

Abstract
Nitrone spin traps such as 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) are commonly used for free radical detection. Though proven examples are rare, artifact formation must be considered. For example, the Forrester-Hepburn mechanism yields the same radical adduct as formed by genuine radical trapping. A hydroxylamine is formed by nucleophilic attack of the substrate to DMPO and subsequently oxidized to the respective nitroxide radical. One potential candidate for this artifact is the sulfur trioxide radical adduct (DMPO/·SO3−), as detected in spin-trapping experiments with horseradish peroxidase and sulfite. It has previously been shown by NMR experiments that the hydroxylamine intermediate does indeed form, but no direct proof for the ESR artifact has been provided. Here we used isotopically labeled DMPO with horseradish peroxidase and ferricyanide to test for the Forrester-Hepburn artifact directly in a spin-trapping experiment. Besides sulfite, we investigated other nucleophiles such as cyanide, cysteine and glutathione. Neither sulfite nor biological thiols produced detectable spin-trapping artifacts, but with cyanide the relatively weak signal originated almost entirely from the nucleophilic reaction. The hydroxylamine intermediate, which is more abundant with cyanide than with sulfite, was identified as cyano-hydroxylamine by means of 2D NMR experiments.
Although our study found that spin trapping provided authentic free radical signals with most of the substrates, the occurrence of the Forrester Hepburn mechanism artifact with cyanide emphasizes the importance of isotope measurements with nucleophile substrates.
Introduction
Electron spin resonance spectroscopy (ESR) is a selective and direct method for detecting free radicals.1 Aside from a few exceptions such as the tyrosyl radical in proteins,2 it is not possible to use this straightforward approach to directly detect radicals of biochemical interest. Radical concentrations are generally too low for detection (in the sub-nanomolar range) because their half-lives are short compared to the rate at which they are produced in biological systems. Hence, the ESR spin-trapping technique3 has been extensively used. Typically an ESR-silent spin trap such as the commonly used nitrone 5,5-dimethyl-1-pyrroline N-oxide (DMPO) reacts with the reactive free radicals to form more stable radical adducts, which can be detected by ESR spectroscopy. This method has been used to investigate processes with free radicals involved in various fields of research; however, it is known that under harsh conditions, spin-trapping is susceptible to artifacts,4 and where milder conditions prevail, examples of proven artifacts are rare but still of concern.4-8 Two alternate reactions of DMPO leading to false-positive results have been discovered: inverted spin trapping9-11 and the Forrester-Hepburn mechanism.5 With both mechanisms, artifacts result from nucleophilic addition and one-electron oxidation - in either order (Scheme 1b and c). The oxidation potential required for oxidation of DMPO to the cation radical is high (DMPO+·/DMPO = 1.63 mV in acetonitrile versus a calomel electrode)12 In fact, the redox potential of the spin trap exceeds the decomposition potential of water at the platinum electrode.12
Scheme 1.

Mechanism of genuine spin-trapping (a), inverted spin trapping (b) and the Forrester-Hepburn mechanism (c).
In biological systems, the Forrester-Hepburn mechanism is considered the more serious artifact source.13 In this reaction, a radical adduct supposedly generated by genuine spin-trapping of free radicals actually originates from nucleophilic chemistry and subsequent one-electron oxidation. Indeed, it has been shown by NMR experiments that the corresponding hydroxylamine is formed by nucleophilic attack of the sulfite anion to DMPO at the 2-position.14 Similar hydroxylamine formation has been reported from the reaction of cysteine and glutathione with another spin trap, DEPMPO.15 Next, the hydroxylamine, a non-radical and ESR-silent compound, may be oxidized even under mild conditions to the corresponding radical adduct. This reaction is insidious, because the nitroxide radicals formed in this non-free radical reaction are the same as the radical adducts formed with genuine spin-trapping (Scheme 1b).
Timmins et al.16, 17 established a technique using isotope-labeled spin traps that allows detection of this kind of artifact in a simple ESR experiment (Scheme 2). The experiment is split into a preincubation and a radical generation phase, with different isotopes of the same spin trap employed for each phase. Since the isotope-labeled spin traps have distinct ESR spectra, the origin of the signal can be correctly identified. Previous experiments of this type with DMPO used deuterated DMPO, which decreased the resolution.17 In order to improve the resolution of the mixed isotope radical adduct spectra, we synthesized 15N-labeled DMPO. In the case of 15N-labeled DMPO, the nuclear spin is I = ½, which results in a doublet splitting, while with 14N-DMPO the spin is I = 1, which results in a triplet splitting with a 40% smaller hyperfine coupling constant. In a typical experiment, the 14N-DMPO and the nucleophile are present during the preincubation phase, and the radical generation phase is started concomitantly with the addition of 15N-DMPO. Then, the corresponding experiment is carried out with the isotope-labeled spin traps reversed (preincubation with 15N-DMPO, followed by 14N-DMPO addition and radical initiation) and the spectra are compared. If no artifact is generated during the preincubation phase, the spectra will be identical, whereas a dependency on the order of isotope addition would indicate that the signal originates, at least in part, from the Forrester-Hepburn artifact mechanism.
Scheme 2.

Forrester-Hepburn artifact detection - Experimental procedure and expected reactions.
Several in vitro and in vivo ESR spin-trapping studies have been carried out to investigate the oxidation of sulfite to free radical species in various systems.18-23 However, the concern was raised in recent publications14, 24 that spin-trapping with nitrone traps may not be a reliable method of detecting sulfur trioxide radicals (·SO3−). It was suggested that the ESR signal may actually originate from the Forrester-Hepburn mechanism and be misinterpreted as radical trapping.24 Since the sulfite does react with DMPO to form the hydroxylamine corresponding to the reduced radical adduct, this is a credible possibility.14 In this study we have investigated the role of the Forrester-Hepburn mechanism in ESR spin-trapping experiments with horseradish peroxidase/H2O2 with the method suggested by Timmins et al.16 In addition to sodium sulfite, we reinvestigated several other good nucleophiles known to form free radicals that may play a role in biochemistry and toxicology, i.e., cyanide,25 L-cysteine26 and glutathione.27 1H-NMR experiments were carried out in order to estimate the extent of hydroxylamine formation with each nucleophile. It is important to note that hydroxylamine formation is a necessary but not sufficient condition for radical adduct generation by the nucleophilic mechanism of Forrester and Hepburn.5
Materials and methods
Chemicals
Sodium sulfite (Na2SO3), potassium cyanide (KCN), potassium ferricyanide K3[Fe(CN)6], reduced L-glutathione (GSH), cysteine, hydrogen peroxide (30%), horseradish peroxidase (type VI), diethylenetriaminepentaacetic acid (DTPA), and potassium phosphate were purchased from Sigma-Aldrich (St. Louis, MO). Chelex-100 resin was purchased from Biorad (Hercules, CA). 14N-DMPO was purchased from Dojindo (Rockville, MD). 15N-DMPO was synthesized and purified by vacuum sublimation, which was carried out twice.
