

Abstract
Duchenne muscular dystrophy (DMD) is a fatal, X-linked muscle disease caused by mutations in the dystrophin gene. Adeno-associated viral (AAV) vector-mediated gene replacement strategies hold promise as a treatment. Studies in animal models and human trials suggested that immune responses to AAV capsid proteins and transgene products prevented efficient gene therapy. In this study, we used widespread intramuscular (i.m.) injection to deliver AAV6-canine micro-dystrophin (c-µdys) throughout a group of skeletal muscles in dystrophic dogs given a brief course of commonly used immunosuppressants. Robust c-µdys expression was obtained for at least two years and was associated with molecular reconstitution of the dystrophin-glycoprotein complex (DGC) at the muscle membrane. Importantly, c-µdys expression was maintained for at least 18 months after discontinuing immunosuppression. The results obtained in a relevant preclinical model of DMD demonstrate feasibility of widespread AAV-mediated muscle transduction and transgene expression in the presence of transient immunosuppression to achieve molecular reconstitution that can be directly translated to human trials.
Introduction
Duchenne muscular dystrophy (DMD), the most common form of muscular dystrophy, is caused by the lack of dystrophin.1 Dystrophin is the key effector in assembling the dystrophin-glycoprotein complex (DGC), which serves to link the subsarcolemmal cytoskeleton to the extracellular matrix in muscle cells.2 In the absence of dystrophin, the complex fails to assemble on the sarcolemma, leading to wasting of skeletal and cardiac muscle, and the gradual replacement of muscle tissues by fibrotic and adipose tissue.3 The disease is lethal, and there is no curative treatment at present. A number of emerging experimental strategies for treating DMD have focused on dystrophin gene replacement or transcript modification. Among these, adeno-associated viral vector (AAV)-mediated delivery of engineered mini- or micro (µ)-dystrophins (dys) into dystrophin-deficient mice with DMD has shown remarkable efficiency,4,5 which has led to the initiation of early-phase clinical trials.6,7
However, a growing body of studies in animal models and human trials show that immune responses can be elicited against AAV capsid proteins or the transgene products, which compromises long-term therapeutic efficacy.8 In a clinical trial of hepatic AAV2-mediated delivery of the factor IX gene in hemophilia B patients, transient transaminitis developed after vector administration, which was accompanied by detection of AAV2 capsid-specific CD8+ T-cells, and a decline in factor IX transgene expression to sub-therapeutic levels by 8 weeks.9,10 T-cell responses specific to AAV1 capsid proteins were observed in half of the patients in a clinical trial for lipoprotein lipase deficiency.11 Recent human trials of AAV-mediated gene delivery to muscle in patients with either limb-girdle6,12 or DMD7 documented humoral and/or cellular immune responses to AAV or/and transgene. In the DMD trial, no mini-dystrophin transgene expression was detected in any of the patients enrolled.7 Four out of six patients had detectable dystrophin-specific T cells, including CD4+ and CD8+ T cells in peripheral blood, with two patients having dystrophin-specific T cells before vector treatment. The authors speculated that T cells targeting self or non-self dystrophin epitopes may have eliminated mini-dystrophin-expressing muscle fibers in the study. This same mechanism might hold true for treatment strategies based on using exon-skipping and stop codon read-through strategies or using stem cells for expressing a wild-type dystrophin;13,14,15,16,17 however, it is also possible that revertant fibers expressing endogenous dystrophin might provide tolerance to the therapeutically delivered dystrophin. In line with reports in humans, cellular immunity to AAV vectors was also observed to various degrees when AAV1, 2, 6, 8 or 9 were injected into dog muscle,18,19,20,21,22,23 which resulted in reduced or short-lived transgene expression. By incorporating a regimen of immunosuppression, we and others demonstrated that immune responses can be averted and transgene expression can be enhanced by either intramuscular (i.m.) or intravenous delivery of vectors into dog muscles.22,24
The ultimate goal of AAV-mediated gene therapy is to achieve persistent transgene expression at therapeutic levels. Our previous study24 demonstrated an immune response to AAV capsid proteins following an i.m. injection of AAV carrying either human or canine micro-dystrophin (c-µdys), whereas a transient immunosuppression with anti-thymocyte globulin (ATG), cyclosporine (CSP) and mycophenolate mofetil (MMF) was sufficient to prevent the immune responses and achieve transgene expression. Following discontinuation of immunosuppression, c-µdys expression persisted for at least 6 weeks. In contrast, human µdys expression was eliminated with the detection of marked T-cell infiltration. Here, we investigated the efficacy of sustaining long-term transgene expression following large-scale AAV delivery to multiple muscles via i.m. injection in dogs with DMD. By using a species-specific c-µdys in combination with a transient immunosuppression with ATG, CSP and MMF, long-term robust transgene expression for at least 2 years was achieved in dystrophic dog muscles. The sustained c-µdys expression improved muscle histology with molecular reconstitution of the DGC. More impressively, c-µdys expression was well maintained for greater than 1 year after immunosuppression was discontinued. This proof of principle study in a clinically relevant dog model of DMD establishes a meaningful approach for successful therapeutic gene delivery in DMD patients.
Results
Transient immunosuppression and c-µdys expression
Two cxmd dogs, G844 and H222, received widespread, dispersed i.m. injections of AAV6-CMV-c-µdys vector at the age of 5 months. The three-drug immunosuppression regimen was applied, including 5 days of ATG from day −2 to day 2 (1 mg/kg/day), 24 weeks of CSP (7.5 mg/kg) twice per day from days −1 to 184, and MMF (5 mg/kg) twice per day from days 0 to 184 (Figure 1). Our previous studies demonstrated robust cellular immune responses following i.m. AAV injection in the absence of immunosuppression and enhanced transgene delivery with a transient immunosuppression;18,23,24 therefore, we used the transient immunosuppression protocol in this study. A total of 1013 vg of the therapeutic vectors were delivered into the gastrocnemius (GAS, 64 injections of 1011 vg/injection), tibialis cranialis (TC) and extensor digitorum longus muscles (a total of 36 injections of 1011 vg/injection) of the left hind limb in an attempt to saturate the three muscles with vector (Figure 2a). The same muscle groups in the right hind limb remained untreated and served as a control. The dogs had no obvious signs of drug toxicities during and after the course of immunosuppression. A biopsy sample was taken from AAV-treated TC muscle from dog G844 15 months after treatment, which was 10 months after discontinuation of all immunosuppressants. Extensive c-µdys expression was detected without obvious cellular infiltration (Figure 2b).
Figure 1.

