for GluN2B and β-actin. For the non-PSD, lower and higher exposures were used for cleaved and full-length GluN2B quantification, respectively. (D and E) In the vehicle condition, YAC128 non-PSD cleaved and total GluN2B expression was significantly higher than WT and was significantly reduced by calpeptin treatment. Quantification of vehicle- and calpeptin-treated WT and YAC128 non-PSD calpain-cleaved (D) and total (E) GluN2B expression relative to the protein loading control, β-actin. A two-way ANOVA revealed significant differences for non-PSD cleaved GluN2B for treatment (F(1,7) = 100.3, ***P < 0.001), genotype (F(1,7) = 14.76, **P < 0.01), interaction (F(1,7) = 49.63, ***P < 0.001), subjects (matching, F(7,7) = 22.11, ***P < 0.001); ***P < 0.001 by Bonferroni's post hoc tests; n = 4 (D). Significant differences were also detected for non-PSD total GluN2B for treatment (F(1,7) = 19.94, **P < 0.01), genotype (F(1,7) = 12.90, **P < 0.01), interaction (F(1,7) = 10.07, *P < 0.05), subjects (matching, F(7,7) = 5.38, *P < 0.05); **P < 0.01, ***P < 0.001 by Bonferroni's post hoc tests; n = 4 (E). (F) Calpeptin treatment had no effect on synaptic total GluN2B expression in either WT or YAC128 mice. Quantification of vehicle- and calpeptin-treated WT and YAC128 PSD total GluN2B expression relative to β-actin; no significance detected for either total (F) or cleaved GluN2B expression (quantification not shown) with a two-way ANOVA (P > 0.05, n = 4).
To determine the effect of calpain cleavage on GluN2B expression, parasagittal slices were incubated with the calpain inhibitor, calpeptin. For accurate quantification of non-PSD GluN2B, lower and higher exposure blots were used for cleaved and full-length GluN2B analysis, respectively (Fig. 1C). Similar to whole striatum fractionations (Fig. 1A and B), cleaved and total GluN2B expression were significantly increased in the vehicle [dimethyl sulfoxide (DMSO)]-treated YAC128 non-PSD fraction relative to the WT (Fig. 1C–E). Non-PSD full-length GluN2B expression was also significantly increased in the vehicle-treated YAC128 slices relative to the WT (Fig. 1C; quantification not shown). Calpeptin treatment significantly reduced YAC128 non-PSD cleaved (33% decrease) and total (30% decrease) GluN2B expression relative to the vehicle control, bringing GluN2B down to WT levels (Fig. 1C–E). Since calpeptin had a similar effect on both cleaved and full-length GluN2B expression (33 and 30% reduction, respectively), cleaved/full-length GluN2B ratios were not directly compared between treatments. In contrast, calpain inhibition had no effect on total (Fig. 1C and F), cleaved or full-length GluN2B (Fig. 1C) expression in the synaptic fraction of either WT or YAC128 mice (cleaved GluN2B/β-actin ratios in PSD for vehicle: 0.50 ± 0.09 and 0.60 ± 0.12 for WT and YAC128, respectively; for calpeptin: 0.68 ± 0.14 and 0.63 ± 0.14 for WT and YAC128, respectively; no significant difference detected with a two-way analysis of variance (ANOVA); P > 0.05, n = 4). Together, these results suggest that calpain cleavage of GluN2B is increased early in this excitotoxin-sensitive mouse model, and that cleaved GluN2B-containing receptors are relatively stable at extrasynaptic sites.
Increase in GluN2B-containing NMDAR surface expression and whole-cell current is rescued by calpain inhibition in YAC128 co-cultured striatal neurons
We next determined the effect of calpain inhibition on surface NMDAR levels in mhtt-expressing medium-sized spiny striatal neurons (MSNs) co-cultured with cortical neurons after 14 days in vitro (DIV14). Yellow fluorescent protein (YFP)-GluN2B was transfected into WT and YAC128 co-cultured MSNs and treated with either vehicle or calpeptin. After treatment, co-cultures were first live stained for surface YFP-GluN2B (green), and then fixed, permeabilized and stained for internal YFP-GluN2B (red) and the presynaptic glutamatergic marker, vesicular glutamate transporter 1 (VGLUT1, blue). We found a significant increase in the surface/internal GluN2B intensity in YAC128 versus WT MSNs in the vehicle condition (Fig. 2A and B). Calpain inhibition significantly reduced the YAC128 but not WT surface/internal GluN2B intensity ratio compared with the vehicle-treated control (Fig. 2A and B). These results suggest that enhanced calpain activation in YAC128 versus WT MSNs favours a shift towards increased surface expression of NMDARs.
Figure 2.

Calpain inhibition reduces the YAC128 surface/internal YFP-GluN2B intensity ratio with no effect on VGLUT1-GluN2B puncta colocalization. (A) Representative embryonic YFP-GluN2B-transfected co-cultured MSNs live stained with a chicken anti-GFP antibody (surface GluN2B, green), fixed, permeabilized and stained again with a rabbit anti-GFP (internal GluN2B, red). Cultures were treated with either vehicle (DMSO) or calpeptin (1 h). (B) In the vehicle condition, the surface/internal YFP-GluN2B intensity ratio was significantly increased in YAC128 MSNs relative to WT and was significantly reduced to WT levels after calpain inhibition. Quantification of vehicle- and calpeptin-treated surface/internal YFP-GluN2B fluorescence intensities for WT and YAC128. With a two-way ANOVA, there was a significant effect of treatment (F(1,40) = 20.79, ***P < 0.001), genotype (F(1,40) = 9.79, **P < 0.01), interaction (F(1,40) = 17.69, ***P < 0.001); ***P < 0.001 by Bonferroni's post hoc tests; cell n = 21 from four cultures. (C) Representative images of vehicle- and calpeptin-treated WT and YAC128 YFP-GluN2B-transfected co-cultured MSNs live stained with an anti-GFP antibody (surface GluN2B, green), fixed, permeabilized and stained with an anti-VGLUT1 antibody (blue). Magnified images are example dendrites with VGLUT1-GluN2B puncta colocalization. (D) Calpain inhibition does not alter VGLUT1-GluN2B colocalization in either WT or YAC128 MSNs. The percentage of VGLUT1 puncta colocalized with surface YFP-GluN2B is quantified. No significance was obtained with either a two-tailed unpaired t-test or a two-way ANOVA.
To investigate the effect of calpain inhibition on synaptic GluN2B, puncta analysis was performed on thresholded images. Calpeptin treatment had no significant effect on the percentage puncta colocalization of the presynaptic marker VGLUT1 with surface GluN2B (Fig. 2C and D), or on the size and density of VGLUT1 and GluN2B puncta in either WT or YAC128 MSNs [GluN2B puncta size (in μm2) for vehicle: WT, 0.09 ± 0.003; YAC128, 0.09 ± 0.004; calpeptin: WT, 0.08 ± 0.005; YAC128, 0.08 ± 0.005; GluN2B puncta density (per μm) for vehicle: WT, 1.31 ± 0.08; YAC128, 1.49 ± 0.07; calpeptin: WT, 1.38 ± 0.08; YAC128, 1.50 ± 0.09; no significant difference detected with either unpaired t-tests or two-way ANOVAs, P > 0.05, cell n = 21 from four cultures. Data for VGLUT1 are not shown]. Consistent with biochemistry results (Fig. 1C and F), these data suggest that calpain inhibition does not affect synaptic GluN2B expression. Together, these results indicate that increased calpain activation in the YAC128 striatum may promote elevated striatal Ex-NMDAR expression, since preventing calpain cleavage reduces the surface/internal GluN2B ratio (Fig. 2A and B) and decreases extrasynaptic GluN2B expression (Fig. 1C–E).
To examine the effect of calpain inhibition on striatal NMDAR function, whole-cell current density was recorded from YFP-labelled postnatal MSNs in co-culture. In the vehicle condition, the mean NMDA-evoked peak current density of YAC128 MSNs was significantly greater than that of WT MSNs, indicating increased NMDAR activation at the cell surface (Fig. 3A–C). Calpain inhibition by calpeptin had no significant effect on NMDA-induced current in WT MSNs. In contrast, NMDA-induced current was significantly reduced by calpeptin in YAC128 MSNs. Overall, these results indicate that elevated calpain activity promotes increased extrasynaptic expression and activation of NMDARs in the YAC128 striatum.
Figure 3.

