

Abstract
The agricultural fungicide N-(3,5-dichlorophenyl)succinimide (NDPS) can induce marked nephrotoxicity in rats following a single intraperitoneal (ip) administration of 0.4 mmol/kg or greater. Although NDPS induces direct renal proximal tubular toxicity, a role for renal vascular effects may also be present. The purpose of this study was to examine the possible role of vasoconstrictor leukotrienes in NDPS and NDPS metabolite nephrotoxicity. Male Fischer 344 rats (4 rats/group) were administered diethylcarbamazine (DEC; 250 or 500 mg/kg, ip), an inhibitor of LTA4 synthesis, 1h before NDPS (0.4 mmol/kg, ip), N-(3,5-dichlorophenyl)-2-hydroxysuccinimide (NDHS, 0.1, 0.2, or 0.4 mmol/kg, ip), or N-(3,5-dichlorophenyl)-2-hydroxysuccinamic acid (2-NDHSA, 0.1 mmol/kg, ip) or vehicle. In a separate set of experiments, the LTD4 receptor antagonist LY171883 (100 mg/kg, po) was administered 0.5 h before and again 6 h after NDHS (0.1 mmol/kg, ip) or 2-NDHSA (0.1 mmol/kg, ip) or vehicle. Renal function was monitored for 48 h post-NDPS or NDPS metabolite. DEC markedly reduced the nephrotoxicity induced by NDPS and its metabolites, while LY171883 treatments provided only partial attenuation of NDHS and 2-NDHSA nephrotoxicity. These results suggest that leukotrienes contribute to the mechanisms of NDPS nephrotoxicity.
Keywords: Leukotrienes; Kidney; Rats; Nephrotoxicity; N-(3, 5-Dichlorophenyl)succinimide
1. Introduction
N-(3,5-Dichlorophenyl)succinimide (NDPS) was introduced during the early 1970s as a broad spectrum antifungal agent against a number of plant pathogenic fungi including Sclerotinia sclerotiorum and Botrytis cinerea (Fujinami et al., 1971; 1972). Currently, NDPS is available commercially as dimetachlone in China and the United Kingdom for use in agricultural, and it is patented for use in many countries, including the United States, either alone or in various combinations with other pesticides. NDPS also possess antiandrogenic activity (Zhang et al., 2007).
NDPS induces marked acute nephrotoxicity in male rats at ip doses of 0.4 mmol/kg or greater (Rankin, 1982; 2004; Rankin et al., 1984; 1985). Acute NDPS nephrotoxicity is characterized as polyuric renal failure with marked proximal tubular necrosis (Rankin; 2004). NDPS can also induce chronic nephrotoxicity when administered daily in food at concentrations of 5000 ppm or greater, which is seen as early proximal tubular toxicity followed by the formation of renal interstitial nephritis (Barrett et al., 1983; Sugihara et al., 1975).
Acute NDPS nephrotoxicity requires bioactivation of NDPS to toxic metabolites (Rankin, 2004; Rankin et al., 1988). Two NDPS metabolites, N-(3,5-dichlorophenyl)-2-hydroxysuccinimide (NDHS) and N-(3,5-dichlorophenyl)-2-hydroxysuccinamic acid (2-NDHSA, are four times more potent than NDPS as a nephrotoxicant in vivo indicating that the ultimate nephrotoxicant species is derived from NDHS and/or 2-NDHSA (Rankin, 2004). The finding that NDPS, NDHS and 2-NDHSA are not nephrotoxicants in vitro (Aleo et al., 1991; Rankin et al., 1988) suggested that additional bioactivation was required to form this ultimate nephrotoxicant species or that NDPS metabolites induced nephrotoxicity via indirect mechanisms (e.g. renal vasoconstriction). Recent findings have identified O-sulfate and O-glucuronide conjugates of NDHS as the most likely candidates for the ultimate/penultimate direct nephrotoxic species formed from NDPS, with the O-sulfate conjugate of NDHS being a nephrotoxicant in vitro (Cui et al., 2005; Hong et al., 1999a,b,c,d; Rankin et al., 2001). In addition, studies in anesthetized Fischer 344 rats administered NDHS have determined that direct proximal tubular toxicity appear to precede significant changes in renal hemodynamics (Beers and Rankin, 1993). Collectively, these results have supported formation and renal accumulation of a direct tubular nephrotoxicant metabolite as a primary mechanism for initiating NDPS nephrotoxicity.
It has been proposed that NDPS nephrotoxicity involves alkylation of key renal macromolecules by reactive NDPS metabolites (Rankin, 2004). Henesey et al. (1999) found covalently bound NDPS-derived radioactivity in higher concentrations in kidney than in liver or plasma, which supported the renal accumulation of hepatic-derived reactive NDPS metabolites. Surprisingly, NDPS administration at nephrotoxic doses does not markedly deplete renal or hepatic glutathione (Yang et al., 1987) and depletion of glutathione with diethyl maleate (DEM) (Yang et al., 1987) or inhibition of glutathione synthesis with buthionine sulfoximine (BSO) is protective rather than potentiating NDPS and NDPS metabolite nephrotoxicity (Rankin et al., 1990; 1991b). It does not appear that decreasing the formation of a nephrotoxic glutathione conjugate of NDPS can explain the mechanisms for how reduced glutathione concentrations attenuates NDPS or NDPS metabolite nephrotoxicity (Rankin et al., 1991a). Thus, the role glutathione plays in NDPS nephrotoxicity is unclear.
Although direct tubular toxicity is key to the basic mechanisms of NDPS nephrotoxicity, altered renal hemodynamics can also play a role. Indomethacin, a nonsteroidal anti-inflammatory drug (NSAID), which inhibits cyclooxgenase-1 and -2 (COX-1 and COX-2), potentiates NDPS nephrotoxicity, suggesting that inhibition of the formation of renal vasodilatory prostaglandins removes a protective renal mechanism against NDPS nephrotoxicity (Rankin et al., 1991c). Thus, renal vasoconstrictor mechanisms may play a contributory role in enhancing NDPS nephrotoxicity in vivo. However, the mediator of these vasoconstrictor effects does not appear to be a thromboxane, since the thromboxane synthase inhibitor dazmegrel did not alter NDPS metabolite-induced nephrotoxicity (Rankin et al., 1991c).
One possible explanation for these observations and the attenuating effects of glutathione depletion is that a portion of the mechanism of NDPS nephrotoxicity involves the formation of vasoconstrictor leukotrienes (LTC4 and/or LTD4, Fig. 1) via the 5-lipoxygenase pathway. The synthesis of LTC4 and LTD4 begins with arachadonic acid being converted to 5-S-hydroperoxy eicosatetraenoic acid [5(S)-HPETE] which forms the epoxide, LTA4 (Brash, 1999; Smyth et al., 2011). LTA4 is then conjugated with glutathione to form the vasoconstrictor LTC4. Metabolism of the glutathionyl group by γ-glutamyl transpeptidase (γ-GT) yields the cysteinylglycine conjugate vasoconstrictor LTD4 which can be converted by dipeptidase to the cysteine conjugate LTE4 (Brash, 1999). Both LTC4 and LTD4 are renal vasoconstrictors, although LTC4 is more potent in rat kidney (Rosenthal and Pace-Asciak, 1983). In addition, depletion of glutathione with BSO attenuated LTC4-mediated hypoxic pulmonary vasoconstriction (Peters-Golden et al., 1989), similar to the attenuating effect of BSO on NDPS and NDPS metabolite nephrotoxicity. Thus, formation of vasoconstrictor leukotrienes might play a role in NDPS nephrotoxicity. However, the role of leukotrienes in NDPS nephrotoxicity has not been explored.
Figure 1.