ESR spectroscopy
ESR spectra were recorded with a modified Bruker ElexSys E-500 spectrometer equipped with an ER 4122 SHQ cavity. For measurements with HRP, the instrument operates at 9.87 GHz (X-Band). For ESR spectroscopic measurements, we used 20 mW microwave power, 4 × 105 receiver gain, a modulation frequency of 100 kHz and modulation amplitude of 1.0 G. The time constant and conversion time were 81.92 ms for the measurements with sodium sulfite and 163.84 ms for all other experiments. The field scan range was 80 G, with a resolution of 1024 points and a center field of 3505 G. With ferricyanide, the receiver gain was 1 × 104 and the time constant and conversion time were 40.96 ms. Four scans were added. For preincubation experiments, 50 mM 14N-DMPO (100 mM for the KCN experiments) was mixed with 10 mM Na2SO3, KCN, glutathione or cysteine. After 15 min of incubation at room temperature, 50 mM 15N-DMPO (100 mM for the KCN measurements) was added and the radical generation was initiated with HRP (1 mg/mL) and 10 μM hydrogen peroxide (350 μM for the KCN measurements). The solution was mixed, transferred into capillaries and measured immediately. For the control experiments, 50 mM 14N-DMPO or 15N-DMPO (100 mM for the KCN measurements) and 10 mM Na2SO3, KCN, glutathione or cysteine were added to phosphate buffer, pH 7.4. Radical generation was initiated with 1 mg/mL HRP and 10 μM hydrogen peroxide (350 μM for the KCN measurements), with a total sample volume of 50 μL. Additionally, the enzymatic system (HRP and H2O2) was replaced by 1 mM K3[Fe(CN)6] for the experiments with sulfite and cyanide. The samples were mixed, transferred to capillaries and measured immediately, except for the measurements with KCN, were the sample was incubated for 15 min before initiation of the ESR scan. The buffer contained phosphate (100 mM) treated with Chelex-100 to reduce metal contamination, and 100 μM DTPA (25 μM with CN) was added as a transition metal chelator. For low oxygen measurements, all buffers and stock solutions were degassed by bubbling with N2 gas for 20 min. Incubation and measurements (flat cell) were carried out under exclusion of oxygen28 and compared to a measurement under the same conditions without degassing. All concentration details reflect the final concentrations during the EPR measurement. The measurement was repeated with the order of addition of the DMPO isotopes reversed: 15N-DMPO was present during the preincubation phase and 14N-DMPO was added just before the radical initiation. To calculate the difference spectrum, the magnetic field shift from changing samples was corrected manually (Bruker Win-EPR software) and the spectrum with 15N-DMPO during the preincubation was subtracted from the spectrum with 14N-DMPO during preincubation. Each set of measurements was carried out 3 times and representative experiments are shown. The simulation software WinSim was employed to determine the coupling constants.29
NMR spectroscopy
100 mM Na2SO3, KCN, glutathione or cysteine was mixed with 50 mM 14N-DMPO in deuterated phosphate buffer, incubated for 15 min at room temperature and the 1H-NMR spectrum measured. Also, a reference spectrum of DMPO incubated for 15 min in the deuterated buffer was recorded. The deuterated phosphate buffer contained 100 mM Na3PO4 and 100 μM DTPA in D2O and the pD was adjusted to 7.4 with DCl and NaOD. 1H NMR spectra were acquired at 25°C on a Varian INOVA 600 spectrometer operating at 1H frequency of 599.763 MHz, with a Varian triple resonance, actively shielded Z-gradient probe. All 1D 1H NMR spectra were acquired using a 1 s presaturation delay, 1 s acquisition time, and 30° excitation pulse to allow adequate recovery of the proton resonances for quantitative integration. In addition, to assign the 13C shifts and confirm the structure of the cyano-adduct, 2D NMR experiments (1H/13C heteronuclear single quantum coherence and heteronuclear multiple bond coherence experiments) were obtained on the sample containing KCN. The 2D spectra were processed using NMRPipe30 and assigned using NMRView31, and the 1H and 13C shifts were referenced to DSS using an external sample in similar buffer. The 1D 1H spectra were calibrated with the water peak (4.79 ppm) as internal standard after application of a Lorentzian window function (1 Hz line broadening), Fourier transformation and polynomial baseline correction (least square). The integrals were calculated and normalized to the allyl protons (2.72 ppm, 2 protons).
Results
ESR spin-trapping experiments with sodium sulfite
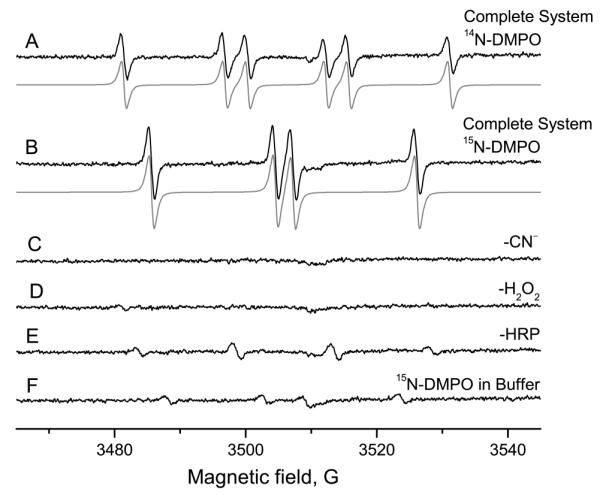
ESR measurement of 50 mM 14N-DMPO, 10 mM sodium sulfite, 1 mg/mL HRP and 10 μM H2O2 gave a six-line signal with a good signal-to-noise ratio (Figure 1A). Spectral simulation revealed coupling constants of aN = 14.5 G and aH = 15.95 G, which were assigned to the DMPO/·SO −3 adduct (Lit.: aN = 14.7 G and aH = 15.9 G).18 With 15N-DMPO as the spin trap, the constants were determined to be aN = 20.28 G and aH = 15.95 G for the 15N-DMPO/·SO −3 adduct (Figure 1 B). The increase in the nitrogen hyperfine coupling constant is as expected from the gyromagnetic ratio of 15N:14N of 1.40.32, 33 Although the signal amplitude appears to be somewhat higher for the 15N analog, the double integrals of the two spectra are equal (not shown). Since the signal of the 14N analog consists of six lines, but the 15N analog of four, the amplitude is 1.5 times greater for the same double integral assuming identical line widths. For controls, each compound was omitted separately (Figure 1C-E). Without sodium sulfite (Figure 1C), no signal was detected. In the spectra without hydrogen peroxide (Figure 1D) or horseradish peroxidase (Figure 1E), signals of much lower intensity were detected. Those findings are in accordance with the literature.18, 22 These control experiments cannot distinguish radical adduct formation by nucleophilic addition from genuine spin trapping because DMPO, the substrate, HRP and hydrogen peroxide must all be present to form the artifact as well as the genuine signal. 15N-DMPO showed no ESR-detectable impurities (Figure 1F).
Figure 1.