Calpain inhibition decreases the YAC128 NMDAR current. (A and B) Representative NMDAR currents from postnatal YFP-transfected WT (A) or YAC128 (B) co-cultured MSNs evoked by NMDA (solid line) in the vehicle condition (grey trace) or following treatment with calpeptin (black trace). (C) Mean peak current density is not changed by calpain inhibition in WT MSNs (vehicle n = 14 cells, calpeptin n = 9). In the vehicle condition, YAC128 MSNs (n = 9) show a greater peak current density than WT MSNs (*P < 0.05), which is decreased by calpeptin treatment (n = 6) to WT levels. A minimum of three independent cultures was used per condition. Differences between groups were tested by a one-way ANOVA followed by Bonferroni's post hoc tests.
Increased synaptic STEP activity and reduced GluN2B Y1472 phosphorylation in YAC128 striatal tissue
The results so far suggest that calpain, which removes most of the GluN2B C-terminus, does not affect synaptic GluN2B expression but modulates Ex-NMDAR expression in YAC128 striatal neurons. Next, we investigated the role of another Ca2+-dependent enzyme, STEP61, which downregulates NMDAR synaptic retention by dephosphorylating a single residue (Y1472) in the GluN2B subunit (29–31,35). Therefore, we postulated that STEP61 activation might influence the distribution of GluN2B to extrasynaptic sites.
Subcellular fractionation was performed from whole striatum dissected from WT and YAC128 mice. Non-PSD and PSD fractions were subsequently probed to assess GluN2B expression and Y1472 phosphorylation (pY1472; Fig. 4A), as well as STEP61 expression and phosphorylation (pSTEP61; Fig. 4B). The pY1472/GluN2B ratio was significantly lower in YAC128 non-PSD and PSD fractions compared with WT mice (Fig. 4A and C). As previously observed in the hippocampus and cortex (36,37), STEP61 was specifically enriched at extrasynaptic membranes compared with the PSD (Fig. 4B). NMDAR-mediated Ca2+ influx leads to activation of the Ca2+-dependent phosphatase, calcineurin, which dephosphorylates STEP61 at its kinase domain serine 221 residue and thereby allows it to associate with and dephosphorylate substrates (33). Thus, a significant reduction in YAC128 synaptic pSTEP61 levels indicates that STEP61 is more active in the YAC128 PSD (Fig. 4B and C). These results suggest that elevated synaptic STEP activity is associated with GluN2B Y1472 dephosphorylation and reduced synaptic stability of GluN2B-containing NMDARs.
Figure 4.

GluN2B Y1472 and STEP phosphorylation levels. (A and B) Representative immunoblots of WT and YAC128 striatal non-PSD and PSD samples probed with anti-GluN2B (N-terminal), pY1472 and β-actin antibodies (A) or with anti-STEP, pSTEP and β-actin antibodies (B). (C) YAC128 non-PSD and PSD GluN2B-pY1472 is significantly reduced compared with WT, and YAC128 PSD pSTEP61 is also significantly lower than WT levels. YAC128 pY1472/full-length GluN2B and pSTEP61/STEP61 levels are normalized to WT for both PSD and non-PSD fractions. Significance tested using one-sample t-tests [n = 4 (non-PSD) and n = 5 (PSD), *P < 0.05, **P < 0.01].
STEP inactivation increases synaptic GluN2B-containing NMDARs
In several studies, a substrate-trapping STEP C-S protein has been used to determine the effect of STEP inactivation on NMDAR and AMPA receptor trafficking (31,38,39). This construct has a point mutation in the catalytic domain that renders it inactive. However, it still binds to its substrates and thereby protects them from dephosphorylation by endogenous STEP. We used trans-activating transduction (TAT)-linked STEP C-S to determine whether STEP modulates synaptic and/or extrasynaptic GluN2B expression and whether it is correlated with changes in GluN2B-pY1472 in WT and YAC128 striata. WT and YAC128 parasagittal slices were incubated with artificial cerebrospinal fluid (aCSF) containing either TAT-myc (negative control) or TAT-STEP C-S fusion proteins. It has been shown in several studies that the fusion proteins efficiently enter cells due to the presence of the HIV-1 TAT protein transduction domain (31,38,39).
As previously observed in Figure 1, immunoblots show increased non-PSD cleaved, full-length and total GluN2B expression in the YAC128 control condition relative to the WT (Fig. 5A). TAT-STEP C-S increases total, particularly full-length, GluN2B expression in the YAC128 PSD but not non-PSD fraction (Fig. 5A–C). Quantification indicates a significant increase (58%) in YAC128 but not WT PSD total GluN2B expression after TAT-STEP C-S treatment compared with TAT-myc (Fig. 5C). Moreover, immunoblots indicate increased pY1472 levels after STEP inactivation in the YAC128 but not WT PSD (Fig. 5A; quantification not shown; PSD TAT-STEP C-S/TAT-myc pY1472 ratios for WT: 1.37 ± 0.19 and YAC128: 1.49 ± 0.06; significance obtained for YAC128 using a one-sample t-test, **P < 0.01, n = 5). The pY1472/GluN2B ratios were not compared between treatments since YAC128 STEP inactivation produced a similar increase in both synaptic full-length GluN2B (46%) and pY1472 (49%) levels. Together, these data suggest that elevated STEP activity in the YAC128 PSD may contribute to synaptic instability of GluN2B-containing NMDARs.
Figure 5.

STEP inactivation increases YAC128 synaptic GluN2B and pY1472 levels. (A) Representative immunoblots of striatal non-PSD and PSD fractions isolated from parasagittal slices treated with either TAT-myc or TAT-STEP C-S fusion proteins (1 h). Samples were probed with anti-GluN2B (N-terminal), pY1472 and β-actin antibodies. Low and high exposure blots were used for cleaved and full-length GluN2B quantification, respectively. (B) STEP inactivation has no effect on either WT or YAC128 non-PSD total GluN2B expression levels. Quantification of TAT-myc and TAT-STEP C-S-treated WT and YAC128 non-PSD total GluN2B expression relative to β-actin; no significance detected with a two-way ANOVA (P > 0.05, n = 4). (C) STEP inactivation significantly increases YAC128 but not WT total GluN2B expression in the PSD. The effect of TAT-myc and TAT-STEP C-S on total GluN2B levels is quantified for both WT and YAC128 in the PSD fraction. A two-way ANOVA revealed significant differences in synaptic total GluN2B for treatment (F(1,8) = 15.14, **P < 0.01) and subjects (matching, F(8,8) = 8.72, **P < 0.01); **P < 0.01 by Bonferroni's post hoc tests; n = 5. As indicated in the immunoblots (A), this was associated with increased Y1472 phosphorylation in the PSD of YAC128 but not WT striata (quantification not shown).
To determine the effect of STEP inactivation on NMDAR internalization and/or lateral diffusion, YFP-GluN2B was transfected into co-cultured embryonic MSNs followed by treatment with either TAT-myc (control) or TAT-STEP C-S fusion proteins. Puncta analysis indicates that TAT-STEP C-S treatment significantly increased the size and density of surface GluN2B puncta in YAC128, while having no detectable effect in WT MSNs [YAC128 GluN2B puncta size (in μm2) for TAT-myc: 0.05 ± 0.002; TAT-STEP C-S: 0.06 ± 0.002;*P < 0.05 with a two-tailed unpaired t-test, cell n = 30 from four cultures. YAC128 GluN2B puncta density (per μm) for TAT-myc: 1.30 ± 0.07; TAT-STEP C-S: 1.56 ± 0.08;*P < 0.05 with a two-tailed unpaired t-test]. There was no effect of STEP C-S on VGLUT1 puncta quantification (data not shown). Fluorescence images and colocalization analysis also indicate that the STEP C-S fusion protein significantly increased the percentage of VGLUT1 colocalized with surface GluN2B (by 30%) (Fig. 6A and B). This suggests that STEP inactivation has a larger effect on YAC128 synaptic GluN2B localization compared with the WT, as also indicated by subcellular fractionation experiments (Fig. 5A and C).
Figure 6.
