

Abstract
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a leading monogenic neurodegenerative disorder affecting premutation carriers of the fragile X (FMR1) gene. To investigate the underlying cellular neuropathology, we produced induced pluripotent stem cell-derived neurons from isogenic subclones of primary fibroblasts of a female premutation carrier, with each subclone bearing exclusively either the normal or the expanded (premutation) form of the FMR1 gene as the active allele. We show that neurons harboring the stably-active, expanded allele (EX-Xa) have reduced postsynaptic density protein 95 protein expression, reduced synaptic puncta density and reduced neurite length. Importantly, such neurons are also functionally abnormal, with calcium transients of higher amplitude and increased frequency than for neurons harboring the normal-active allele. Moreover, a sustained calcium elevation was found in the EX-Xa neurons after glutamate application. By excluding the individual genetic background variation, we have demonstrated neuronal phenotypes directly linked to the FMR1 premutation. Our approach represents a unique isogenic, X-chromosomal epigenetic model to aid the development of targeted therapeutics for FXTAS, and more broadly as a model for the study of common neurodevelopmental (e.g. autism) and neurodegenerative (e.g. Parkinsonism, dementias) disorders.
INTRODUCTION
Premutation CGG-repeat expansions (55–200 repeats) within the 5′ non-coding portion of the fragile X (FMR1) gene give rise to the neurodegenerative disorder, fragile X-associated tremor/ataxia syndrome (FXTAS) (1–3). Approximately 1 in 130 women and 1 in 300 men are carriers of premutation FMR1 alleles (4–6), and many of these carriers will develop features of FXTAS in late adulthood. FXTAS arises through a toxic gain of function of the expanded CGG-repeat FMR1 mRNA (7). However, the lack of human neuronal models has impeded our understanding of the detailed mechanism underlying the disorder, in part because the mouse models do not fully recapitulate the clinical (FXTAS) phenotype (8).
From the perspective of the potential development of useful cellular models, induced pluripotent stem cell (iPSC)-based reprogramming of fibroblasts presents many advantages over the use of either post-mortem neural progenitor cells or human embryonic stem cells (hESCs), in particular due to the larger number of subjects available for study. Patient-specific iPSCs are emerging as a powerful tool for disease phenotype investigation and drug screening (9,10). However, population-based studies are still limited by background gene effects in any groupwise comparison. Additionally, in the study of X-linked diseases, an important advantage exists in the ability to generate cellular subclones from single individuals in which exclusively either the maternal or the paternal X allele is active. In the case of the FMR1 gene, female premutation carriers are mosaic for the active allele, with individual cells expressing either normal or mutant (expanded-CGG) FMR1 alleles.
To exploit the advantages afforded by the iPSC technology and an X-linked gene, we have generated multiple fibroblast subclones of individual primary fibroblast lines, with the subclones differing exclusively at the X chromosome. We have subsequently reprogrammed the fibroblast subclones into iPSCs, followed by differentiation into neurons (Fig. 1, graphical summary). In this manner, we have successfully established isogenic, epi-isoautosomal (allelic differences elsewhere in the two X chromosomes) neuron pairs. Using this model system, we show that the premutation-active neurons have defective synapses and neurite outgrowth. Moreover, functional aberrations reflected by activity-dependent calcium transients were also observed in these neurons, indicating that our model is able to recapitulate major features of the morphological and functional disease phenotype. Importantly, we have demonstrated that the morphological and functional abnormalities do not arise as a consequence of lowered fragile X mental retardation protein (FMRP), the levels of which are identical between normal-active and premutation- active neurons.
Figure 1.
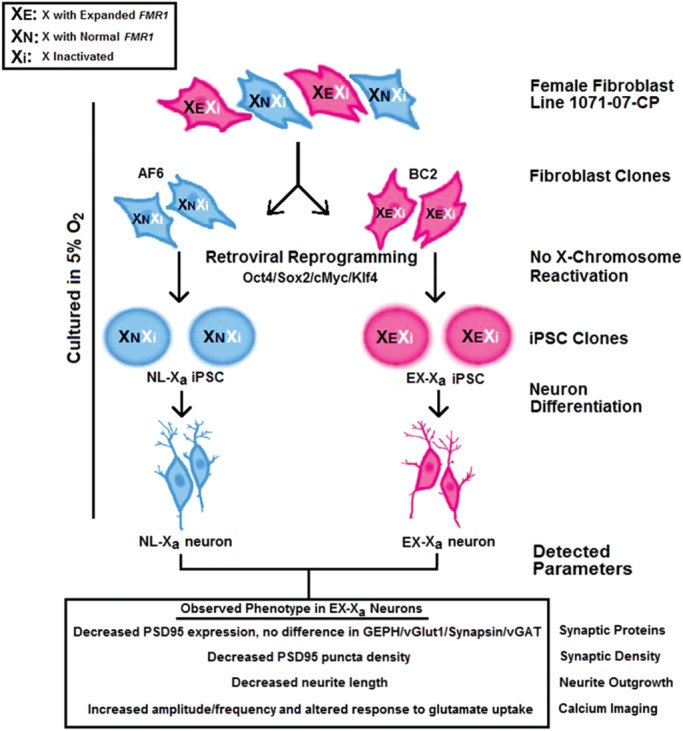
Schematic outline of epi-isoautosomal neuron generation from cloned fibroblasts. A female fibroblast line, 1071-07-CP, heterozygous for FMR1 premutation, was cloned to generate multiple lines expressing exclusively either the normal allele (e.g. AF6 clone) or the expanded allele (e.g. BC2 clone). NL-Xa and EX-Xa iPSCs were then generated using retroviruses expressing four transcription factors (Oct3/4, Sox2, Klf4 and c-Myc) from AF6 and BC2 fibroblast clones, respectively. During the reprogramming, the inactive X chromosome did not reactivate, thus the NL-Xa and EX-Xa iPSCs retained the same FMR1 allele as the donor AF6 and BC2 clones, respectively. Both types of iPSCs were differentiated into NPCs and thence into mature neurons, which were used to identify the FMR1 premutation-specific cellular phenotypes. Decreased postsynaptic density protein 95 (PSD95) protein expression, shortened neurite length and alterations in calcium oscillation and response to glutamate were observed in EX-Xa neurons. The culture protocol was performed under normoxic (physiological) oxygen concentration (5%).