Generation and ESR spin-trapping of the sulfite radical. A: Complete system and simulation. The sample contained 50 mM 14N-DMPO, 10 mM Na2SO3, 1 mg/ml HRP, 10 μM H2O2, in 100 mM Chelex-treated phosphate buffer, pH = 7.4, with 100 μM DTPA. The sample was measured immediately (scan time: 83.89 s). B: As in A, but 15N-DMPO was used. C: As in A, but without sulfite. No ESR signal emerged. D: As in A, but in the absence of hydrogen peroxide. A background signal of low amplitude was detected. E: As in A, but no horseradish peroxidase. A minor signal was detected. F: 15N-DMPO in phosphate buffer as used in A. No paramagnetic impurities are visible.
To assess the susceptibility to artifacts formed by the Forrester-Hepburn Mechanism, we carried out preincubation experiments. 10 mM Na2SO3 were incubated for 15 min with 50 mM 14N-DMPO at room temperature. After this, 50 mM 15N-DMPO was added, the radical generation was initiated with 1 mg/mL horseradish peroxidase and 10 μM hydrogen peroxide, and the resulting spectrum was measured within 5 min. The ESR signal was a superposition of the 15N- and 14N-DMPO sulfite radical adducts in about equal concentrations (Figure 2A). The corresponding experiment in which the order of isotope addition was reversed (15N-DMPO during preincubation and 14N immediately prior to radical initiation) gave a very similar spectrum (Figure 2B), with the difference spectrum giving merely noise (Figure 2C). If nucleophilic addition had contributed to the nitroxide adduct formation, it would have been revealed in the difference spectrum.
Figure 2.
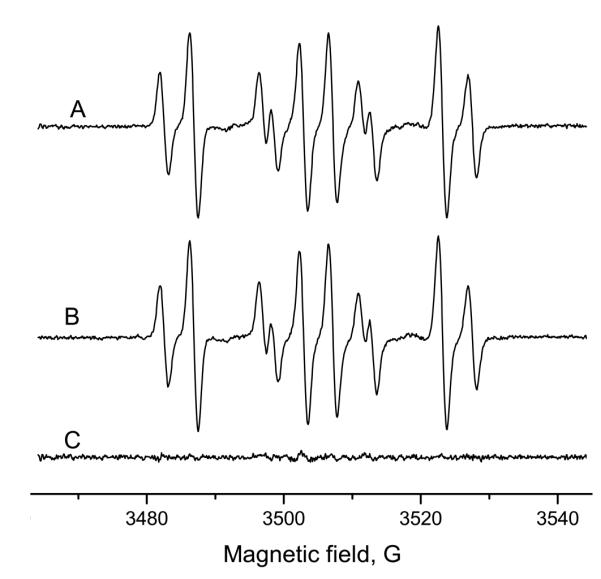
Preincubation experiment with sodium sulfite and horseradish peroxidase/H2O2. 10 mM Na2SO3 were preincubated (15 min, room temperature) with 50 mM DMPO. Then, 50 mM the other DMPO isotope, 1 mg/ml HRP and 10 μM H2O2 were added and the ESR spectrum was recorded immediately. A: Preincubation with 14N-incubation and addition of 15N-DMPO, enzyme and H2O2. B: Preincubation with 15N-DMPO; thereafter, addition of 14N-DMPO, HRP and H2O2. Both signals are almost identical and the differential spectrum (C; A minus B) shows no significant signal. The artifact formed according to the Forrester-Hepburn mechanism does not contribute to the ESR signal in a significant manner.
ESR artifact formation was also investigated with potassium ferricyanide as the oxidizing agent. ESR measurements with 10 mM sulfite, 100 mM 14N- or 15N-DMPO and 1 mM K3[Fe(CN)6]) again resulted in strong signals (Figure 3). Without sulfate and ferricyanide, no signal was detected (data not shown). The preincubation experiments (10 mM sulfate and 50 mM of either 14N-DMPO (Figure 3A) or 15N-DMPO (Figure 3B) for 15 min, then addition of the other isotope of DMPO (50 mM) and 1 mM K3[Fe(CN)6]) resulted in almost identical signals (difference, Figure C), indicating predominantly authentic signals.
Figure 3.
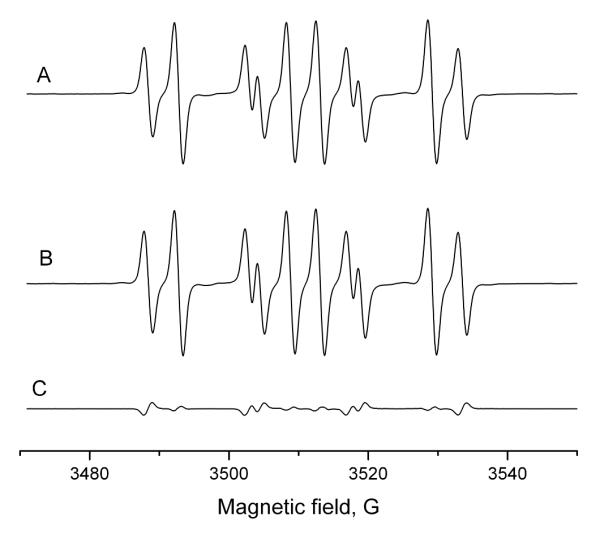
Preincubation experiment with sulfite and potassium ferricyanide. A: 10 mM Na2SO3, and 50 mM 14N-DMPO were incubated for 15 minutes. Then 50 mM 15N-DMPO and 1 mM K3[Fe(CN)6] were added and the ESR spectrum was recorded. B: As in A, but the order of spin trap addition was reversed. Both experiments have similar spectra, a mixture of 14N- and 15N-DMPO/·SO −3). C: Difference spectrum of A and B. Only a minor signal was detected, indicating that there is no significant contribution from the Forrester-Hepburn mechanism.
ESR experiments with cysteine or glutathione
Strong ESR signals of similar intensity were obtained from samples with 10 mM either L-cysteine or glutathione as well as 50 mM 14N-DMPO, 1 mg/mL HRP and 10 μM H2O2. A six-line signal was obtained with cysteine (Figure 4A) and a pseudo-quartet with glutathione (not shown). Spectral simulation revealed the coupling constants to be aN = 15.16 G and aβH = 17.39 G in the presence of L-cysteine (DMPO/·SCys, Lit.: aN = 15.3 G and aβH = 17.0 G)26 and aN = 15.10 G and aβH = 16.15 in the presence of glutathione (DMPO/·SG, Lit.: aN = 15.4 G and aβH = 16.2 G).27 With 15N-DMPO as the spin trap, the coupling constants were aN = 21.23 G and aβH = 17.39 G for the 15N-DMPO/·SCys adduct (Figure 4 B) and aN = 21.13 G and aβH = 16.18 G for the 15N-DMPO/·SG adduct (not shown). No signal was detected in the absence of either Cys (Figure 4C) or GSH (not shown), but a weaker signal was detected without hydrogen peroxide in experiments with both cysteine (Figure 4D) and glutathione (data not shown). In the absence of HRP there was no ESR signal in either system. These findings are in accordance with the literature.26, 27
Figure 4.