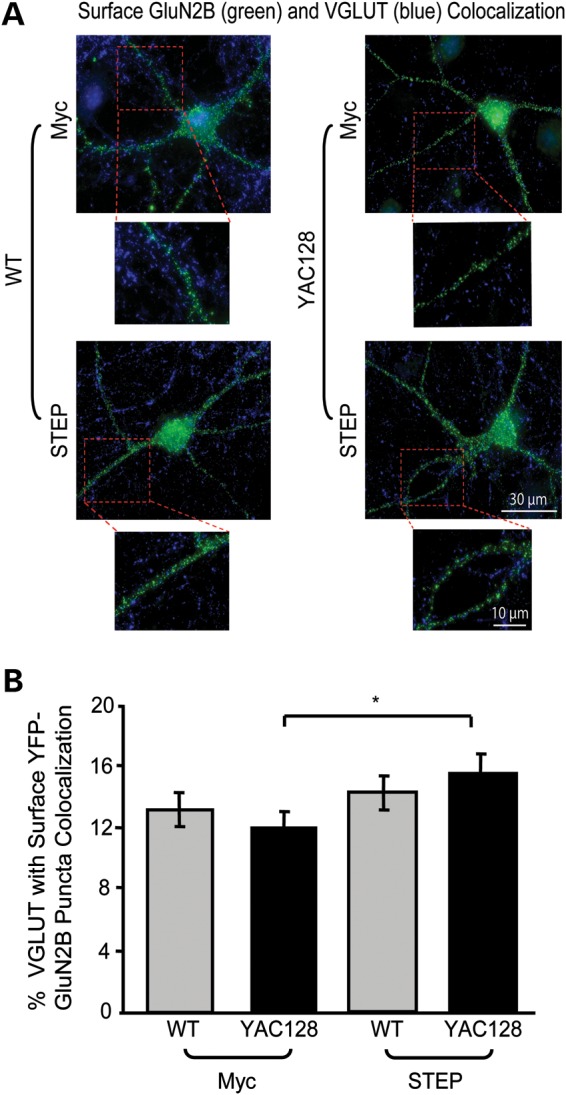
Inactivating STEP increases YAC128 VGLUT1-GluN2B colocalization. (A) Representative images of TAT-myc and TAT-STEP C-S treated (1 h) YFP-GluN2B-transfected MSNs, live stained with an anti-GFP antibody (surface GluN2B, green), fixed, permeabilized and stained with anti-VGLUT1 antibody (blue). Magnified images are example dendrites of VGLUT1-GluN2B puncta colocalization. (B) STEP inactivation significantly increases YAC128 but not WT VGLUT1-GluN2B colocalization. The percentage of VGLUT1-GluN2B puncta colocalization is quantified for TAT-myc and TAT-STEP C-S-treated WT and YAC128 MSNs. Significance obtained for treatment (F(1,58) = 4.14, *P < 0.05) with both a two-way ANOVA and a two-tailed unpaired t-test (*P < 0.05; cell n = 30 from four different cultures).
Consistent with results shown in Figure 2, TAT-myc (control) treated YAC128 MSNs showed significantly higher surface/internal GluN2B fluorescence intensity compared with the WT. However, TAT-STEP C-S had no significant effect on WT or YAC128 surface/internal GluN2B fluorescence intensities [quantification not shown; the surface/internal GluN2B ratio for TAT-myc: WT, 0.67 ± 0.04; YAC128, 0.82 ± 0.05; for TAT-STEP C-S: WT, 0.77 ± 0.06; YAC128, 0.93 ± 0.08; significant difference detected between WT and YAC128 TAT-myc conditions with a two-tailed unpaired t-test (*P < 0.05), cell n = 30 from four cultures]. Together, our data suggest that enhanced synaptic STEP activation in the YAC128 striatum induces a shift in surface NMDAR distribution from the PSD to extrasynaptic sites. This is consistent with no significant change in the YAC128 surface to internal GluN2B ratio as well as increased density and size of GluN2B puncta and elevated colocalization of VGLUT1 and GluN2B upon STEP inactivation (Fig. 6A and B).
Decreasing both calpain and STEP activity alters both synaptic and extrasynaptic NMDAR localization in the YAC128 striatum
To determine the effect of combined calpain inhibition and STEP inactivation, parasagittal slices were incubated with either vehicle (DMSO) plus TAT-myc (control) or calpeptin plus TAT-STEP C-S. Combined treatment with calpeptin and TAT-STEP C-S significantly reduced non-PSD total GluN2B levels (by 24%; Fig. 7A and B) and also increased synaptic total GluN2B expression (by 42%; Fig. 7A and C) in the YAC128 striatum. As illustrated in the representative immunoblots (Fig. 7A), the increase in total GluN2B in the YAC128 PSD was associated with elevated GluN2B-pY1472 levels (quantification not shown; PSD calpeptin plus TAT-STEP C-S/vehicle plus TAT-myc pY1472 ratios for WT: 1.47 ± 0.31 and YAC128: 1.55 ± 0.18; significance obtained for YAC128 using a one-sample t-test, *P < 0.05, n = 4). In summary, it is apparent that combined calpain inhibition and STEP inactivation is sufficient to decrease non-PSD GluN2B levels and increase PSD GluN2B expression in the YAC128 striatum.
Figure 7.

Combined calpain inhibition and STEP inactivation normalizes YAC128 NMDAR expression. (A) Representative immunoblots of striatal non-PSD and PSD fractions isolated from parasagittal slices treated with either vehicle (Veh; DMSO) and TAT-myc or calpeptin and TAT-STEP C-S (1 h). Samples were probed with anti-GluN2B (N-terminal), pY1472 and β-actin antibodies. Low and high exposure blots were used for cleaved and full-length GluN2B quantification, respectively. (B and C) Combined calpain inhibition and STEP inactivation significantly decreases non-PSD YAC128 total GluN2B expression (B), while significantly increasing YAC128 PSD total GluN2B levels (C). The effect of the vehicle plus TAT-myc and calpeptin plus TAT-STEP C-S on non-PSD (B) and PSD (C) total GluN2B levels is quantified relative to β-actin. The decrease in YAC128 non-PSD GluN2B after combined calpain inhibition and STEP inactivation was significant with a two-tailed unpaired t-test [*P < 0.05, n = 4 (B)]. A two-way ANOVA revealed significant differences in synaptic total GluN2B for treatment (F(1,8) = 10.60, *P < 0.05); *P < 0.05 by Bonferroni's post hoc tests; n = 5 (C). As indicated in the immunoblots (A), increased YAC128 synaptic total GluN2B expression was accompanied by increased YAC128 pY1472 after treatment with both calpeptin and TAT-STEP C-S (quantification not shown).
DISCUSSION
Early cognitive decline in HD is associated with synaptic dysfunction and enhanced sensitivity to NMDAR-mediated excitotoxicity (reviewed in 40,41). Previously, it has been reported that acute brain slices and mono-cultured MSNs expressing polyQ-expanded htt exhibit enhanced striatal neuronal NMDAR activation, current and surface expression due to increased forward trafficking of receptors (7–12,14,42). Supporting this, we have shown that YAC128 co-cultured MSNs have increased NMDAR whole-cell current as well as an elevated ratio of surface to internal GluN2B receptors compared with WT MSNs. However, the mechanism(s) underlying altered receptor localization were unknown. This study determined that activation of calpain and STEP contributes to increased expression of Ex-NMDARs, while inhibition of these two enzymes attenuates the disrupted receptor localization. The data suggest that elevated STEP activity in the YAC128 PSD reduces synaptic NMDAR retention and facilitates movement to extrasynaptic sites, and that increased calpain cleavage of GluN2B promotes Ex-NMDAR expression in HD (see proposed model in Fig. 8). Elevated Ex-NMDAR localization and signalling is linked to activation of downstream apoptotic signalling cascades that facilitate further synaptic dysfunction and neurodegeneration (41).
Figure 8.

Model summarizing consequences of elevated calpain and STEP activation for GluN2B expression in YAC128 MSNs. Elevated calpain activity preferentially promotes YAC128 Ex-NMDAR expression, as calpain inhibition significantly reduced both surface and extrasynaptic GluN2B-containing NMDAR expression and function. Increased STEP activation in the YAC128 PSD could mediate reduced synaptic NMDAR retention. Since receptors laterally diffuse on the plasma membrane, enhanced STEP61-mediated GluN2B Y1472 dephosphorylation may facilitate increased NMDAR lateral diffusion from the PSD to extrasynaptic sites. Alternatively, STEP activation could trigger internalization of synaptic NMDARs followed by rapid recycling to the extrasynaptic membrane (not shown in model). GluN2B-containing NMDARs may then be more stable extrasynaptically because of enhanced calpain-mediated cleavage of the AP-2 α and β2 subunits and reduced clathrin-mediated endocytosis. NMDAR mislocalization could contribute to altered activation of cell survival and apoptotic signalling cascades in HD.