RESULTS
Generation of iPSCs from isogenic, premutation FMR1 fibroblast subclones
Since the FMR1 gene is located on the X chromosome, females generally harbor two alleles, only one of which is active in any given cell. Thus, for female premutation carriers, individual cells express either the normal or the premutation allele; this feature can be exploited to generate, through single-cell subcloning, populations of cells that express exclusively one or the other of the parental alleles.
To obtain pure fibroblast clones for iPSC generation, skin fibroblasts from a 54-year-old female FMR1 premutation carrier (30 and 94 CGG repeats) were subcloned to generate multiple derivative lines, each with either the normal or the expanded FMR1 allele exclusively active (Fig. 2A). Clonality was confirmed for each line by methylation-sensitive restriction digestion followed by a CGG-repeat (genotyping) PCR, as shown for AF6, with an active normal allele (30 CGG repeats; NL-Xa); and for BC2, with an active premutation allele (94 CGG repeats; EX-Xa) (Fig. 3A and B). Interestingly, 42% of the clones generated were EX-Xa (data not shown), suggesting that there was no strong bias against the growth of the expanded clones. AF6 and BC2 were then reprogrammed using retroviruses carrying Oct3/4, Sox2, Klf4 and c-Myc transcription factors (11), resulting in six to seven iPSC clones from each parent fibroblast line, each continuously expressing markers of pluripotency and maintaining a normal karyotype (Fig. 2B, C and E). A teratoma assay demonstrated the in vivo differentiation capacity of iPSCs into three germ layers for each line (Fig. 2D; Supplementary Material, Fig. S1C). Retroviral silencing of all four transgenes was confirmed by qPCR using primers specific for retroviral transcripts (Supplementary Material, Fig. S1C–F) (11).
Figure 2.

iPSC generation from cloned fibroblasts. (A) Schematic procedure for obtaining subclones of a primary fibroblast line to generate clones expressing exclusively either the normal allele (NL-Xa) or the premutation (expanded) allele (EX-Xa) from a female carrier of a premutation CGG-repeat expansion of the FMR1 gene. (B and C) Representative NL-Xa and EX-Xa iPSCs, derived from the corresponding fibroblast clones, show expression of pluripotent markers by RT-PCR and immunostaining. (D) Teratoma assay demonstrated the in vivo differentiation capacity of the iPSCs. (E) Representative karyotyping results reveal normal karyotypes of the iPSCs used in this study. Scale bars, 100μm. See also Supplementary Material, Fig. S1, for more characterization of iPSC clones.
Figure 3.

iPSCs retain the same active (X-chromosomal) FMR1 allele as the donor fibroblast clone. (A) Single-allele expression of normal FMR1 in the donor fibroblast clone AF6, and resulting iPSC clones, revealed by the methylation-sensitive restriction digestion followed by PCR amplification of the CGG repeat. (B) The presence of methylated and unmethylated forms of both normal and expanded FMR1 alleles in the parent fibroblast (Fib) line; however, only the expanded allele is active in the donor fibroblast clone BC2 and its derived iPSCs. (C and D) FISH for XIST RNA in NL-Xa iPSCs and EX-Xa iPSCs, showing focal nuclear signals. Scale bars, 25 μm; +, digested by HpaII; −, no digestion.
Retention of allele-specific X-inactivation in iPSCs
It is crucial to determine whether X-inactivation remains stable during reprogramming; that is, whether the active FMR1 allele in the fibroblast clone remains exclusively active throughout the process of iPSC generation and subsequent redifferentiation. Such stability would enable the production of pure control- and premutation-active isogenic subclone pairs (iPSCs and neurons) from previously subcloned fibroblast lines with selected epigenotypes, which is far more efficient than selecting and expanding allele-specific neural precursor cells (NPCs). We first assessed allele-specific methylation patterns using methylation-sensitive restriction digestion followed by CGG-repeat PCR, which consistently demonstrated that all iPSC clones derived from the NL-Xa fibroblast subclone maintained the active, normal allele (NL-Xa iPSC), and that all iPSCs from the EX-Xa subclone uniformly maintained the active premutation allele (EX-Xa iPSC; Fig. 3A and B). Therefore, all iPSC clones maintained the same active X chromosome as the original fibroblast. As noted, the inactive alleles for lines NL-Xa 7 and 8 showed a slightly smaller CGG-repeat length, which was likely caused by a minor deletion that may have occurred during the subcloning process. RNA fluorescence in situ hybridization (FISH) of XIST, a non-coding RNA associated with the inactive X chromosome, validated the maintenance of X-chromosomal inactivation (XCI) (12) in the current iPSC model; over 80% of iPSCs showed intense staining of intranuclear foci (Fig. 3C and D), resembling the pattern observed in the female fibroblasts (data not shown). The lack of XIST RNA coating on some of the iPSC cells was also observed in other studies (13,14), which may reflect methylation of its promoter during in vitro culture, and/or the release of XIST RNA from the Xi during mitosis (13,14).