Generation and ESR spin-trapping of the cysteine radical. A: Complete system and simulation. The sample contained 50 mM 14N-DMPO, 10 mM cysteine, 1 mg/ml horseradish peroxidase, 10 μM H2O2, in 100 mM Chelex-treated phosphate buffer, pH = 7.4, with 100 μM DTPA. The sample was measured immediately (scan time: 167.77 s). B: As in A, but 15N-DMPO was used. C: As in A, but without cysteine. No EPR signal detected. D: As in A, but in the absence of hydrogen peroxide. A significant background signal was detected. E: As in A, but no horseradish peroxidase. No signal appeared. F: 15N-DMPO in phosphate buffer as used in A. No paramagnetic impurities are visible.
In order to detect the Forrester-Hepburn artifact, either cysteine or GSH (both 10 mM) was incubated for 15 min with 50 mM 14N-DMPO at room temperature. After this, 50 mM 15N-DMPO was added and the radical generation was initiated with 1 mg/ml horseradish peroxidase and 10 μM hydrogen peroxide and the spectrum measured within 5 min. In both cases, the ESR signal was a superposition of the 15N- and 14N-DMPO thiol radical adduct, in nearly equal concentrations (Figure 5A, cysteine; GSH not shown). The corresponding experiment with the order of isotope addition reversed (15N-DMPO during preincubation and 14N prior to radical initiation) gave very similar spectra (Figure 5B, cysteine; GSH not shown). In both cases, the difference spectra (A-B) showed only noise (Figure 5C). Thus, neither cysteine nor glutathione produced a signal attributable to the Forrester-Hepburn mechanism. With similar experiments, DMPO/·SG was demonstrated to form authentic glutathione thiyl radicals during photolysis.17
Figure 5.
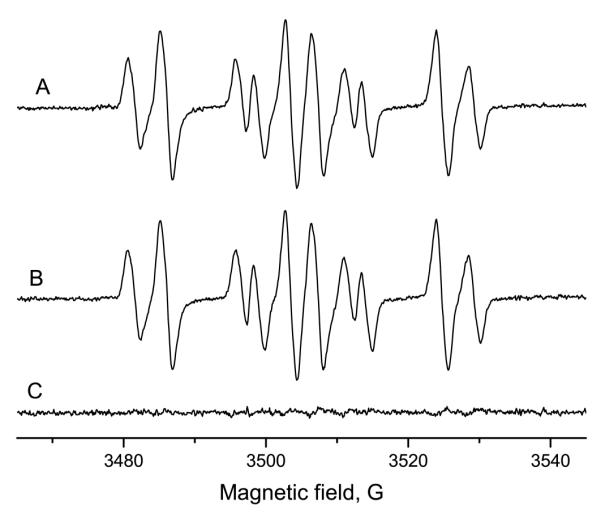
Preincubation experiment with cysteine and horseradish peroxidase/H2O2. 10 mM cysteine were preincubated (15 min, room temperature) with 50 mM one DMPO isotope. Then, 50 mM the other DMPO isotope, 1 mg/ml HRP and 10 μM H2O2 were added and the ESR spectrum was recorded. A: Preincubation with 14N-DMPO, incubation, addition of 15N-DMPO, enzyme and H2O2. B: Preincubation with 15N-DMPO, thereafter addition of 14N-DMPO, HRP and H2O2. Both signals are almost identical and the residual (C; A minus B) consists of noise. This demonstrates that no significant Forrester-Hepburn artifact contributed to the spectrum.
ESR spin-trapping experiments with potassium cyanide
With KCN, the ESR experiment under conditions similar to those of the sodium sulfite measurements exhibited an unsatisfactory signal-to-noise ratio (not shown). Thus, the 14N-DMPO and hydrogen peroxide concentrations were increased to 100 mM and 350 μM, as used by Moreno et al.,25 and the spectrum was recorded after an incubation time of 15 min (Figure 6A), which still resulted in a much lower signal-to-noise ratio in comparison to the sulfite system. Spectral simulation showed coupling constants of aN = 15.42 G and aβH = 18.90 G, which were assigned to the DMPO/·CN adduct (Lit.: aN = 15.5 G and aβH = 18.9 G).25 With 15N-DMPO as the spin trap (Figure 6 B), aN = 21.58 G and aβH = 18.89 G were observed as coupling constants for the 15N-DMPO/·CN adduct. Control experiments in the absence of each component were recorded (Figure 6C-E). Without cyanide (Figure 6C), no signal was observed. In the spectra without hydrogen peroxide (Figure 6D) or horseradish peroxidase (Figure 6E), no significant signals were detected. Those findings are in accordance with the literature.25 Figure 6F shows that minor paramagnetic impurities are detected with 100 mM 15N-DMPO in buffer.
Figure 6.
Generation and ESR spin trapping of the cyanide radical. A: Complete system and simulation. 100 mM 14N-DMPO, 10 mM KCN, 1 mg/ml horseradish peroxidase and 350 μM H2O2 were dissolved in 100 mM Chelex-treated phosphate buffer, pH = 7.4, with 25 μM DTPA. To achieve an acceptable signal/noise ratio, the sample was incubated for 15 min (room temperature) and then measured (scan time: 167.77 s). B: As in A, but in the presence of 100 mM 15N-DMPO. C: As in A, but no KCN. D: As in A, but without H2O2. E: As in A, but in the absence of HRP. No significant signals are visible in traces C-E. F: 15N-DMPO in phosphate buffer as used in A. Due to the higher concentration of DMPO and the more sensitive EPR parameters, a slight background signal was detected.
For artifact detection, 10 mM KCN was incubated with 100 mM 14N-DMPO. After 15 min at room temperature, 100 mM 15N-DMPO was added, and the radical generation was initiated with 1 mg/mL horseradish peroxidase and 350 μM hydrogen peroxide. The spectrum consisted predominantly of the 14N-DMPO cyanide radical adduct (Figure 7A). The corresponding experiment with the order of isotope addition reversed (15N-DMPO during preincubation and 14N prior radical initiation) gave the 15N-DMPO cyanide adduct in large excess (Figure 7B). In contrast to the other experiments, the two spectra are markedly different and the difference consists of the positive 14N-DMPO/·CN and the inverted 15N-DMPO/·CN signals (Figure 7C). Thus, under these conditions, almost all of the signal must originate from Forrester-Hepburn chemistry and represents a spin-trapping artifact.
Figure 7.