Role of calpain activation in altered NMDAR localization
In the presence of mhtt, impairments in metabolic pathways and mitochondrial function lead to increased cytosolic Ca2+ levels and activation of downstream apoptotic signalling cascades (43). Depolarization of mitochondrial membranes at lower intracellular Ca2+ concentrations, interaction of mhtt with voltage-dependent anion channel 1, reduced expression of genes such as peroxisome proliferator-activated receptor-γ coactivator-1α involved in mitochondrial biogenesis, increased inositol-1,4,5-trisphosphate receptor sensitivity and elevated Ca2+ release from intracellular stores may all contribute to early Ca2+ dyshomeostasis (reviewed in 44–46). In particular, striatal neurons cultured from new-born YAC128 mice display increased mitochondrial sensitivity to Ca2+ and reduced cytosolic Ca2+ clearance (47), which likely contributes to the elevated calpain activity observed in striatal tissue from 1- to 2-month-old YAC128 mice (27).
Increased calpain activation may contribute to early neuronal dysfunction and subsequent degeneration in HD (27,28,48,49). Although calpain can cleave other proteins including mhtt, previous studies have reported that calpain cleavage of the GluN2B subunit is important in modulating surface NMDAR distribution and that this cleavage is increased in HD (25–28). Extending previous work (17,27), we observed that striatal tissue from young YAC128 mice has elevated non-PSD full-length and cleaved GluN2B expression, as well as increased synaptic cleaved GluN2B expression, compared with WT mice. Thus, increased cleavage of GluN2B did not decrease either synaptic or extrasynaptic full-length GluN2B expression. This could be because increased forward NMDAR trafficking to the cell surface in YAC128 MSNs (8) replenishes full-length GluN2B-containing receptors to compensate for enhanced calpain cleavage. Although calpain may be active at both synaptic and extrasynaptic sites, the high ratio of cleaved to full-length GluN2B in WT and YAC128 non-PSD fractions compared with the PSD implies that calpain preferentially cleaves Ex-NMDARs, in agreement with previous studies (50,51).
Our data suggest that elevated calpain activity contributes to increased Ex-NMDAR expression in HD. Calpain inhibition had no effect on WT or YAC128 synaptic GluN2B expression, GluN2B puncta size or density, or VGLUT1-GluN2B colocalization, indicating that this protease does not play an important role in regulating synaptic GluN2B in striatal neurons. However, calpain inhibition significantly reduced YAC128 surface GluN2B expression, NMDAR whole-cell current and non-PSD total and cleaved GluN2B expression. It is interesting to note that increased Ex-NMDAR expression and activity could enhance calpain activity (51) and facilitate further increases in Ex-NMDAR localization, in a feed-forward cycle.
Although the non-PSD fraction contains endosomal, presynaptic and extrasynaptic membranes, data using three different techniques suggest that Ex-NMDAR expression and function is enhanced in YAC128 mice by increased calpain activity. This is consistent with studies reporting that calpain-cleaved NMDARs remain functional on the plasma membrane (25,50,52), and that synaptic receptor retention may be reduced after removal of C-terminal protein-interaction motifs (3). Although it has been shown that calpain-cleaved NMDARs are internalized and degraded in cortical neurons (53) due to unmasking of membrane proximal motifs (54), this does not seem to reduce Ex-NMDAR expression in the YAC128 striatum.
Increased calpain activation may facilitate retention of surface GluN2B-containing Ex-NMDARs by cleaving the clathrin adaptor protein-2 (AP-2) complex α and β2 subunits and downregulating clathrin-mediated endocytosis (55). This could explain why calpain inhibition results in a net loss of surface Ex-NMDARs and no significant increase in either synaptic or extrasynaptic total GluN2B expression in YAC128 MSNs. Previously, we reported that incubating YAC128 mono-cultured MSNs with calpain inhibitor I for 12–24 h slows lysosomal degradation of surface NMDARs (27), whereas here we show that 1 h of calpeptin treatment increases NMDAR internalization in YAC128 co-cultured MSNs. Possible explanations for this apparent discrepancy include lack of cortical input in mono-cultured MSNs, different inhibitors utilized and different effects of short- and long-term calpain inhibition.
Altered STEP activity prevents accumulation of synaptic GluN2B-containing NMDARs in HD
Previously, we showed that delivery of GluN2B-containing NMDARs to the surface of MSNs is accelerated in neuronal cultures (8); however, GluN2B surface expression is enhanced only at extrasynaptic sites in YAC128 mouse brain tissue and acute parasagittal slices, whereas synaptic NMDAR current and expression is similar to WT levels (17). The current study suggests that enhanced STEP61 activity results in reduced retention of synaptic NMDARs and limits accumulation of synaptic GluN2B despite accelerated surface delivery.
This is the first time that STEP regulation has been investigated in an excitotoxin-sensitive HD mouse model. In excitoxin-resistant models, STEP61 expression and activity is downregulated because of altered STEP61 transcription and phosphorylation levels, via dysregulation of PKA and calcineurin (32). In those models, increased pSTEP61 may be due to decreased calcineurin activity and/or increased PKA activity in response to elevated D-1 and D-2 dopamine receptor activation (56–58). In excitotoxin-sensitive models, NMDAR over-activation and excessive intracellular Ca2+ may activate calcineurin (34,59,60), which increases STEP61 dephosphorylation and activation (33). Although elevated Ex-NMDAR activation can facilitate calpain cleavage of STEP61 (51), this does not seem to diminish synaptic STEP activity in the YAC128 striatum.
The role of STEP in NMDAR trafficking has not been determined in the striatum, as previous studies were performed in heterologous cells and other brain areas (29–31). In the YAC128 striatum, STEP inactivation resulted in increased synaptic GluN2B and pY1472 levels, increased surface GluN2B puncta size and density and elevated VGLUT1-GluN2B colocalization without changing the surface to internal GluN2B ratio. This was in contrast to WT mice, in which STEP inhibition had no effect on synaptic GluN2B expression, pY1472 levels or VGLUT1-GluN2B puncta colocalization, possibly because STEP basal activity was lower in the WT striatum compared with YAC128 mice. Although STEP inactivation did not reduce YAC128 non-PSD GluN2B levels, trafficking to and from internal pools may have been altered as the non-PSD fraction included these receptors.
In both WT and YAC128 striatal neurons, STEP C-S treatment had no significant effect on the surface to internal GluN2B ratio. This suggests that STEP may facilitate lateral diffusion of GluN2B-containing NMDARs from synaptic to extrasynaptic sites and/or internalization of synaptic NMDARs followed by rapid recycling to the extrasynaptic membrane. This is consistent with the finding that a mutation in the AP-2 binding motif (YEKL domain containing the Y1472 residue) specifically disrupts synaptic GluN2B retention without affecting overall NMDAR surface expression (35). Thus in the HD striatum, elevated synaptic STEP activity and increased GluN2B Y1472 dephosphorylation may facilitate increased Ex-NMDAR expression by triggering either lateral diffusion and/or internalization followed by recycling. In future studies, single molecule imaging experiments that track individual surface NMDAR diffusion patterns would help to distinguish these possibilities (61–63).
Notably, in the non-PSD compartment, there was no correlation between STEP activity or GluN2B-pY1472 with alterations in GluN2B expression, suggesting that pY1472 specifically stabilizes synaptic GluN2B-containing NMDAR expression in the striatum. Other studies support this idea, since STEP inhibition or YEKL domain mutations increase synaptic GluN2B expression (30,35). Our results are also consistent with a previous hippocampal study indicating that pY1472 and pY1336 modulate synaptic and Ex-NMDAR localization, respectively (36). However, the latter study also showed that Src-family tyrosine kinases and STEP modulate synaptic and Ex-NMDARs, respectively, which is not in agreement with our findings but could be explained by differences between brain areas. Differential phosphorylation of GluN2B at synaptic versus extrasynaptic sites could contribute to increased calpain cleavage of Ex-NMDARs in HD, since GluN2B pY1336 but not pY1472 potentiates calpain cleavage of the GluN2B subunit (50).
We found that the effects of calpain inhibition and STEP inactivation are additive, resulting in increased synaptic and decreased Ex-NMDAR expression in YAC128 mice. These data support the idea that enhanced calpain activation preferentially promotes Ex-NMDAR localization, whereas STEP specifically modulates synaptic NMDAR expression. Thus, combined treatment with calpeptin and STEP C-S shifts NMDAR distribution towards WT levels. Future studies will determine whether cell survival and apoptotic signalling is also normalized in response to corrected GluN2B localization.
Conclusions
We report mechanisms contributing to enhanced Ex-NMDAR localization in an excitotoxin-sensitive HD mouse model. Increased activation of calpain and STEP facilitate altered localization of GluN2B-containing NMDARs to extrasynaptic sites. Future studies will investigate these mechanisms further in both excitotoxin-sensitive and resistant HD mouse models. The ability to rectify the abnormal distribution of NMDAR may be a powerful therapeutic approach to improve early NMDAR function and signalling, synaptic dysfunction and cognitive decline in HD. Specific inhibitors that prevent extrasynaptic calpain cleavage and synaptic STEP-mediated GluN2B dephosphorylation may avoid adverse side effects associated with NMDAR inhibition.
