

Abstract
While numerous lines of evidence point to increased levels of oxidative stress playing a causal role in a number of neurodegenerative conditions, our current understanding of the specific role of oxidative stress in the genesis and/or propagation of neurodegenerative diseases remains poorly defined. Even more challenging to the “oxidative stress theory of neurodegeneration” is the fact that many antioxidant-based clinical trials and therapeutic interventions have been largely disappointing in their therapeutic benefit. Together, these factors have led researchers to begin to focus on understanding the contribution of highly localized structures, and defined anatomical features, within the brain as the sites responsible for oxidative stress-induced neurodegeneration. This review focuses on the potential for oxidative stress within the cerebrovascular architecture serving as a modulator of neurodegeneration in a variety of pathological settings. In particular, this review highlights important implications for vascular-derived oxidative stress in the initiating and promoting pathophysiology in the brain, identifying new roles for cerebrovascular oxidative stress in a variety of brain disorders.
Keywords: Oxidative Stress, Antioxidants, Cerebral Endothelial Cells, Neurodegeneration, Blood Brain Barrier, Vascularization
1. Introduction
The brain accounts for ∼2% of total body mass, but requires ∼20% of the total blood flow from the heart to supply the cells of the brain with continual fresh oxygen and energy for the maintenance of homeostasis [1]. The exquisite vascular network within the brain accomplishes this delivery of oxygen and gases, as well as the removal of numerous potentially toxic molecules, via a continual and seamless perfusion of the entire brain [2]. The selectivity of the blood brain barrier (BBB) allows some molecules to move between the peripheral circulation into the brain, and vice versa, all the while assisting in maintaining homeostasis for the neurons, glia, and vascular cells in the immediate area of the BBB [2, 3].
As pointed out in numerous reviews within this collection, there is little doubt that increased levels of oxidative stress occurs in conditions where neurodegeneration is observed. There are almost an unlimited number of studies, in a plethora of settings, which firmly establish evidence for oxidation to proteins, lipids, sugars, and nucleic acids occurring in most (if not all) neurodegenerative conditions. An equally large number of studies have demonstrated oxidative stress occurs in the earliest stages of these conditions, and tightly correlates with the development of motor and cognitive disturbances [4-6]. In contrast to this strong level of association, many questions remain in regards to whether oxidative stress is a cause or consequence of neurodegenerative processes. This is largely based on the relatively nonspecific and ubiquitous nature of oxidative stress in these different settings. Furthermore, current antioxidant-based therapies for the treatment and/or management of neurodegenerative diseases in clinical settings (randomized and epidemiological) and rodent studies, have identified antioxidant based therapies to be largely ineffective [4, 5].
The premise of this review is that the cerebrovasculature may be an important source, and target, of oxidative stress in the brain. We believe that the very factors which allow the vasculature to conduct its vital functions for the brain predispose it to oxidative stress. For example, while only accounting for small global elevations in oxidative stress, localized increases in the levels of oxidative damage within the vasculature may be sufficient to promote deleterious changes in blood-flow, BBB integrity, and serve as an initiator for numerous lines of pathogenesis in the brain. If our model is correct, such data would have important implications for not only understanding oxidative stress, but may lead to the development of novel antioxidant strategies for numerous disorders.
This review will define oxidative stress, as well as outline the numerous neurodegenerative disorders associated with oxidative stress and vascular pathogenesis. Additionally, this review will outline what is known in regards to the efficacy of a variety of antioxidant-based interventions in regards to their use in treatment of neurodegenerative disorders and modulation of pathological processes. The review will then go into greater detail as to the underlying basis for the genesis of oxidative stress within the cerebrovasculature, and the specific contribution of how oxidative stress in the vascular components of the brain mediates disease pathogenesis. We will then attempt to explain the importance of targeting the vasculature in the design and deployment of antioxidant-based therapies for the brain in the future.
1.1 Oxidative Stress
An imbalance between cellular pro-oxidants and anti-oxidants can occur during aging as well as during many age-related diseases of the central nervous system [7]. This phenomenon is referred to as oxidative stress, and is induced by highly reactive oxygen species (ROS) and reactive nitrogen species (RNS) that possess an unpaired electron [4]. All macromolecules in the cell are vulnerable to oxidative damage, including lipids, proteins, and nucleic acids [8]. However, cells are equipped to counteract this oxidative attack with numerous cellular antioxidant defenses such as glutathione (GSH), superoxide dismutases, and catalase [9]. During aging and various disease states, these antioxidant defense systems can be altered leading to progressive oxidative damage and subsequent cell death and/or significant loss of function [6]. Changes in antioxidant status within specific cells and structures of the brain may be particularly useful in dictating whether oxidative stress is benign or pathological.
1.2 Diseases of the Nervous System: Oxidative Stress
Oxidative stress has been implicated in numerous diseases of the nervous system such as (but not limited to): Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), HIV-associated neurocognitive disorder (HAND or “Neuro AIDS”), cerebral ischemia/reperfusion injury (I/R), and traumatic brain injury (TBI). In these disease states, oxidative stress has been implicated in playing an early, initiating role as well as a potentially late, by-product of neurodegeneration.
In the case of AD, oxidative stress is proposed to be an early event that aids in progressive neurodegeneration and subsequent cognitive impairment [10-12]. One of the contributing factors that is common to many neurodegenerative diseases involving oxidative stress pathways is mitochondrial dysfunction [12-16]. It has previously been shown that neurons in the AD brain exhibit damaged mitochondria, specifically altered α-ketoglutarate dehydrogenase complex (KGDHC) and pyruvate dehydrogenase complex (PDHC) which leads to increased ROS production [17-19]. In addition, components of the β-amyloid (Aβ) processing machinery are found in mitochondria and it has been shown that Aβ accumulates in the mitochondrial matrix leading to toxicity and increased release of ROS [20, 21]. The interaction between Aβ toxicity and increased oxidative stress has a large impact on AD pathology [16]. Not only does Aβ accumulate in mitochondria, but as it deposits throughout certain areas of the brain, microglia and astrocytes also cluster at these sites leading to “hot spots” of inflammation and production of ROS/RNS [22]. Specifically, the Aβ peptide can activate microglial NADPH oxidase (NOX) which increases production of superoxide radicals and hydrogen peroxide [23, 24]. Aβ also interferes with the proteasome by decreasing proteolytic activity [25-27]; it has been shown in our lab [27] as well as others [25, 28] that inhibition of the proteasome leads to increased oxidative stress. In addition to the inflammatory and oxidative stress reactions occurring in the AD brain which have profound effects on neuronal health, these detrimental effects are also occurring in the brain microvasculature [3]. The combined effects of reactivity from glial cells, cerebral endothelial cells, neurodegeneration, and BBB disruption during this process provide an environment for further activation of inflammatory and oxidative stress responses [3, 29].