Immunosuppression regimen using ATG, CSP and MMF. ATG, anti-thymocyte globulin; BID, twice daily; CSP, cyclosporine; MMF, mycophenolate mofetil; PO, oral administration; QD, once daily; SQ, subcutaneous.
Figure 2.

Large-scale intramuscular injection. (a) Muscles of hind limb targeted during large scale intramuscular injection of AAV6-CMV-c-µdys. (b) Muscle histology at 15 months after vector treatment (9 months after discontinuation of immunosuppression). b1, c-µdys expression in green; b2, H&E staining; b3, staining of CD8 T-cell marker in green. Blue shows DAPI staining for nuclei. Bar = 100 µm. AAV, Adeno-associated viral vector; c-µdys, canine micro-dystrophin; DAPI, 4′, 6-diamidino-2-phenylindole; H&E, hematoxylin and eosin.
Sustained expression of c-µdys and improved muscle structure
Dogs G844 and H222 were euthanized at 2 years and 19 months, respectively, after initial AAV treatment, which was 18 and 13 months, respectively, after discontinuation of all immunosuppression. An increase in the circumference of the lower limb around the belly of the GAS muscle was observed on the treated sides compared to the untreated sides, 5 cm versus 4.5 cm for G844, and 5.2 cm versus 4.6 cm for H222. The gross weight of the GAS and TC muscles from the treated sides was more than that from the untreated sides: 29.4 g versus 27 g, and 15.9 g versus 13.4 g, respectively, for G844; 36.2 g versus 34.5 g and 15.7 g versus 13.5 g, respectively, for H222.
Muscles were dissected into 1 cm3 pieces and further cut into 10 µm sections for histology and transgene expression analysis. Figure 2 shows representative data from G844 revealing near uniform c-µdys expression throughout the treated GAS muscle at the 2-year time point (Figure 3a-a1) along with much improved muscle histology (Figure 3a-a2). No obvious cellular infiltration was detected (Figure 3a-a3). The untreated GAS from the right limb remained dystrophin negative with dystrophic histology (Figure 3a-a4, a5, and a6). Sustained c-µdys expression was also detected in H222 at 19 months after treatment, although the expression was not as uniform as that in G844 (Figure 3b). By western blot, c-µdys was detected in the treated GAS muscles from both dogs, but not in the untreated muscle (Figure 3c). These findings demonstrated the feasibility of achieving persistent expression of canine µdys following large-scale AAV injections in cxmd dogs given ATG/CSP/MMF.
Figure 3.

