

Abstract
Complex I (CI, NADH ubiquinone oxidoreductase), the largest complex of the respiratory chain, is composed of 45 structural subunits, 7 of which are encoded in mtDNA. At least 10 factors necessary for holoenzyme assembly have been identified; however, the specific roles of most of them are not well understood. We investigated the role of NDUFAF3, NDUFAF4, C8orf38 and C20orf7, four early assembly factors, in the translation of the mtDNA-encoded CI structural subunits. Transient, or stable, siRNA-mediated knock-down of any of these factors abrogated the assembly of CI, and resulted in a specific decrease in the labeling of the ND1 subunit in a pulse translation experiment, whereas knock-down of NDUFAF2, a late assembly factor, did not affect ND1 translation. Pulse-chase experiments in cells knocked down for NDUFAF3 showed that the half-life of ND1 in the chase was reduced 4-fold, fully accounting for the decrease in pulse labeling. Transient, short-term knock-down of the m-AAA protease AGF3L2 in cells that had been depleted of any of the early CI assembly factors completely rescued the ND1 labeling phenotype, confirming that it is not a synthesis defect, but rather results from rapid proteolysis of newly synthesized ND1. NDUFAF3 co-immunoprecipitated with NDUFAF4, and three matrix arm structural subunits (NDUFS2, NDUFA9, NDUFS3) that are found in a 400 kDa assembly intermediate containing ND1. These data suggest that the four early CI assembly factors have non-redundant functions in the assembly of a module that docks and stabilizes newly synthesized ND1, nucleating assembly of the holoenzyme.
INTRODUCTION
Deficiencies in the activity of complex I (CI) are reported to be the most common cause of mitochondrial disease in infants (1,2). They are associated with a wide variety of clinical phenotypes, most commonly affecting the brain, cardiac and skeletal muscles (reviewed in 3). CI is composed of 38 nuclear-encoded and 7 mtDNA-encoded subunits that assemble to form an active holoenzyme of ∼980 kDa. Mutational analyses have identified defects in all of the core structural subunits (those with bacterial orthologues) and several of the so-called supernumerary subunits of the complex (3); however, these mutations explain <50% of the cases of CI deficiency (4), implicating assembly factors as an important cause of disease.
In the past several years, mutations in a large number of CI assembly factor genes have been identified, including NDUFAF1 (5), NDUFAF2 (B17.2L) (6), NDUFAF3 (7), NDUFAF4 (8), C8orf38 (9), C20orf7 (10), FOXRED1 and NUBPL (4,11) and ACAD9 (12). Additionally, the deficiencies in AIF (13) and Ecsit (14) have also been shown to impair the assembly of the CI holoenzyme. The precise molecular role of most of these assembly factors is not well understood, and it is likely that many more factors remain to be identified. As a result, we have an incomplete model for the assembly of the holoenzyme.
CI biogenesis has been investigated in many cell lines from patients with isolated CI deficiency (15,16), and by studying the incorporation of tagged or radio-labeled structural subunits in the assembly pathway (17–19). These approaches have identified a number of what appear to be bonefide assembly intermediates, and a consensus model for CI assembly incorporating these data has been proposed (3). The assembly pathway is thought to start with two separate intermediates, one containing hydrophilic nuclear-encoded subunits (NDUFS2, NDUFS3, NDUFS7, NDUFS8 and NDUFA9), which assembles with a membrane-associated intermediate containing the mtDNA-encoded subunit ND1 to form a 400 kDa subcomplex (15,19). This nucleates the assembly process and the rest of the assembly pathway continues in a stepwise manner in which, principally, two other modules, one comprising an NADH dehydrogenase module (6,19) and the other the distal proton pumping membrane module (20) are added to complete assembly of the holoenzyme. There is also some evidence that individual subunits can be exchanged in the fully assembled enzyme, either as individual proteins, or by modular exchange (19).
Four of the CI assembly factors that have been identified in patient studies: NDUFAF3 (C3orf60), NDUFAF4 (C6orf66), C8orf38 and C20orf7, are thought to play a role in the early steps of the assembly pathway, but their precise roles remain unknown. These nuclear-encoded proteins are targeted to mitochondria where they associate with the inner mitochondrial membrane, but none appear to be integral membrane proteins. NDUFAF3 has been shown to co-immunoprecipitate with NDUFAF4, and with the CI structural subunits NDUFS2 and NDUFS3, consistent with a role in early assembly (7). Pulse-chase radiolabeling of mtDNA-encoded polypeptides followed by two-dimensional gel analysis of CI assembly in C20orf7 and C8orf38 patient lines showed undetectable ND1 at early chase times, also indicative of early assembly defects (9,10). The block in CI assembly due to mutations in C8orf38 was proposed to result from an inhibition of the synthesis of the ND1 subunit, suggesting that it plays a role in the synthesis of the ND1 subunit, perhaps as a translational activator (9).
In this study, we show that siRNA-mediated knock-down of all four early CI assembly factors (NDUFAF3, NDUFAF4, C8orf38 and C20orf7), but not the late assembly factor NDUFAF2, strongly reduces ND1 radiolabeling in a pulse mitochondrial translation experiment. Chase experiments showed that this was due to rapid proteolysis of the newly synthesized ND1 subunit. This phenotype could be rescued by transient siRNA-mediated knock-down of the mitochondrial inner membrane m-AAA protease AFG3L2, the enzyme responsible for quality control of mitochondrial membrane proteins (21).