Consistent with previous reports on the generation of human iPSCs (13,14), our findings confirm that global X chromosome reactivation does not occur during reprogramming, in contrast to reported reactivation in murine fibroblasts (15). However, our observations do not preclude the possible heterogeneity and dynamics of XCI in human iPSCs demonstrated in other cases (9,16). Moreover, we did not observe any difference in the reprogramming efficiency of normal and mutant fibroblast clones (∼0.017%); in particular, the X-chromosome skewing reported previously (17) was not observed in the current work. Interestingly, ground-state hESCs (18) containing two active X chromosomes were reported to be established under physiological oxygen concentration, although chronic exposure to atmospheric oxygen was sufficient to induce irreversible XCI (19); however, we did not observe the occurrence of XCI in our protocol, which utilized the physiological (normoxic, 5%) oxygen concentration throughout the stages of fibroblast expansion, reprogramming and neuronal differentiation.
Neural differentiation of iPSCs
In the current work, iPSCs were differentiated into NPCs and thence into neurons by co-culturing with human astrocytes, which was reported to facilitate neuronal maturation (10) (Fig. 4A). EX-Xa iPSCs did not manifest any deficits in NPC generation, nor was there any apparent alteration in the cellular proliferation of EX-Xa NPCs (Supplementary Material, Fig. S2). As expected, iPSC-derived neurons were positive for MAP2 (microtubule-associated protein 2) and β-tubulin III, and were outlined with synaptic puncta of synapsin and vesicular glutamate transporter 1 (vGLUT1) (Fig. 4B–D). Comparable fractions of NL-Xa and EX-Xa neurons were labeled by MAP2 following differentiation from NPCs (Fig. 4E), indicating that there is no significant defect in the NPC-to-neuron transition for the EX-Xa allele. In accordance with previous reports (9), the derived neuronal culture was a heterogeneous population (i.e. not a specific neuronal subtype), which we feel is more reflective of the disease process in FXTAS, which involves a variety of neuronal cell types.
Figure 4.

Differentiation of iPSCs to generate neurons. (A) Schematic of neural differentiation, reflecting EB formation, rosette dissection and dissociation, and neuronal differentiation through co-culture with human astrocytes to facilitate neuronal maturation. Composition of EB, NP, NPC and DN media are presented in the Experimental procedure section. (B) The protocol outlined in (A) produced high yields of neurons positive for MAP2 staining; scale bar, 50 µm. (C) Representative images of synapsin outlining the MAP2 positive neurons; scale bar, 50 µm. (D) Representative images of vGLUT1 co-stained with MAP2; scale bar, 20 µm. (E) Neural differentiation efficiency reflected by percentage of MAP2 positive cells. See also Supplementary Material, Fig. S2, for proliferation of NPCs.
Relative FMR1 mRNA and FMRP levels in EX-Xa iPSCs and neurons
To assess the relative expression levels between the normal and premutation-expanded alleles, FMR1 mRNA was measured at several points during the reprogramming process (Fig. 5). Although the mRNA levels for the BC2 fibroblast and EX-Xa neuronal subclones were both elevated relative to their normal counterparts (Fig. 5A and E, respectively), the difference was not large. Such a mild increase of FMR1 mRNA was also observed in EX-Xa iPSCs (Fig. 5C). This behavior is similar to that observed in human brains, where the difference between the normal and premutation mRNA levels is much smaller than in lymphocytes, although the mRNA levels of both normal and premutation FMR1 mRNA are dramatically elevated in brain relative to lymphocytes (20). Nevertheless, the important feature of the EX-Xa neuron is that its FMR1 mRNA carries the expanded CGG repeat necessary for toxicity. Of fundamental importance for the interpretation of the pathogenesis of the premutation cellular phenotype (see Discussion) is the fact that the FMRP levels are essentially identical for normal-active and premutation-active alleles in both the fibroblast and derived neuronal subclones (Fig. 5B and F, respectively). Human astrocytes in the co-culture did not affect the expression differences of FMRP and FMR1 mRNA in derived neurons, as reported above, since similar trends were detected in neurons differentiated at a low ratio of astrocytes (1:10, instead of 1:1, initial plating, Supplementary Material, Fig. S3; additional data not shown). Finally, the expression status of the alleles remained stable through the entire reprogramming of fibroblasts into neurons, with derived neurons retaining, exclusively, the initial X-inactivation status of the original NL-Xa and EX-Xa fibroblast subclones (Fig. 5D).
Figure 5.

FMR1 and FMRP expression in various stages of reprogramming. (A) FMR1 mRNA levels in the female fibroblast line, 1071-07-CP, and its subclones, AF6 (normal) and BC2 (expanded). (B) Comparable FMRP expression levels in BC2 and AF6 fibroblast subclones used in In-cell western analysis. (C) Representative FMR1 transcript expression levels in derived NL-Xa and EX-Xa iPSCs, normalized to a control male fibroblast. (D) Representative genotyping results (CGG-repeat sizes) in neurons, showing that NL-Xa neurons inherited the normal allele, whereas EX-Xa neurons inherited the expanded allele. HpaII digestion (+) cleaves the active (unmethylated) FMR1 allele, so that only the inactive allele is amplified by PCR. (E) FMR1 mRNA levels in neurons harboring the normal-active (NL-Xa-neuron) and expanded-active (EX-Xa-neuron) alleles. (F) FMRP levels in NL-Xa and EX-Xa neurons determined by western blot (representative blots above bar graph). Data in (E) and (F) were obtained from the neurons derived from two different iPSC clones for each NL-Xa and EX-Xa, and from two to three batches of differentiation for each iPSC clone. Data are presented as mean ± SEM; * denotes P< 0.05. See also Supplementary Material, Fig. S3.