MATERIALS AND METHODS
Transgenic mice, neuron culture preparation and transfection
WT and YAC128 (line 55) mice bred on the FVB/N mouse strain were housed, cared for and used according to Canadian Council on Animal Care regulations at the University of British Columbia (UBC) Faculty of Medicine Animal Resource Unit. For embryonic culture preparation, pregnant (embryonic days 17–18) WT and YAC128 mice were deeply anesthetized with halothane vapour and killed by decapitation. Embryos were removed, brains extracted and the striatum and cortex dissected on ice in Hank's Balanced Salt Solution [HBSS, GIBCO; described in detail in reference (16). For postnatal cultures, striatal and cortical tissue was dissected from postnatal days 0–1 WT and YAC128 mice. More than 85% of striatal cells cultured from embryonic and/or postnatal day 0–1 mice are GABAergic MSNs (12).
In experiments using cortico-striatal co-cultured neurons, striatal cells were labelled by transfection on the day of plating (DIV0). For patch-clamp recording, postnatal cells were labelled with YFP on a β-actin promoter (a gift from K. She and A. M. Craig, UBC, Vancouver, originally from S. Kaech and G. Banker, Oregon Health Sciences University, Portland), which was previously linked to a CAG promoter [gift from J. Miyazaki (64,65)]. For immunocytochemical localization of surface GluN2B-containing NMDARs, we used an N-terminally tagged YFP-GluN2B plasmid on a synapsin promoter to label embryonic striatal cells [gift from K. She and A. M. Craig, originally from M. Sheng, Genentech, San Francsco (66)]. Site-directed mutagenesis and polymerase chain reaction was used to insert YFP and a linker LVPRGSRSR into rat GluN2B between amino acids 2 and 4 in a modified pLentiLox 3.7 vector. The coding region sequence was subsequently verified. YFP-GluN2B transfections were performed in order to assess surface GluN2B levels due to unavailability of sensitive N-terminal anti-GluN2B antibodies suitable for immunocytochemistry. Furthermore, biotinylation of co-cultured neurons was not suitable for surface GluN2B labelling since the cultures contained a mixture of both cortical and striatal neurons.
Cells were suspended in electroporation buffer (Mirus Bio) with DNA and nucleofected (AMAXA nucleofector I). Transfected striatal cells (MSNs) were resuspended with non-transfected cortical neurons (1:1 ratio) in D-minimum essential medium (DMEM) (GIBCO) plus 10% fetal bovine serum (DMEM+). Cells were then plated on glass cover slips pre-coated with poly-D-lysine (250 μg/ml) in 24-well plates, at a total density of 2 × 105 cells per well. 1-2 h after plating, the DMEM+ was replaced with 500 μl plating medium [PM; B27, penicillin/streptomycin, α-glutamine, neurobasal medium (GIBCO)]. At DIV3, another 500 μl PM was added, after which half of the media was replaced every 3–4 days. The cultures were kept in a humidified 37°C incubator with 5% CO2 until day of use (DIV12–15). Images from YFP-transfected MSNs indicated that striatal neurons were relatively mature and spiny at this age.
Striatal dissections and brain slice preparation
Male mice between 1 and 2 months of age were halothane-anesthetized and decapitated. To look at basal protein expression, brains were rapidly removed and immersed in ice-cold protease and phosphatase inhibitor-containing sucrose buffer (0.32 m sucrose, 10 mm HEPES, pH 7.4). The striatum was dissected on ice in sucrose buffer and homogenized for subcellular fractionation. For parasagittal slice preparations, brains were immersed in oxygenated (95% O2–5% CO2), chilled aCSF (in mm: 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 6 MgCl2, 2 CaCl2, 10 glucose, pH 7.3–7.4, 300–310 mosmol l−1). A 3:1 Mg2+:Ca2+ ratio was used for cutting aCSF to minimize NMDAR activation. A vibratome (Leica VT1000) was used to cut 300 μm parasagittal slices, which were placed into a 1:2 Mg2+:Ca2+ containing aCSF to rest [1 h, room temperature (RT)].
Parasagittal slice treatment
For calpain inhibition, we used calpeptin (Calbiochem), which inhibits calpain I, calpain II and papain. For STEP inactivation, a dominant-negative STEP fusion protein (TAT-STEP C-S) was used, and a TAT-myc protein [containing the myelocytomatosis virus (myc) tag] served as a control. The TAT-STEP C-S fusion protein has a point mutation in the catalytic domain that inactivates the tyrosine phosphatase. The STEP fusion protein still binds to its substrates and thereby acts as a substrate-trapping construct. TAT-STEP C-S has been well characterized and readily transduces into slices and neuronal cultures (31,38,39). For calpain inhibition experiments, parasagittal slices were incubated in aCSF containing DMSO (vehicle control) or 15 μm calpeptin (1 h, RT) (51). For STEP inactivation, slices were incubated in aCSF containing either 2 μm TAT-myc (negative control) or 2 μm TAT-STEP C-S (1 h, RT) (51). For combined calpain inhibition and STEP inactivation, slices were incubated in aCSF containing DMSO plus 2 μm TAT-myc or 15 μm calpeptin plus 2 μm TAT-STEP C-S (1 h, RT). Slices were oxygenated throughout. After treatment, the striatum was dissected on ice in sucrose buffer and homogenized in 0.32 m sucrose buffer for subcellular fractionation.
Subcellular fractionation
The subcellular fractionation protocol was followed as described previously (17). Striatal PSD and non-PSD fractions were isolated based on the principle that the PSD fractions are Triton X-100 insoluble, while non-PSD fractions are soluble (17,36,67). All buffers contained ‘complete’ protease and ‘phosSTOP’ phosphatase inhibitor cocktails (Roche diagnostics) in addition to 40 mm β-glycerophosphate, 20 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 30 mm NaF, 1 mm EDTA, 1 mm EGTA and 15μm calpeptin. Fractions were stored at −80°C until use.
Western blotting
Western blotting was performed as described previously (17). In brief, protein concentration was normalized, samples heated (4.5 min, 95°C) in 3× sample buffer, separated via 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membrane using semi-dry electrophoresis (BioRad). After transfer, membranes were blocked in 3% BSA or 5% milk in 0.5% Tween-20 in TBS (TBST) (1 h, RT), and incubated in primary antibody (3% BSA or 3% milk in TBST, overnight, 4°C). After washing, blots were incubated with horseradish peroxidase (HRP)-coupled secondary antibodies (1% BSA or 3% milk in TBST, 2 h, RT), washed and visualized using enhanced chemiluminescence substrate (Amersham) and exposed to film (Amersham). β-Actin was visualized using an anti-goat alkaline phosphatase (AP)-conjugated secondary antibody and a Lumi-phos WB chemiluminescent substrate detection system (Pierce).
Primary antibodies used were rabbit N-terminal anti-GluN2B (AGC-003; Alomone, 1:500), rabbit anti-pY1472 (p1516-1472, Phosphosolutions, 1:500), goat anti-β-actin (sc-1616, Santa Cruz, 1:1500), mouse anti-synaptophysin (S5768; Sigma, 1:1000), mouse anti-PSD-95 (MA1-045, Affinity BioReagents, 1:500), mouse anti-STEP (23E5, 1:500) (68) and rabbit anti-pSTEP61 (serine 221, 1:500) (33). Secondary antibodies used were anti-mouse HRP-conjugated (NA931V, Amersham, 1:5000–1:10 000), anti-rabbit HRP-conjugated (NA934V, Amersham; 1:5000–1:10 000) and anti-goat AP-conjugated (sc-2037; Santa Cruz, 1:5000). All antibodies were added to BSA-containing TBST, except for anti-pSTEP61 that required milk.