RESULTS
Knock-down of NDUFAF3 impairs CI assembly
To investigate the function of NDUFAF3 (C3orf60), we transiently knocked it down in immortalized human skin fibroblasts using two different RNAi targets (KD-A and KD-B in Fig 1A). NDUFAF3 was undetectable by immunoblot analysis after 6 days of this treatment, resulting in decreased levels of several nuclear-encoded CI structural subunits including NDUFS2 and NDUFA9, and the mtDNA-encoded subunit ND1 (Fig. 1A). We performed a Blue-Native PAGE (BN-PAGE) analysis on the same cells (siRNA construct 1094. KD-B) to determine the steady-state levels of each of the five complexes in the oxidative phophorylation system. The assembly and in-gel activity of CI were specifically decreased to almost undetectable levels in these cells (Fig. 1B). A second dimension (denaturing) gel showed an overall decrease in the ND1 and NDUFS2 subunits, suggesting that NDUFAF3 knock-down abolishes CI assembly at an early stage (Fig. 1C) as had previously been suggested (7).
Figure 1.

Analysis of NDUFAF3 knock-down cell lines. (A) SDS–PAGE immunodetection of NDUFAF3 and CI subunits (NDUFS2, NDUFA9 and ND1) in control (MCH64), Alexa (fluorescent control siRNA), NDUFAF3-KD-A and NDUFAF3-KD-B (double-stranded siRNA duplexes stealth-957 and stealth-1094, respectively) cell lines. The SD70 subunit of complex II is shown as a loading control. (B) BN-PAGE analysis of NDUFAF3-KD-B cell line and controls. OXPHOS complexes were detected with subunit-specific antibodies. In-gel activity (IGA) of CI. (C) A second dimension denaturing gel of NDUFAF3-KD-B cell line and controls were immunobloted with antibodies against the CI subunits NDUFS2 and ND1. Co I and Co II lines refer to fully assembled complexes. Co II is shown as a loading control.
In order to identify the specific stage of CI assembly at which NDUFAF3 works, NDUFAF3 was C-terminal tagged with a hemagglutinin epitope (NDUFAF3-HA), stably expressed in human skin fibroblasts and analyzed by BN-PAGE (Fig. 2A). NDUFAF3 was detected in an early assembly intermediate of 200 kDa, consistent with the 2D-PAGE result. NDUFAF3 was also detected as part of a 700 kDa subcomplex containing the mtDNA-encoded subunit ND1, and in the 830 kDa subcomplex just before CI gets fully assembled, supporting a role for NDUFAF3 as a CI assembly factor (Fig. 2A).
Figure 2.

Analysis of NDUFAF3-HA association with CI intermediates. (A) BN-PAGE of NDUFAF3-HA. Immunoblotting against the mitochondrially encoded subunit ND1 (left) and hemagglutinin epitope (right). Co II is shown as a loading control. (B) NDUFAF3-HA immunoprecipitation analysis. Input (IN) and elution (E) fractions were analyzed by SDS–PAGE analysis. NDUFAF3 was detected using an anti-hemagglutinin antibody and CI structural subunits NDUFS2, NDUFA9 and NDUFS3 were identified with specific antibodies.
To further investigate the role of NDUFAF3 in early CI assembly, we performed immunoprecipitation experiments on a cell line expressing NDUFAF3-HA, and used mass spectrometry and immunoblotting to identify co-immunoprecipitating proteins. This experiment identified several nuclear-encoded structural subunits of the matrix arm of CI: NDUFS2, NDUFA9 and NDUFS3 (Fig. 2B), and the assembly factor NDUFAF4 (which appeared in the mass spectrometry analysis, but for which we do not have an antibody). Similar results were reported by using a TAP-tagged version of NDUFAF3 (7).
Knock-down of NDUFAF3 causes a specific decrease in ND1 labeling in a pulse translation experiment
To test if NDUFAF3 is involved in the translation of mtDNA-encoded polypeptides, cells transiently transfected with siRNA were pulse-labeled with [35S]methionine/cysteine for 60 min, and then chased for 10 min. NDUFAF3 knock-down caused a specific decrease in the labeling of the ND1 subunit of CI in the pulse translation experiment, while the synthesis of all other CI subunits, as well as subunits of complexes III (cyt b), IV (COX subunits) and V (ATP subunits), was indistinguishable from controls (Fig. 3). The decrease in ND1 labeling correlated with the severity of the CI assembly defect (data not shown). Northern blot analysis of NDUFAF3 knocked down cells showed normal levels of the ND1 transcript when compared with controls, demonstrating that this defect was not due to impaired transcription or processing of the ND1 mRNA (data not shown).
Figure 3.