Synaptic alterations of EX-Xa neurons
Both reduced dendritic growth and complexity were observed in the premutation mouse model using in vitro hippocampal cultures (21). Moreover, mice expressing premutation Fmr1 alleles show defects in early developmental programs of brain formation, including reduced numbers and lengths of neurites (22). In the current work, although the average number of neurites did not differ between the genotypes (Fig. 6C), EX-Xa neurons consistently had shorter neurite extensions compared with those elaborated by the NL-Xa neurons (Fig. 6D). Moreover, EX-Xa neurons showed a significantly reduced postsynaptic density protein 95 (PSD95) protein expression (Fig. 6E and F) and puncta density (Fig. 6A, B and G) compared with their normal neuronal counterparts, whereas other synaptic proteins, such as GEPH, synapsin and vGLUT1, were expressed at comparable levels.
Figure 6.

Alterations of synaptic proteins and neurites in EX-Xa neurons. (A) Representative images of PSD95 co-stained with β-tubulin III; scale bar, 20 µm. (B) Representative images of synaptic puncta co-localization between PSD95 and vGLUT1 on neurites; scale bar, 20 µm. (C) No change in the numbers of neurites from the soma in EX-Xa neurons. (D) Decreased neurite length in EX-Xa neurons. (E) Quantified synaptic protein expression showing the decreased level of PSD95 in EX-Xa neurons. (F) Representative immunoblot of synaptic proteins (PSD95, synapsin, vGLUT1, gephyrin and vesicular GABA transporter) detected in NL-Xa and EX-Xa neurons. (G) Decreased PSD95 synaptic density in EX-Xa neurons. All data were generated from neurons derived from two different iPSC clones for each NL-Xa and EX-Xa, and from two to three batches of differentiation for each iPSC clone. Data were presented as mean ± SEM, * denotes P< 0.05.
Aberrant calcium activity of EX-Xa neurons
To examine the consequences of the premutation allele on neuronal function, we used calcium imaging to measure spontaneous Ca2+ oscillatory behavior and glutamate-evoked neuronal activity. In the iPSC-derived neurons from Rett syndrome patients, Ca2+ oscillations exhibited a reduced frequency and transient amplitude compared with the iPSC-derived neurons from unaffected individuals, suggesting that those mutant neurons have an impaired neuronal network activity (9). In contrast, the iPSC-derived neurons from schizophrenic patients show Ca2+ oscillatory activity similar to those of unaffected individuals (10). Primary hippocampal neuronal cultures from a FXTAS mouse model display an enhanced neuronal electric activity characterized by clustered burst neuronal firing and clustered spontaneous Ca2+ oscillations (23). Here, we found that EX-Xa neurons displayed more spontaneous Ca2+ transients than NL-Xa neurons (Fig. 7A and 7B), despite there being comparable fractions (~20%) of the NL-Xa and EX-Xa neurons that exhibited spontaneous Ca2+ oscillations within each randomly chosen microscopic field. Moreover, the mean amplitude of spontaneous Ca2+ transients was significantly larger in the EX-Xa neurons. These spontaneous Ca2+ transients were quickly abolished upon the addition of tetrodotoxin (TTX), indicating that spontaneous oscillatory activity was dependent on the activation of Na+ channels (Fig. 7C). In addition, although both NL-Xa and EX-Xa neurons respond to glutamate, the response to glutamate was much more profound in the EX-Xa population; essentially, all EX-Xa neurons displayed a sustained elevation of intracellular Ca2+ concentration following application of 150 µm of glutamate, whereas only about one-quarter of the NL-Xa neurons showed a sustained Ca2+ response (Fig. 7D and E; Supplementary Material, Fig. S4). In the remaining NL-Xa neurons, glutamate produced a transient rise in intracellular Ca2+ that recovered to baseline within 5 min.
Figure 7.

Altered action potential-dependent Ca2+ transients and glutamate response in EX-Xa neurons. (A) Representative traces for spontaneous calcium transients; (B) EX-Xa neurons displayed a higher frequency of Ca2+ spikes (left axis) and spike amplitude (right axis)(n= 56 neurons for NL-Xa; n= 80 neurons for EX-Xa); (C) A representative trace of TTX (1 μM) abrogated spontaneous Ca2+ transients. (D) Averaged Ca2+ response of NL-Xa and EX-Xa neurons to glutamate exposure; (E) quantification of the percentage of cells with sustained elevation of intracellular Ca2+ concentration ([Ca2+]i). 26.6 ± 7.8% NL-Xa neurons (n = 5 wells) displayed sustained elevation on [Ca2+]i after 150 µM glutamate exposure, whereas this percentage increased to 98.8 ± 1.3% (n= 8 wells) in EX-Xa neurons. These data were collected from three batches of neurons derived from two different iPSC clones for each NL-Xa and EX-Xa. Data are presented as mean ± SEM, ** denotes P < 0.01. See also Supplementary Material, Fig. S4.
DISCUSSION
This study is the first to observe functional differences in iPSC-derived neuronal cells from an individual with an expanded (premutation) FMR1 CGG-repeat allele. A unique strength of the current approach is that the isogenic, epi-isoautosomal background should largely eliminate the influences of differing genetic backgrounds on the neuronal phenotype (except for residual effects of allelic differences in other X-chromosomal genes). By reprogramming from clonal fibroblasts, our study provides convincing evidence that no reactivation of the inactive X chromosome occurred during the reprogramming. This epigenetic stability, throughout the process of neuronal differentiation, has permitted the generation of epi-isoautosomal neurons (with differing Xa epigenotypes) to study the influences of the premutation allele on morphological and functional abnormalities of the derived neurons. Other groups (13,24) have generated epi-isoautosomal neurons [referred to as ‘isogenic’ (13)] from mixed fibroblasts of Rett syndrome patients by taking advantage of non-random XCI. However, those studies did not explore functional neuronal phenotypes.