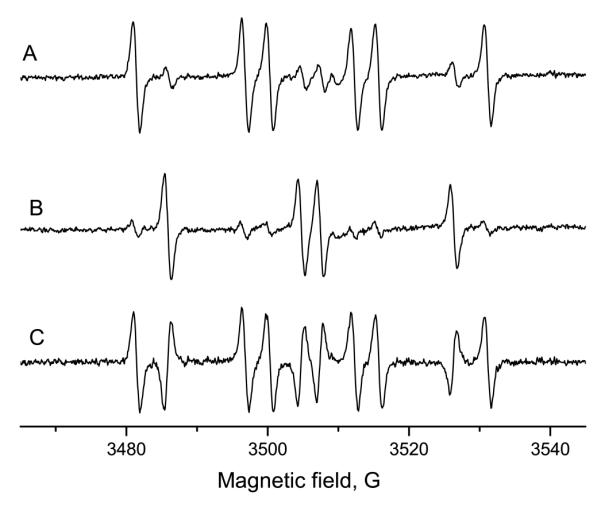
Preincubation experiment with potassium cyanide and horseradish peroxidase/H2O2. 10 mM KCN were preincubated (15 min, room temperature) with 100 mM of one DMPO isotope. Then, 100 mM the other DMPO isotope, 1 mg/ml HRP and 350 μM H2O2 were added and the ESR spectrum recorded. A: Preincubation with 14N-DMPO, incubation, addition of 15N-DMPO, enzyme and H2O2. B: Preincubation with 15N-DMPO; thereafter, addition of 14N-DMPO, HRP and H2O2. The signals consist mainly of the DMPO isotope that was present in the preincubation phase. C: Difference of A and B. The signal consists of the positive 14N-DMPO/·CN and the inverted 15N-DMPO/·CN signal. The detected ESR signal, thus, is a Forrester-Hepburn artifact.
The preincubation experiment was also carried out with 1mM potassium ferricyanide as oxidant. The spectrum consisted predominantly of the DMPO/·CN adduct formed during preincubation (Figure 8 A: Preincubation of 14N-DMPO and cyanide, 15 min preincubation, addition of ferricyanide and ESR measurement; Figure 8 B: Order of isotopes reversed: 15N-DMPO during preincubation and 14N prior radical initiation) . The difference is the superimposition of the 14N-DMPO/·CN and the inverted 15N-DMPO/·CN signal (Figure 8C). As with HRP/H2O2, the signal is attributed to the Forrester-Hepburn mechanism. In the respective control experiments, no signal was detectable if cyanide or ferricyanide were omitted (data not shown). In order to exclude the possibility that atmospheric oxygen is responsible for nitroxide radical formation, measurements under nitrogen were carried out. The signal of the complete system with an air atmosphere (100 mM 14N-DMPO, 10 mM KCN, 1 mg/ml HRP and 350 μM H2O2; Figure 9) does not significantly differ from the measurement under nitrogen (Figure 9B; Difference: 9C).
Figure 8.
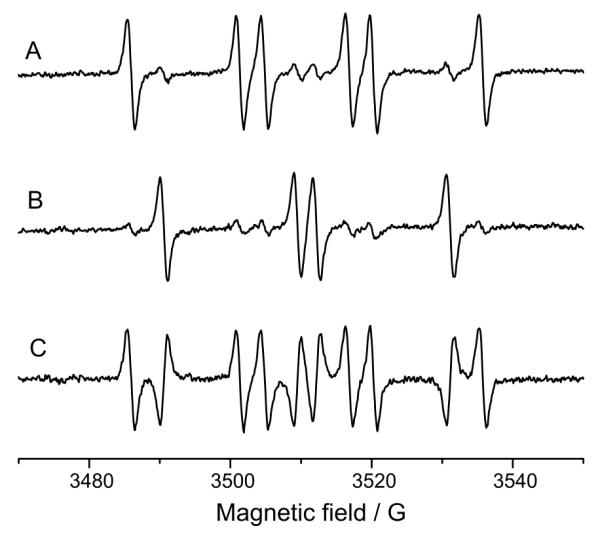
Preincubation experiment with potassium cyanide and potassium ferricyanide as the oxidant. 10 mM KCN were preincubated (15 min, room temperature) with 100 mM of one DMPO isotope. Then 100 mM of the other DMPO isotope and 1 mM K3[Fe(CN)6] were added and the ESR spectrum recorded. A: Preincubation with 14N-DMPO, incubation, addition of 15N-DMPO and ferricyanide. B: Preincubation with 15N-DMPO; thereafter, addition of 14N-DMPO, and ferricyanide. The signals consist mainly of the DMPO isotope that was present in the preincubation phase. C: Difference spectrum of A and B. The signal consists of the positive 14N-DMPO/·CN and the inverted 15N-DMPO/·CN signal. The detected ESR signal is thus a Forrester-Hepburn artifact.
Figure 9.
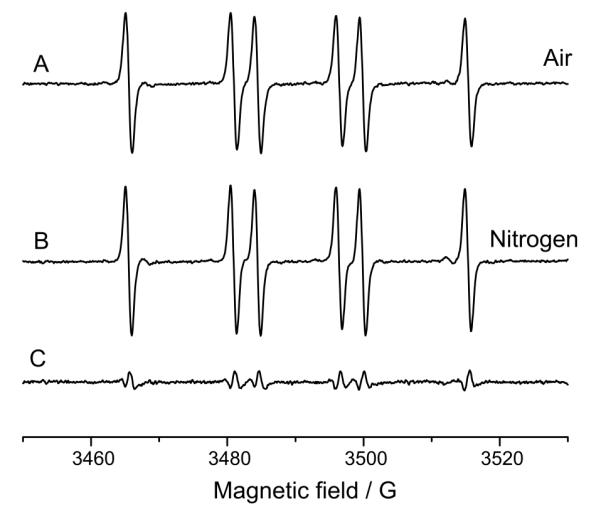
Influence of dissolved oxygen on artifact formation. A: Aerobic measurement. 100 mM 14N-DMPO, 10 mM KCN, 1 mg/ml horseradish peroxidase and 350 μM H2O2 were dissolved in 100 mM Chelex-treated phosphate buffer, pH = 7.4, with 25 μM DTPA, incubated for 15 min (room temperature), and then measured. B: as in A, but under anaerobic conditions. C: Difference spectrum of A and B. The spectra are almost identical, suggesting that dissolved oxygen does not significantly contribute to nitroxide formation from the hydroxylamine.
NMR measurements
The 1H NMR spectrum of 50 mM DMPO was recorded as a reference spectrum after 15 min incubation in deuterated phosphate buffer (Figure 10A). Then samples with 50 mM DMPO and 100 mM (10 times the concentration used in ESR experiments) of sulfite, cyanide, cysteine or glutathione were preincubated for 15 min at room temperature and the 1H NMR spectrum was measured. In the experiment with sulfite (Figure 10B), a doublet of doublets was formed at 4.05 ppm (H2) as well as two methyl group singlets at 1.21 and 1.09 ppm (5-Me and 5-Me’), due to addition of the sulfite group at C2, as reported previously.14 The reduction in intensity of the vinylic proton resonance at 7.23 ppm and the appearance of the resonance at 4.05 ppm (H2) is caused by the loss of the double bond upon addition of the sulfite group. This addition also results in the loss of the magnetic equivalence of the methyl protons in DMPO resonating at 1.41 ppm (5-Me), giving rise to the two singlets at 1.21 and 1.09 ppm (5-Me and 5-Me’) in the sulfite adduct.
Figure 10A.