Mitochondrial defects are also observed in PD [16]; specifically, reduced activity of the mitochondrial respiratory complex I (NADPH-quinone oxidoreductase) in the substantia nigra [30] as well as the frontal cortex [31, 32]. It has been shown that oxidative stress causes damage to the complex I subunits, altering assembly, leading to mitochondrial dysfunction and consequently reducing electron transfer rates and increasing ROS production [31, 33]. It is well known that Parkinson's disease pathology includes the selective loss of dopamine neurons [34], and it is hypothesized that a main cause of this loss is the high levels of ROS in dopamine neurons due to dopamine metabolism and their high iron content [35]. In support of the role of mitochondrial complex I inhibition in PD, various disease models for PD also involve inhibition of complex I. For example, accidental formation of the drug 1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine (MPTP) in the late 1970's led to an important animal model of PD after the drug was self-administered in a number of humans that quickly displayed parkinsonian behaviors and pathology [36]. It was later determined that MPTP gets converted to MPP+ by glial cells in the brain which is neurotoxic, and it is taken up specifically by the dopamine transporter in the substantia nigra dopamine neurons [37]. Interestingly, MPP+ binds to the mitochondrial complex I and inhibits its activity leading to mitochondrial dysfunction, oxidative stress and cell loss [34]. Two other PD models that also have effects on complex I and lead to parkinsonian behavior/pathology include the pesticides rotenone and paraquat [38-41]. In addition to mitochondrial defects due to complex I, it has been proposed that the parkin gene (PRKN) plays an important role in PD pathogenesis [32, 42, 43]. In juvenile PD especially, a large number of patients exhibit mutations in PRKN which affects the proteasome and mitochondrial activity [43]. In experiments with parkin-deficient mice, increased protein oxidation, increased lipid peroxidation and decreased mitochondrial respiratory capacity have been observed [44]. On the other hand, overexpression of parkin reduces ROS, enhances complex I activity and increases the mitochondrial membrane potential, pointing to an important role of parkin in mitochondrial performance [45]. Lastly, and similar to findings for Aβ in AD, the aggregated protein, α-synuclein found presynaptically in PD brains affects mitochondria and increases oxidative stress [46]. While PD exhibits some similar pathways for degeneration as AD, the role of oxidative stress at cerebral endothelial cells has not yet been explored for PD pathogenesis; we argue this is an important source of oxidative stress in neurodegenerative diseases including PD.
Similar to AD and PD, mitochondrial injury plays a role in MS progression [47]. However, the major source for free radical production is thought to be stimulated by chronic inflammation and the release of toxic radicals by activated microglia [48, 49]. ROS and RNS can pecifically affect mitochondrial complex IV of the respiratory chain, which has been shown in axons, oligodendrocytes and astrocytes of MS patients [50]. When this occurs in axons, transport is disturbed due to a loss of energy and when this occurs in oligodendrocytes, apoptosis follows [51]. Results are controversial but some laboratories have shown oxidized DNA and lipids within active lesions, also pointing to the role of oxidative stress in MS [47, 52, 53]. The large focus of MS research is at axons and oligodendrocytes due to their obvious role in disease progression, however we argue the potential role of cerebral endothelial cells in contributing to inflammation and oxidative stress at these sites. This must be further investigated as another potential source for the observed damage.
Much less is known about the source of oxidative stress found in ALS and HAND. In 20% of familial ALS (about 2% of all cases), a mutation in the copper-zinc superoxide dismutase (SOD1) gene was found and hypothesized to alter axonal transport of mitochondria, especially to distal nerve terminals [54, 55]. Expression of mutant SOD1 has been observed in the cytoplasm, nucleus, endoplasmic reticulum and intramembrane space of mitochondria, providing sites where oxidative damage likely occurs [56]. Unfortunately, sporadic ALS (the majority of cases) is not well understood. Some data for these cases reveal dense clusters of mitochondria that have accumulated in the anterior horn of the lumbar spinal cord, which may contribute to oxidative stress and subsequent degeneration [54, 57, 58]. However, more studies must be performed to understand the mechanisms for oxidative stress production in ALS, especially the sporadic cases. The etiology of HAND is also not currently well understood. However, similar to MS, it is thought that increased inflammation and ensuing activated microglia play a key role in stimulating production of ROS; inflammation and oxidative stress together can contribute to neurodegeneration and subsequent cognitive decline [59, 60]. As with the other neurodegenerative diseases briefly reviewed herein, the role of cerebral endothelial cells in promoting oxidative stress and subsequent disease progression has not yet been well explored for ALS or HAND; we hypothesize it to play a role in these neurodegenerative diseases and it must be further examined.
Lastly, I/R and TBI have clear mechanisms for ROS production, which also contribute to BBB disruption. In both cases, a large influx of hemoglobin (during reperfusion in I/R and due to mechanical injury in TBI) leads to the increased presence of iron to stimulate free radical production [61-63]. Both cases also involve mitochondrial dysfunction and increased microglial activation, as previously described [64, 65]. Another interesting factor in both of these injuries is the breakdown of the BBB which provides a cycle of damage: increased production of ROS and then further breakdown of the BBB from oxidative stress [65, 66]. The unique features of the BBB that can protect against oxidative stress and those features that in fact cause it to be susceptible to oxidative stress are described below.
1.3 The Blood Brain Barrier and Oxidative Stress
Capillaries found in the central nervous system are distinct from those found in the rest of the body due to the BBB, an important filter that protects the brain [67, 68]. Features of the BBB include tight junction proteins that “glue” together the cerebral endothelial cells, such as occludin, claudins, and junction adhesion molecules (JAMs) [69]. These proteins are then anchored in the endothelial cells by scaffolding proteins such as the zona occludens (ZO-1, ZO-2, ZO-3) [69]. Another protective layer is provided by a thick basement membrane and astrocytic end feet [69].
In addition to these protective layers, cerebral endothelial cells are equipped with a defense system against oxidative stress including increased GSH, glutathione peroxidase, glutathione reductase and catalase compared to the rest of the brain [9]. GSH in particular has been shown to play an important role in maintenance of BBB integrity [70].
All areas of the healthy brain also consist of antioxidants such as superoxide dismutase (SOD) in order to provide a balance against the high concentration of ROS and RNS produced [71]. Manganese-containing SOD (MnSOD) is found in the mitochondrial matrix while copper-and zinc- containing SOD (CuZnSOD) is found mostly in the cell cytosol [72]. These enzymes aid in the conversion of O2- to H2O2, which is also a ROS, but it is removed by catalase (and other enzymes) that are found in most tissues [71]. Through this conversion, the production of ONOO- (peroxynitrite) is reduced. Without this system, peroxynitrite can rapidly form due to high concentrations of NO, particularly at the endothelial cells [73].
Lastly, throughout the body, NF-erythroid 2-related factor 2 (Nrf2) plays a key role in defense against oxidative stress. It has previously been shown that Nrf2 is important for ischemic lesion repair and new studies are focusing on this transcription factor for therapies against neurodegenerative diseases such as Parkinson's disease [74]. Under basal conditions, Nrf2 is anchored to the cytoplasm by Kelch-like ECH-associated protein 1 (Keap1) [75]. However, when oxidative stress occurs, the cysteine residues of Keap1 become oxidized, releasing Nrf2 to enter the nucleus. There, it binds to the antioxidant response element (ARE) of many different genes, allowing transcription of antioxidants [74, 75]. It is also believed to increase anti-inflammatory mediators, activity of the proteasome and other transcription factors involved in mitochondrial biogenesis [74]. Therefore, the brain is well-equipped with a number of antioxidant properties to stave off oxidative stress.
On the other hand, there are a number of factors inherent to the brain and cerebral endothelial cells that increase opportunities for oxidative stress. First, the brain uses a lot of oxygen (20% of the body's basal levels) [1]. Neurons require a high amount of ATP for maintaining their membrane potential, packaging neurotransmitters and releasing neurotransmitters; this requires a high level of activity from mitochondria and therefore, high oxygen consumption [4]. With an increased presence of oxygen, comes an increased chance for reactive oxygen species production. The brain also contains nitric oxide, increasing the opportunity for RNS production, such as peroxynitrite [76]. Nitric oxide (NO) or endothelium-derived relaxing factor (EDRF) is not only found in the brain for neurotransmission, but at the cerebral endothelial cells for vasodilation as well [77]. Next, the neuronal membrane is largely made up of polyunsaturated fatty acids (PUFAs), especially docosahexaenoic acid [4]. PUFAs in particular are highly susceptible to lipid peroxidation. For example, 4-hydroxynonenal (HNE) is produced from lipid peroxidation and has been shown to be cytotoxic to neurons [78]. High levels of HNE have been reported in numerous neurodegenerative diseases [78-80]. Cerebral endothelial cells also have a high concentration of mitochondria, providing an increased opportunity for generating oxidative stress [3]. However, this high concentration of mitochondria is also believed to play a role in BBB maintenance. For example, the BBB performs energy-dependent transport mechanisms that require the function of many mitochondria [3]. With aging, a decrease in mitochondrial number at cerebral endothelial cells and subsequent loss of BBB integrity has been reported [81]. Therefore, a number of features that have evolved for the high-level functioning of the BBB and cerebral endothelial cells themselves also come at a price, with the prospect for producing oxidative stress. Some of these factors are displayed in Figure 1.