We also observed that there was no CGG-repeat length instability throughout the entire reprogramming process (data not shown). The observed repeat length stability is in contrast to a previous report (25), in which iPSCs derived from FMR1 full mutation fibroblasts appeared to carry different repeat sizes than the original lines; however, that observation is likely to reflect cryptic allele-size mosaicism, which is common among full mutation carriers. An earlier study of iPSC generation with full mutation alleles (26) did not observe any repeat instability.
Since the influence of neuronal activity on dendritic growth is primarily mediated by spatial and temporal changes in intracellular Ca2+ (27,28), the higher frequency of amplified Ca2+ transients observed in iPSC-derived, premutation-expressing neurons is likely to influence not only the rate and the extent of neurite growth, but also the refinement of synaptic connections, as evidenced by reduced PSD95 puncta density. Moreover, the growth of neuronal dendrites is strongly influenced by Ca2+ oscillatory activity; with low-to-moderate intracellular Ca2+ signals promoting dendritic growth, and large increases causing dendritic retraction (27,28).
Results from our recent work (23) with cultured premutation mouse (preCGG) neurons show that neither vesicular GABA transporter (VGAT) nor vGLUT1 expression (normalized to β-actin) differ in preCGG compared with wildtype (WT) at 7 days in vitro (DIV; no differences early in development at 7DIV)—a stage of development similar to the human neurons studied in the current work. Even though at 21DIV the preCGG mouse neurons have lower vGLUT1 and VGAT markers by western blotting, their ratio does not differ from WT. Collectively, these data suggest that the composition of glutamatergic and GABAergic neurons does not differ between preCGG and WT neurons in early (in vitro) development, and that even mature neurons do not show a significant shift in the ratio of vGlut and VGAT between the genotypes. The mouse data suggest that the differences observed in the human neurons reflect functional differences in vGlut responses due to the FMR1 mutation rather than a shift in the fraction of glutamatergic and GABAergic neuronal populations; however, additional studies would be needed to fully resolve this issue.
Although the principal mode of pathogenesis in the premutation-associated disorders appears to be through the toxic gain of function of the expanded CGG-repeat FMR1 mRNA (7), the slight-to-moderate decreases in FMRP levels in the upper portion of the premutation range observed in both humans and preCGG mouse models have raised the possibility that the reduced FMRP levels may contribute to FXTAS and/or developmental involvement (e.g. autism; 21,22,29,30). Indeed, the decreased levels of FMRP could also play a modifying role in the altered neuronal migration in the embryonic neocortex of the premutation mouse (150 CGG repeats) (22). Thus, the sufficiency of mRNA toxicity for the abnormal premutation neuronal phenotype has remained unresolved. In this regard, we observed no diminution in FMRP levels, either between NL-Xa and EX-Xa fibroblasts or between the corresponding NL-Xa and EX-Xa neurons. Thus, our results establish that reduced FMRP levels are not necessary for the development of the abnormal neuronal phenotype and, consequently, that the altered neuronal morphology, Ca2+ oscillatory behavior and the response to glutamate observed in the affected (EX-Xa) neurons arise through CGG-repeat RNA toxicity.
We note that a recent study using iPSC-derived neurons with full mutation FMR1 alleles failed to observe FMRP in a mosaic fibroblast line (166 and 800 CGG repeats), and greatly reduced FMRP in premutation and mixed iPSC lines, despite greatly elevated mRNA (25). This result may reflect a specific feature of the derived lines, since no other instance of the profound reduction of FMRP for a premutation allele has been reported in either human (31) or mouse (21) culture models.
Our discovery of dysregulated Ca2+ signals in iPSC-derived FMR1 premutation neurons has already revealed important clues regarding the link between the expanded CGG-repeat FMR1 gene (mRNA- versus protein-based pathogenesis) and the progression of premutation carriers to clinical FXTAS. Although the more specific mechanisms for the amplified Ca2+ activity reported here remain to be elucidated, numerous reports have demonstrated that both ionotropic and metabotropic glutamate receptors regulate the properties of Ca2+ oscillation in neurons (32,33). A demonstrated consequence of the imbalance of excitatory and inhibitory neuronal transmission, primarily through enhanced type I metabotropic glutamate receptors activity and decreased presynaptic gamma-aminobutyric acid release, is the dysregulation of Ca2+ and neuronal network electric activity in premutation hippocampal neuronal cultures (23). Thus, from the clinical perspective, our observation that glutamate modulates the behavior of the Ca2+ signals in the iPSC-derived neurons suggests that mGluR inhibitors, currently in clinical trials for fragile X syndrome (34,35), may have broader applicability to the premutation-associated disorders as well.
Here we have generated isogenic, neuronal clonal lines from a female patient who is heterozygous for a premutation FMR1 allele. Since X-inactivation is maintained during the process of generating the iPSC-derived neuronal lines, each line expresses (exclusively) either the normal or the premutation FMR1 allele. Due to anticipated differences in the expression of other genes on the paternal and maternal X chromosomes, the current study should more properly be considered an isoautosomal comparison; follow-up studies on additional patients are warranted to test for secondary effects of other X-linked functional polymorphisms. Nevertheless, our study provides both a greater understanding of the pathogenesis of FXTAS and the basis (and a novel model system) for the development of targeted therapeutic approaches for FXTAS and other premutation-associated disorders, including autism spectrum disorders and premature ovarian insufficiency. Our approach represents a unique isogenic, X-chromosomal epigenetic model to aid the study of neuronal pathology of X-linked disease, thereby facilitating the development of targeted therapeutic interventions for additional disorders.