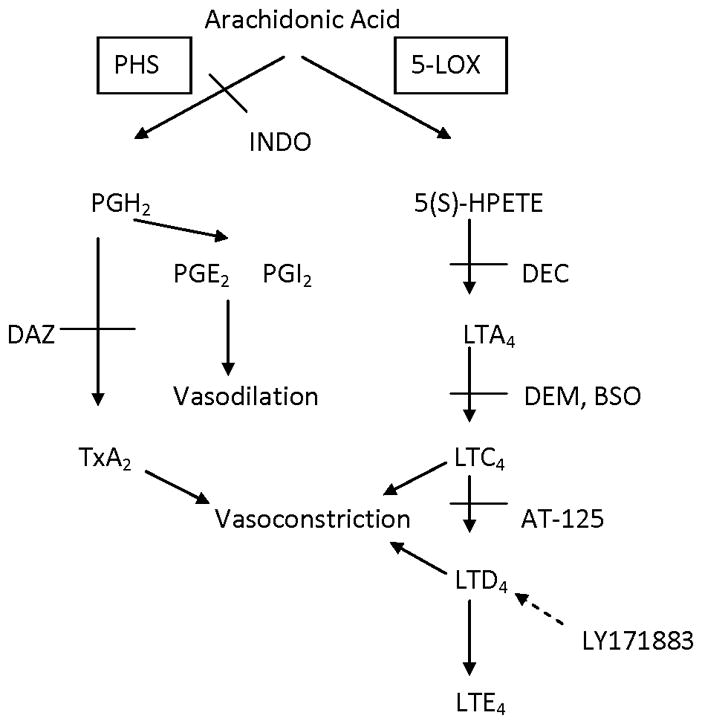
Pathway production of prostaglandins, thromboxanes and leukotrienes from arachidonic acid and sites of potential inhibitors. BSO = buthionine sulfoximine; DAZ = dazmegrel; DEC = diethylcarbamazine; DEM = diethyl maleate; 5-(S)-HPETE = 5-(S)-hydroperoxyeicosatetraenoic acid; INDO = indomethacin; 5-LOX = 5-lipoxygenase; LTA4 (C4, D4, E4) = leukotriene A4 (C4, D4, E4); PGH2 (E2; I2) = prostaglandin H2 (E2; I2); PHS = prostaglandin H synthase; TxA2 = thromboxane A2.
The purpose of this study was to examine the possibility that vasoconstrictor leukotrienes might contribute to mediating NDPS nephrotoxicity by examining the effects of two inhibitors of the 5-lipoxygenase pathway. Diethylcarbamazine (DEC) was selected as an inhibitor of LTA4 formation (Fig. 1) with the dose used based on the work of Balaa and Subramony (1990). LY171883 was chosen as an inhibitor of the LTD4 receptor (Fig. 1) with the dose selected based on the work of Tabuchi et al. (1994).
2. Materials and methods
2.1 Experimental animals
Male Fischer 344 rats (200–250 g) were obtained from Hilltop Lab Animals, Inc. (Scottdale, PA). All animals were kept in a controlled environment with a regulated light cycle (on 06.00 h, off 18.00 h), temperature (21–23°C), and humidity (40–55%). Food (Purina Rat Chow) and water were available ad libitum. Rats were allowed at least one week to acclimate to the facilities prior to use in any experiments. Animal use for these experiments was reviewed and approved by the Marshall University Institutional Animal Care and Use Committee.
2.2 Chemicals
NDPS, NDHS and 2-NDHSA were synthesized, purified and characterized as previously described (Rankin et al., 1984; 1988; Shih and Rankin, 1989). Dimethyl sulfoxide (DMSO) was obtained from Fisher Scientific Co. (Pittsburgh, PA, USA) and was certified A.C.S. grade. Coomassie brilliant blue G-250 dye was purchased from Calbiochem Corp. (La Jolla, CA, USA). LY171883 was a generous gift from Eli Lilly and Co. Diethylcarbamazine (DEC) citrate was obtained from Sigma (St. Louis, MO, USA). All other chemicals were obtained from Sigma or Aldrich (St. Louis, MO, USA) and were of the highest purity available. p-[14C]Aminohippurate (PAH; specific activity = 40.4 mCi/mmol) and [14C]tetraethylammonium (TEA; specific activity = 4.0 mCi/mmol) were obtained from New England Nuclear Corp. (Boston, MA).
2.3 Animal treatments
Following the acclimatization period, rats (four rats/group) were placed singly in stainless steel metabolism cages to allow for the separation of urine from feces. After one acclimatization day in the metabolism cages, control day (day 0) values were obtained. On the following day, rats were administered a single intraperitoneal (ip) injection of NDPS, NDHS or 2-NDHSA (0.4, 0.1 or 0.1 mmol/kg, respectively) or vehicle (sesame oil, 2.5 ml/kg for NDPS; 25% DMSO in sesame oil, 2.5 ml/kg for NDHS and 2-NDHSA). These doses represent the minimum dose of NDPS, NDHS or 2-NDHSA in male Fischer 344 rats which induces marked nephrotoxicity (Rankin, 2004). In a second set of experiments, rats (four rats/group) were administered DEC (250 or 500 mg/kg, 1 ml/kg in 0.9%saline, ip) 1 h prior to NDPS (0.4 mmol/kg, ip), NDHS (0.1, 0.2 or 0.4 mmol/kg, ip) or 2-NDHSA (0.1 mmol/kg, ip) or the appropriate vehicle. In a third set of experiments, rats (four rats/group) were administered LY171883 (100 mg/kg in 0.9% saline, ip) 0.5 h before and 6 h after NDHS (0.1 mmol/kg, ip), 2-NDHSA (0.1 mmol/kg, ip) or NDPS metabolite vehicle. In all experiments, renal function was monitored at 24 and 48 h after NDPS, NDPS metabolite or the appropriate vehicle.
Urine volume, food and water intake and body weight measurements were made at 24 h intervals. On the control and both post-treatment days, food was withheld for 6 h to obtain a urine sample without food contamination for urinary protein excretion measurements. Urinary protein excretion was determined spectrophotometrically at 595 nm using Coomassie blue with bovine albumin (fraction V) serving as the protein standard (Bradford, 1976).
At 24 or 48 h post-treatment with NDPS, an NDPS metabolite or vehicle, rats were anesthetized (diethyl ether) and laparotomized. A blood sample was withdrawn from the dorsal aorta into a heparinized syringe, and plasma was obtained by centrifugation (3,000 x g, 5 min). Plasma was stored at −20°C for blood urea nitrogen (BUN) concentration determination. In some cases, a portion of the plasma was stored at 4°C for ALT activity measurements. A sample of blood was obtained from the tail of each rat prior to placement in a metabolism cage to allow for the determination of the day 0 BUN concentration.
After obtaining the aortic blood sample, the kidneys and liver from each rat were rapidly removed and weighed. Kidneys were then placed in ice-cold Krebs-Ringer buffer (pH 7.4). The left kidney was quartered, renal cortical slices prepared freehand (~0.5 mm thickness) with a razor blade, and the accumulation of [14C]-PAH and [14C]-TEA by these slices (~75–100 mg/beaker) determined as previously described (Rankin et al., 1988). Lactate (10 mM) was added in some PAH experiments to measure lactate-stimulated PAH accumulation. Renal accumulation of PAH and TEA was expressed as the slice-to-medium (S/M) ratio where S equaled the radioactivity (dpm) per gram of tissue and M equaled the radioactivity (dpm) per ml of medium. The right kidney was quartered and placed in 10% neutral buffered formalin. Tissues from NDPS metabolite treated and control rats were examined using light microscopy for evidence of chemically induced morphological changes as previously described (Yang et al., 1987).
In all experiments, control rats were pair-fed to the appropriate treatment group to ensure that any changes observed were chemically induced rather than the result of altered food intake.
2.4 Statistics
Data are presented as the mean ± S.E. for four rats per group. The data were analyzed using an analysis of variance (ANOVA) test followed by a Dunnett’s or Newman-Keuls analysis. All statistical tests were run at a 95% confidence interval and significance was denoted as P < 0.05.
3. Results
3.1 Effects of DEC on NDPS and NDPS metabolite nephrotoxicity
To determine if leukotrienes might play a role in mediating NDPS nephrotoxicity, experiments were first conducted with DEC, an inhibitor of LTA4 formation. Pretreatment of rats with DEC was very effective in attenuating NDPS (0.4 mmol/kg) nephrotoxicity, nephrotoxicity. Diuresis induced by the minimal nephrotoxic dose of NDPS was markedly reduced by DEC on both post-treatment days (Table 1). In the DEC + NDPS treatment group, some diuresis was still observed on post-treatment day 1, but not on day 2. DEC also markedly attenuated NDPS-induced proteinuria on post-treatment days 1 and 2 (Table 2). While the DEC + NDPS treatment group exhibited increased proteinuria on post-treatment day 1, this proteinuria was markedly lower that in the NDPS only treatment groups. DEC pretreatment prevented any NDPS-induced increase in proteinuria on post-treatment day 2.