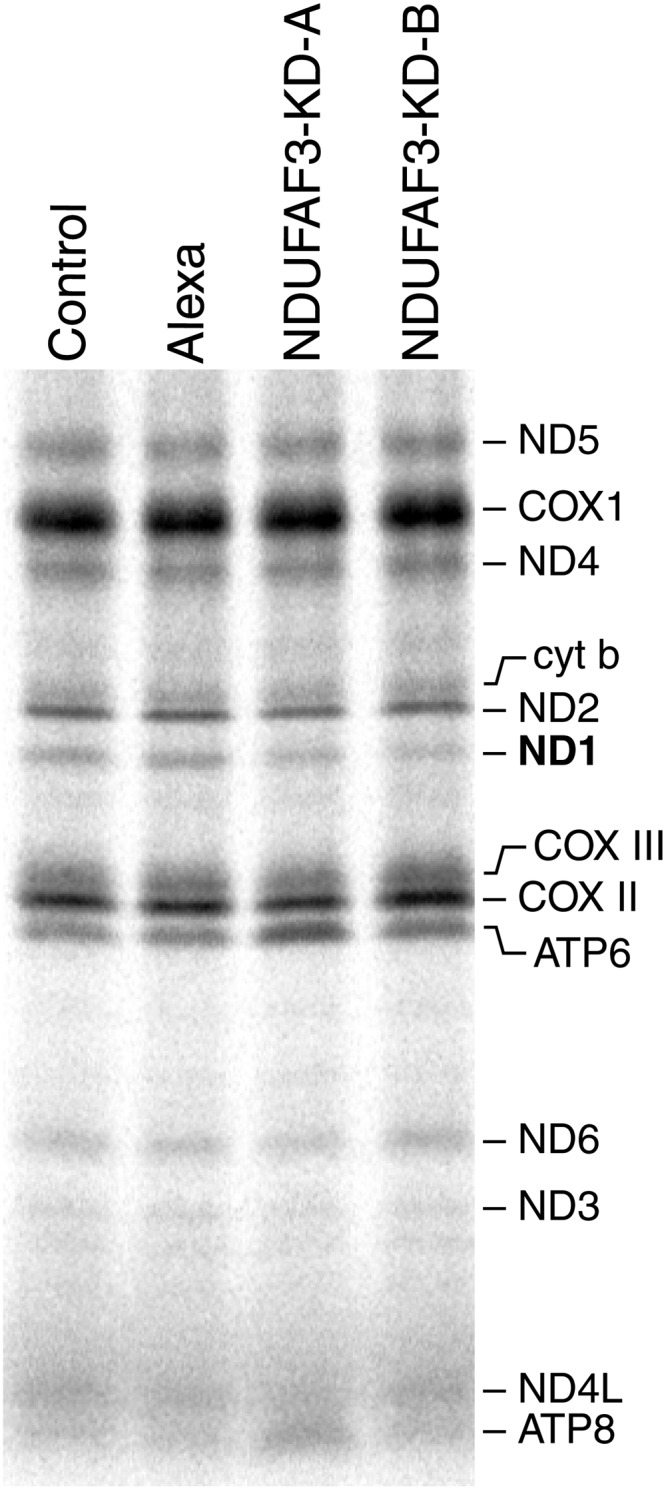
Mitochondrial translation assay in NDUFAF3 knock-down cell line. Pulse labeling of mitochondrially encoded polypeptides in control (MCH64), Alexa (fluorescent control siRNA), NDUFAF3-KD-A and NDUFAF3-KD-B (double-stranded siRNA duplexes stealth-957 and stealth-1094) cell lines. The positions of the ND subunits of CI; COX, subunits of complex IV, cyt b, subunit of complex III; ATP, subunits of complex V are indicated on the right.
The ND1 labeling defect results from rapid turnover of the newly synthesized polypeptide
To investigate whether the observed decrease in ND1 labeling was due to decreased synthesis or to rapid degradation of the ND1 polypeptide, cells in which NDUFAF3 had been knocked down were labeled with [35S]methionine/cysteine for 15, 30 or 60 min to assess the rate of synthesis, and then chased for 10, 20 and 40 min to examine the rate of degradation (Fig. 4A). Quantification of the rate of synthesis (pulse) and degradation (chase) showed that the decrease in ND1 translation is largely the result of increased degradation (Fig. 4B). The half-life of ND1 in NDUFAF3 knock-down cells was ∼4-fold lower (29.4 min) than in the Alexa control (115.8 min), indicating that the labeling defect observed in ND1 after a 15 min pulse could be largely explained by the rapid degradation of ND1. A similar analysis of ATP6 synthesis and degradation showed no difference between control and knock-down cells consistent with a specific defect in ND1 turnover (Fig. 4B).
Figure 4.

Pulse and chase experiment in NDUFAF3 knock-down cell line. (A) Alexa (fluorescent control siRNA) and NDUFAF3-KD-B (double-stranded siRNA duplex stealth-1094) cell lines were label for 15, 30 and 60min (pulse). Cells labeled for 60min were chased for 10, 20 or 40min (chase). (B) Analysis of ND1 and ATP6 total counts using ImageQuant5.2 software. Synthesis (pulse) and degradation (chase) of ND1 and ATP6 are shown in minutes.