Electrophysiology
Whole-cell patch-clamp recordings were performed on DIV12-15 co-cultured YFP-transfected postnatal MSNs. Electrophysiology experiments were conducted under voltage clamp at a holding potential of −70 mV, as described previously (47). Briefly, neurons were superfused at RT with extracellular solution (ECS, in mm: 167 NaCl, 2.4 KCl, 0.010 MgCl2, 10 glucose, 10 HEPES, 2 CaCl2, pH 7.2). Tetrodotoxin (0.3 µm), glycine (10 µm) and picrotoxin (100 µm) were added to ECS before use. Calpeptin (10 µm) was applied for 1–2 h in ECS before and during recording. In the control condition, an equal volume of the solvent DMSO was applied with ECS. Rapid switching between ECS and NMDA solutions was achieved using a pressure-driven solenoid system controlled by pClamp (Harvard Apparatus). NMDA (1 mm) was applied for 3 s and repeated 5× at intervals of 60 s. Intracellular recording solution contained in mm: 130 CsMe, 5 CsCl, 4 NaCl, 1 MgCl, 10 HEPES, 5 EGTA, 5 lidocaine, 0.5 GTP, 10 Na-phosphocreatine, 5 MgATP, pH 7.2. Data were acquired using the Axopatch 200B amplifier and analysed using the pClamp 10.2 software (Molecular Devices). Whole-cell current density was calculated as peak NMDA current (pA)/cell capacitance (pF).
Immunocytochemistry
Embryonic YFP-GluN2B-transfected WT and YAC128 MSNs co-cultured with cortical neurons were treated at DIV14 with conditioned media containing either DMSO (vehicle) or 10 μm calpeptin (1 h, 37°C) (51). For STEP inactivation experiments, MSNs were treated with conditioned media containing either 1 μm TAT-myc or 1 μm TAT-STEP C-S (1 h, 37°C).
Similar to methods in reference (69), cover slips were subsequently live stained with chicken anti-green fluorescent protein (GFP) antibody (cross reactive with eYFP; ab13970, AbCam, 1:1000, 10 min, 37°C) in conditioned media, washed (3×, PBS, RT) and fixed with 4% paraformaldehyde (PFA) plus 4% sucrose (15 min, RT). After washing (PBS), cover slips were incubated with anti-chicken Alexa-488-conjugated (green fluorescent) secondary antibody (A11039, Molecular Probes, 1:1000, 30 min, RT) in PBS. Cover slips were washed again (PBS), permeabilized with filtered ice-cold methanol (5 min, −20°C), washed (PBS) and incubated in PBS containing 0.03% Triton X-100 (PBST, 5 min, RT). Cover slips were blocked in 10% normal goat serum (NGS) in PBS (30 min, RT) and incubated in guinea-pig anti-VGLUT1 (AB5905, Chemicon, 1:4000) and rabbit anti-GFP (132-002, Synaptic Systems, 1:500) primary antibodies in 2% NGS containing PBST (1 h, RT) followed by gentle shaking (overnight, 4°C). Cover slips were washed (PBST) and incubated in anti-rabbit Alexa 568 (red fluorescent; A21069, Molecular Probes, 1:1000) and anti-guinea-pig AMCA (blue fluorescent; 706-155-148, Jackson Laboratories, 1:100) conjugated secondary antibodies (1 h, RT). Cover slips were finally washed (PBST) and mounted onto slides using fluoromount (Southern Biotech).
To confirm the specificity of surface (green) and internal (red) GluN2B staining, control experiments were performed on non-transfected and YFP-GluN2B-transfected MSNs (data not shown). In non-transfected MSNs (with and without permeabilization), no significant dendritic green or red fluorescent signal was observed. Red fluorescent signal was also minimal for transfected cells without the permeabilization step. Under normal staining conditions, minimal overlap of green and red puncta was observed.
Microscopy
A Zeiss Axiovert 200M fluorescence microscope was used for image acquisition. To obtain optimal images, approximately six to ten 0.45 μm z-stacks were taken at ×63 magnification (NA: 1.4). The same exposure time was used for green, red and blue channels throughout the experiment. An extended focus projection function (Axiovision 4.6A) was utilized to create flattened images from stacks with soma and dendrites in focus and 16 bit images were exported for data analysis.
Data analysis and statistics
For western blots, the optical density of immunoreactive bands was quantified using Image J (NIH). The protein loading control β-actin was used for band density normalization apart for pY1472 blots where β-actin reprobes were not possible. For microscope images, Image J was used to quantify mean intensities of surface (green) and internal (red) YFP-GluN2B fluorescence in a particular area (mask) of dendrite. Background intensity measures were subtracted from dendrite intensities and an average of three dendrites per cell was calculated. Cell averages from four different cultures were subsequently assessed.
The same cells and dendrites were used for VGLUT1 and surface YFP-GluN2B puncta analysis. Images were blinded and manually thresholded for puncta size, density and colocalization analysis per micron of dendrite. An Image J colocalization plugin [http://rsb.info.nih.gov/ij/plugins/colocalization.html (70)] was utilized to quantify the percentage of VGLUT1 puncta colocalized with surface YFP-GluN2B puncta.
Group analysis and statistics was performed in GraphPad Prism and Microsoft Excel. Data are presented as the mean ± SEM. Significance was tested using two-tailed unpaired t-tests, one-sample t-tests, one-way and two-way ANOVAs and Bonferroni's post hoc tests as indicated; P-values < 0.05 were deemed significant.
FUNDING
This study was supported by the Canadian Institutes of Health Research (CIHR MOP-12699 to L.A.R.; a joint CIHR-Huntington Society of Canada postdoctoral fellowship to C.M.G.), the Cure Huntington's Disease Initiative (CHDI, to L.A.R.) and the National Institutes of Health (NIH MH01527 and MH52711 to P.J.L.). C.M.G. is also a Hereditary Disease Foundation (HDF) Scholar.
ACKNOWLEDGEMENTS
We would like to thank M. Parsons and K. Kolodziejczyk for critically reading the manuscript and A. M. Kaufman and L. Nam for experimental advice and input. We would also like to thank L. Wang for technical support and A. M. Craig and K. She for the YFP and YFP-GluN2B plasmids.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Bliss T.V., Collingridge G.L. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. doi:10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 2.Arundine M., Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34:325–337. doi: 10.1016/s0143-4160(03)00141-6. doi:10.1016/S0143-4160(03)00141-6. [DOI] [PubMed] [Google Scholar]
- 3.Gladding C.M., Raymond L.A. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol. Cell Neurosci. 2011;48:308–320. doi: 10.1016/j.mcn.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Lau C.G., Zukin R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 5.Graveland G.A., Williams R.S., DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science. 1985;227:770–773. doi: 10.1126/science.3155875. doi:10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- 6.HDCRG. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. doi:10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 7.Cepeda C., Ariano M.A., Calvert C.R., Flores-Hernandez J., Chandler S.H., Leavitt B.R., Hayden M.R., Levine M.S. NMDA receptor function in mouse models of Huntington disease. J. Neurosci. Res. 2001;66:525–539. doi: 10.1002/jnr.1244. doi:10.1002/jnr.1244. [DOI] [PubMed] [Google Scholar]
- 8.Fan M.M., Fernandes H.B., Zhang L.Y., Hayden M.R., Raymond L.A. Altered NMDA receptor trafficking in a yeast artificial chromosome transgenic mouse model of Huntington's disease. J. Neurosci. 2007;27:3768–3779. doi: 10.1523/JNEUROSCI.4356-06.2007. doi:10.1523/JNEUROSCI.4356-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graham R.K., Pouladi M.A., Joshi P., Lu G., Deng Y., Wu N.P., Figueroa B.E., Metzler M., Andre V.M., Slow E.J., et al. Differential susceptibility to excitotoxic stress in YAC128 mouse models of Huntington disease between initiation and progression of disease. J. Neurosci. 2009;29:2193–2204. doi: 10.1523/JNEUROSCI.5473-08.2009. doi:10.1523/JNEUROSCI.5473-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L., Murphy T.H., Hayden M.R., Raymond L.A. Enhanced striatal NR2B-containing N-methyl-D-aspartate receptor-mediated synaptic currents in a mouse model of Huntington disease. J. Neurophysiol. 2004;92:2738–2746. doi: 10.1152/jn.00308.2004. doi:10.1152/jn.00308.2004. [DOI] [PubMed] [Google Scholar]