Figure 1. Sources of Oxidative Stress at Cerebral Endothelial cells.
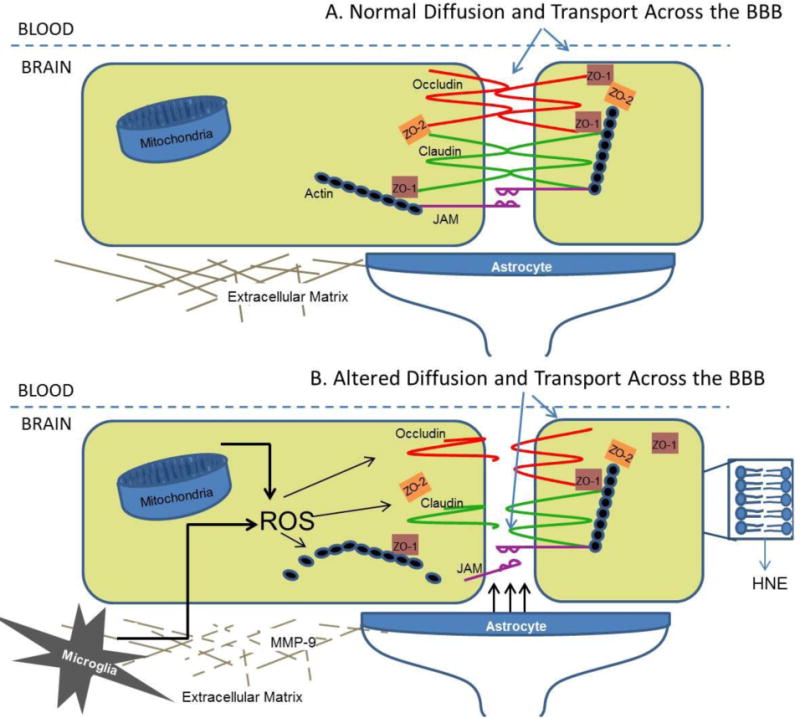
A) In the healthy brain, the BBB is intact with the help of claudins, occludin, and junctional adhesion molecules (JAMs) to create the tight junction between endothelial cells. Actin and the zona occludens (ZO-1 and ZO-2, etc.) also create a scaffold for the tight junction. Further support for the BBB comes from the extracellular matrix and astrocytic end feet. B) In the unhealthy brain, ROS accumulate from various sources including mitochondria, microglia, the lipid bilayer, and astrocytes. This causes altered assembly of the tight junctions, breakdown of the extracellular matrix by MMPs and subsequent loss of BBB integrity.
Lastly, the properties described herein that can increase oxidative stress not only lead to neurodegeneration and breakdown of the BBB via disruption of tight junction proteins, but can alter blood flow as well [82]. This interaction is complex, particularly for O2- which can cause relaxation as well as contraction of cerebral blood vessels depending on the concentration and presence of other species [82]. For example, in the presence of NADPH, contraction occurs but when O2- is produced from xanthine, dilatation occurs [82]. When O2- inactivates NO, a loss of vasodilation is observed [83]. H2O2 also has varying effects: in lower concentrations, dilation occurs [84] and in high concentrations, the vasodilation is preceded by a period of vasoconstriction [82, 85]. Altogether, the brain, particularly the cerebrovascular system, features many sources of producing oxidative stress as well as targets vulnerable to this stress allowing for a cycle of damage.
2.1 Therapies Designed to Combat Oxidative Stress: Pharmaceutical Approaches
As more evidence points to the role of oxidative stress in neurodegenerative disorders, a number of antioxidant-based therapies have been developed. Unfortunately, there has not been a significant amount of success with these antioxidant treatments in humans. For this review, we will recount a number of pharmaceutical approaches and nutritional approaches that have been attempted. However, this list is not exhaustive for all antioxidant-based therapies.
Selegiline ((R)-N-methyl-N-(1-phenylpropan-2-yl)prop-1-yn-3-amine) is a selective, irreversible monoamine oxidase B (MAO-B) inhibitor, when given in low doses [86]. It has been prescribed mostly for Parkinson's disease patients [86], however, it has also been prescribed to treat Alzheimer's disease, dementia and depression [87, 88]. In addition to its effects on the monoamine oxidase system, the drug has also exhibited antioxidant and neuroprotective effects [89]. The main mechanisms of action for its antioxidant properties include reducing oxidative stress products formed during dopamine catabolism and increasing the activity of superoxide dismutase and catalase [89]. Unfortunately, the drug (as for all monoamine oxidase inhibitors) has negative side effects including withdrawal, numerous drug interactions and interactions with foods that can cause hypertensive crisis [88]. For PD treatment, it has not shown a significant effect when given alone, but in combination with levodopa (L-DOPA), some improved effects have been observed (reviewed by [90]). However, this is mainly used to treat early symptoms of PD and is not curative. Another drug that has been developed mainly for treatment of PD is nitecapone (OR-462), a selective catechol-O-methyltransferase (COMT) inhibitor. It also has antioxidant properties including scavenging of free radicals such as peroxy radicals and subsequent inhibition of lipid peroxidation [91]. Again, nitecapone has not displayed significant treatment effects for PD alone, but it has shown efficacy in combination with L-DOPA [5, 92]. While selegiline and nitecapone exert effects particularly on the dopamine metabolism pathway, they both exhibit neuroprotection and antioxidant effects. In considering whether these drugs may also be applied specifically at the BBB for reducing cerebral endothelial cell oxidative stress, selegiline may be the only promising target; selegiline has been shown to cross the BBB [89], however nitecapone does not [93]. Intead, nitecapone exerts its effects mainly on L-DOPA in the periphery [93]. On the other hand, targeting oxidative stress at the BBB may require action on both sides of the endothelial cells (within the vasculature and within the brain). Pharmaceutical antioxidants that can target these areas must be further investigated in future studies.
N-acetylcysteine (NAC) has many proposed uses including treatment of paracetamol overdose [94], drug addiction [95], obsessive-compulsive disorder [96], diabetic neuropathy [97], as well as many neurodegenerative disorders: AD, PD, and MS [98]. NAC is a derivative of cysteine which also exerts antioxidant effects [99]. The mechanisms of action include generation of intracellular GSH [97] and it is reported to directly scavenge H2O2 and OH- in vitro [100]. It has also been shown to decrease the lipid peroxidation product, HNE [101]. While many studies have investigated the various actions and potential uses of NAC, no successful clinical trials for the treatment of neurodegenerative disease have been reported at this time. However, N-acetylcysteine administration has been proposed for various diseases, and it is able to cross the BBB [102], making it an interesting candidate for application at the cerebral endothelial cells.
Recent clinical trials with edavarone (3-Methyl-1-phenyl-2-pyrazolin-5-one), a free radical scavenger, have shown some success for ischemic stroke patients. In a randomized, controlled clinical trial in India, treatment with edavarone compared to placebo resulted in improved functional outcome following acute ischemic stroke [103]. In a randomized, controlled clinical trial in Japan, edavarone treatment reduced the volume of infarct and improved neurological deficits following acute ischemic stroke [104]. It has been proposed that edavarone may be a successful antioxidant therapy in PD as well since experiments performed in mice have shown protection against MPTP-induced damage in the substantia nigra. However, these effects were not observed in the striatum [105]. In comparison to the other pharmaceutical antioxidants reviewed herein, edavarone appears to be the most promising for administration at cerebral endothelial cells and subsequent improvement to BBB disruption. In fact, recent studies have demonstrated improved BBB integrity following edavarone treatment particularly due to its effects on inhibiting MMP-9 [106-108]. Therefore, future studies investigating the direct effects of edavarone on the BBB are necessary.