To directly test whether the decrease in ND1 pulse labeling was due to rapid turnover of the newly synthesized ND1 polypeptide, we knocked down the m-AAA protease AFG3L2 using siRNA and repeated the translation experiment. This experiment is complicated by the fact that the protease is thought to regulate mitochondrial translation by processing one of the structural subunits of the mitochondrial ribosome (MRPL32) (22). Consistent with this, siRNA-mediated knock-down of AFG3L2 for 6 days resulted in a marked translation defect (Fig. 5A), and a concomitant decrease in the oxidative phosphorylation complexes (data not shown). We observed, however, that knocking down AFG3L2 for 2 days had no measureable effect on mitochondrial translation, so we investigated ND1 labeling in cells in which NDUFAF3 had been knocked down for 6 days, and AFG3L2 for 2 days (Fig. 5B). This resulted in a complete rescue of the ND1 labeling defect, supporting the idea that ND1 undergoes rapidly proteolysis in the absence of this early assembly factor, completely preventing the assembly of CI.
Figure 5.

Mitochondrial translation in NDUFAF3-AFG3L2 knock-down cell line. (A) Pulse labeling of mitochondrially encoded polypeptides in control Alexa (fluorescent control siRNA) and AFG3L2-KD (double-stranded siRNA duplex stealth-b1N) cell lines after 6 days of siRNA treatment. (B) Pulse labeling of mitochondrially encoded polypeptides in Alexa, AFG3L2-KD, NDUFAF3-KD-B and NDUFAF3-KD-B-AFG3L2-KD (2 and 6 days of siRNA treatment for AFG3L2 and NDUFAF3, respectively). The positions of the ND subunits of CI; COX, subunits of complex IV, cyt b, subunit of complex III; ATP, subunits of complex V are indicated on the right. SDS–PAGE immunodetection of AFG3L2 and SD70 as loading control in pulse-labeled cell lines: Alexa, AFG3L2-KD, NDUFAF3-KD-B and NDUFAF3-KD-B-AFG3L2-KD.
Rapid turnover of ND1 is a common feature of early CI assembly failure
After observing that NDUFAF3 knock-down increased the turnover of ND1, we tested whether other early CI assembly factors had a similar effect on any of the mtDNA-encoded CI polypeptides. To this end, we used siRNAs to transiently knock-down NDUFAF4, and C20orf7, and created a stable knock-down line for C8orf38 using a lentiviral construct expressing an shRNA. Analysis of all of the knocked down cell lines using BN-PAGE and mitochondrial translation assays showed a decrease in the steady-state levels of CI and in the labeling of ND1 in the pulse translation experiment (Fig. 6). In contrast, mitochondrial translation was indistinguishable from control in a patient cell line carrying a null mutation in the late assembly factor NDUFAF2, which is necessary for the addition of the NADH dehydrogenase module to an 830 kDa subcomplex in the last stage of holoenzyme assembly (Fig. 6). To investigate if the decrease in ND1 labeling was due to rapid turnover of the newly synthesized polypeptide in the NDUFAF4, C20orf7 and C8orf38 depleted cell lines, we knocked down the m-AAA protease AFG3L2 for 2 days using siRNA and repeated the translation experiment. ND1 labeling was rescued in all the three early assembly factors, consistent with the previous result observed in NDUFAF3 (Fig. 7).
Figure 6.

The ND1 translation phenotype of CI assembly factors. BN-PAGE analysis was used to assess the assembly of complex I by immunodetection of NDUFA9 subunit (left) in NDUFAF4-KD, C20orf7-KD (double-stranded siRNA duplexes stealth-226 and stealth-187996, respectively), C8orf38-KD (shRNA stable knock-down) and NDUFAF1 patient cell line. The ND1 translation phenotype is shown after mitochondrial translation assays (right).
Figure 7.

Mitochondrial translation in NDUFAF4, C8orf38 and C20orf7-AFG3L2 knock-down cell lines. Pulse labeling of mitochondrially encoded polypeptides in fibroblast cell lines in which the early assembly factors had been knocked down for 6 days (NDUFAF4, C20orf7), or in a stable knock-down line (C8orf38), and in the same lines in which AFG3L2 was knocked down for 2 days. The positions of the ND subunits of complex I; COX, subunits of complex IV, cyt b, subunit of complex III; ATP, subunits of complex V are indicated on the right. SDS–PAGE immunodetection of AFG3L2 and SD70 as loading control is shown below the translation gels.
To further characterize the roles of NDUFAF4, C8orf38 and C20orf7 in the CI assembly pathway, NDUFAF4 and C8orf38 knocked down cell lines were analyzed on a second dimension denaturing gel. The NDUFS2 subunit was undetectable, and ND1 was almost completely depleted in NDUFAF4 knock-down cells. No intermediate assemblies were detected, indicative of an early CI assembly defect (Fig. 8A). This phenotype is very similar to that observed in NDUFAF3 knock-down, consistent with the fact that they form a complex that can be immunoprecipitated. The C8orf38 knock-down second dimension experiment showed reduced levels of the NDUFS2, NDUFA9 and ND1 subunits compared with control cell line (Fig. 8C), and again no subcomplexes were detected. Over-exposure of the membrane showed that the nuclear subunit NDUFS2 accumulates as a monomer, suggesting that C8orf38 is also involved in the very first steps of the assembly, pathway probably assisting assembly of NDUFS2 (Fig. 8B). Similar results were obtained after knock-down of C20orf7 (Fig. 8B).