- 11.Milnerwood A.J., Raymond L.A. Corticostriatal synaptic function in mouse models of Huntington's disease: early effects of huntingtin repeat length and protein load. J. Physiol. 2007;585:817–831. doi: 10.1113/jphysiol.2007.142448. doi:10.1113/jphysiol.2007.142448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shehadeh J., Fernandes H.B., Zeron Mullins M.M., Graham R.K., Leavitt B.R., Hayden M.R., Raymond L.A. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol. Dis. 2006;21:392–403. doi: 10.1016/j.nbd.2005.08.001. doi:10.1016/j.nbd.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 13.Tang T.S., Slow E., Lupu V., Stavrovskaya I.G., Sugimori M., Llinas R., Kristal B.S., Hayden M.R., Bezprozvanny I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc. Natl. Acad. Sci. USA. 2005;102:2602–2607. doi: 10.1073/pnas.0409402102. doi:10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeron M.M., Hansson O., Chen N., Wellington C.L., Leavitt B.R., Brundin P., Hayden M.R., Raymond L.A. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. doi:10.1016/S0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 15.Chen N., Luo T., Wellington C., Metzler M., McCutcheon K., Hayden M.R., Raymond L.A. Subtype-specific enhancement of NMDA receptor currents by mutant huntingtin. J. Neurochem. 1999;72:1890–1898. doi: 10.1046/j.1471-4159.1999.0721890.x. doi:10.1046/j.1471-4159.1999.0721890.x. [DOI] [PubMed] [Google Scholar]
- 16.Fan J., Cowan C.M., Zhang L.Y., Hayden M.R., Raymond L.A. Interaction of postsynaptic density protein-95 with NMDA receptors influences excitotoxicity in the yeast artificial chromosome mouse model of Huntington's disease. J. Neurosci. 2009;29:10928–10938. doi: 10.1523/JNEUROSCI.2491-09.2009. doi:10.1523/JNEUROSCI.2491-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milnerwood A.J., Gladding C.M., Pouladi M.A., Kaufman A.M., Hines R.M., Boyd J.D., Ko R.W., Vasuta O.C., Graham R.K., Hayden M.R., et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron. 2010;65:178–190. doi: 10.1016/j.neuron.2010.01.008. doi:10.1016/j.neuron.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Song C., Zhang Y., Parsons C.G., Liu Y.F. Expression of polyglutamine-expanded huntingtin induces tyrosine phosphorylation of N-methyl-D-aspartate receptors. J. Biol. Chem. 2003;278:33364–33369. doi: 10.1074/jbc.M304240200. doi:10.1074/jbc.M304240200. [DOI] [PubMed] [Google Scholar]
- 19.Hardingham G.E., Fukunaga Y., Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- 20.Leveille F., El Gaamouch F., Gouix E., Lecocq M., Lobner D., Nicole O., Buisson A. Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. Faseb J. 2008;22:4258–4271. doi: 10.1096/fj.08-107268. doi:10.1096/fj.08-107268. [DOI] [PubMed] [Google Scholar]
- 21.Okamoto S., Pouladi M.A., Talantova M., Yao D., Xia P., Ehrnhoefer D.E., Zaidi R., Clemente A., Kaul M., Graham R.K., et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. doi:10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groc L., Bard L., Choquet D. Surface trafficking of N-methyl-D-aspartate receptors: physiological and pathological perspectives. Neuroscience. 2009;158:4–18. doi: 10.1016/j.neuroscience.2008.05.029. doi:10.1016/j.neuroscience.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 23.Hardingham G.E., Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. doi:10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petralia R.S., Al-Hallaq R.A., Wenthold R.J. Trafficking and Targeting of NMDA Receptors. In: Van Dongen A.M., editor. Biology of the NMDA Receptor. CRC Press; 2009. Chapter 8. [PubMed] [Google Scholar]
- 25.Guttmann R.P., Baker D.L., Seifert K.M., Cohen A.S., Coulter D.A., Lynch D.R. Specific proteolysis of the NR2 subunit at multiple sites by calpain. J. Neurochem. 2001;78:1083–1093. doi: 10.1046/j.1471-4159.2001.00493.x. doi:10.1046/j.1471-4159.2001.00493.x. [DOI] [PubMed] [Google Scholar]
- 26.Guttmann R.P., Sokol S., Baker D.L., Simpkins K.L., Dong Y., Lynch D.R. Proteolysis of the N-methyl-d-aspartate receptor by calpain in situ. J. Pharmacol. Exp. Ther. 2002;302:1023–1030. doi: 10.1124/jpet.102.036962. doi:10.1124/jpet.102.036962. [DOI] [PubMed] [Google Scholar]
- 27.Cowan C.M., Fan M.M., Fan J., Shehadeh J., Zhang L.Y., Graham R.K., Hayden M.R., Raymond L.A. Polyglutamine-modulated striatal calpain activity in YAC transgenic huntington disease mouse model: impact on NMDA receptor function and toxicity. J. Neurosci. 2008;28:12725–12735. doi: 10.1523/JNEUROSCI.4619-08.2008. doi:10.1523/JNEUROSCI.4619-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gafni J., Ellerby L.M. Calpain activation in Huntington's disease. J. Neurosci. 2002;22:4842–4849. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braithwaite S.P., Adkisson M., Leung J., Nava A., Masterson B., Urfer R., Oksenberg D., Nikolich K. Regulation of NMDA receptor trafficking and function by striatal-enriched tyrosine phosphatase (STEP) Eur. J. Neurosci. 2006;23:2847–2856. doi: 10.1111/j.1460-9568.2006.04837.x. doi:10.1111/j.1460-9568.2006.04837.x. [DOI] [PubMed] [Google Scholar]
- 30.Pelkey K.A., Askalan R., Paul S., Kalia L.V., Nguyen T.H., Pitcher G.M., Salter M.W., Lombroso P.J. Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron. 2002;34:127–138. doi: 10.1016/s0896-6273(02)00633-5. doi:10.1016/S0896-6273(02)00633-5. [DOI] [PubMed] [Google Scholar]
- 31.Snyder E.M., Nong Y., Almeida C.G., Paul S., Moran T., Choi E.Y., Nairn A.C., Salter M.W., Lombroso P.J., Gouras G.K., et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. doi:10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 32.Saavedra A., Giralt A., Rue L., Xifro X., Xu J., Ortega Z., Lucas J.J., Lombroso P.J., Alberch J., Perez-Navarro E. Striatal-enriched protein tyrosine phosphatase expression and activity in Huntington's disease: a STEP in the resistance to excitotoxicity. J. Neurosci. 2011;31:8150–8162. doi: 10.1523/JNEUROSCI.3446-10.2011. doi:10.1523/JNEUROSCI.3446-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paul S., Nairn A.C., Wang P., Lombroso P.J. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat. Neurosci. 2003;6:34–42. doi: 10.1038/nn989. doi:10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- 34.Xifro X., Garcia-Martinez J.M., Del Toro D., Alberch J., Perez-Navarro E. Calcineurin is involved in the early activation of NMDA-mediated cell death in mutant huntingtin knock-in striatal cells. J. Neurochem. 2008;105:1596–1612. doi: 10.1111/j.1471-4159.2008.05252.x. doi:10.1111/j.1471-4159.2008.05252.x. [DOI] [PubMed] [Google Scholar]
- 35.Prybylowski K., Chang K., Sans N., Kan L., Vicini S., Wenthold R.J. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. doi:10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goebel-Goody S.M., Davies K.D., Alvestad Linger R.M., Freund R.K., Browning M.D. Phospho-regulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience. 2009;158:1446–1459. doi: 10.1016/j.neuroscience.2008.11.006. doi:10.1016/j.neuroscience.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 37.Jiang X., Knox R., Pathipati P., Ferriero D. Developmental localization of NMDA receptors, Src and MAP kinases in mouse brain. Neurosci. Lett. 2011;503:215–219. doi: 10.1016/j.neulet.2011.08.039. doi:10.1016/j.neulet.2011.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paul S., Olausson P., Venkitaramani D.V., Ruchkina I., Moran T.D., Tronson N., Mills E., Hakim S., Salter M.W., Taylor J.R., et al. The striatal-enriched protein tyrosine phosphatase gates long-term potentiation and fear memory in the lateral amygdala. Biol. Psychiatry. 2007;61:1049–1061. doi: 10.1016/j.biopsych.2006.08.005. doi:10.1016/j.biopsych.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y., Venkitaramani D.V., Gladding C.M., Kurup P., Molnar E., Collingridge G.L., Lombroso P.J. The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J. Neurosci. 2008;28:10561–10566. doi: 10.1523/JNEUROSCI.2666-08.2008. doi:10.1523/JNEUROSCI.2666-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan M.M., Raymond L.A. N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington's disease. Prog. Neurobiol. 2007;81:272–293. doi: 10.1016/j.pneurobio.2006.11.003. doi:10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 41.Milnerwood A.J., Raymond L.A. Early synaptic pathophysiology in neurodegeneration: insights from Huntington's disease. Trends Neurosci. 2010;33:513–523. doi: 10.1016/j.tins.2010.08.002. doi:10.1016/j.tins.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 42.Zhang H., Li Q., Graham R.K., Slow E., Hayden M.R., Bezprozvanny I. Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington's disease. Neurobiol. Dis. 2008;31:80–88. doi: 10.1016/j.nbd.2008.03.010. doi:10.1016/j.nbd.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cowan C.M., Raymond L.A. Selective neuronal degeneration in Huntington's disease. Curr. Top. Dev. Biol. 2006;75:25–71. doi: 10.1016/S0070-2153(06)75002-5. doi:10.1016/S0070-2153(06)75002-5. [DOI] [PubMed] [Google Scholar]