2. 2 Therapies Designed to Combat Oxidative Stress: Dietary Approaches
A large collection of antioxidants are available from dietary sources including: carotenoids, vitamins A, C and E, and polyphenols/flavonoids [109]. Antioxidants are mainly found in fresh fruits, vegetables, nuts and oils; some of the most well-known antioxidants include: blueberries, spinach, grapes, red wine, curcumin, and olive oil; there is an abundant source in the everyday diet. With many findings on the role of oxidative stress in disease, both pharmacological (as described above) and dietary antioxidants have become prospects for therapeutic development. Unfortunately, most clinical trials have not been successful; in fact, many trials have even proved harmful. As reviewed by Edeas [110], numerous clinical trials with dietary antioxidants for disorders such as cancer (reviewed by [111]), cardiovascular disease (reviewed by [112]), and incidence of stroke (reviewed by [113]) have not shown clinical efficacy to date. Clinical trials with antioxidants for neurodegenerative diseases such as AD and PD have provided some optimism, but they are also not currently at optimal efficacy. As reviewed by Grundman and Delaney [114], it was first determined that certain dietary antioxidants in serum or plasma are at lower levels in AD patients compared to healthy controls such as vitamin E [115-117], vitamin C [115, 116, 118], and vitamin A [115-117, 119]. However, these are controversial results as other studies have found no difference between AD patients and healthy controls for plasma vitamin E [118], vitamin C [120], and vitamin A [120]. The subsequent experiments and clinical trials to increase levels of these vitamins have also provided conflicting results. In the Rotterdam study, as reported by Engelhart [121, 122], subjects were interviewed for dietary/lifestyle factors and follow-up tests included various neurological, metabolic, and cardiovascular exams. It was determined that subjects with a high dietary intake of vitamin E and vitamin C, had a decreased risk of developing AD. In a clinical trial of vitamin C supplements over a four year period, subjects were found to exhibit improved cognitive performance [123]. On the other hand, a recent clinical trial for vitamin E supplementation in stroke did not show improvement [124]. Furthermore, in a review by Boothby and Doering [125], it was concluded that vitamin C and vitamin E supplementation not only showed a lack of improvement for Alzheimer's disease prevalence and incidence, but also increased risk for cardiovascular issues and mortality [125].
It has recently been determined that a necessary balance in dosage and opportunity for synergism with other antioxidants (endogenous or given) must be found in antioxidant treatments [126]. For clinical trials that have actually led to damage, it is believed that the dosage of antioxidant not only reduces ROS and/or RNS, but can halt proper cellular activities as well. Oxidative stress is defined as an imbalance between pro-oxidants and antioxidants [5]; therefore, delivery of too much antioxidant can still tip the scale to an unfavorable level, leading to more oxidative stress. As researchers have now determined this “tightrope” to walk in antioxidant therapy, more attention will be given towards site-specific delivery, regulated dosing, and cocktails of various antioxidants (dietary and/or pharmacological) that may work synergistically for the proper effect. We argue that the cerebral endothelial cells are an important site for new therapies to be designed.
Proposed Mechanisms for Damage at the BBB from Oxidative Stress
One of the most well-studied mechanisms for disruption of the BBB from oxidative stress is via matrix metalloproteinase (MMP) activation. The MMPs are zinc-containing enzymes that degrade extracellular matrix around cerebral blood vessels and neurons [66]. Free radicals activate MMPs which then stimulates their degradation activity; this can occur at the tight junctions and therefore disrupt the vital barrier [65]. Loss of BBB integrity due to oxidative stress via MMP activation is a main issue of cerebral ischemia – reperfusion injury [65]. Therefore, this may be an important target for antioxidant therapies. As mentioned above, the beneficial effects of edavarone as a free radical scavenger, but also a potential MMP9 inhibitor make it an interesting candidate for delivery at the BBB. Other pharmaceutical and/or dietary antioxidants with similar effects must also be explored.
NADPH oxidases (NOXs) also play an important role in vascular oxidative stress as well as BBB disruption [127, 128]. The NOX family (i.e. NOX1, NOX2, NOX3, NOX4) provide a major source of ROS in arterial walls and are implicated in the pathogenesis of hypertension, hypercholesterolemia, diabetes, aging, cerebral amyloid angiopathy (CAA) and Alzheimer's disease [128-133]. On one hand, NOXs are necessary for the regulation of vascular smooth muscle tone, controlling endothelium-dependent dilation responses, and cerebrovascular tone [132]. On the other hand, they are also responsible for disruption of the BBB particularly during stroke as one of the most powerful producers of O2- in the brain and cardiovascular system [64]. Specifically, these enzymes transfer electrons from NADPH to oxygen which creates the superoxide radical [134]. Antioxidants that can act at the BBB to scavenge these free radicals or pharmaceuticals that can block the excessive transfer of electrons to oxygen from NADPH could help to protect the integrity of the BBB.
Another feature described in stroke pathogenesis, but one that likely plays a role in neurodegenerative conditions as well, is the toxicity of circulating free iron. Similar to the features described above, iron has an essential role in proper brain functioning such as energy production, protein function, and mitochondrial enzyme function [135]. In fact, iron is taken up through nutrition and then transported across the BBB, mainly through the transferrin pathway [63]. However, when free iron is circulating, it binds oxygen to produce hydroxyl radicals [4]. During stroke, when the BBB is compromised, free plasma iron can leak into the brain leading to increased oxidative stress and neuronal death [62]. This provides a damaging cycle of cell death and the opportunity for further degradation of the BBB. Therefore, therapies targeted at scavenging free iron as well as the hydroxyl radicals produced would be useful for maintaining the BBB.
Arguably, all neurodegenerative conditions involve inflammation and therefore, glial (microglial and/or astrocyte) activation. As described for the neurodegenerative disorders above, the activation of glia acts as a stimulator for oxidative stress. Together, breakdown of the BBB can occur. Upon activation, glia are capable of releasing O2-, H2O2, and NO- in addition to cytokines [4]. In fact, certain cytokines may lead to increased production of ROS and RNS during stimulation. All of the mechanisms involved in BBB disruption are not currently understood, however, MMP degradation of tight junction proteins and lack of astrocytic end feet for support are two ways that glial activation contribute. As mentioned previously, drug cocktails are likely an important method for treating the multifactorial neurodegenerative conditions; therapeutics that include anti-inflammatory and antioxidant action seem to be a promising technique.
There are a few neurodegenerative conditions that have already been described to involve loss of BBB integrity due, at least in part, to oxidative stress. In hypoxia-reoxygenation, ROS production increases particularly during the reoxygenation phase which can alter tight junction protein assembly and function leading to leakage of the BBB [2, 136]. As mentioned previously, in Alzheimer's disease, production of ROS from various sources including activated microglia and Aβ plaques also causes a breakdown of the BBB. Finally, in CAA, which is characterized by Aβ in the blood vessel wall, oxidative stress is implicated in disease pathogenesis and BBB breakdown, with the contribution of MMP activation by ROS [133].