Table 1.
| Treatment | Urine Volume (ml)b
|
||
|---|---|---|---|
| Day 0 | Day 1 | Day 2 | |
| Vehicle | 14.5 ± 0.6 | 10.5 ± 1.2c | 6.7 ± 1.3c |
| NDPS | 14.2 ± 0.5 | 40.0 ± 4.9c,d | 31.5 ± 1.4c,d |
| DEC + Vehicle | 10.9 ± 1.0 | 10.5 ± 1.0 | 7.2 ± 0.9c |
| DEC + NDPS | 9.3 ± 1.0 | 21.0 ± 3.2c,d | 16.8 ± 4.1 |
| Vehicle | 13.1 ± 0.5 | 10.1 ± 1.7 | 3.1 ± 0.4c |
| NDHS | 14.6 ± 0.6 | 29.5 ± 1.0c,d | 25.9 ± 1.6c,d |
| DEC + Vehicle | 11.3 ± 0.8 | 9.9 ± 0.9 | 8.5 ± 1.3 |
| DEC + NDHS | 12.4 ± 1.0 | 14.7 ± 2.5 | 10.2 ± 0.7 |
| LY171883 + Vehicle | 12.5 ± 0.5 | 12.4 ± 2.3 | N.D.e |
| LY171883 + NDHS | 10.4 ± 1.2 | 32.1 ± 2.6c,d | N.D.e |
| Vehicle | 11.7 ± 0.9 | 11.9 ± 1.7 | 7.0 ± 0.7c |
| 2-NDHSA | 12.2 ± 0.9 | 32.6 ± 2.4c,d | 26.7 ± 0.8c,d |
| DEC + Vehicle | 12.3 ± 0.9 | 9.4 ± 1.4c | 8.1 ± 1.0c |
| DEC + 2-NDHSA | 10.6 ± 0.8 | 21.6 ± 6.0 | 18.4 ± 4.2 |
| LY171883 + Vehicle | 11.0 ± 0.3 | 12.6 ± 0.4 | N.D.e |
| LY171883 + 2-NDHSA | 11.6 ± 0.7 | 31.9 ± 1.0c,d | N.D.e |
Values are means ± S.E. for N=4 rats per group. NDPS (0.4 mmol/kg), NDHS (0.1 mmol/kg), 2-NDHSA (0.1 mmol/kg) or vehicle was administered i.p. on day 1. DEC (250 mg/kg, i.p.) was given 1 hr prior to NDPS, NDHS, 2-NDHSA, or vehicle. LY171883 (100 mg/kg, p.o.) was given 0.5 hr before and 6 hr after the administration of NDHS, 2-NDHSA, or vehicle.
Urine volume was measured at 24 hr intervals.
Significantly different from day 0 value within a group, P<0.05.
Significantly different from the appropriate pair-fed control group value for that day’s measurement, P<0.05.
Not determined. LY171883 experiments were only conducted for 24 hr.
Table 2.
Effect of DEC or LY171883 Treatment Prior to NDPS, NDHS, or 2-NDHSA Administration on Urinary Protein Excretiona
| Treatment | Proteinuria (6 hr)b
|
||
|---|---|---|---|
| Day 0 | Day 1 | Day 2 | |
| Vehicle | 0.9 ± 0.1 | 2.1 ± 0.2c | 1.1 ± 0.2 |
| NDPS | 0.7 ± 0.1 | 25.5 ± 5.0c,d | 10.2 ± 1.2c,d |
| DEC + Vehicle | 1.3 ± 0.1 | 1.8 ± 0.3 | 1.1 ± 0.5 |
| DEC + NDPS | 1.2 ± 0.2 | 11.9 ± 4.4c | 2.5 ± 0.5 |
| Vehicle | 2.9 ± 0.2 | 2.8 ± 0.3 | 1.1 ± 0.1c |
| NDHS | 2.2 ± 0.1 | 28.7 ± 1.4c,d | 15.9 ± 2.1c,d |
| DEC + Vehicle | 2.4 ± 0.4 | 4.0 ± 0.3c | 1.1 ± 0.2c |
| DEC + NDHS | 2.1 ± 0.4 | 6.2 ± 1.2c | 2.0 ± 0.7 |
| LY171883 + Vehicle | 3.0 ± 0.4 | 3.5 ± 0.7 | N.D.e |
| LY171883 + NDHS | 2.2 ± 0.3 | 43.2 ± 5.7c,d | N.D.e |
| Vehicle | 3.4 ± 0.2 | 4.5 ± 0.4 | 2.2 ± 0.3 |
| 2-NDHSA | 2.5 ± 0.1d | 32.5 ± 6.5c,d | 6.9 ± 1.6d |
| DEC + Vehicle | 2.2 ± 0.4 | 2.4 ± 0.2 | 1.6 ± 0.2 |
| DEC + 2-NDHSA | 1.5 ± 0.2 | 16.5 ± 6.1c | 3.6 ± 1.2 |
| LY171883 + Vehicle | 2.9 ± 0.3 | 3.9 ± 0.7 | N.D.e |
| LY171883 + 2-NDHSA | 2.7 ± 0.3 | 34.0 ± 3.1c,d | N.D.e |
Values are means ± S.E. for N=4 rats per group. NDPS (0.4 mmol/kg), NDHS (0.1 mmol/kg), 2-NDHSA (0.1 mmol/kg) or vehicle was administered i.p. on day 1. DEC (250 mg/kg, i.p.) was given 1 hr prior to NDPS, NDHS, 2-NDHSA, or vehicle. LY171883 (100 mg/kg, p.o.) was given 0.5 hr before and 6 hr after the administration of NDHS, 2-NDHSA, or vehicle.
Urine was collected from 09:00 hr to 15:00 hr each day.
Significantly different from day 0 value within a group, P<0.05.
Significantly different from the appropriate pair-fed control group value for that day’s measurement, P<0.05.
Not determined. LY171883 experiments were only conducted for 24 hr.
At 48 hr post-treatment, kidney weight was not significantly increased in the DEC + NDPS group, while NDPS alone significantly increased kidney weight (Table 3). Also, TEA, but not PAH, accumulation was significantly reduced by NDPS (Table 4). The reduction in TEA accumulation induced by NDPS was completely prevented by DEC pretreatment (Table 4). BUN concentration was significantly increased at 48 hr following NDPS administration (Fig. 2). Pretreatment with DEC completely attenuated the increase in BUN concentration induced by NDPS administration (Fig. 2) and returned BUN concentrations in the DEC-pretreated NDPS group to the control group level.
Table 3.
Effect of DEC or LY171883 Treatment Prior to NDPS, NDHS, or 2-NDHSA Administration on Kidney Weight.a
| Treatment | Kidney Weight (g/100 g body wt.)b
|
|
|---|---|---|
| 24 hr | 48 hr | |
| Vehicle | N.D.c | 0.33 ± 0.01 |
| NDPS | N.D.c | 0.49 ± 0.03d |
| DEC + Vehicle | N.D.c | 0.37 ± 0.01 |
| DEC + NDPS | N.D.c | 0.44 ± 0.04 |
| Vehicle | 0.36 ± 0.01 | 0.36 ± 0.1 |
| NDHS | 0.47 ± 0.04d | 0.63 ± 0.03d |
| DEC + Vehicle | N.D.c | 0.39 ± 0.01 |
| DEC + NDHS | N.D.c | 0.36 ± 0.01d |
| LY171883 + Vehicle | 0.35 ± 0.02 | N.D.c |
| LY171883 + NDHS | 0.43 ± 0.01d | N.D.c |
| Vehicle | 0.42 ± 0.01 | 0.31 ± 0.01 |
| 2-NDHSA | 0.49 ± 0.01d | 0.45 ± 0.02d |
| DEC + Vehicle | N.D.c | 0.38 ± 0.01 |
| DEC + 2-NDHSA | N.D.c | 0.44 ± 0.04 |
| LY171883 + Vehicle | 0.33 ± 0.01 | N.D.c |
| LY171883 + 2-NDHSA | 0.45 ± 0.01d | N.D.c |
Values are means ± S.E. for N=4 rats per group. NDPS (0.4 mmol/kg), NDHS (0.1 mmol/kg), 2-NDHSA (0.1 mmol/kg) or vehicle was administered i.p. on day 1. DEC (250 mg/kg, i.p.) was given 1 hr prior to NDPS, NDHS, 2-NDHSA, or vehicle. LY171883 (100 mg/kg, p.o.) was given 0.5 hr before and 6 hr after the administration of NDHS, 2-NDHSA, or vehicle.
Kidneys were obtained at 24 hr or 48 hr post-treatment.
Not determined. LY171883 experiments were only conducted for 24 hr. In NDPS only and DEC pretreatment experiments, kidney weight was only determined at 48hr.
Significantly different from the appropriate pair-fed control group value for that day’s measurement, P<0.05.
Table 4.