Figure 8.

Characterization of CI assembly factors by 2D gel electrophoresis. Knocked down (NDUFAF4, C8orf38 and C20orf7) cell lines were subjected to second dimension denaturing (2D) experiments. A cocktail of antibodies directed against the indicated proteins was used to probe the immunoblots. (A) Alexa (fluorescent control siRNA) and NDUFAF4-KD (double-stranded siRNA duplex stealth-226). (B) Alexa (fluorescent control siRNA) and C20orf7-KD (double-stranded RNAi duplex stealth-187996). (C) Control (MCH64) and C8orf38-KD (shRNA stable knock-down). SD70 of Co II is shown as a loading control.
DISCUSSION
In this study, we investigated four different CI assembly factors viz., NDUFAF3, NDUFAF4, C8orf38 and C20orf7, to elucidate their role in the assembly pathway and in the synthesis of mtDNA-encoded CI structural subunits. Knock-down of any of the factors abrogated the assembly of CI, without the obvious accumulation of subcomplexes, consistent with previous suggestions that all act early in the assembly pathway. Pulse-chase radiolabeling of mtDNA-encoded polypeptides showed that this was associated in all cases with an apparent decrease in the synthesis of the ND1 subunit, as evidenced by a marked decrease in ND1 labeling intensity after a short pulse, and normal synthesis of all other CI subunits. However, longer chase experiments in NDUFAF3 knock-down cells showed that the half-life of newly synthesized ND1 was reduced 4-fold, largely explaining the decrease in the intensity of ND1 labeling, and demonstrating that the labeling defect was not due to decreased synthesis, but rather rapid turnover of newly synthesized ND1. Transient knock-down of the inner membrane AAA protease AFG3L2 also rescued ND1 labeling in cells in which the early assembly factors had also been individually knocked down, showing that the mitochondrial quality-control machinery rapidly degrades newly synthesized ND1 that is prevented from associating with a matrix module.
NDUFAF3 is associated with a 200 kDa subcomplex and immunoprecipitation experiments showed that this complex contains, minimally, the nuclear-encoded subunits NDUFS2, NDUFS3 and NDUFA9 and the assembly factor NDUFAF4. Similar experiments, using a TAP-tagged version of NDUFAF3, identified three of these proteins, but failed to identify NDUFA9 (7). NDUFA9 is a 39 kDa protein that is thought to bridge the peripheral matrix and membrane arms of CI, and is thus likely crucial in stabilizing the assembly intermediate containing ND1. The second dimension denaturing gel analysis of both C8orf38 and C20orf7 knock-down cells showed that NDUFS2 accumulated as a monomer, suggesting that these factors might be assisting the assembly of the earliest subcomplex that brings together NDUFS2 and NDUFS3. Our results rule out the suggestion that C8orf38 acts by inhibiting the synthesis of ND1 (9).
Bacterial proteins belonging to the Sec translocation complex are involved in the transport of nascent polypeptides across, or their integration into, the cytoplasmic membrane, and it has been suggested that NDUFAF3 (and C8orf38) could couple mitochondrial translation to membrane insertion based on the gene order conservation of NDUFAF3 with the SecD/SecF/YajC gene cluster (7,9). Some NDUFAF3 also appears to associate with larger CI subcomplexes of 700 and 830 kDa, but we could never co-immunoprecipitate ND1 with NDUFAF3, even using chemical cross-linking conditions. While neither NDUFAF3 nor NDUFAF4 have predicted transmembrane domains, a significant fraction of NDUFAF3 is associated with the inner mitochondrial membrane in alkaline carbonate extraction experiments (data not shown), suggesting that it shuttles between a soluble form and a form that is tightly associated with another protein in the membrane. Thus, it could act as a dock between the matrix subcomplex containing NDUFS2, NDUFA9, NDUFS3 and the eight transmembrane ND1 protein, which is likely co-translationally inserted into the inner mitochondrial membrane.
AFG3L2 is a mitochondrial inner membrane m-AAA protease with dual roles in mitochondrial protein quality control and in processing specific proteins that have cellular regulatory functions (21).The yeast homologue of AFG3L2 has been described to play a role in protein synthesis by regulating ribosome assembly through the processing of MRPL32, a function which is conserved in mammals (22). This occurs by a novel processing mechanism that depends upon the MRPL32 mitochondrial targeting presequence (23). Our data show that depletion of AFG3L2 after 6 days of siRNA treatment dramatically reduces the translation of all mitochondrial-encoded polypeptides, demonstrating a role for AFG3L2 in the regulation of human mitochondrial translation. The brief (2-day) depletion of AFG3L2 that we used to investigate its role in the proteolysis of ND1 substantially reduced the level of the protein, but had no impact on mitochondrial translation, presumably because such an effect would require the turnover of already functioning mitochondrial ribosomes. This allowed us to evaluate the two different roles of AFG3L2 independently. The observation that knock-down of the protease rescued the rapid ND1 turnover phenotype in all of the cell lines with early assembly defects is consistent with its well-studied role in the turnover of inner membrane protein that are either mis-folded or that fail to assemble into productive complexes.