- 44.Bezprozvanny I., Hayden M.R. Deranged neuronal calcium signaling and Huntington disease. Biochem. Biophys. Res. Commun. 2004;322:1310–1317. doi: 10.1016/j.bbrc.2004.08.035. doi:10.1016/j.bbrc.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 45.Giacomello M., Hudec R., Lopreiato R. Huntington's disease, calcium, and mitochondria. Biofactors. 2011;37:206–218. doi: 10.1002/biof.162. doi:10.1002/biof.162. [DOI] [PubMed] [Google Scholar]
- 46.Oliveira J.M. Nature and cause of mitochondrial dysfunction in Huntington's disease: focusing on huntingtin and the striatum. J. Neurochem. 2010;114:1–12. doi: 10.1111/j.1471-4159.2010.06741.x. [DOI] [PubMed] [Google Scholar]
- 47.Fernandes H.B., Baimbridge K.G., Church J., Hayden M.R., Raymond L.A. Mitochondrial sensitivity and altered calcium handling underlie enhanced NMDA-induced apoptosis in YAC128 model of Huntington's disease. J. Neurosci. 2007;27:13614–13623. doi: 10.1523/JNEUROSCI.3455-07.2007. doi:10.1523/JNEUROSCI.3455-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gafni J., Hermel E., Young J.E., Wellington C.L., Hayden M.R., Ellerby L.M. Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J. Biol. Chem. 2004;279:20211–20220. doi: 10.1074/jbc.M401267200. doi:10.1074/jbc.M401267200. [DOI] [PubMed] [Google Scholar]
- 49.Kim Y.J., Yi Y., Sapp E., Wang Y., Cuiffo B., Kegel K.B., Qin Z.H., Aronin N., DiFiglia M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc. Natl. Acad. Sci. USA. 2001;98:12784–12789. doi: 10.1073/pnas.221451398. doi:10.1073/pnas.221451398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu H.Y., Hsu F.C., Gleichman A.J., Baconguis I., Coulter D.A., Lynch D.R. Fyn-mediated phosphorylation of NR2B Tyr-1336 controls calpain-mediated NR2B cleavage in neurons and heterologous systems. J. Biol. Chem. 2007;282:20075–20087. doi: 10.1074/jbc.M700624200. doi:10.1074/jbc.M700624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu J., Kurup P., Zhang Y., Goebel-Goody S.M., Wu P.H., Hawasli A.H., Baum M.L., Bibb J.A., Lombroso P.J. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J. Neurosci. 2009;29:9330–9343. doi: 10.1523/JNEUROSCI.2212-09.2009. doi:10.1523/JNEUROSCI.2212-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simpkins K.L., Guttmann R.P., Dong Y., Chen Z., Sokol S., Neumar R.W., Lynch D.R. Selective activation induced cleavage of the NR2B subunit by calpain. J. Neurosci. 2003;23:11322–11331. doi: 10.1523/JNEUROSCI.23-36-11322.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuen E.Y., Ren Y., Yan Z. Postsynaptic density-95 (PSD-95) and calcineurin control the sensitivity of N-methyl-D-aspartate receptors to calpain cleavage in cortical neurons. Mol. Pharmacol. 2008;74:360–370. doi: 10.1124/mol.108.046813. doi:10.1124/mol.108.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scott D.B., Michailidis I., Mu Y., Logothetis D., Ehlers M.D. Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J. Neurosci. 2004;24:7096–7109. doi: 10.1523/JNEUROSCI.0780-04.2004. doi:10.1523/JNEUROSCI.0780-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rudinskiy N., Grishchuk Y., Vaslin A., Puyal J., Delacourte A., Hirling H., Clarke P.G., Luthi-Carter R. Calpain hydrolysis of alpha- and beta2-adaptins decreases clathrin-dependent endocytosis and may promote neurodegeneration. J. Biol. Chem. 2009;284:12447–12458. doi: 10.1074/jbc.M804740200. doi:10.1074/jbc.M804740200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paoletti P., Vila I., Rife M., Lizcano J.M., Alberch J., Gines S. Dopaminergic and glutamatergic signaling crosstalk in Huntington's disease neurodegeneration: the role of p25/cyclin-dependent kinase 5. J. Neurosci. 2008;28:10090–10101. doi: 10.1523/JNEUROSCI.3237-08.2008. doi:10.1523/JNEUROSCI.3237-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spektor B.S., Miller D.W., Hollingsworth Z.R., Kaneko Y.A., Solano S.M., Johnson J.M., Penney J.B., Jr, Young A.B., Luthi-Carter R. Differential D1 and D2 receptor-mediated effects on immediate early gene induction in a transgenic mouse model of Huntington's disease. Brain Res. Mol. Brain Res. 2002;102:118–128. doi: 10.1016/s0169-328x(02)00216-4. doi:10.1016/S0169-328X(02)00216-4. [DOI] [PubMed] [Google Scholar]
- 58.Tang T.S., Chen X., Liu J., Bezprozvanny I. Dopaminergic signaling and striatal neurodegeneration in Huntington's disease. J. Neurosci. 2007;27:7899–7910. doi: 10.1523/JNEUROSCI.1396-07.2007. doi:10.1523/JNEUROSCI.1396-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pardo R., Colin E., Regulier E., Aebischer P., Deglon N., Humbert S., Saudou F. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J. Neurosci. 2006;26:1635–1645. doi: 10.1523/JNEUROSCI.3706-05.2006. doi:10.1523/JNEUROSCI.3706-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosenstock T.R., de Brito O.M., Lombardi V., Louros S., Ribeiro M., Almeida S., Ferreira I.L., Oliveira C.R., Rego A.C. FK506 ameliorates cell death features in Huntington's disease striatal cell models. Neurochem. Int. 2011;59:600–609. doi: 10.1016/j.neuint.2011.04.009. doi:10.1016/j.neuint.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 61.Groc L., Choquet D. AMPA and NMDA glutamate receptor trafficking: multiple roads for reaching and leaving the synapse. Cell Tissue Res. 2006;326:423–438. doi: 10.1007/s00441-006-0254-9. doi:10.1007/s00441-006-0254-9. [DOI] [PubMed] [Google Scholar]
- 62.Groc L., Heine M., Cognet L., Brickley K., Stephenson F.A., Lounis B., Choquet D. Differential activity-dependent regulation of the lateral mobilities of AMPA and NMDA receptors. Nat. Neurosci. 2004;7:695–696. doi: 10.1038/nn1270. doi:10.1038/nn1270. [DOI] [PubMed] [Google Scholar]
- 63.Groc L., Heine M., Cousins S.L., Stephenson F.A., Lounis B., Cognet L., Choquet D. NMDA receptor surface mobility depends on NR2A-2B subunits. Proc. Natl. Acad. Sci. USA. 2006;103:18769–18774. doi: 10.1073/pnas.0605238103. doi:10.1073/pnas.0605238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaech S., Banker G. Culturing hippocampal neurons. Nat. Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. doi:10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- 65.Niwa H., Yamamura K., Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. doi:10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 66.Kim M.J., Dunah A.W., Wang Y.T., Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. doi:10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 67.Pacchioni A.M., Vallone J., Worley P.F., Kalivas P.W. Neuronal pentraxins modulate cocaine-induced neuroadaptations. J. Pharmacol. Exp. Ther. 2009;328:183–192. doi: 10.1124/jpet.108.143115. doi:10.1124/jpet.108.143115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boulanger L.M., Lombroso P.J., Raghunathan A., During M.J., Wahle P., Naegele J.R. Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. J. Neurosci. 1995;15:1532–1544. doi: 10.1523/JNEUROSCI.15-02-01532.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suh Y.H., Terashima A., Petralia R.S., Wenthold R.J., Isaac J.T., Roche K.W., Roche P.A. A neuronal role for SNAP-23 in postsynaptic glutamate receptor trafficking. Nat. Neurosci. 2010;13:338–343. doi: 10.1038/nn.2488. doi:10.1038/nn.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tapia L., Milnerwood A., Guo A., Mills F., Yoshida E., Vasuta C., Mackenzie I.R., Raymond L., Cynader M., Jia W., et al. Progranulin deficiency decreases gross neural connectivity but enhances transmission at individual synapses. J. Neurosci. 2011;31:11126–11132. doi: 10.1523/JNEUROSCI.6244-10.2011. doi:10.1523/JNEUROSCI.6244-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]