MATERIALS AND METHODS
Fibroblast cloning
The female fibroblast line (1071-07-CP) from a 54-year-old asymptomatic FMR1 premutation carrier (30 and 94 CGG repeats) was subcloned by low-density (single-cell) seeding and subsequent expansion of a single cell into a full population. Culture was performed at 37°C in the 5% CO2 and 5% O2 atmosphere using initially AmnioMAX-C100 Basal Medium with 15% AmnioMAX-C100 Supplement (Invitrogen, Carlsbad, CA, USA), with 20 ng/ml of human basic fibroblast growth factor (bFGF; Invitrogen); and later a 1:1 solution of AmnioMAX-C100 and RPMI-1640 Basal Medium supplemented with 10% fetal bovine serum (Invitrogen), 1× penicillin/streptomycin/glutamine (Invitrogen), 1% non-essential amino acids (Invitrogen), and 1:250 fungizone (J R Scientific, Woodland, CA, USA).
iPSC generation and characterization
Two fibroblast clones (AF6 and BC2) were transduced with retroviruses expressing the transcription factors Oct3/4, Sox2, Klf4 and c-Myc (11). Transduced fibroblasts were then split onto mouse embryonic fibroblasts and cultured in iPSC medium containing 80% knockout Dulbecco's modified eagle medium (DMEM)/F12 (Invitrogen), 20% knockout serum replacement (Invitrogen), 10 ng/ml bFGF, 1 mm glutamax (Invitrogen), 0.1 mm β-mercaptoethanol (Invitrogen) and 1% non-essential amino acid solution. Stem cell-like colonies were selected and manually passaged 3–4 weeks later. After establishment, iPSCs were maintained in mTeSR medium (Stem Cell Technologies, Vancouver, British Columbia) with matrigel. Teratoma assays were performed by injecting the iPSC subcutaneously into the hind limb of NOD SCID gamma (NSG) mice. Tumors were harvested, sectioned and stained with hematoxylin and eosin.
Neural progenitor cell-derivation and neuron differentiation
iPSCs cultured in mTeSR medium were detached using dispase (1 mg/ml, Stem Cell Technologies), and cultured in suspension as embryoid bodies in iPSC medium minus bFGF (EB medium) for 4 days. To enhance the neural differentiation, 500 ng/ml noggin (StemRD, Burlingame, CA, USA) and 10 μm SB431542 (Sigma, St Louis, MO, USA) were added (36). Embryoid bodies were plated on petri dishes coated with growth factor-reduced matrigel (BD Biosciences, San Jose, CA, USA), with medium containing DMEM/F12 with Glutamax, NEAA, 1% N2 (Invitrogen) and 20 ng/ml bFGF (NP medium) for 1 week. Neural rosettes were manually dissected for further dissociation into single NPCs using accutase (Stem Cell Technologies). The NPC population was expanded in NPC medium [DMEM/F12 with Glutamax, NEAA, 1% N2, 20 ng/ml bFGF with 10 ng/ml EGF (R&D Systems, Minneapolis, MN, USA) and 0.1% B27].
For neuronal differentiation, NL-Xa and EX-Xa NPCs at the same passages were co-cultured with human fetal astrocytes (ScienCell, Carlsbad, CA, USA) in medium consisting of neurobasal medium, 1% B27, 20 ng/ml GDNF, 20 ng/ml BDNF, 0.2 mm ascorbic acid and 0.5% FBS (DN medium) for 4–5 weeks. Human astrocytes (passage 5–6) were plated in 6-well plates and acid-etched glass-bottom dishes at 1.5 × 104 cells/cm2 coated with polyornithine/laminin. The following day, NPCs were seeded on top of astrocytes at 1 × 104 cells/cm2. NL-Xa1, NL-Xa5, EX-Xa3 and EX-Xa7 NPCs were used to generate four batches of NL-Xa and EX-Xa neurons.
Genotyping/X-inactivation analysis
To determine the repeat length, CGG-repeat PCR was performed as described previously (37). The activation status of the FMR1 allele was assessed by digesting 1 µg of genome DNA (gDNA) with HpaII restriction endonuclease (New England Biolabs, Ipswich, MA, USA) in a 20 µl, 6-h reaction, using 3 µl of the digest as a template for the PCR. The allele that failed to amplify after nuclease digestion was designated the active allele. We note that the presence of background astrocyte DNA would have no effect on the conclusions drawn from derived neurons (Fig. 5D). In particular, the normal CGG-repeat male astrocyte gDNA, migrating identically to the female normal allele, is present in only a single copy and is exclusively active (always digested by HpaII). Thus, it would never contribute to the intensities of the premutation alleles, active or inactive. For the inactive expanded allele (HpaII resistance), the absence of any normal allele establishes exclusivity—this would not be influenced by any normal allele from the astrocyte. For the inactive normal allele, only the female gDNA would remain following HpaII treatment. Thus, the presence of astrocyte gDNA will not have any influence on the assignment of epigenotypes (active versus inactive alleles).
Immunostaining
Cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton-X. After blocking with 10% goat serum, cells were incubated with the following primary antibodies at 4°C overnight: rabbit anti-Sox1 (AB15766, Millipore, Billerica, MA, USA), mouse anti-nestin (MAB1259, R&D Systems), rabbit anti-TuJ1 (ab24629, Abcam, Cambridge, MA, USA), mouse anti-MAP2 (ab11267, Abcam), rabbit anti-synapsin (AB1543P, Millipore) and mouse anti-PSD95 (75-028, NeuroMab, Davis, CA, USA). Appropriate fluorescence-labeled secondary antibodies were applied, and nuclei were counterstained with 4′,6-diamidino-2-phenylindole (H1200, Vector Labs, Burlingame, CA, USA). Images were captured on a Nikon fluorescence microscope or Zeiss confocal microscope.