Effect of DEC or LY171883 Treatment Prior to NDPS, NDHS, or 2-NDHSA Administration on Organic Ion Accumulation by Renal Cortical Slicesa
| Treatment | S/M Ratio
|
||
|---|---|---|---|
| PAH | PAH + LAC | TEA | |
| 48 hrb | |||
| Vehicle | 2.2 ± 0.2 | 6.8 ± 0.5 | 17.7 ± 0.8 |
| NDPS | 2.3 ± 0.5 | 5.6 ± 1.4 | 13.6 ± 0.6c |
| DEC + Vehicle | 3.5 ± 0.1 | 10.0 ± 0.2 | 16.4 ± 0.4 |
| DEC + NDPS | 3.9 ± 0.2 | 9.3 ± 1.7 | 17.4 ± 2.1 |
| Vehicle | 2.3 ± 0.1 | 5.1 ± 0.3 | 13.4 ± 0.7 |
| NDHS | 2.4 ± 0.3 | 3.5 ± 0.3c | 7.2 ± 0.4c |
| DEC + Vehicle | 5.1 ± 0.2 | 10.8 ± 0.4 | 20.5 ± 1.0 |
| DEC + NDHS | 5.0 ± 0.1 | 10.7 ± 0.6 | 23.7 ± 1.0 |
| Vehicle | 3.0 ± 0.2 | 7.6 ± 0.5 | 18.4 ± 0.7 |
| 2-NDHSA | 2.5 ± 0.2 | 4.1 ± 0.3c | 12.8 ± 0.9c |
| DEC + Vehicle | 4.2 ± 0.2 | 10.9 ± 0.5 | 17.6 ± 0.7 |
| DEC + 2-NDHSA | 4.6 ± 0.3 | 10.3 ± 1.2 | 18.9 ± 0.5 |
| 24 hrd | |||
| Vehicle | 4.3 ± 0.2 | 10.1 ± 0.3 | 20.5 ± 0.5 |
| NDHS | 2.8 ± 0.3c | 6.9 ± 1.1c | 19.0 ± 0.6 |
| LY171883 + Vehicle | 2.7 ± 0.1 | 6.7 ± 0.4 | 18.6 ± 0.4 |
| LY171883 + NDHS | 2.6 ± 0.1 | 6.5 ± 0.6 | 17.8 ± 0.4 |
| Vehicle | 5.3 ± 0.1 | 13.7 ± 0.5 | 18.4 ± 0.3 |
| 2-NDHSA | 3.2 ± 0.2c | 7.5 ± 0.4c | 18.4 ± 0.8 |
| LY171883 + Vehicle | 4.0 ± 0.3 | 9.0 ± 0.3 | 18.0 ± 0.5 |
| LY171883 + 2-NDHSA | 3.4 ± 0.2 | 7.2 ± 0.4c | 18.9 ± 0.3 |
Values are mean S/M ratios ± S.E. for N=4 rats per group. NDPS (0.4 mmol/kg), NDHS (0.1 mmol/kg), 2-NDHSA (0.1 mmol/kg) or vehicle was administered i.p. on day 1. DEC (250 mg/kg, i.p.) was given 1 hr prior to NDPS, NDHS, 2-NDHSA, or vehicle. LY171883 (100 mg/kg, p.o.) was given 0.5 hr before and 6 hr after the administration of NDHS, 2-NDHSA, or vehicle.
Kidneys used in this study were obtained at 48 hr post-treatment.
Significantly different from the appropriate pair-fed control group value for that day’s measurement, P<0.05.
Kidneys were obtained at 24 hr post-treatment.
Figure 2.
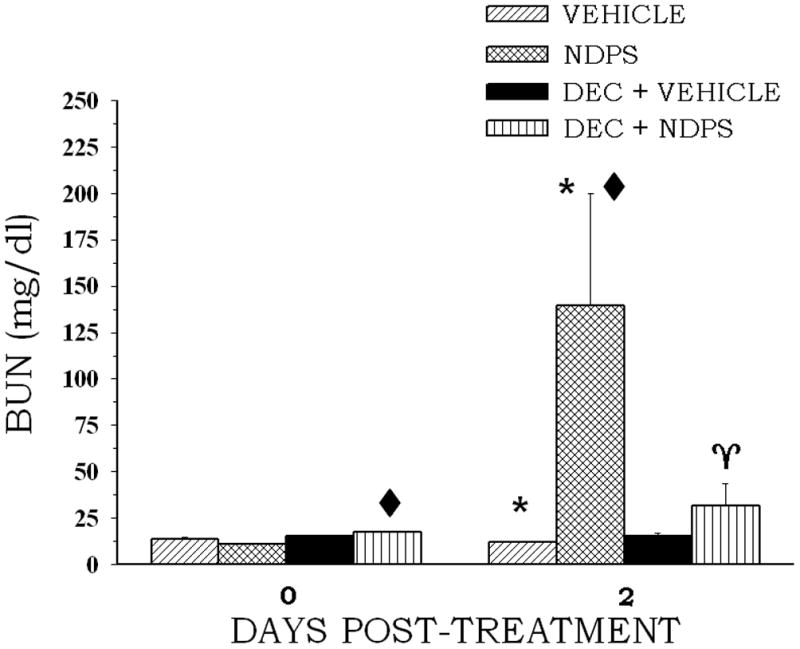
The effect of DEC pretreatment on NDPS-induced changes in BUN concentration. Rats (4 per group) were administered DEC (250 mg/kg, ip) 1 h before an ip injection of NDPS (0.4 mmol/kg) (treated) or the appropriate vehicle (control). Blood samples were obtained at 48 post-treatment following NDPS or vehicle administration. Values are means ± S.E. A diamond indicates significantly different from the appropriate control group value for that day’s measurement, p < 0.05. An asterisk indicates significantly different from the corresponding day 0 value, p < 0.05. A gamma indicates significantly different from the NDPS only value for the day 2 measurement, p < 0.05.
To ensure that the protective effect of DEC was not due to inhibiting the metabolism of NDPS to nephrotoxic metabolites, the effects of DEC on NDPS metabolite nephrotoxicity was also examined. NDHS (0.1 mmol/kg) or 2-NDHSA (0.1 mmol/kg) treatment induced marked nephrotoxicity (Tables 1–4, Fig. 3), as previously reported (Rankin et al., 2004). DEC pretreatment markedly reduced all aspects of NDHS and 2-NDHSA nephrotoxicity. Diuresis induced by the minimal nephrotoxic dose of NDHS or 2-NDHSA was significantly reduced by DEC on both post-treatment days (Table 1). In addition, DEC pretreatment was effective at attenuating increased proteinuria (Table 2) or kidney weight (Table 3) induced by NDHS or 2-NDHSA. The greatest protection on kidney weight appeared to be in the DEC + NDHS treatment group where kidney weight was slightly, but significantly decreased.
Figure 3.