In summary, our data suggest that the four early CI assembly factors have non-redundant functions in the assembly of a subcomplex containing several soluble matrix structural subunits. This subcomplex acts to dock and stabilize newly synthesized ND1 in a 400 kDa intermediate that nucleates assembly of the holoenzyme complex. Consistent with this conclusion, the ND1 translation labeling phenotype was not observed in a patient cell line mutant for the late assembly factor NDUFAF2, where CI assembly is stalled at an 830 kDa subcomplex. If any of the early factors are missing, ND1 cannot be stabilized in the inner mitochondrial membrane and CI assembly stalls. The exact molecular function of each of the early assembly factors, however, remains to be determined.
MATERIALS AND METHODS
Cell culture
Primary cell lines were established from subject skin fibroblasts and immortalized by transduction with a retroviral vector expressing the HPV-16 E7 gene plus a retroviral vector expressing the catalytic component of human telomerase (htert) (24). The fibroblasts were grown at 37°C in an atmosphere of 5% CO2 in high-glucose DMEM supplemented with 10% FBS.
Mitochondrial isolation
Fibroblasts were resuspended in ice-cold 250 mm sucrose/10 mm Tris–HCl/1 mm EDTA (pH 7.4) and homogenized with 10 passes through a pre-chilled, zero clearance homogenizer (Kimble/Kontes). The homogenized cellular extract was then centrifuged twice for 10 min at 600g to obtain a postnuclear supernatant. Mitochondria were pelleted by centrifugation for 10 min at 10 000g, and washed once in the same buffer.
NDUFAF3 antibody production
A polyclonal antibody against two peptides (Ac-LYQRTRISLLQREAAQAC-amide and Ac-CLQAMRQRGIAVEVQDTP-amide) from the human NDUFAF3 protein was prepared by 21st Century Biochemicals (Marlboro, MA, USA). Crude serum and affinity-purified antibodies were tested on cell lines overexpressing NDUFAF3-HA protein and detected a band of ∼20 kDa. The C-terminal affinity-purified antibody was used for further experiments.
RNA interference
For the knock-down experiments, two Stealth RNA interference (RNAi) duplex constructs for NDUFAF3 (Invitrogen, Carlsbad, CA, USA) were used; Stealth A: CACCUUCAACUUCCUGUGUCAUGAA and Stealth B: CAGGCUUCCCAAUGCUUUCACUCUU. NDUFAF4 was knocked down with the Stealth RNAi duplex: AGUUAAAGGAGAGAUUGCUCGUAAA and C20orf7 with the Stealth RNAi duplex: CCGUGUAUAUGACAUACCCAGAAAU. Stealth RNAi duplexes or the fluorescent oligo control Block-iT Alexa FluorRed (Invitrogen) were transiently transfected at a final concentration of 12 nm using Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer's specifications. Knocked down cell lines were analyzed after 6 days of Stealth treatment. C8orf38 was stable knocked down using the lentiviral vector (pLKO.1) containing the specific small hairpin sequence: CAGAATCCCGAAGCTCTGTTT, obtained from the Broad RNAi Consortium (http://www.broadinstitute.org/genome_bio/trc/protocols/trcLentiVirusProd.pdf). The control fibroblast cell line was transduced with viral supernatant as described elsewhere (25).
Epitope-tagged construct
To generate C-terminal HA-tag (NDUFAF3-HA) construct, cDNA was amplified by OneStep RT–PCR (Qiagen) using specific primers modified for cloning into Gateway vectors (Invitrogen) as previously described (26). Retroviral constructs were transiently transfected into the Phoenix packaging cell line using the HBS/Ca3(PO4)2 method (25). Control fibroblasts were infected 48 h later by exposure to the virus-containing medium in the presence of 4 g/ml of polybrene as described elsewhere (25).
Electrophoresis and immunobloting
Blue-Native PAGE was used to separate samples in the first dimension on 6–15% polyacrylamide gradient gels, as previously described (25). Mitoplasts were prepared from fibroblasts by treatment with 0.8 mg of digitonin/mg of protein and solubilized with 1% lauryl maltoside and 20 μg of the solubilized proteins were used for electrophoresis. For the two-dimensional analysis, the BN-PAGE/SDS–PAGE was done as previously described (27). Individual structural subunits of complexes I, II, III, IV and V were detected by immunoblot analysis using commercially available monoclonal antibodies (Molecular Probes), except for CI, where a polyclonal antibody against subunit ND1 (a gift of A. Lombes, INSERM U582, Hôpital de La Salpêtrière, Paris) was used. In gel, qualitative assay of CI activity was performed as described elsewhere (6). SDS–PAGE was used to separate denatured whole cell extracts or isolated mitochondria using 12% polyacrylamide gels followed by immunoblot analysis with indicated antibodies. The AFG3L2 antibody was a kind gift of Dr Thomas Langer, Cologne, Germany.