1H-NMR spectrum of DMPO, preincubated for 15 min with deuterated phosphate buffer (100 mM) and 100 uM DTPA, pH 7.4. The integrals are normalized to the allyl hydrogen (2 H). The spectrum shows no additional peaks generated in decomposition reactions. B: 1H-NMR spectrum of DMPO, preincubated for 15 min with 100 mM sodium sulfite in deuterated phosphate buffer as in Fig. 10A. In respect to the pure DMPO spectrum, additional peaks appeared [4.05, ppm (H2, double doublet), 1.21 and 1.09 ppm (5-Me and 5Me’, each singlet)]. According to the literature, these lines are assigned to hydroxylamine formation. By means of integration, the hydroxylamine partition is estimated to be 3%. C: 1H-NMR spectrum of DMPO, preincubation experiment as in Fig. 10b, but with 100 mM potassium cyanide. A double doublet at 4.09 ppm (2-H) and singlets at 1.23 and 1.07 ppm (5-Me) appeared, and were assigned to the respective hydroxylamines. Comparison of the integrals gave 10 % as the hydroxylamine partition. This proportion is higher than that from the sulfide preincubation experiment.
The incubation of DMPO with cyanide gave a similar result (Figure 10C). The newly formed doublet of doublets at 4.09 ppm is again assigned to the β-proton (H2); The corresponding 13C shifts of the cyano-adduct are obtained from the 1H/13C heteronuclear single quantum coherence spectrum, which correlates the proton chemical shifts with the shifts of the carbon atoms to which they are directly attached, shown in Figure 11A. The C2, C3, C4, and C-methyl shifts are 56.92, 25.80, 36.21, and 27.12 and 21.20, respectively. The unprotonated 13C shifts were assigned from the 1H/13C heteronuclear multiple bond coherence spectrum, which correlates the proton shifts with the shifts of the carbon atoms that are 2-4 bonds removed, shown in Figure 11B. The cyano group 13C shift appears at 123.78 ppm and is coupled to the H2 methine proton and both of the H3 methylene protons (Figure 11B). The C5 shift appears at 67.25 ppm and is correlated to both sets of methyl protons and the H4 methylene protons (Figure 11B). The remaining long-range proton carbon couplings appearing in the heteronuclear multiple bond coherence spectrum are consistent with addition of the cyano group at the C2 carbon position.
Figure 11A.

1H-13C heteronuclear single quantum coherence NMR spectrum of DMPO and the cyano adduct showing the assignments of the protonated carbons of the adduct. B: 1H-13C heteronuclear multiple bond coherence NMR spectrum of DMPO and the adduct showing long-range proton-carbon correlations from the adduct. The correlations arising from coupling of the H2 proton and the H3 methylene protons with the cyano carbon (CN) prove that the cyano group has been added to the C2 position in the adduct. The C5 carbon is also assigned from this spectrum.
By means of integration of the 1H spectra, the percentage of hydroxylamine formed was estimated. With cyanide (Figure 10C) the hydroxylamine is estimated to be 10% of DMPO, whereas with sulfite (Figure 10B), only 3% of the DMPO was converted to hydroxylamine. This implies higher nucleophilic reactivity with cyanide. With the sulfur nucleophiles, cysteine and GSH, no evidence of hydroxylamine formation was found. It is possible, however, that hydroxylamine formation with thiols may occur at higher pH values where the thiolate form is more abundant. In the literature it has been reported that 5-diethoxyphosphoryl-5-methyl-1-pyrroline-N-oxide (DEPMPO) undergoes reversible nucleophilic reactions with cysteine and glutathione, as determined by 31P-NMR.15 The calculated positive charge on the β-carbon is much more positive on DEPMPO than on DMPO (0.043 e and 0.019 e, respectively),34 which will promote nucleophilic attack, making a higher reactivity for DEPMPO plausible.
Discussion
Even though the hydroxylamine intermediate of the Forrester-Hepburn mechanism has been detected by NMR and mass spectroscopy,14, 35 this is only a necessary but not sufficient condition for artifactual formation of radical adducts. In the past, kinetic ESR and oxygen consumption studies have been used to test for artifactual radical adduct formation.8 Timmins et al. have developed an ESR approach which does not require direct kinetic measurements,16, 17 but is dependent on the relative rates of radical adduct formation from either mechanism inherent in the steady-state spectrum. The nucleophilic reaction is revealed in a preincubation experiment where the hydroxylamine DMPO adduct will accumulate as an isotopically distinct DMPO radical adduct. In principle, the technique could be done without isotope labeling by comparing radical adduct concentrations with and without preincubation. This approach would best be done by simulation and double integration to give the ratio of the relative concentrations. A ratio of one would indicate no radical adduct formation during the preincubation. In the Timmins et al. approach, an isotopically distinct spin trap is employed in the preincubation experiment in order to assign the origin of the radical adduct, as outlined in scheme 2. 14N-DMPO is preincubated with the nucleophile. After that, 15N-DMPO is added just prior to the initiation of radical generation with HRP and H2O2 or K3[Fe(CN)6]. Compared to the preincubation phase, the time from addition of the second trap to the end of the ESR experiment was short (5 versus 15 min), in order to make a potentially occurring artifact more apparent. In genuine spin-trapping, no reactions occur during preincubation. During radical generation, the nucleophile is oxidized to a radical species, which forms a stable, ESR-detectable nitroxide radical adduct by its reaction with DMPO. Since the isotopic distribution is 50/50 during this phase, the radical adducts have the same proportion. If artifact formation takes place, the nucleophile reacts with 14N-DMPO in a non-radical reaction to form a hydroxylamine during the preincubation phase. The latter is oxidized after addition of the enzyme and oxidant to the nitroxide radical (14N isotope exclusively). So, the signal of the 14N isotope would predominate. When the order of isotope addition is reversed, the identical composite spectrum of the two isotopically labeled radical adducts will be obtained in authentic spin trapping. Any difference in the spectra obtained from the two experiments must be due to artifactual radical adduct formation. This is best discerned by a difference spectrum.
With sodium sulfite, no significant artifacts originating from the reaction of DMPO with Na2SO3 were detected in ESR preincubation experiments with HRP and with potassium ferricyanide. Under the present experimental conditions, the 14N/15N ratio of the signal is independent of the sequence of isotope addition (Figures 2 and 3, A and B). The identical spectra demonstrate that the radical adduct is being formed during the radical generation phase. This is strikingly apparent in the difference spectrum (Figure 2 and 3, C). However, in 1H-NMR experiments (Figure 10), peaks have been detected after preincubation which have been assigned to the respective hydroxylamines14 and confirmed here. Apparently, the rate of genuine radical formation and trapping greatly exceed hydroxylamine oxidation in the radical adduct (Scheme 1a). No significant Forrester-Hepburn artifacts were detected in ESR preincubation experiments with either cysteine or glutathione. The 14N/15N ratio of the signal is not dependent on the isotope present during preincubation, implying that the signal originates only from genuine spin trapping. In NMR experiments, no indications for hydroxylamine formation were observed with thiols, suggesting that DMPO does not easily react with thiols as DEPMPO does.15
In contrast, with cyanide, nucleophilic chemistry totally dominates radical adduct formation (Scheme 1c). In the ESR preincubation experiment, the signal consisted almost entirely of the isotope chosen for the preincubation. The radical adduct concentration was also much lower than with the other substrates. Working under anaerobic conditions did not avert artifact formation, suggesting that the hydroxylamine is oxidized by the enzyme or ferricyanide and not by dissolved atmospheric oxygen. Additionally, the NMR experiment found a more robust hydroxylamine formation with cyanide than with sulfite. The new peaks formed during the reaction of DMPO with cyanide were about three times larger than in the corresponding sulfite experiment. With 1H/13C heteronuclear single quantum coherence and 1H/13C heteronuclear multiple bond coherence measurements, we demonstrated that those peaks indeed represent the respective protons in the structure of β-cyano hydroxylamine.