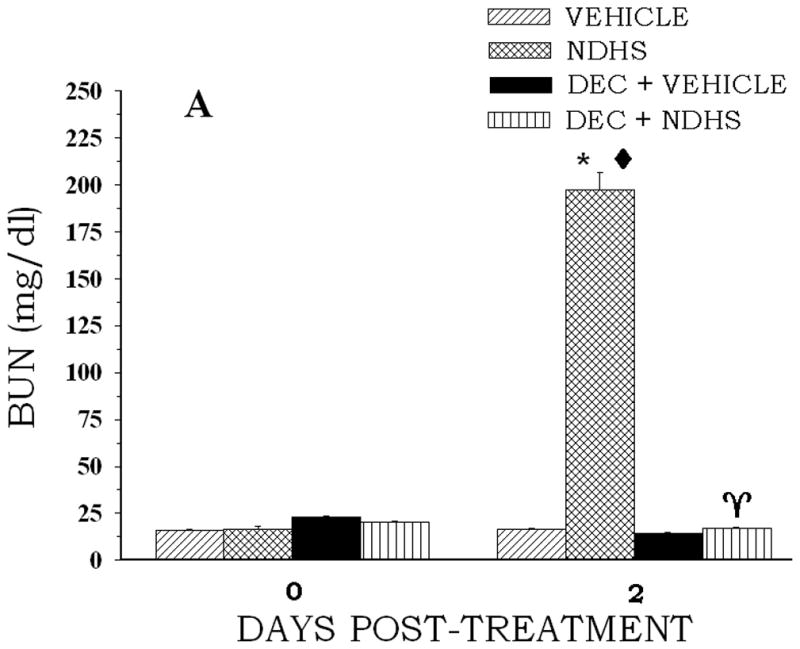
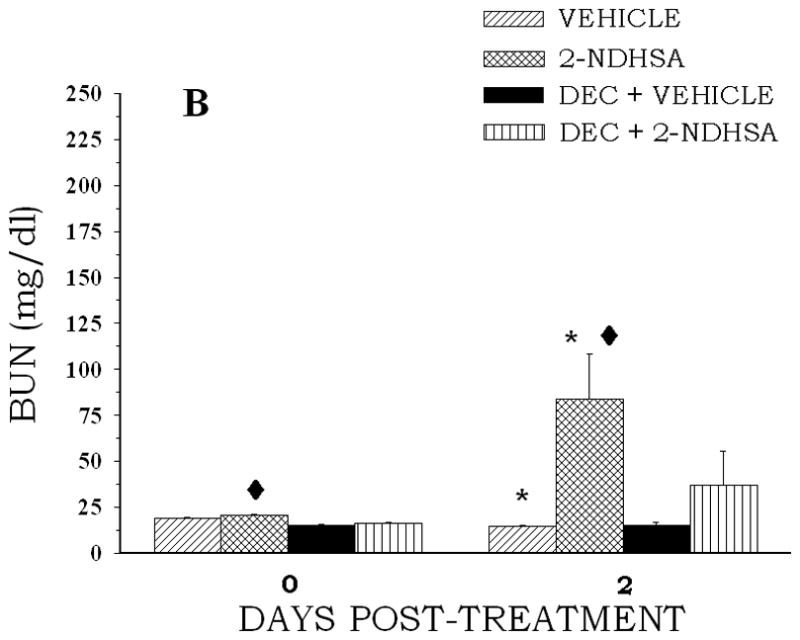
The effect of DEC pretreatment on NDPS metabolite-induced changes in BUN concentration. Rats (4 per group) were administered DEC (250 mg/kg, ip) 1 h before an ip injection of NDHS (0.1 mmol/kg)(A) or 2-NDHSA (0.1 mmol/kg)(B) (treated) or the appropriate vehicle (control). Blood samples were obtained at 48 post-treatment following the NDPS metabolite or vehicle administration. Values are means ± S.E. A diamond indicates significantly different from the appropriate control group value for that day’s measurement, p < 0.05. An asterisk indicates significantly different from the corresponding day 0 value, p < 0.05. A gamma indicates significantly different from the NDPS metabolite only value for the day 2 measurement, p < 0.05.
NDHS or 2-NDHSA treatment reduced both lactate-stimulated PAH and TEA accumulation (Table 4). NDHS treatment produced the largest decrease in TEA accumulation (47%), while 2-NDHSA treatment induced the largest reduction in lactate-stimulated PAH accumulation (46%). The reductions in organic anion and/or cation accumulation induced by the NDPS metabolites were completely prevented by DEC pretreatment (Table 4). BUN concentration was also significantly increased at 48 hr following NDPS metabolite administration (Fig. 3). Pretreatment with DEC attenuated the increase in BUN concentration induced by NDHS or 2-NDHSA administration (Fig. 3). In addition, DEC pretreatment was effective in reducing morphological changes induced by NDPS metabolites. The proximal tubular necrosis induced by NDHS and 2-NDHSA was markedly reduced by DEC pretreatment. Figure 4 illustrates the proximal tubular necrosis induced by 2-NDHSA (0.1 mmol/kg) at 48 hr and the marked reduction in necrosis provided by DEC pretreatment. Similar results were seen with DEC pretreatment before NDHS (0.1 mmol/kg) administration (data not shown).
Figure 4.
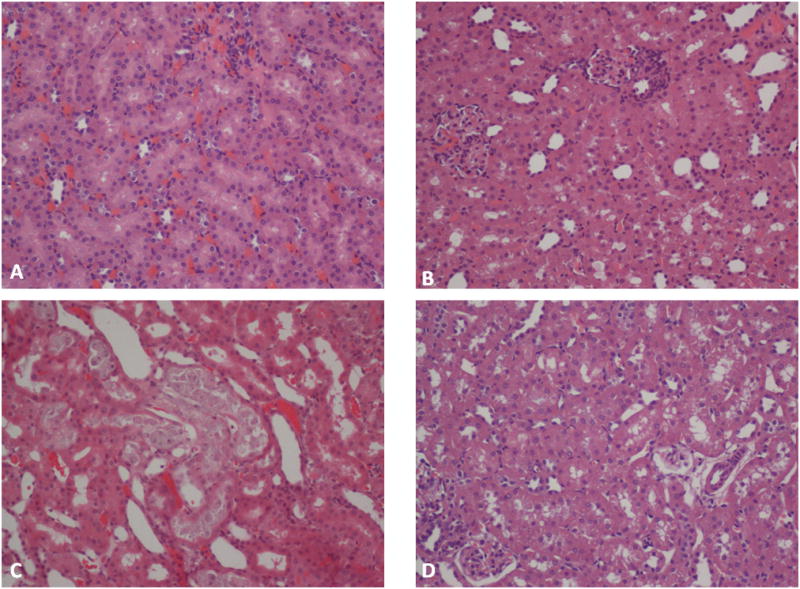
The effect of DEC pretreatment on 2-NDHSA-induced changes on proximal tubule morphology at 48 hr post 2-NDHSA treatment. Panel A shows renal cortex from a rat administered 2-NDHSA vehicle (control); panel B shows renal cortex from a rat administered DEC (250mg/kg); panel C shows renal cortex from a rat administered 2-NDHSA (0.1 mmol/kg), note the marked proximal tubular necrosis; and panel D shows renal cortex from a rat administered DEC (250 mg/kg) prior to 2-NDHSA (0.1 mmol/kg). Magnification was x200.
To determine if the marked attenuation of NDPS and NDPS metabolite nephrotoxicity could be overcome by increasing the dose of the nephrotoxicant, experiments were conducted in DEC pretreated animals receiving NDHS 0.2 or 0.4 mmol/kg or vehicle (Table 5). DEC pretreatment was able to prevent some NDHS-induced effects as seen by the lack of a decrease in PAH or TEA accumulation, proteinuria was not significantly increased at 24 or 48 hr, and BUN concentration was not significantly elevated at 48 hr (Table 5). The effects on proteinuria and BUN concentration were mainly due to the variability in the protective ability of DEC pretreatment. However, when the NDHS was increased to 0.4 mmol/kg, the protective effects of DEC pretreatment were markedly reduced (Table 5), as the DEC + NDHS 0.4 mmol/kg treatment group exhibited diuresis, proteinuria at 24 hr, elevated kidney weight and BUN concentration, and decreased PAH and TEA accumulation. Increasing the pretreatment dose of DEC to 500 mg/kg had no effect on NDHS 0.2 or 0.4 mmol/kg nephrotoxicity (data not shown).
Table 5.
Effect of DEC Treatment Prior to High Doses of NDHS (0.2 or 0.4 mmol/kg) Administration on Renal Parametersa
| Renal Parameters | DEC/NDHS 0.2 mmol/kg
|
DEC/NDHS 0.4 mmol/kg
|
||
|---|---|---|---|---|
| − NDHS | + NDHS | − NDHS | + NDHS | |
| Urine Volume (ml) | ||||
| Day 0 | 9.2 ± 1.7 | 8.3 ± 0.2 | 10.9 ± 1.0 | 11.0 ± 1.8 |
| Day 1 | 8.8 ± 1.6 | 27.3 ± 1.7b,c | 8.5 ± 0.4 | 24.0 ± 1.8b,c |
| Day 2 | 6.3 ± 0.5 | 34.4 ± 5.0b,c | 6.4 ±0.8b | 28.2 ± 5.8b,c |
| Proteinuria (mg/6 hr) | ||||
| Day 0 | 3.4 ± 0.9 | 2.7 ± 0.2 | 3.2 ± 0.1 | 2.9 ± 0.4 |
| Day 1 | 3.6 ± 0.5 | 59.5 ± 36.5 | 3.1 ± 0.2 | 22.9 ± 6.9b,c |
| Day 2 | 1.9 ± 0.1 | 6.5 ± 2.0 | 2.4 ± 0.2b | 5.2 ± 1.3 |
| Kidney Weight (g/100 g body wt.) | 0.38 ± 0.01 | 0.51 ± 0.01c | 0.38 ± 0.01 | 0.55 ± 0.03c |
| BUN (mg/dl) | ||||
| Day 0 | 20.0 ± 0.4 | 18.4 ± 1.0 | 22.1 ± 1.0 | 20.8 ± 1.4 |
| Day 2 | 12.1 ± 0.8b | 72.2 ± 36.0 | 11.0 ± 0.1b | 125.6 ± 43.6c |
| S/M Ratio | ||||
| PAH | 3.2 ± 0.2 | 3.5 ± 0.1 | 5.5 ± 0.3 | 2.4 ± 0.4c |
| PAH + LAC | 8.3 ± 0.3 | 8.5 ± 0.9 | 14.9 ± 0.4 | 6.2 ± 0.5c |
| TEA | 16.1 ± 0.9 | 15.8 ± 0.9 | 23.3 ± 0.4 | 14.4 ± 0.6c |
Values are means ± S.E. for N=4 rats per group. DEC (250 mg/kg, i.p.) was given 1 hr prior to NDHS (0.2, 0.4 mmol/kg) or vehicle administration.