3. Summary and Future Directions
Taken together, these data point to the cerebrovasculature as both a source, and target of oxidative stress in a variety of neurodegenerative conditions. The causative role of oxidative stress in stroke and mediating role in stroke pathogenesis has been well described. However, relatively little is known in regards to the contribution of these factors to other neurodegenerative disorders. Studies in the near future, from our laboratory and others, will identify whether oxidative stress in the vasculature serves as an essential role in propagating neuropathogenesis in AD and other disorders. In particular, studies are needed to selectively target the cerebrovasculature for antioxidant-based interventions. Taking advantage of the unique microenvironment of the BBB and cerebrovasculature will likely be key to identifying the most effective antioxidant-based interventions for numerous age-related neurodegenerative conditions. Preservation of the function of the vasculature via vessel-targeted antioxidants will ensure sufficient tissue perfusion, maintenance of BBB integrity, and the promotion of brain health in a variety of neurodegenerative disorders.
Highlights.
Review of the role of oxidative stress in neurodegenerative disease
Sources of cerebral endothelial cell oxidative stress are discussed
Effects of oxidative stress on cerebral endothelial cells are discussed
Some of the current antioxidant treatments are reviewed (pharmaceutical and dietary)
The potential use of antioxidants targeted to cerebrovascular oxidative stress is discussed
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shulman RG, Rothman DL, Behar KL, Hyder F. Energetic basis of brain activity: implications for neuroimaging. Trends Neurosci. 2004;27:489–495. doi: 10.1016/j.tins.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Lochhead JJ, McCaffrey G, Quigley CE, Finch J, DeMarco KM, Nametz N, Davis TP. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010;30:1625–1636. doi: 10.1038/jcbfm.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grammas P, Martinez J, Miller B. Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseases. Expert Rev Mol Med. 2011;13:e19. doi: 10.1017/S1462399411001918. [DOI] [PubMed] [Google Scholar]
- 4.Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18:685–716. doi: 10.2165/00002512-200118090-00004. [DOI] [PubMed] [Google Scholar]
- 5.Halliwell B. Antioxidants and human disease: a general introduction. Nutr Rev. 1997;55:S44–49. doi: 10.1111/j.1753-4887.1997.tb06100.x. discussion S49-52. [DOI] [PubMed] [Google Scholar]
- 6.Halliwell B. Antioxidant defence mechanisms: from the beginning to the end (of the beginning) Free Radic Res. 1999;31:261–272. doi: 10.1080/10715769900300841. [DOI] [PubMed] [Google Scholar]
- 7.Mariani E, Polidori MC, Cherubini A, Mecocci P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;827:65–75. doi: 10.1016/j.jchromb.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 8.Halliwell B. Oxygen and nitrogen are pro-carcinogens. Damage to DNA by reactive oxygen, chlorine and nitrogen species: measurement, mechanism and the effects of nutrition. Mutat Res. 1999;443:37–52. doi: 10.1016/s1383-5742(99)00009-5. [DOI] [PubMed] [Google Scholar]
- 9.Tayarani I, Chaudiere J, Lefauconnier JM, Bourre JM. Enzymatic protection against peroxidative damage in isolated brain capillaries. J Neurochem. 1987;48:1399–1402. doi: 10.1111/j.1471-4159.1987.tb05677.x. [DOI] [PubMed] [Google Scholar]
- 10.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 11.Good PF, Werner P, Hsu A, Olanow CW, Perl DP. Evidence of neuronal oxidative damage in Alzheimer's disease. Am J Pathol. 1996;149:21–28. [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu X, Lee HG, Casadesus G, Avila J, Drew K, Perry G, Smith MA. Oxidative imbalance in Alzheimer's disease. Mol Neurobiol. 2005;31:205–217. doi: 10.1385/MN:31:1-3:205. [DOI] [PubMed] [Google Scholar]
- 13.Schapira AH. Mitochondrial dysfunction in neurodegenerative disorders. Biochim Biophys Acta. 1998;1366:225–233. doi: 10.1016/s0005-2728(98)00115-7. [DOI] [PubMed] [Google Scholar]
- 14.Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, Zhu X. Impaired Mitochondrial Biogenesis Contributes to Mitochondrial Dysfunction in Alzheimer's Disease. J Neurochem. 2011 doi: 10.1111/j.1471-4159.2011.07581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swerdlow RH, Burns JM, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis. J Alzheimers Dis. 2010;20(2):S265–279. doi: 10.3233/JAD-2010-100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swerdlow RH. The neurodegenerative mitochondriopathies. J Alzheimers Dis. 2009;17:737–751. doi: 10.3233/JAD-2009-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandrasekaran K, Giordano T, Brady DR, Stoll J, Martin LJ, Rapoport SI. Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease. Brain Res Mol Brain Res. 1994;24:336–340. doi: 10.1016/0169-328x(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 18.Maurer I, Zierz S, Moller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. 2000;21:455–462. doi: 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 19.Nagy Z, Esiri MM, LeGris M, Matthews PM. Mitochondrial enzyme expression in the hippocampus in relation to Alzheimer-type pathology. Acta Neuropathol. 1999;97:346–354. doi: 10.1007/s004010050997. [DOI] [PubMed] [Google Scholar]
- 20.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 21.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 22.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Muiswinkel FL, Veerhuis R, Eikelenboom P. Amyloid beta protein primes cultured rat microglial cells for an enhanced phorbol 12-myristate 13-acetate-induced respiratory burst activity. J Neurochem. 1996;66:2468–2476. doi: 10.1046/j.1471-4159.1996.66062468.x. [DOI] [PubMed] [Google Scholar]
- 24.Klegeris A, McGeer PL. beta-amyloid protein enhances macrophage production of oxygen free radicals and glutamate. J Neurosci Res. 1997;49:229–235. doi: 10.1002/(sici)1097-4547(19970715)49:2<229::aid-jnr11>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 25.Favit A, Grimaldi M, Alkon DL. Prevention of beta-amyloid neurotoxicity by blockade of the ubiquitin-proteasome proteolytic pathway. J Neurochem. 2000;75:1258–1263. doi: 10.1046/j.1471-4159.2000.0751258.x. [DOI] [PubMed] [Google Scholar]
- 26.Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- 27.Keller JN, Hanni KB, Markesbery WR. Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech Ageing Dev. 2000;113:61–70. doi: 10.1016/s0047-6374(99)00101-3. [DOI] [PubMed] [Google Scholar]
- 28.Gregori L, Hainfeld JF, Simon MN, Goldgaber D. Binding of amyloid beta protein to the 20 S proteasome. J Biol Chem. 1997;272:58–62. doi: 10.1074/jbc.272.1.58. [DOI] [PubMed] [Google Scholar]
- 29.Dorheim MA, Tracey WR, Pollock JS, Grammas P. Nitric oxide synthase activity is elevated in brain microvessels in Alzheimer's disease. Biochem Biophys Res Commun. 1994;205:659–665. doi: 10.1006/bbrc.1994.2716. [DOI] [PubMed] [Google Scholar]