In retrospect, the artifact formation of DMPO/·CN is not surprising because not only is cyanide an inhibitor of HRP, but the oxidation potential of cyanide (CN·/CN−) is 1.9 volts36-38, which is significantly greater than the oxidation potential of horseradish peroxidase Compound I/Compound II (1.0 V)39 and potassium ferricyanide (0.43 V).40 Partial inhibition of HRP with cyanide may also contribute to this result. 25 In this study we have shown that spin-trapping with DMPO can indeed be compromised by the Forrester-Hepburn mechanism, but only under special conditions. In the presence of a large excess of possible substrate over spin trap, if rapid addition occurred, it might be envisaged that both spin-traps might react completely,16 hence giving identical spectra in the reverse addition experiment. However, our NMR experiments show that the hydroxylamine adduct is never more than 10 % of the DMPO even if the substrates are 100 mM.
Conclusions
Preincubation experiments with isotope-labeled spin traps allowed direct and unambiguous detection of the Forrester-Hepburn artifact. With the chosen conditions typical for enzymatic generation of the investigated free radicals, no such ESR artifact was detectable in DMPO spin-trapping experiments with the potentially nucleophilic substrates sulfite, cysteine and glutathione with the horseradish peroxidase/hydrogen peroxide system. Again, little or no Forrester-Hepburn artifact was found for sulfite oxidation by potassium ferricyanide. However, the ESR signal in the presence of cyanide originated almost entirely from Forrester-Hepburn chemistry. The hydroxylamine intermediate, which is necessary but not sufficient evidence for the Forester Hepburn artifact, is formed with sulfite and, more abundantly with cyanide, as shown by NMR spectroscopy. The structure of the cyano-hydroxylamine has been proven by 2D NMR experiments, but only with cyanide does the ESR-detectable Forrester-Hepburn artifact occur. Our investigations support the conclusions of the original publications except for the cyanide radical. According to our experiments, ESR spin trapping of biochemically generated radicals provide reliable results in most cases. These isotope experiments should be performed wherever possible, since we have shown that artifacts from the Forrester-Hepburn mechanism can indeed occur.
Acknowlegement
We would like to thank Dr. Ann G. Motten, Ms. Mary J. Mason and Ms. Jean Corbett for their help with the manuscript.
Funding sources This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Abbreviations
- DEPMPO
[5-(diethoxyphosphoryl)-5-methyl-1-pyrroline N-oxide
- DMPO
5,5-dimethyl-1-pyrroline N-oxide
- DTPA
diethylenetriaminepentaacetic acid
- HRP
horseradish peroxidase
- SCE
Standard Calomel Electrode
References
- (1).Weil JA, Bolton JR, Wertz JE, editors. Electron paramagnetic resonance: Elementary theory and practical applications. Wiley-Interscience; New York, NY: 1994. [Google Scholar]
- (2).Svistunenko DA. Reaction of haem containing proteins and enzymes with hydroperoxides: The radical view. BBA - Bioenergetics. 2005;1707:127–155. doi: 10.1016/j.bbabio.2005.01.004. [DOI] [PubMed] [Google Scholar]
- (3).Janzen EG. Spin trapping. Acc. Chem. Res. 1971;4:31–40. [Google Scholar]
- (4).Eberson L. Inverted spin trapping - reactions between the radical cation of a-phenyl-n-tert-butylnitrone and ionic and neutral nucleophiles. J. Chem. Soc. Perkin Trans. 1992;2:1807–1813. [Google Scholar]
- (5).Forrester AR, Hepburn SP. Spin traps. A cautionary note. J. Chem. Soc. C. 1971:701–703. [Google Scholar]
- (6).Ionita P, Gilbert BC, Whitwood AC. Authentic versus alternative mechanisms in spin trapping. Formation of azide spin-adducts in biphasic and non-aqueous systems by the oxidation of azide anion with a variety of hydrazyl radicals. J. Chem. Soc. Perkin Trans. 2000;2:2436–2440. [Google Scholar]
- (7).Timmins GS, Liu KJ. Spin trapping in vivo: Facts and artifacts. In: Berliner LJ, editor. In vivo EPR (ESR): Theory and application. Kluwe Academic/Plenum Publishers; New York: 2003. pp. 285–309. [Google Scholar]
- (8).Ranguelova K, Mason RP. The fidelity of spin trapping with DMPO in biological systems. Magn. Reson. Chem. 2011;49:152–158. doi: 10.1002/mrc.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chandra H, Symons MCR. Hydration of spin-trap cations as a source of hydroxyl adducts. J. Chem. Soc. Chem. Comm. 1986:1301–1302. [Google Scholar]
- (10).Eberson L. Inverted spin trapping part III. Further studies on the chemical and photochemical oxidation of spin traps in the presence of nucleophiles. J. Chem. Soc. Perkin Trans. 1994;2:171–176. [Google Scholar]
- (11).Cerri V, Frejaville C, Vila F, Allouche A, Gronchi G, Tordo P. Synthesis, redox behavior, and spin-trap properties of 2,6-di-tert-butylnitrosobenzene (DTBN) J. Org. Chem. 1989;54:1447–1450. [Google Scholar]
- (12).McIntire GL, Blount HN, Stronks HJ, Shetty RV, Janzen EG. Spin trapping in electrochemistry .2. Aqueous and non-aqueous electrochemical characterizations of spin traps. J. Phys. Chem.-US. 1980;84:916–921. [Google Scholar]
- (13).Eberson L. Formation of hydroxyl spin adducts via nucleophilic addition-oxidation to 5,5-dimethyl-1-pyrroline N-oxide (DMPO) Acta Chem. Scand. 1999;53:584–593. [Google Scholar]
- (14).Potapenko DI, Bagryanskaya EG, Reznikov VV, Clanton TL, Khramtsov VV. NMR and EPR studies of the reaction of nucleophilic addition of (bi)sulfite to the nitrone spin trap DMPO. Magn. Reson. Chem. 2003;41:603–608. [Google Scholar]
- (15).Potapenko DI, Bagryanskaya EG, Tsentalovich YP, Reznikov VA, Clanton TL, Khramtsov VV. Reversible reactions of thiols and thiyl radicals with nitrone spin traps. J. Phys. Chem. B. 2004;108:9315–9324. [Google Scholar]