Synaptic protein expression
iPSC-derived neurons were harvested 4–6 weeks in RIPA buffer (89901, Thermo, Rochester, NY, USA) with proteinase inhibitor cocktail (1861281, Thermo). Thirty micrograms of total protein were loaded in 4–20% Tris–HCl gel (456-1093, Bio-Rad, Hercules, CA, USA) and separated by electrophoresis. The blot was probed with mouse anti-MAP2 (ab11267, Abcam), rabbit anti-synapsin (AB1543P, Millipore), rabbit anti-VGAT (131002, Synaptic Systems, Goettingen, Germany), mouse anti-VGLUT1 (75-066, NeuroMab), mouse anti-PSD95 (75-028, NeuroMab) and mouse anti-gephyrin (147-111, Synaptic Systems). Detection was performed with Supersignal West Pico Chemiluminescent substrate (34078, Thermo). MAP2 and β-actin served as the internal controls. Data were normalized to MAP2.
Calcium imaging
At 4–6 weeks, cultured neurons were incubated with dye-loading buffer containing 4 μm Fluo-4 (Invitrogen) and 0.5 mg/ml BSA in Locke's buffer (8.6 mm HEPES, 5.6 mm KCl, 154 mm NaCl, 5.6 mm glucose, 1.0 mm MgCl2, 2.3 mm CaCl2 and 0.0001 mm glycine, pH 7.4), and incubated for 30 min. After washing with Locke's buffer, the cells were excited at 488 nm with a fluorescein isothiocyanate (FITC) filter; the fluorescence signals were recorded at 33 frames/s using a charge-coupled device camera (model 512B; Photometrics, Tucson, AZ) under a ×40 objective lens attached to an IX-71 inverted microscope (Olympus, Center Valley, PA, USA) controlled by EasyRatioPro software (Photon Technologies International, Birmingham, NJ, USA). The amplitude of fluorescence signals for each region of interest was presented as relative fluorescence changes (ΔF/F) after background subtraction. A fluorescence change (ΔF/F) exceeding 10% was considered a positive Ca2+ transient. The Ca2+ transient frequency and amplitude were manually counted over a 10-min period.
Statistical analysis
Two different iPSC clones for each NL-Xa and EX-Xa were differentiated into neurons, in two to three independent experiments, to generate the data for morphological and functional analysis on derived neurons. Data are presented as mean ± SEM and t-test was performed for the comparisons between groups.
SUPPLEMENTARY MATERIAL
FUNDING
This project is funded by a National Institutes of Health Challenge (ARRA) grant [RC1 AG036022 to P.J.H.] and by a National Institutes of Health Interdisciplinary Research Consortium (IRC) grant [UL1 DE019583 to P.J.H., RL1 AG032119 to P.J.H. and I.N.P., RL1 AG032115 to R.J.H]. J.L. is supported by the Shriners Hospital Fellowship and is the 2011 Wing Kai Fat Memorial Fund scholar; N.G. is a scholar of the California Institute for Regenerative Medicine (CIRM) Bridges Program at CSUS and UC Davis [CIRM TB1-01184]; J.N. is the recipient of an NIH Director's Transformative Grant [R01 GM099688]. This research is also supported by an unrestricted research grant from J.B. Johnson Foundation (I.N.P.), and by facilities from the UC Davis MIND Institute.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to express their thanks to the families who have supported their fragile X research program.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Hagerman R.J., Leehey M., Heinrichs W., Tassone F., Wilson R., Hills J., Grigsby J., Gage B., Hagerman P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- 2.Berry-Kravis E., Abrams L., Coffey S.M., Hall D.A., Greco C., Gane L.W., Grigsby J., Bourgeois J.A., Finucane B., Jacquemont S., et al. Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov. Disord. 2007;22:2018–2030. doi: 10.1002/mds.21493. quiz 2140. [DOI] [PubMed] [Google Scholar]
- 3.Jacquemont S., Hagerman R.J., Leehey M.A., Hall D.A., Levine R.A., Brunberg J.A., Zhang L., Jardini T., Gane L.W., Harris S.W., et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291:460–469. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- 4.Hagerman P.J. The fragile X prevalence paradox. J. Med. Genet. 2008;45:498–499. doi: 10.1136/jmg.2008.059055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez-Revenga L., Madrigal I., Pagonabarraga J., Xuncla M., Badenas C., Kulisevsky J., Gomez B., Mila M. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur. J. Hum. Genet. 2009;17:1359–1362. doi: 10.1038/ejhg.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song F.J., Barton P., Sleightholme V., Yao G.L., Fry-Smith A. Screening for fragile X syndrome: a literature review and modelling study. Health Technol. Assess. 2003;7:1–106. doi: 10.3310/hta7160. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Arocena D., Hagerman P.J. Advances in understanding the molecular basis of FXTAS. Hum. Mol. Genet. 2010;19:R83–R89. doi: 10.1093/hmg/ddq166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berman R.F., Willemsen R. Mouse Models of Fragile X-Associated Tremor Ataxia. J. Investig. Med. 2009;57:837–841. doi: 10.231/JIM.0b013e3181af59d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marchetto M.C., Carromeu C., Acab A., Yu D., Yeo G.W., Mu Y., Chen G., Gage F.H., Muotri A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brennand K.J., Simone A., Jou J., Gelboin-Burkhart C., Tran N., Sangar S., Li Y., Mu Y., Chen G., Yu D., et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 12.Lyon M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.) Nature. 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- 13.Cheung A.Y., Horvath L.M., Grafodatskaya D., Pasceri P., Weksberg R., Hotta A., Carrel L., Ellis J. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum. Mol. Genet. 