Significantly different from day 0 value within a group, P<0.05.
Significantly different from the appropriate pair-fed control group value for that day’s measurement, P<0.05.
None of the treatment or control groups experienced elevated ALT concentrations at any time tested.
3.2 Effects of LY171883 on 24 hr NDPS metabolite nephrotoxicity
To determine the role of LTD4 in mediating NDPS metabolite nephrotoxicity, rats were pretreated with the LTD4 receptor blocker LY171883. For these experiments, nephrotoxicity was examined over a 24 hr period rather than 48 hr. Since NDPS nephrotoxicity is evident as early as 1–3 hr post-NDPS administration (Rankin et al., 1984), it was decided to only look at the 24 hr time frame for these studies since multiple LY171883 administrations would be required. However, LY171883 treatments were not very effective in attenuating NDHS or 2-NDHSA nephrotoxicity.
Marked diuresis and increased proteinuria (6 hr) were observed on post-treatment day 1 in both the NDHS and 2-NDHSA treatment groups that was not attenuated by LY171883 treatment (Table 1). LY171883 also did not attenuate NDHS or 2-NDHSA increases in kidney weight (Table 3) or BUN concentration (data not shown). LY171883 pretreatment did attenuate NDHS and 2-NDHSA-induced decreases in PAH accumulation and NDHS-induced decreases in lactate-stimulated PAH accumulation, but did not alter 2-NDHSA-induced decreases in lactate-stimulated PAH accumulation (Table 4). In addition, no differences were noted in renal morphology between NDPS metabolite treated rats with or without LY171883 (data not shown).
4. Discussion
Leukotrienes are recognized as playing a role in various disease states including inflammation, endotoxic shock, asthma, atherosclerosis, sepsis, and possibly cancer (Young and Passmore, 1990; Badr, 1992; Riccioni et al., 2010; Jawein and Korbut, 2010; Matsuyama and Yoshimura, 2010; Oi et al., 2010; Smyth et al. 2011). Leukotrienes are also contributory to drug and chemical induced toxicity (Gonzalez et al., 1994; Scuito and Hurt, 2004; Aleo et al., 2008) with renal vasoconstrictor leukotrienes playing a key role in cyclosporine A nephrotoxicity (Butterly et al., 2000; Atakan et al., 2008). The results of this study provide additional evidence that leukotrienes contribute to NDPS nephrotoxicity, at least at minimal nephrotoxic doses.
Results of previous studies on NDPS nephrotoxicity suggested that leukotrienes might play a role in NDPS nephrotoxicity. Although NDPS induces nephrotoxicity via reactive NDPS metabolites arising from NDHS and/or 2-NDHSA (Rankin, 2004; Cui et al., 2005), glutathione depletion, which would be expected to potentiate the toxicity induced by reactive metabolites, is protective against NDPS nephrotoxicity (Rankin et al., 1990; 1991b). These results indicated that glutathione plays a role in mediating NDPS nephrotoxicity, and we have found this response is not related to the formation of nephrotoxic NDPS metabolites (Rankin et al., 1991a). However, the ability of pretreatments such as DEM or BSO, pretreatments that would reduce the formation of vasoconstrictor leukotrienes, to reduce NDPS and NDPS metabolite nephrotoxicity (Yang et al., 1987; Rankin 1990; 1991b) suggested that the renal vasoconstrictor leukotrienes LTC4 and/or LTD4 might be contributing to the mechanisms of NDPS nephrotoxicity. The ability of DEC, a 5-lipoxgenase inhibitor, to markedly attenuate NDPS nephrotoxicity adds further evidence that leukotrienes contribute to NDPS-induced nephropathy. The finding that DEC also blocks NDHS and 2-NDHSA nephrotoxicity provides evidence that DEC was not merely blocking the metabolism of NDPS as seen with some other protective agents (Rankin et al., 1991a; Rankin et al., 1995).
The possibility that DEC could be inhibiting NDPS and NDPS metabolite nephrotoxicity via mechanisms other than inhibiting the formation of leukotrienes should be considered. Although DEC is considered to be a relatively specific inhibitor of the 5-lipoxygenase pathway (Bach, 1986), Balaa and Subramony (1990) found that not only does DEC (250mg/kg) inhibit LTC4 formation in rats, but it also inhibits formation of PGE2. This finding suggests that DEC (250 mg/kg) has the ability to inhibit cyclooxygenase as well. However, inhibition of cyclooxygenase alone with indomethacin potentiates NDPS nephrotoxicity (Rankin et al., 1991c), indicating that inhibition of the 5-lipoxygenase rather than the cyclooxygenase pathway by DEC is more important for attenuating NDPS nephrotoxicity. This observation would also support the formation of vasodilatory prostaglandins as a partially compensatory response for the formation of vasoconstrictor leukotrienes following NDPS or NDPS metabolite administration. Recently, LY171883 has been shown to be a peroxisome-proliferator-activated receptor alpha and gamma (PPARα and PPARγ) agonist (Grau et al., 2006; Kim et al., 2011), however, the inability of LY171883 to alter NDHS or 2-NDHSA nephrotoxicity suggests that PPAR-mediated actions of LY171883 do not assist any actions that LY171883 might have exhibited.
While leukotrienes may contribute to NDPS nephrotoxicity, other mechanisms (e.g. direct proximal tubular cell toxicity) for inducing nephrotoxicity are present at doses above the minimal nephrotoxic dose that are responsible for the majority of the observed renal effects. This conclusion is supported by the observation that increasing the dose of NDHS from 0.1 mmol/kg to 0.2 or 0.4 mmol/kg markedly reduced or reversed the protective effects of DEC. In addition, increasing the dose of DEC to 500 mg/kg provided no further protective effect on NDHS 0.2 or 0.4 nephrotoxicity. Thus, leukotrienes are more important mechanistically for NDPS-induced renal effects at minimally toxic doses of NDPS (or its nephrotoxic metabolites) than at NDPS doses greater than the minimally nephrotoxic dose.
It also appears that of the potential leukotrienes that might contribute to NDPS nephrotoxicity, LTC4 is more likely to be the key renal vasoconstrictor leukotriene than LTD4. This conclusion is based on the previous finding that pretreatments (DEM, BSO) that would reduce renal LTC4 formation (Yang et al., 1987; Rankin et al., 1990; 1991b) and the current work with the 5-lipoxygenase synthesis inhibitor DEC all produced marked attenuation of NDPS and NDPS metabolite nephrotoxicity, while the LTD4 receptor blocker LY171883 was fairly ineffective at attenuating NDPS metabolite nephrotoxicity. In addition, the γ-glutamyl transpeptidase inhibitor, AT-125 (acivicin), which would be expected to reduce the conversion of LTC4 to LTD4 did not attenuate NDPS nephrotoxicity (Rankin et al., 1991a). Thus, LTC4 rather than LTD4 appears to be the most like vasoconstrictor leukotriene contributing to the mechanism of NDPS or NDPS metabolite nephrotoxicity. In addition, there is no evidence that NDPS, NDHS or 2-NDHSA directly stimulate the synthesis of leukotrienes. Although the origin of the LTC4 and the initial activation mechanism for its production following NDPS or a nephrotoxic NDPS metabolite are unknown, it is possible that the proximal tubule cell damage and/or altered renal urinary production induced by these compounds is responsible for initiating leukotriene synthesis. Further studies are needed to ascertain the origin and nature of the initiating event for increased leukotriene effects in the kidney following NDPS administration.
In summary, the results of these studies support a role for leukotrienes in NDPS and NDPS metabolite nephrotoxicity. The primary leukotriene that contributes to the mechanism of NDPS and NDPS metabolite nephrotoxicity is most likely to be LTC4 based on this study and previous studies that demonstrated the ability of LTC4 synthesis inhibitors (DEC, DEM and BSO) to attenuate NDPS nephrotoxicity, while inhibitors of LTD4 synthesis (AT-125) or receptors (LY171883) had little effect on NDPS or NDPS metabolite-induced renal effects. The importance of leukotrienes in NDPS nephrotoxicity decreases as the toxic dose increases, since increasing the dose of NDHS was able to overcome the effects of DEC, while increasing the dose of DEC could not further attenuate NDHS-induced nephrotoxicity.