- (16).Timmins GS, Barlow GK, Silvester JA, Wei X, Whitwood AC. Use of isotopically labelled spin-traps to determine definitively the presence or absence of non-radical addition artefacts in EPR spin-trapping systems. Redox Rep. 1997;3:125–133. doi: 10.1080/13510002.1997.11747099. [DOI] [PubMed] [Google Scholar]
- (17).Silvester JA, Wei XD, Davies MJ, Timmins GS. A study of photochemically-generated protein radical spin adducts on bovine sevum albumin: The detection of genuine spin-trapping and artefactual, non-radical addition in the same molecule. Redox Rep. 1997;3:225–231. doi: 10.1080/13510002.1997.11747114. [DOI] [PubMed] [Google Scholar]
- (18).Mottley C, Mason RP. Sulfate anion free radical formation by the peroxidation of (bi)sulfite and its reaction with hydroxyl radical scavengers. Arch. Biochem. Biophys. 1988;267:681–689. doi: 10.1016/0003-9861(88)90077-x. [DOI] [PubMed] [Google Scholar]
- (19).Sun X, Shi X, Dalal NS. Xanthine oxidase/hydrogen peroxide generates sulfur trioxide anion radical (SO3·−) from sulfite (so32−) FEBS Lett. 1992;303:213–216. doi: 10.1016/0014-5793(92)80522-i. [DOI] [PubMed] [Google Scholar]
- (20).Jiang J, Liu KJ, Shi X, Swartz HM. Detection of short-lived free radicals by low-frequency electron paramagnetic resonance spin trapping in whole living animals. Arch. Biochem. Biophys. 1995;319:570–573. doi: 10.1006/abbi.1995.1332. [DOI] [PubMed] [Google Scholar]
- (21).Constantin D, Bini A, Meletti E, Moldeus P, Monti D, Tomasi A. Age-related differences in the metabolism of sulphite to sulphate and in the identification of sulphur trioxide radical in human polymorphonuclear leukocytes. Mech. Ageing Dev. 1996;88:95–109. doi: 10.1016/0047-6374(96)01728-9. [DOI] [PubMed] [Google Scholar]
- (22).Ranguelova K, Mason RP. New insights into the detection of sulfur trioxide anion radical by spin trapping: Radical trapping versus nucleophilic addition. Free Radic. Biol. Med. 2009;47:128–134. doi: 10.1016/j.freeradbiomed.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ranguelova K, Chatterjee S, Ehrenshaft M, Ramirez DC, Summers FA, Kadiiska MB, Mason RP. Protein radical formation resulting from eosinophil peroxidase-catalyzed oxidation of sulfite. J. Biol. Chem. 2010;285:24195–24205. doi: 10.1074/jbc.M109.069054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Potapenko DI, Clanton TL, Bagryanskaya EG, Gritsan NP, Reznikov VA, Khramtsov VV. Nonradical mechanism of (bi)sulfite reaction with DEPMPO: Cautionary note for SO3−· radical spin trapping. Free Radic. Biol. Med. 2003;34:196–206. doi: 10.1016/s0891-5849(02)01194-2. [DOI] [PubMed] [Google Scholar]
- (25).Moreno SNJ, Stolze K, Janzen EG, Mason RP. Oxidation of cyanide to the cyanyl radical by peroxidase/H2O2 systems as determined by spin trapping. Arch. Biochem. Biophys. 1988;265:267–271. doi: 10.1016/0003-9861(88)90127-0. [DOI] [PubMed] [Google Scholar]
- (26).Harman LS, Mottley C, Mason RP. Free radical metabolites of l-cysteine oxidation. J. Biol. Chem. 1984;259:5606–5611. [PubMed] [Google Scholar]
- (27).Harman LS, Carver DK, Schreiber J, Mason RP. One- and two-electron oxidation of reduced glutathione by peroxidases. J. Biol. Chem. 1986;261:1642–1648. [PubMed] [Google Scholar]
- (28).Mason RP. Assay of in situ radicals by electron spin resonance. Methods in enzymology. 1984;105:416–422. doi: 10.1016/s0076-6879(84)05058-8. [DOI] [PubMed] [Google Scholar]
- (29).Duling DR. Simulation of multiple isotropic spin-trap EPR-spectra. J. Magn. Reson. Ser. B. 1994;104:105–110. doi: 10.1006/jmrb.1994.1062. [DOI] [PubMed] [Google Scholar]
- (30).Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe - a multidimensional spectral processing system based on unix pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- (31).Johnson BA, Blevins RA. NMR view: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- (32).Jiang JJ, Jordan SJ, Barr DP, Gunther MR, Maeda H, Mason RP. In vivo production of nitric oxide in rats after administration of hydroxyurea. Mol. Pharmacol. 1997;52:1081–1086. doi: 10.1124/mol.52.6.1081. [DOI] [PubMed] [Google Scholar]
- (33).Wertz JE, Bolton JR, editors. Electron spin resonance: Elementary theory and practical applications. McGraw-Hill; New York, NY: 1972. [Google Scholar]
- (34).Villamena FA, Xia S, Merle JK, Lauricella R, Tuccio B, Hadad CM, Zweier JL. Reactivity of superoxide radical anion with cyclic nitrones: Role of intramolecular H-bond and electrostatic effects. J. Am. Chem. Soc. 2007;129:8177–8191. doi: 10.1021/ja0702622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Triquigneaux M, Tuccio B, Lauricella R, Charles L. Nucleophile addition of reduced glutathione on 2-methyl-2-nitroso compound: A combined electron paramagnetic resonance spectroscopy and electrospray tandem mass spectrometry study. J. Am. Soc. Mass Spectr. 2009;20:2013–2020. doi: 10.1016/j.jasms.2009.07.018. [DOI] [PubMed] [Google Scholar]
- (36).Berdnikov VM, Bazhin NM. Oxidation-reduction potentials of certain inorganic radicals in aqueous solutions. Russ. J. Phys. Ch. USSR. 1970;44:395–398. [Google Scholar]
- (37).Wardman P. Reduction potentials of one-electron couples involving free radicals in aqueous solution. J. Phys. Chem. Ref. Data. 1989;18:1637–1755. [Google Scholar]
- (38).Eberson L. Spin trapping and electron transfer. Adv. Phys. Org. Chem. 1998;31:91–141. [Google Scholar]
- (39).Hayashi Y, Yamazaki I. Oxidation-reduction potentials of compound I/compound II and compound II/ferric couples of horseradish peroxidases a2 and c. J. Biol. Chem. 1979;254:9101–9106. [PubMed] [Google Scholar]
- (40).O’Reilly JE. Oxidation-reduction potential of the ferro-ferricyanide system in buffer solutions. Biochim. Biophys. Acta. 1973;292:509–515. doi: 10.1016/0005-2728(73)90001-7. [DOI] [PubMed] [Google Scholar]