- 30.Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 31.Parker WD, Jr, Parks JK, Swerdlow RH. Complex I deficiency in Parkinson's disease frontal cortex. Brain Res. 2008;1189:215–218. doi: 10.1016/j.brainres.2007.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bueler H. Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson's disease. Exp Neurol. 2009;218:235–246. doi: 10.1016/j.expneurol.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 33.Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 35.Chinta SJ, Andersen JK. Redox imbalance in Parkinson's disease. Biochim Biophys Acta. 2008;1780:1362–1367. doi: 10.1016/j.bbagen.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 37.Kopin IJ. MPTP: an industrial chemical and contaminant of illicit narcotics stimulates a new era in research on Parkinson's disease. Environ Health Perspect. 1987;75:45–51. doi: 10.1289/ehp.877545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 39.Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson's disease. J Neurosci. 2003;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem. 2002;277:1641–1644. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
- 41.McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA. Environmental risk factors and Parkinson's disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10:119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- 42.Kuroda Y, Mitsui T, Akaike M, Azuma H, Matsumoto T. Homozygous deletion mutation of the parkin gene in patients with atypical parkinsonism. J Neurol Neurosurg Psychiatry. 2001;71:231–234. doi: 10.1136/jnnp.71.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 44.Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 45.Kuroda Y, Mitsui T, Kunishige M, Matsumoto T. Parkin affects mitochondrial function and apoptosis in neuronal and myogenic cells. Biochem Biophys Res Commun. 2006;348:787–793. doi: 10.1016/j.bbrc.2006.06.201. [DOI] [PubMed] [Google Scholar]
- 46.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 47.Lassmann H. Mechanisms of neurodegeneration shared between multiple sclerosis and Alzheimer's disease. J Neural Transm. 2011;118:747–752. doi: 10.1007/s00702-011-0607-8. [DOI] [PubMed] [Google Scholar]
- 48.Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Lassmann H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132:1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lassmann H. Pathophysiology of inflammation and tissue injury in multiple sclerosis: what are the targets for therapy. J Neurol Sci. 2011;306:167–169. doi: 10.1016/j.jns.2010.07.023. [DOI] [PubMed] [Google Scholar]
- 50.Mahad D, Lassmann H, Turnbull D. Review: Mitochondria and disease progression in multiple sclerosis. Neuropathol Appl Neurobiol. 2008;34:577–589. doi: 10.1111/j.1365-2990.2008.00987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veto S, Acs P, Bauer J, Lassmann H, Berente Z, Setalo G, Jr, Borgulya G, Sumegi B, Komoly S, Gallyas F, Jr, Illes Z. Inhibiting poly(ADP-ribose)polymerase: a potential therapy against oligodendrocyte death. Brain. 2010;133:822–834. doi: 10.1093/brain/awp337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Horssen J, Schreibelt G, Drexhage J, Hazes T, Dijkstra CD, van der Valk P, de Vries HE. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic Biol Med. 2008;45:1729–1737. doi: 10.1016/j.freeradbiomed.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 53.van Horssen J, Witte ME, Schreibelt G, de Vries HE. Radical changes in multiple sclerosis pathogenesis. Biochim Biophys Acta. 2011;1812:141–150. doi: 10.1016/j.bbadis.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 54.Shi P, Wei Y, Zhang J, Gal J, Zhu H. Mitochondrial dysfunction is a converging point of multiple pathological pathways in amyotrophic lateral sclerosis. J Alzheimers Dis. 2010;20(2):S311–324. doi: 10.3233/JAD-2010-100366. [DOI] [PubMed] [Google Scholar]
- 55.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 56.Higgins CM, Jung C, Ding H, Xu Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci. 2002;22:RC215. doi: 10.1523/JNEUROSCI.22-06-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 58.Dupuis L, de Tapia M, Rene F, Lutz-Bucher B, Gordon JW, Mercken L, Pradier L, Loeffler JP. Differential screening of mutated SOD1 transgenic mice reveals early up-regulation of a fast axonal transport component in spinal cord motor neurons. Neurobiol Dis. 2000;7:274–285. doi: 10.1006/nbdi.2000.0292. [DOI] [PubMed] [Google Scholar]
- 59.Turchan-Cholewo J, Dimayuga FO, Gupta S, Keller JN, Knapp PE, Hauser KF, Bruce-Keller AJ. Morphine and HIV-Tat increase microglial-free radical production and oxidative stress: possible role in cytokine regulation. J Neurochem. 2009;108:202–215. doi: 10.1111/j.1471-4159.2008.05756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Turchan-Cholewo J, Dimayuga VM, Gupta S, Gorospe RM, Keller JN, Bruce-Keller AJ. NADPH oxidase drives cytokine and neurotoxin release from microglia and macrophages in response to HIV-Tat. Antioxid Redox Signal. 2009;11:193–204. doi: 10.1089/ars.2008.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jung JE, Kim GS, Chen H, Maier CM, Narasimhan P, Song YS, Niizuma K, Katsu M, Okami N, Yoshioka H, Sakata H, Goeders CE, Chan PH. Reperfusion and neurovascular dysfunction in stroke: from basic mechanisms to potential strategies for neuroprotection. Mol Neurobiol. 2010;41:172–179. doi: 10.1007/s12035-010-8102-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davalos A, Castillo J, Marrugat J, Fernandez-Real JM, Armengou A, Cacabelos P, Rama R. Body iron stores and early neurologic deterioration in acute cerebral infarction. Neurology. 2000;54:1568–1574. doi: 10.1212/wnl.54.8.1568. [DOI] [PubMed] [Google Scholar]
- 63.Qian ZM, Shen X. Brain iron transport and neurodegeneration. Trends Mol Med. 2001;7:103–108. doi: 10.1016/s1471-4914(00)01910-9. [DOI] [PubMed] [Google Scholar]
- 64.Chrissobolis S, Miller AA, Drummond GR, Kemp-Harper BK, Sobey CG. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front Biosci. 2011;16:1733–1745. doi: 10.2741/3816. [DOI] [PubMed] [Google Scholar]
- 65.Gu Y, Zheng G, Xu M, Li Y, Chen X, Zhu W, Tong Y, Chung SK, Liu KJ, Shen J. Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J Neurochem. 2011 doi: 10.1111/j.1471-4159.2011.07542.x. [DOI] [PubMed] [Google Scholar]
- 66.Gu Y, Dee CM, Shen J. Interaction of free radicals, matrix metalloproteinases and caveolin-1 impacts blood-brain barrier permeability. Front Biosci (Schol Ed) 2011;3:1216–1231. doi: 10.2741/222. [DOI] [PubMed] [Google Scholar]
- 67.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 68.Liebner S, Czupalla CJ, Wolburg H. Current concepts of blood-brain barrier development. Int J Dev Biol. 2011;55:467–476. doi: 10.1387/ijdb.103224sl. [DOI] [PubMed] [Google Scholar]
- 69.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 70.Agarwal R, Shukla GS. Potential role of cerebral glutathione in the maintenance of blood-brain barrier integrity in rat. Neurochem Res. 1999;24:1507–1514. doi: 10.1023/a:1021191729865. [DOI] [PubMed] [Google Scholar]
- 71.Vatassery GT. Vitamin E and other endogenous antioxidants in the central nervous system. Geriatrics. 1998;53(1):S25–27. [PubMed] [Google Scholar]
- 72.Miao L, St Clair DK. Regulation of superoxide dismutase genes: implications in disease. Free Radic Biol Med. 2009;47:344–356. doi: 10.1016/j.freeradbiomed.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Forstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 2010;459:923–939. doi: 10.1007/s00424-010-0808-2. [DOI] [PubMed] [Google Scholar]