Highlights.
NDPS nephrotoxicity was decreased by DEC, an inhibitor of leukotriene synthesis.
LTD4 receptor inhibition didn’t decrease NDPS metabolite nephrotoxicity.
The results support a role for leukotrienes in NDPS nephrotoxicity.
LTC4 is the most likely leukotriene contributing to NDPS nephrotoxicity.
Acknowledgments
This work was supported by NIH grants DK31210 and RR016477 to G.O.R.
Abbreviations
- BSO
buthionine sulfoximine
- DAZ
dazmegrel
- DEC
diethylcarbamazine
- DEM
diethyl maleate
- 5-(S)-HPETE
5-(S)-hydroperoxyeicosatetraenoic acid
- INDO
indomethacin
- 5-LOX
5-lipoxygenase
- LTA4 (C4D4, E4)
leukotriene A4 (C4, D4, E4)
- NDHS
N-(3,5-dichlorophenyl)-2-hydroxysuccinimide
- 2-NDHSA
N-(3,5-dichlorophenyl)-2-hydroxysuccinamic acid
- NDPS
N-(3,5-dichlorophenyl)succinimide
- PGH2 (E2I2)
prostaglandin H2 (E2; I2)
- PHS
prostaglandin H synthase
- TxA2
thromboxane A2
Footnotes
Conflict of Interest
None of the authors have any conflicts of interest related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Gary O. Rankin, Department of Pharmacology, Physiology & Toxicology, Joan C. Edwards School of Medicine, Marshall University, Huntington, WV 25755, USA
Suk K. Hong, Department of Pharmacology, Physiology & Toxicology, Joan C. Edwards School of Medicine, Marshall University, Huntington, WV 25755, USA
Dianne K. Anestis, Department of Pharmacology, Physiology & Toxicology, Joan C. Edwards School of Medicine, Marshall University, Huntington, WV 25755, USA
John G. Ball, Department of Pharmacology, Physiology & Toxicology, Joan C. Edwards School of Medicine, Marshall University, Huntington, WV 25755, USA
Monica A. Valentovic, Department of Pharmacology, Physiology & Toxicology, Joan C. Edwards School of Medicine, Marshall University, Huntington, WV 25755, USA
Vincent A. Graffeo, Department of Anatomy and Pathology, Joan C. Edwards School of Medicine, Marshall University, Huntington, WV 25755, USA
References
- Aleo MD, Doshna CM, Fritz CA. An underlying role for hepatobiliary dysfunction in cyclosporine A nephrotoxicity. Toxicol Appl Pharmacol. 2008;230:126–134. doi: 10.1016/j.taap.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Aleo MD, Rankin GO, Cross TJ, Schnellmann RG. Toxicity of N-(3,5-dichlorophenyl)succinimide and metabolites to rat renal proximal tubules and mitochondria. Chem-Biol Interact. 1991;78:109–121. doi: 10.1016/0009-2797(91)90107-i. [DOI] [PubMed] [Google Scholar]
- Atakan A, Arikan H, Macunlunoglu B, Tuglular S, Ulfer G, Cakalagaoglu F, Ozener C, Akoglu E. Renal protective effects of leukotrienes receptor blockers in an experimental model of cyclosporine nephrotoxicity. Transplant Proc. 2008;40:279–284. doi: 10.1016/j.transproceed.2007.11.026. [DOI] [PubMed] [Google Scholar]
- Bach MK. Inhibition of the leukotriene synthase of rat basophil leukemia cells by diethylcarbamazine, and synergism between diethylcarbamazine and piriprost, a 5-lipoxygenase inhibitor. Biochem Pharmacol. 1986;35:425–433. doi: 10.1016/0006-2952(86)90215-7. [DOI] [PubMed] [Google Scholar]
- Badr KF. Sepsis-associated renal vasoconstriction: potential targets for future study. Am J Kidney Dis. 1992;20:207–213. doi: 10.1016/s0272-6386(12)80692-5. [DOI] [PubMed] [Google Scholar]
- Balaa MA, Subramony C. Diethylcarbamazine decreases ethanol-injury to the gastric mucosa in the rat. Eicosanoids. 1990;3:201–203. [PubMed] [Google Scholar]
- Barrett MC, Cashman SJ, Moss J. Experimental interstitial renal fibrosis in rats: Nephritis induced by N-(3,5-dichlorophenyl)succinimide. Br J Exp Pathol. 1983;64:425–435. [PMC free article] [PubMed] [Google Scholar]
- Beers KW, Rankin GO. Effect of N-(3,5-dichlorophenyl)2-hydroxysuccinimide on renal function and hemodynamics in the anesthetized rat. Toxicology. 1993;79:139–148. doi: 10.1016/0300-483x(93)90127-e. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brash AR. Lipoxygenases: Occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- Butterly DW, Spurney RF, Ruiz P, Griffiths R, Albrightson C, Coffman TM. A role for leukotrienes in cyclosporine nephrotoxicity. Kidney Int. 2000;57:2586–2593. doi: 10.1046/j.1523-1755.2000.00118.x. [DOI] [PubMed] [Google Scholar]
- Cui D, Rankin GO, Harvison PJ. Metabolism of the nephrotoxicant N-(3,5-dichlorophenyl)succinimide in rats: Evidence for bioactivation through alcohol-O-glucuronidation and O-sulfation. Chem Res Toxicol. 2005;18:991–1003. doi: 10.1021/tx0496587. [DOI] [PubMed] [Google Scholar]
- Fujinami A, Ozaki T, Yamamoto S. Studies on biological activity of cyclic imide compounds. Part I Antimicrobial activity of 3-phenyloxazolidine-2,4-diones and related compounds. Agric Biol Chem. 1971;35:1707–1719. [Google Scholar]
- Fujinami A, Ozaki T, Nodera K, Tanaka K. Studies on biological activity of cyclic imide compounds. Part II Antimicrobial activity of 1-phenylpyrrolidine-2,5-diones and related compounds. Agric Biol Chem. 1972;36:318–323. [Google Scholar]
- Gonzalez R, Ancheta O, Marquez M, Rodriguez S. Heptoprotective effects of diethylcarbamazine in acute liver damage induced by carbon tetrachloride in rats. Zhongguo Yao Li Xue Bao. 1994;15:495–495. [PubMed] [Google Scholar]
- Grau R, Punzon C, Fresno M, Iniguez MA. Peroxisome-proliferator-activated receptor alpha agonists inhibit cylco-oxygenase 2 and vascular endothelial growth factor transcriptional activation in human colorectal carcinoma cells via inhibition of activator protein-1. Biochem J. 2006;395:81–88. doi: 10.1042/BJ20050964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henesey CM, Kellner-Weibel GL, Tarloff JB, Harvison PJ. Comparative disposition of the nephrotoxicant N-(3,5-dichlorophenyl)succinimide and the non-nephrotoxicant N-(3,5-difluorophenyl)succinimide in Fischer 344 rats. Drug Metab Dispos. 1999;27:674–680. [PubMed] [Google Scholar]
- Hong SK, Anestis DK, Ball JG, Valentovic MA, Brown PI, Rankin GO. Sodium sulfate potentiates N-(3,5-dichlorophenyl)-2-hydroxysuccinimide (NDHS) and N-(3,5-dichlorophenyl)-2-hydroxysuccinamic acid (2-NDHSA) nephrotoxicity in the Fischer 344 rat. Toxicology. 1999a;138:165–174. doi: 10.1016/s0300-483x(99)00102-x. [DOI] [PubMed] [Google Scholar]
- Hong SK, Anestis DK, Brown PI, Rankin GO. Effect of glucuronidation substrates/inhibitors on N-(3,5-dichlorophenyl)succinimide nephrotoxicity in Fischer 344 rats. Toxicology. 1999b;132:43–55. doi: 10.1016/s0300-483x(98)00140-1. [DOI] [PubMed] [Google Scholar]
- Hong SK, Anestis DK, Kennedy S, Rankin GO, Brown PI. Effect of sulfation substrates/inhibitors on N-(3,5-dichlorophenyl)succinimide (NDPS) nephrotoxicity in Fischer 344 rats. J Toxicol Environ Health Part A. 1999c;57:47–62. doi: 10.1080/009841099157854. [DOI] [PubMed] [Google Scholar]