2011;20:2103–2115. doi: 10.1093/hmg/ddr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tchieu J., Kuoy E., Chin M.H., Trinh H., Patterson M., Sherman S.P., Aimiuwu O., Lindgren A., Hakimian S., Zack J.A., et al. Female human iPSCs retain an inactive X chromosome. Cell Stem Cell. 2010;7:329–342. doi: 10.1016/j.stem.2010.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maherali N., Sridharan R., Xie W., Utikal J., Eminli S., Arnold K., Stadtfeld M., Yachechko R., Tchieu J., Jaenisch R., et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 16.Bruck T., Benvenisty N. Meta-analysis of the heterogeneity of X chromosome inactivation in human pluripotent stem cells. Stem Cell Res. 2011;6:187–193. doi: 10.1016/j.scr.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 17.Pomp O., Dreesen O., Leong D.F., Meller-Pomp O., Tan T.T., Zhou F., Colman A. Unexpected X chromosome skewing during culture and reprogramming of human somatic cells can be alleviated by exogenous telomerase. Cell Stem Cell. 2011;9:156–165. doi: 10.1016/j.stem.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Nichols J., Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4:487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 19.Lengner C.J., Gimelbrant A.A., Erwin J.A., Cheng A.W., Guenther M.G., Welstead G.G., Alagappan R., Frampton G.M., Xu P., Muffat J., et al. Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell. 2010;141:872–883. doi: 10.1016/j.cell.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 20.Tassone F., Hagerman R.J., Garcia-Arocena D., Khandjian E.W., Greco C.M., Hagerman P.J. Intranuclear inclusions in neural cells with premutation alleles in fragile X-associated tremor/ataxia syndrome. J. Med. Genet. 2004;41:e43. doi: 10.1136/jmg.2003.012518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y., Tassone F., Berman R.F., Hagerman P.J., Hagerman R.J., Willemsen R., Pessah I.N. Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum. Mol. Genet. 2010;19:196–208. doi: 10.1093/hmg/ddp479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cunningham C.L., Martinez Cerdeno V., Navarro Porras E., Prakash A.N., Angelastro J.M., Willemsen R., Hagerman P.J., Pessah I.N., Berman R.F., Noctor S.C. Premutation CGG-repeat expansion of the Fmr1 gene impairs mouse neocortical development. Hum. Mol. Genet. 2011;20:64–79. doi: 10.1093/hmg/ddq432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao Z., Hulsizer S., Tassone F., Tang H.T., Hagerman R.J., Rogawski M.A., Hagerman P.J., Pessah I.N. Clustered burst firing in FMR1 premutation hippocampal neurons: amelioration with allopregnanolone. Hum. Mol. Genet. 2012 doi: 10.1093/hmg/dds118. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim K.Y., Hysolli E., Park I.H. Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA. 2011;108:14169–14174. doi: 10.1073/pnas.1018979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheridan S.D., Theriault K.M., Reis S.A., Zhou F., Madison J.M., Daheron L., Loring J.F., Haggarty S.J. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS ONE. 2011;6:e26203. doi: 10.1371/journal.pone.0026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urbach A., Bar-Nur O., Daley G.Q., Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lohmann C., Wong R.O. Regulation of dendritic growth and plasticity by local and global calcium dynamics. Cell Calcium. 2005;37:403–409. doi: 10.1016/j.ceca.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Segal I., Korkotian I., Murphy D.D. Dendritic spine formation and pruning: common cellular mechanisms. Trends Neurosci. 2000;23:53–57. doi: 10.1016/s0166-2236(99)01499-x. [DOI] [PubMed] [Google Scholar]
- 29.Brouwer J.R., Huizer K., Severijnen L.A., Hukema R.K., Berman R.F., Oostra B.A., Willemsen R. CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J. Neurochem. 2008;107:1671–1682. doi: 10.1111/j.1471-4159.2008.05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Entezam A., Biacsi R., Orrison B., Saha T., Hoffman G.E., Grabczyk E., Nussbaum R.L., Usdin K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. 2007;395:125–134. doi: 10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng Y., Zhang F., Lokey L.K., Chastain J.L., Lakkis L., Eberhart D., Warren S.T. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995;268:731–734. doi: 10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- 32.Nunez L., Sanchez A., Fonteriz R.I., Garcia-Sancho J. Mechanisms for synchronous calcium oscillations in cultured rat cerebellar neurons. Eur. J. Neurosci. 1996;8:192–201. doi: 10.1111/j.1460-9568.1996.tb01180.x. [DOI] [PubMed] [Google Scholar]
- 33.Dravid S.M., Murray T.F. Spontaneous synchronized calcium oscillations in neocortical neurons in the presence of physiological [Mg(2+)]: involvement of AMPA/kainate and metabotropic glutamate receptors. Brain Res. 2004;1006:8–17. doi: 10.1016/j.brainres.2004.01.059. [DOI] [PubMed] [Google Scholar]
- 34.Levenga J., de Vrij F.M., Oostra B.A., Willemsen R. Potential therapeutic interventions for fragile X syndrome. Trends Mol. Med. 2010;16:516–527. doi: 10.1016/j.molmed.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L.W., Berry-Kravis E., Hagerman R.J. Fragile X: leading the way for targeted treatments in autism. Neurotherapeutics. 2010;7:264–274. doi: 10.1016/j.nurt.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chambers S.M., Fasano C.A., Papapetrou E.P., Tomishima M., Sadelain M., Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Filipovic-Sadic S., Sah S., Chen L., Krosting J., Sekinger E., Zhang W., Hagerman P.J., Stenzel T.T., Hadd A.G., Latham G.J., et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin. Chem. 2010;56:399–408. doi: 10.1373/clinchem.2009.136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.