- 74.Tufekci KU, Civi Bayin E, Genc S, GEnc K. The Nrf2/ARE Pathway: A Promising Target to Counteract Mitochondrial Dysfunction in Parkinson's Disease. Parkinsons Dis. 2011;2011:314082. doi: 10.4061/2011/314082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Merrill JE, Murphy SP. Inflammatory events at the blood brain barrier: regulation of adhesion molecules, cytokines, and chemokines by reactive nitrogen and oxygen species. Brain Behav Immun. 1997;11:245–263. doi: 10.1006/brbi.1997.0496. [DOI] [PubMed] [Google Scholar]
- 77.Dawson TM, Snyder SH. Gases as biological messengers: nitric oxide and carbon monoxide in the brain. J Neurosci. 1994;14:5147–5159. doi: 10.1523/JNEUROSCI.14-09-05147.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 79.Butterfield DA, Bader Lange ML, Sultana R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer's disease. Biochim Biophys Acta. 2010;1801:924–929. doi: 10.1016/j.bbalip.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zarkovic K. 4-hydroxynonenal and neurodegenerative diseases. Mol Aspects Med. 2003;24:293–303. doi: 10.1016/s0098-2997(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 81.Mooradian AD. Effect of aging on the blood-brain barrier. Neurobiol Aging. 1988;9:31–39. doi: 10.1016/s0197-4580(88)80013-7. [DOI] [PubMed] [Google Scholar]
- 82.Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4:461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- 83.Miller AA, Dusting GJ, Roulston CL, Sobey CG. NADPH-oxidase activity is elevated in penumbral and non-ischemic cerebral arteries following stroke. Brain Res. 2006;1111:111–116. doi: 10.1016/j.brainres.2006.06.082. [DOI] [PubMed] [Google Scholar]
- 84.Paravicini TM, Chrissobolis S, Drummond GR, Sobey CG. Increased NADPH-oxidase activity and Nox4 expression during chronic hypertension is associated with enhanced cerebral vasodilatation to NADPH in vivo. Stroke. 2004;35:584–589. doi: 10.1161/01.STR.0000112974.37028.58. [DOI] [PubMed] [Google Scholar]
- 85.Faraci FM, Sobey CG. Role of potassium channels in regulation of cerebral vascular tone. J Cereb Blood Flow Metab. 1998;18:1047–1063. doi: 10.1097/00004647-199810000-00001. [DOI] [PubMed] [Google Scholar]
- 86.Rascol O, Lozano A, Stern M, Poewe W. Milestones in Parkinson's disease therapeutics. Mov Disord. 2011;26:1072–1082. doi: 10.1002/mds.23714. [DOI] [PubMed] [Google Scholar]
- 87.Riederer P, Lachenmayer L, Laux G. Clinical applications of MAO-inhibitors. Curr Med Chem. 2004;11:2033–2043. doi: 10.2174/0929867043364775. [DOI] [PubMed] [Google Scholar]
- 88.Yamada M, Yasuhara H. Clinical pharmacology of MAO inhibitors: safety and future. Neurotoxicology. 2004;25:215–221. doi: 10.1016/S0161-813X(03)00097-4. [DOI] [PubMed] [Google Scholar]
- 89.Takahata K, Shimazu S, Katsuki H, Yoneda F, Akaike A. Effects of selegiline on antioxidant systems in the nigrostriatum in rat. J Neural Transm. 2006;113:151–158. doi: 10.1007/s00702-005-0309-1. [DOI] [PubMed] [Google Scholar]
- 90.Lew MF. The evidence for disease modification in Parkinson's disease. Int J Neurosci, 121 Suppl. 2011;2:18–26. doi: 10.3109/00207454.2011.620194. [DOI] [PubMed] [Google Scholar]
- 91.Bertolini F, Novaroli L, Carrupt PA, Reist M. Novel screening assay for antioxidant protection against peroxyl radical-induced loss of protein function. J Pharm Sci. 2007;96:2931–2944. doi: 10.1002/jps.20881. [DOI] [PubMed] [Google Scholar]
- 92.Martinez-Martin P, O'Brien CF. Extending levodopa action: COMT inhibition. Neurology. 1998;50:S27–32. doi: 10.1212/wnl.50.6_suppl_6.s27. discussion S44-28. [DOI] [PubMed] [Google Scholar]
- 93.Merello M, Lees AJ, Webster R, Bovingdon M, Gordin A. Effect of entacapone, a peripherally acting catechol-O-methyltransferase inhibitor, on the motor response to acute treatment with levodopa in patients with Parkinson's disease. J Neurol Neurosurg Psychiatry. 1994;57:186–189. doi: 10.1136/jnnp.57.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ferner RE, Dear JW, Bateman DN. Management of paracetamol poisoning. BMJ. 2011;342:d2218. doi: 10.1136/bmj.d2218. [DOI] [PubMed] [Google Scholar]
- 95.Reichel CM, Moussawi K, Do PH, Kalivas PW, See RE. Chronic N-acetylcysteine during abstinence or extinction after cocaine self-administration produces enduring reductions in drug seeking. J Pharmacol Exp Ther. 2011;337:487–493. doi: 10.1124/jpet.111.179317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Camfield DA, Sarris J, Berk M. Nutraceuticals in the treatment of obsessive compulsive disorder (OCD): a review of mechanistic and clinical evidence. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:887–895. doi: 10.1016/j.pnpbp.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 97.Kamboj SS, Chopra K, Sandhir R. Hyperglycemia-induced alterations in synaptosomal membrane fluidity and activity of membrane bound enzymes: beneficial effect of N-acetylcysteine supplementation. Neuroscience. 2009;162:349–358. doi: 10.1016/j.neuroscience.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 98.Deigner HP, Haberkorn U, Kinscherf R. Apoptosis modulators in the therapy of neurodegenerative diseases. Expert Opin Investig Drugs. 2000;9:747–764. doi: 10.1517/13543784.9.4.747. [DOI] [PubMed] [Google Scholar]
- 99.Kerksick C, Willoughby D. The antioxidant role of glutathione and N-acetyl-cysteine supplements and exercise-induced oxidative stress. J Int Soc Sports Nutr. 2005;2:38–44. doi: 10.1186/1550-2783-2-2-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid, Free Radic. Biol Med. 1989;6:593–597. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 101.Hara R, Inomata Y, Kawaji T, Sagara N, Inatani M, Fukushima M, Tanihara H. Suppression of choroidal neovascularization by N-acetyl-cysteine in mice. Curr Eye Res. 2010;35:1012–1020. doi: 10.3109/02713683.2010.500112. [DOI] [PubMed] [Google Scholar]
- 102.Shi R, Huang CC, Aronstam RS, Ercal N, Martin A, Huang YW. N-acetylcysteine amide decreases oxidative stress but not cell death induced by doxorubicin in H9c2 cardiomyocytes. BMC Pharmacol. 2009;9:7. doi: 10.1186/1471-2210-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sharma P, Sinha M, Shukla R, Garg RK, Verma R, Singh MK. A randomized controlled clinical trial to compare the safety and efficacy of edaravone in acute ischemic stroke. Ann Indian Acad Neurol. 2011;14:103–106. doi: 10.4103/0972-2327.82794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nakase T, Yoshioka S, Suzuki A. Free radical scavenger, edaravone, reduces the lesion size of lacunar infarction in human brain ischemic stroke. BMC Neurol. 2011;11:39. doi: 10.1186/1471-2377-11-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kawasaki T, Ishihara K, Ago Y, Baba A, Matsuda T. Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a radical scavenger, prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in the substantia nigra but not the striatum. J Pharmacol Exp Ther. 2007;322:274–281. doi: 10.1124/jpet.106.119206. [DOI] [PubMed] [Google Scholar]
- 106.Isahaya K, Yamada K, Yamatoku M, Sakurai K, Takaishi S, Kato B, Hirayama T, Hasegawa Y. Effects of Edaravone, a Free Radical Scavenger, on Serum Levels of Inflammatory Biomarkers in Acute Brain Infarction. J Stroke Cerebrovasc Dis. 2011 doi: 10.1016/j.jstrokecerebrovasdis.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 107.Nakamura T, Kuroda Y, Yamashita S, Zhang X, Miyamoto O, Tamiya T, Nagao S, Xi G, Keep RF, Itano T. Edaravone attenuates brain edema and neurologic deficits in a rat model of acute intracerebral hemorrhage. Stroke. 2008;39:463–469. doi: 10.1161/STROKEAHA.107.486654. [DOI] [PubMed] [Google Scholar]