Mitochondrial translation essay
In vitro labeling of mitochondrial translation products was performed as previously described (28). For pulse and chase studies, cells were incubated for 23 h in 40 μg/ml chloramphenicol before labeling, then cells were pulse-labeled for 15, 30 or 60 min at 37°C in methionine/cysteine-free DMEM containing 200 Ci/ml of [35S]methionine/cysteine (Perkin Elmer) and 100 μg/ml of anisomycin. Cells labeled for 60min were chased for 10, 20 or 40 min in regular DMEM. Protein extraction was done in labeled cells (50 μg) by resuspension in loading buffer containing 93 mm Tris–HCl, pH 6.7, 7.5% glycerol, 1% SDS, 0.25 mg bromophenol blue/ml and 3% mercaptoethanol, sonicated for 3–8 s, loaded and run on 15–20% polyacrylamide gradient gels. The labeled mitochondrial translation products were detected through direct autoradiography. Quantitative analysis was done using ImageQuant5.2 software.
Immunoprecipitation experiments
Mitochondrial protein was extracted from control or NDUFAF3-HA fibroblasts (400 μg) in 180 μl extraction buffer (50 mm HEPES buffer, pH 7.6, 150 mm NaCl, 1% taurodeoxycholate), supplemented with complete protease inhibitors (Roche), on ice, for 45 min. Protein extract was centrifuged at 25 000g at 4°C, for 40 min, and the supernatant was used to immunoprecipitate NDUFAF3. Immunoprecipitation was performed on HA-coated agarose beads (Sigma) according to the manufacturer's instructions (IP0010), the incubation of the protein extract with the beads was carried out overnight at 4°C. After washing 5× with 0.1 m NaPO4, pH 8, with 0.05% Tween and 0.05% t-doc, bound protein was elute with 100 μl of 0.1 m glycine pH 2.5 with 0.5% DDM at 52°C. The IP fractions were then analyzed by immunoblotting, and the eluates were sent for mass spectrometry analysis (Orbitrap, Thermo Scientific, Waltham, MA, USA) at the Institut de Recherches Cliniques de Montreal.
Conflict of Interest statement. None declared.
FUNDING
This research was supported by a grant from the CIHR (MOP-15460) to E.A.S. O.Z.R. was supported by fellowships from CONACyT (Consejo Nacional de Ciencia y Tecnología) (209378) and PBEEE (Fonds québécois de la recherché sur la nature et les technologies) (140802). E.A.S. is an International Scholar of the HHMI.
References
- 1.Kirby D.M., Crawford M., Cleary M.A., Dahl H.H., Dennett X., Thorburn D.R. Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder. Neurology. 1999;52:1255–1264. doi: 10.1212/wnl.52.6.1255. [DOI] [PubMed] [Google Scholar]
- 2.Triepels R.H., Van Den Heuvel L.P., Trijbels J.M., Smeitink J.A. Respiratory chain complex I deficiency. Am. J. Med. Genet. 2001;106:37–45. doi: 10.1002/ajmg.1397. [DOI] [PubMed] [Google Scholar]
- 3.Mimaki M., Wang X., McKenzie M., Thorburn D.R., Ryan M.T. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta. 2011;1817:851–862. doi: 10.1016/j.bbabio.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Calvo S.E., Tucker E.J., Compton A.G., Kirby D.M., Crawford G., Burtt N.P., Rivas M., Guiducci C., Bruno D.L., Goldberger O.A., et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet. 2010;42:851–858. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunning C.J., McKenzie M., Sugiana C., Lazarou M., Silke J., Connelly A., Fletcher J.M., Kirby D.M., Thorburn D.R., Ryan M.T. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. EMBO J. 2007;11:3227–3237. doi: 10.1038/sj.emboj.7601748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogilvie I., Kennaway N.G., Shoubridge E.A. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J. Clin. Invest. 2005;115:2784–2792. doi: 10.1172/JCI26020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saada A., Vogel R.O., Hoefs S.J., van den Brand M.A., Wessels H.J., Willems P.H., Venselaar H., Shaag A., Barghuti F., Reish O., et al. Mutations in NDUFAF3 (C3ORF60), encoding an NDUFAF4 (C6ORF66)-interacting complex I assembly protein, cause fatal neonatal mitochondrial disease. Am. J. Hum. Genet. 2009;84:718–727. doi: 10.1016/j.ajhg.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saada A., Edvardson S., Rapoport M., Shaag A., Amry K., Miller C., Lorberboum-Galski H., Elpeleg O. C6ORF66 is an assembly factor of mitochondrial complex I. Am. J. Hum. Genet. 2008;82:32–38. doi: 10.1016/j.ajhg.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKenzie M., Tucker E.J., Compton A.G., Lazarou M., George C., Thorburn D.R., Ryan M.T. Mutations in the gene encoding C8orf38 block complex I assembly by inhibiting production of the mitochondria-encoded subunit ND1. J. Mol. Biol. 2011;414:413–426. doi: 10.1016/j.jmb.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Sugiana C., Pagliarini D.J., McKenzie M., Kirby D.M., Salemi R., Abu-Amero K.K., Dahl H.H., Hutchison W.M., Vascotto K.A., Smith S.M., et al. Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. Am. J. Hum. Genet. 2008;83:468–478. doi: 10.1016/j.ajhg.