- Hong SK, Anestis DK, Skaggs C, Brown PI, Rankin GO. The role of glucuronidation in N-(3,5-dichlorophenyl)succinimide (NDPS) nephrotoxicity: nephrotoxic potential of NDPS and NDPS metabolites in Gunn, Wistar, and Fischer 344 rats. Toxicol Appl Pharmacol. 1999d;154:170–180. doi: 10.1006/taap.1998.8554. [DOI] [PubMed] [Google Scholar]
- Jawien J, Korbut R. The current view on the role of leukotrienes in arterogenesis. J Physiol Pharmacol. 2010;61:647–650. [PubMed] [Google Scholar]
- Kim SJ, Nian C, McIntosh CH. Adipocyte expression of the glucose-dependent insulinotropic polypeptide receptor involves gene regulation by PPARγ and histone acetylation. J Lipid Res. 2011;52:759–770. doi: 10.1194/jlr.M012203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama M, Yoshimura R. Cysteinyl-leukotriene1 receptor is a potent target for the prevention and treatment of human urological cancer. Mol Med Report. 2010;3:245–251. doi: 10.3892/mmr_00000247. [DOI] [PubMed] [Google Scholar]
- Oi N, Jeong CH, Nardas J, Cho YY, Pugliese A, Bode AM, Dong Z. Resveratrol, a red wine polyphenol, suppresses pancreatic cancer by inhibiting leukotrienes A4 hydrolase. Cancer Res. 2010;70:9755–9764. doi: 10.1158/0008-5472.CAN-10-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters-Golden M, Shelly C, Morganroth ML. Inhibition of rat lung glutathione synthesis attenuates hypoxic pulmonary vasoconstriction and the associated leukotriene C4 production. Am Rev Respir Dis. 1989;140:1210–1215. doi: 10.1164/ajrccm/140.5.1210. [DOI] [PubMed] [Google Scholar]
- Rankin GO. Nephrotoxicity following acute administration of N-(3,5-dichlorophenyl)succinimide in Sprague-Dawley rats. Toxicology. 1982;23:21–31. doi: 10.1016/0300-483x(82)90038-5. [DOI] [PubMed] [Google Scholar]
- Rankin GO. Nephrotoxicity induced by C- and N-arylsuccinimides. J Toxicol Environ Health, Part B. 2004;7:399–416. doi: 10.1080/10937400490486113. [DOI] [PubMed] [Google Scholar]
- Rankin GO, Beers KW, Nicoll DW, Anestis DK, Ball JG, Valentovic MA, Brown PI. Effect of DMSO on N-(3,5-dichlorophenyl)succinimide (NDPS) and NDPS metabolite nephrotoxicity. Toxicology. 1995;100:79–88. doi: 10.1016/0300-483x(95)93709-d. [DOI] [PubMed] [Google Scholar]
- Rankin GO, Cressey-Veneziano K, Brown PI. Onset and recovery from acute N-(3,5-dichlorophenyl)succinimide-induced nephrotoxicity in Sprague-Dawley rats. Toxicology. 1984;30:209–216. doi: 10.1016/0300-483x(84)90092-1. [DOI] [PubMed] [Google Scholar]
- Rankin GO, Hong SK, Anestis DK, Lash LH, Miles SL. In vitro nephrotoxicity induced by N-(3,5-dichlorophenyl)succinimide (NDPS) metabolites in isolated renal cortical cells from male and female Fischer 344 rats: evidence for a nephrotoxic sulfate conjugate metabolite. Toxicology. 2001;163:73–82. doi: 10.1016/s0300-483x(01)00376-6. [DOI] [PubMed] [Google Scholar]
- Rankin GO, Shih HC, Yang DJ, Richmond CD, Teets VJ, Brown PI. Nephrotoxicity of N-(3,5-dichlorophenyl)succinimide metabolites in vivo and in vitro. Toxicol Appl Pharmacol. 1988;96:405–416. doi: 10.1016/0041-008x(88)90001-4. [DOI] [PubMed] [Google Scholar]
- Rankin GO, Shih HC, Teets VJ, Nicoll DW, Anestis DK, Brown PI. N-(3,5-Dichlorophenyl)succinimide nephrotoxicity: evidence against the formation of nephrotoxic glutathione or cysteine conjugates. Toxicology. 1991a;68:307–325. doi: 10.1016/0300-483x(91)90077-e. [DOI] [PubMed] [Google Scholar]
- Rankin GO, Teets VJ, Nicoll DW, Brown PI. Effect of buthionine sulfoximine on acute N-(3,5-dichlorophenyl)succinimide-induced nephrotoxicity in Fischer 344 rats. Toxicol Lett. 1990;52:91–100. doi: 10.1016/0378-4274(90)90169-m. [DOI] [PubMed] [Google Scholar]
- Rankin GO, Teets VJ, Nicoll DW, Brown PI. Effect of buthionine sulfoximine on N-(3,5-dichlorophenyl)-2-hydroxysuccinimide and N-(3,5-dichlorophenyl)-2-hydroxysuccinamic acid nephrotoxicity. Toxicol Lett. 1991b;57:297–308. doi: 10.1016/0378-4274(91)90204-j. [DOI] [PubMed] [Google Scholar]
- Rankin GO, Valentovic MA, Teets VJ, Nicoll DW, Anestis DK, Brown PI. Effect of autacoid modulation on N-(3,5-dichlorophenyl)succinimide (NDPS) and NDPS metabolite nephrotoxicity. Toxicology. 1991c;70:327–344. doi: 10.1016/0300-483x(91)90007-n. [DOI] [PubMed] [Google Scholar]
- Rankin GO, Yang DJ, Cressey-Veneziano K, Brown PI. N-(3,5-Dichlorophenyl)succinimide nephrotoxicity in the Fischer 344 rat. Toxicol Lett. 1985;24:99–105. doi: 10.1016/0378-4274(85)90146-8. [DOI] [PubMed] [Google Scholar]
- Riccioni G, Back M, Capra V. Leukotrienes and atherosclerosis. Curr Drug Targets. 2010;11:882–887. doi: 10.2174/138945010791320881. [DOI] [PubMed] [Google Scholar]
- Rosenthal A, Pace-Asciak CR. Potent vasoconstriction of the isolated perfused rat kidney by leukotrienes C4 and D4. Can J Physiol Pharmacol. 1983;61:325–328. doi: 10.1139/y83-049. [DOI] [PubMed] [Google Scholar]
- Sciuto AA, Hurt HH. Therapeutic treatments of phosgene-induced lung injury. Inhal Toxicol. 2004;16:565–580. doi: 10.1080/08958370490442584. [DOI] [PubMed] [Google Scholar]
- Shih HC, Rankin GO. Convenient synthesis of N-aryl-2-hydroxysuccinimides and characterization of their hydrolyzed products. Synthesis. 1989;11:866–867. [Google Scholar]
- Smyth EM, Grosser T, Fitzgerald GA. Lipid-derived autacoids: eicosanoids and platelet-activating factor. In: Brunton LL, Chabner BA, Knollmann BC, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 12. Chapter 33. McGraw Hill; New York: 2011. pp. 937–957. [Google Scholar]
- Sugihara S, Shinohara Y, Miyata Y, Inoue K, Ito N. Pathologic analysis of chemical nephritis in rats induced by N-(3,5-dichlorophenyl)succinimide. Lab Invest. 1975;33:219–230. [PubMed] [Google Scholar]
- Tabuchi Y, Kawarabayashi K, Furuhama K. Inhibitory effect of DS-4574, a pepidoleukotriene antagonist with mast cell stabilizing action, on compound 48/80-induced gastric mucosal lesions in rats. Agents Actions. 1994;41:21–24. doi: 10.1007/BF01986388. [DOI] [PubMed] [Google Scholar]
- Yang DJ, Teets VJ, Bolton B, Brown PI, Rankin GO. Role of glutathione in acute N-(3,5-dichlorophenyl)succinimide-induced nephrotoxicity in Sprague-Dawley and Fischer 344 rats. Toxicology. 1987;45:25–44. doi: 10.1016/0300-483x(87)90112-0. [DOI] [PubMed] [Google Scholar]
- Young JS, Passmore JC. Hemodynamic and renal advantages of dual cyclooxygenase and leukotrienes blockade during canine endotoxic shock. Circ Shock. 1990;32:243–255. [PubMed] [Google Scholar]
- Zhang J, Zhang GJ, Zheng YF, Zhu HJ, Yang J, Yao GD, Zhu XQ. The antiandrogenic activity of the fungicide N-(3,5-dichlorophenyl)succinimide in in vivo and in vitro assays. J Reprod Dev. 2007;53:535–543. doi: 10.1262/jrd.18131. [DOI] [PubMed] [Google Scholar]