- 108.Yagi K, Kitazato KT, Uno M, Tada Y, Kinouchi T, Shimada K, Nagahiro S. Edaravone, a free radical scavenger, inhibits MMP-9-related brain hemorrhage in rats treated with tissue plasminogen activator. Stroke. 2009;40:626–631. doi: 10.1161/STROKEAHA.108.520262. [DOI] [PubMed] [Google Scholar]
- 109.Frei B. Efficacy of dietary antioxidants to prevent oxidative damage and inhibit chronic disease. J Nutr. 2004;134:3196S–3198S. doi: 10.1093/jn/134.11.3196S. [DOI] [PubMed] [Google Scholar]
- 110.Edeas M. Strategies to target mitochondria and oxidative stress by antioxidants: key points and perspectives. Pharm Res. 2011;28:2771–2779. doi: 10.1007/s11095-011-0587-2. [DOI] [PubMed] [Google Scholar]
- 111.Halliwell B. Effect of diet on cancer development: is oxidative DNA damage a biomarker? Free Radic Biol Med. 2002;32:968–974. doi: 10.1016/s0891-5849(02)00808-0. [DOI] [PubMed] [Google Scholar]
- 112.Farbstein D, Kozak-Blickstein A, Levy AP. Antioxidant vitamins and their use in preventing cardiovascular disease. Molecules. 2010;15:8098–8110. doi: 10.3390/molecules15118098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Leppala JM, Virtamo J, Fogelholm R, Huttunen JK, Albanes D, Taylor PR, Heinonen OP. Controlled trial of alpha-tocopherol and beta-carotene supplements on stroke incidence and mortality in male smokers. Arterioscler Thromb Vasc Biol. 2000;20:230–235. doi: 10.1161/01.atv.20.1.230. [DOI] [PubMed] [Google Scholar]
- 114.Grundman M, Delaney P. Antioxidant strategies for Alzheimer's disease. Proc Nutr Soc. 2002;61:191–202. doi: 10.1079/PNS2002146. [DOI] [PubMed] [Google Scholar]
- 115.Jeandel C, Nicolas MB, Dubois F, Nabet-Belleville F, Penin F, Cuny G. Lipid peroxidation and free radical scavengers in Alzheimer's disease. Gerontology. 1989;35:275–282. doi: 10.1159/000213037. [DOI] [PubMed] [Google Scholar]
- 116.Foy CJ, Passmore AP, Vahidassr MD, Young IS, Lawson JT. Plasma chain-breaking antioxidants in Alzheimer's disease, vascular dementia and Parkinson's disease. QJM. 1999;92:39–45. doi: 10.1093/qjmed/92.1.39. [DOI] [PubMed] [Google Scholar]
- 117.Bourdel-Marchasson I, Delmas-Beauvieux MC, Peuchant E, Richard-Harston S, Decamps A, Reignier B, Emeriau JP, Rainfray M. Antioxidant defences and oxidative stress markers in erythrocytes and plasma from normally nourished elderly Alzheimer patients. Age Ageing. 2001;30:235–241. doi: 10.1093/ageing/30.3.235. [DOI] [PubMed] [Google Scholar]
- 118.Riviere S, Birlouez-Aragon I, Nourhashemi F, Vellas B. Low plasma vitamin C in Alzheimer patients despite an adequate diet. Int J Geriatr Psychiatry. 1998;13:749–754. doi: 10.1002/(sici)1099-1166(1998110)13:11<749::aid-gps860>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 119.Zaman Z, Roche S, Fielden P, Frost PG, Niriella DC, Cayley AC. Plasma concentrations of vitamins A and E and carotenoids in Alzheimer's disease. Age Ageing. 1992;21:91–94. doi: 10.1093/ageing/21.2.91. [DOI] [PubMed] [Google Scholar]
- 120.Sinclair AJ, Bayer AJ, Johnston J, Warner C, Maxwell SR. Altered plasma antioxidant status in subjects with Alzheimer's disease and vascular dementia. Int J Geriatr Psychiatry. 1998;13:840–845. doi: 10.1002/(sici)1099-1166(1998120)13:12<840::aid-gps877>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 121.Engelhart MJ, Geerlings MI, Ruitenberg A, Van Swieten JC, Hofman A, Witteman JC, Breteler MM. Diet and risk of dementia: Does fat matter?: The Rotterdam Study. Neurology. 2002;59:1915–1921. doi: 10.1212/01.wnl.0000038345.77753.46. [DOI] [PubMed] [Google Scholar]
- 122.Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Witteman JC, Breteler MM. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA. 2002;287:3223–3229. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- 123.Paleologos M, Cumming RG, Lazarus R. Cohort study of vitamin C intake and cognitive impairment. Am J Epidemiol. 1998;148:45–50. doi: 10.1093/oxfordjournals.aje.a009559. [DOI] [PubMed] [Google Scholar]
- 124.Bin Q, Hu X, Cao Y, Gao F. The role of vitamin E (tocopherol) supplementation in the prevention of stroke. A meta-analysis of 13 randomised controlled trials. Thromb Haemost. 2011;105:579–585. doi: 10.1160/TH10-11-0729. [DOI] [PubMed] [Google Scholar]
- 125.Boothby LA, Doering PL. Vitamin C and vitamin E for Alzheimer's disease. Ann Pharmacother. 2005;39:2073–2080. doi: 10.1345/aph.1E495. [DOI] [PubMed] [Google Scholar]
- 126.Halliwell B. Establishing the significance and optimal intake of dietary antioxidants: the biomarker concept. Nutr Rev. 1999;57:104–113. doi: 10.1111/j.1753-4887.1999.tb06933.x. [DOI] [PubMed] [Google Scholar]
- 127.Basuroy S, Bhattacharya S, Leffler CW, Parfenova H. Nox4 NADPH oxidase mediates oxidative stress and apoptosis caused by TNF-alpha in cerebral vascular endothelial cells. Am J Physiol Cell Physiol. 2009;296:C422–432. doi: 10.1152/ajpcell.00381.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Touyz RM, Briones AM, Sedeek M, Burger D, Montezano AC. NOX isoforms and reactive oxygen species in vascular health. Mol Interv. 2011;11:27–35. doi: 10.1124/mi.11.1.5. [DOI] [PubMed] [Google Scholar]
- 129.Bruce-Keller AJ, Gupta S, Knight AG, Beckett TL, McMullen JM, Davis PR, Murphy MP, Van Eldik LJ, St Clair D, Keller JN. Cognitive impairment in humanized APPxPS1 mice is linked to Abeta(1-42) and NOX activation. Neurobiol Dis. 2011;44:317–326. doi: 10.1016/j.nbd.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bruce-Keller AJ, Gupta S, Parrino TE, Knight AG, Ebenezer PJ, Weidner AM, LeVine H, 3rd, Keller JN, Markesbery WR. NOX activity is increased in mild cognitive impairment. Antioxid Redox Signal. 2010;12:1371–1382. doi: 10.1089/ars.2009.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bruce-Keller AJ, White CL, Gupta S, Knight AG, Pistell PJ, Ingram DK, Morrison CD, Keller JN. NOX activity in brain aging: exacerbation by high fat diet. Free Radic Biol Med. 2010;49:22–30. doi: 10.1016/j.freeradbiomed.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Touyz RM, Briones AM. Reactive oxygen species and vascular biology: implications in human hypertension. Hypertens Res. 2011;34:5–14. doi: 10.1038/hr.2010.201. [DOI] [PubMed] [Google Scholar]
- 133.Carrano A, Hoozemans JJ, van der Vies SM, Rozemuller AJ, van Horssen J, de Vries HE. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal. 2011;15:1167–1178. doi: 10.1089/ars.2011.3895. [DOI] [PubMed] [Google Scholar]
- 134.Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sorond FA, Ratan RR. Ironing-out mechanisms of neuronal injury under hypoxic-ischemic conditions and potential role of iron chelators as neuroprotective agents. Antioxid Redox Signal. 2000;2:421–436. doi: 10.1089/15230860050192206. [DOI] [PubMed] [Google Scholar]
- 136.McCaffrey G, Willis CL, Staatz WD, Nametz N, Quigley CA, Hom S, Lochhead JJ, Davis TP. Occludin oligomeric assemblies at tight junctions of the blood-brain barrier are altered by hypoxia and reoxygenation stress. J Neurochem. 2009;110:58–71. doi: 10.1111/j.1471-4159.2009.06113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]