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fassone E., Duncan A.J., Taanman J.W., Pagnamenta A.T., Sadowski M.I., Holand T., Qasim W., Rutland P., Calvo S.E., Mootha V.K., et al. FOXRED1, encoding an FAD-dependent oxidoreductase complex-I-specific molecular chaperone, is mutated in infantile-onset mitochondrial encephalopathy. Hum. Mol. Genet. 2010;19:4837–4847. doi: 10.1093/hmg/ddq414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nouws J., Nijtmans L., Houten S.M., van den Brand M., Huynen M., Venselaar H., Hoefs S., Gloerich J., Kronick J., Hutchin T., et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010;12:283–294. doi: 10.1016/j.cmet.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Vahsen N., Cande C., Briere J.J., Benit P., Joza N., Larochette N., Mastroberardino P.G., Pequignot M.O., Casares N., Lazar V., et al. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vogel R.O., Janssen R.J., van den Brand M.A., Dieteren C.E., Verkaart S., Koopman W.J., Willems P.H., Pluk W., van den Heuvel L.P., Smeitink J.A., et al. Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev. 2007;21:615–624. doi: 10.1101/gad.408407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antonicka H., Ogilvie I., Taivassalo T., Anitori R.P., Haller R.G., Vissing J., Kennaway N.G., Shoubridge E.A. Identification and characterization of a common set of complex I assembly intermediates in mitochondria from patients with complex I deficiency. J. Biol. Chem. 2003;278:43081–43088. doi: 10.1074/jbc.M304998200. [DOI] [PubMed] [Google Scholar]
- 16.Perales-Clemente E., Fernandez-Vizarra E., Acin-Perez R., Movilla N., Bayona-Bafaluy M.P., Moreno-Loshuertos R., Perez-Martos A., Fernandez-Silva P., Enriquez J.A. Five entry points of the mitochondrially encoded subunits in mammalian complex I assembly. Mol. Cell Biol. 2010;30:3038–3047. doi: 10.1128/MCB.00025-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dieteren C.E., Willems P.H., Vogel R.O., Swarts H.G., Fransen J., Roepman R., Crienen G., Smeitink J.A., Nijtmans L.G., Koopman W.J. Subunits of mitochondrial complex I exist as part of matrix- and membrane-associated subcomplexes in living cells. J. Biol. Chem. 2008;283:34753–34761. doi: 10.1074/jbc.M807323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogel R.O., Dieteren C.E., van den Heuvel L.P., Willems P.H., Smeitink J.A., Koopman W.J., Nijtmans L.G. Identification of mitochondrial complex I assembly intermediates by tracing tagged NDUFS3 demonstrates the entry point of mitochondrial subunits. J. Biol. Chem. 2007;282:7582–7590. doi: 10.1074/jbc.M609410200. [DOI] [PubMed] [Google Scholar]
- 19.Lazarou M., McKenzie M., Ohtake A., Thorburn D.R., Ryan M.T. Analysis of the assembly profiles for mitochondrial- and nuclear-DNA-encoded subunits into complex I. Mol. Cell Biol. 2007;27:4228–4237. doi: 10.1128/MCB.00074-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunte C., Zickermann V., Brandt U. Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science. 2010;329:448–451. doi: 10.1126/science.1191046. [DOI] [PubMed] [Google Scholar]
- 21.Gerdes F., Tatsuta T., Langer T. Mitochondrial AAA proteases—towards a molecular understanding of membrane-bound proteolytic machines. Biochim. Biophys. Acta. 2012;1823:49–55. doi: 10.1016/j.bbamcr.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 22.Nolden M., Ehses S., Koppen M., Bernacchia A., Rugarli E.I., Langer T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell. 2005;123:277–289. doi: 10.1016/j.cell.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 23.Bonn F., Tatsuta T., Petrungaro C., Riemer J., Langer T. Presequence-dependent folding ensures MrpL32 processing by the m-AAA protease in mitochondria. EMBO J. 2011;30:2545–2556. doi: 10.1038/emboj.2011.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lochmuller H., Johns T., Shoubridge E.A. Expression of the E6 and E7 genes of human papillomavirus (HPV16) extends the life span of human myoblasts. Exp. Cell Res. 1999;248:186–193. doi: 10.1006/excr.1999.4407. [DOI] [PubMed] [Google Scholar]
- 25.Antonicka H., Ostergaard E., Sasarman F., Weraarpachai W., Wibrand F., Pedersen A.M., Rodenburg R.J., van der Knaap M.S., Smeitink J.A., Chrzanowska-Lightowlers Z.M., et al. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am. J. Hum. Genet. 2010;87:115–122. doi: 10.1016/j.ajhg.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antonicka H., Mattman A., Carlson C.G., Glerum D.M., Hoffbuhr K.C., Leary S.C., Kennaway N.G., Shoubridge E.A. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003;72:101–114. doi: 10.1086/345489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sasarman F., Brunel-Guitton C., Antonicka H., Wai T., Shoubridge E.A. LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Mol. Biol. Cell. 2010;21:1315–1323. doi: 10.1091/mbc.E10-01-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sasarman F., Shoubridge E.A. Radioactive labeling of mitochondrial translation products in cultured cells. Methods Mol. Biol. 2012;837:207–217. doi: 10.1007/978-1-61779-504-6_14. [DOI] [PubMed] [Google Scholar]