Prolonged expression of c-µdys and muscle histology following treatment in dystrophic muscle. (a) a1 to a3 show muscle from the treated side of dog G844, and a4 to a6 were from the untreated side. Insert shows dystrophin expression (green) in muscle from a normal dog. a1 and a4 show c-µdys expression in green; a2 and a5 show H&E staining for muscle histology; a3 and a6 show staining of a CD8 T-cell marker in green. Blue is DAPI staining for nuclei. (b) Dystrophin expression in muscle from dog H222 in green. (c) Detection of c-µdys by western blot. The first four lanes contain lysates from dog muscle and the last 2 from 293 cells. Molecular weight markers (sizes in kD) are shown on the right and detected proteins on the left The high molecular weight band seen in the 293 cells transfected with the plasmid containing c-µdys is an artifact presumably due to plasmid rearrangement during transfection. However, a band of the size expected of c-µdys is clearly seen between the 100 and 150 kD size markers. As a control for the amount of protein in the dog muscle samples, an actin antibody that recognized an alpha actin found in skeletal muscle was used. No actin band is seen in the 293 cells because they make little to none of this form of actin, and there was less protein loaded in the two lanes containing lysates from 293 cell than in the ones from dog muscle (5 versus 30 µg, respectively). Bar = 100 µm. c-µdys, canine micro-dystrophin; DAPI, 4′, 6-diamidino-2-phenylindole; H&E, hematoxylin and eosin.
The absence of dystrophin prevents assembly of the DGC and reduces the levels of all DGC components.25,26 To determine whether the expression of c-µdys was able to restore sarcolemmal assembly of the DGC, the localization of a few components of the complex, β-sarcoglycan and α-dystrobrevin, for example, was analyzed. Compared to untreated dystrophic muscle, increased levels of β-sarcoglycan and α-dystrobrevin were detected at the sarcolemma in treated but not in untreated muscle (Figure 4a showing representative data from G844).
Figure 4.

Expression of DGC components and the phenotype of achilles myotendinous junctions from wild-type, untreated and c-µdys treated cxmd muscle. (a) Expression of β-sarcoglycan (β-SG) and α-dystrobrevin (αDB2) in green in normal muscle (a1 and a2), untreated contra-lateral limb muscle (a3 and a4) and treated dystrophic muscle (a5 and a6) from dog G844. Blue indicates DAPI staining for nuclei. (b) Achilles myotendinous junctions. Note that the tendon extends deep folds into the muscle in wild-type controls, but these are diminished in muscles from cxmd G844 (arrows). Note also the unusually thin filament attachments in the untreated dystrophic muscle from G844 which weren't improved by c-µdys treatment (arrow heads). Bar = 2 µm. c-µdys, canine micro-dystrophin; DAPI, 4′, 6-diamidino-2-phenylindole; DGC, dystrophin-glycoprotein complex.
The myotendinous junctions normally contain folds of tendon that extend deep into the muscle fiber to minimize membrane stress under shear.27 The filament attachment between the myofibrils and the tendon is disrupted in DMD.28 There were many folds in the untreated cxmd myotendinous junction. However, the folds were unusual compared to those in normal muscle (Figure 4b) as they did not extend far into the muscles and had extensive thin filament attachments to the myofibrils (Figure 4b). The extensive thin filament attachments to the myofibrils weren't improved by c-µdys treatment, which is consistent with previous findings in mdx mice.29
Neutralizing antibodies to AAV6 developed and persisted but did not abrogate dystrophin expression
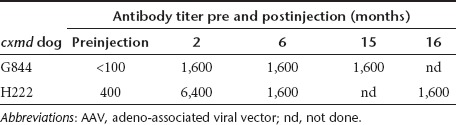
We have previously demonstrated that multiple different epitopes of the AAV capsid protein elicit a T-cell response,23 and here, a virus neutralization assay was used to measure the antibody response against AAV6 following vector delivery. This analysis showed that a significant neutralizing antibody response developed in both dogs during the period of immunosuppression (Table 1). The neutralizing titer was defined as the inverse of the highest dilution that still inhibited vector transduction by 50% or more. Prior to AAV delivery, neutralizing activity was not detected in serum from G844, whereas H222 had a pre-existing neutralizing titer of 400. For both dogs, the post-AAV antibody responses peaked at 2 months after vector administration. The titers did not increase after discontinuing immunosuppression and were maintained for at least 1 year at neutralizing titers of 1,600.
Table 1. Time course of AAV6 neutralizing antibody titers.
A sandwich enzyme-linked immunosorbent assay utilizing two different anti-dystrophin antibodies was employed to detect antibodies to c-µdys in sera from the two dogs. Assay wells were first coated with polyclonal antibodies against dystrophin, proteins from 293 cells or c-µdys expressing 293 cells were added, and nonadherent proteins were removed by washing. Antibodies to c-µdys in sera from the two dogs were measured by comparing binding of serum to bound proteins from the 293 or c-µdys expressing 293 cells. No significant difference in serum binding to c-µdys or control wells was observed for preinjection, 2 m, 6 m, or >12 m serum samples from either dog (data not shown), indicating that antibodies against c-µdys were at least not at a detectable level in either dog.
Discussion
The ultimate goal of AAV-mediated gene therapy is to achieve persistent transgene expression at therapeutic levels. The goal is compromised by host immune responses to either AAV capsid protein and/or transgene product.8 The strategy is more challenging in treating muscular dystrophy with five potential immune barriers to overcome: (i) cellular immune responses to AAV capsid protein or newly introduced µdys; (ii) humoral immune responses to AAV capsid protein or newly introduced µdys; (iii) pre-existing inflamed micro-environment in dystrophic muscle; (iv) pre-existing immunity to AAV; and (v) pre-existing immunity to dystrophin protein. In the current study, we demonstrated in a preclinical dog model of DMD that widespread, sustained transgene expression and molecular reconstitution following large-scale regional AAV delivery is feasible with a clinically acceptable immunosuppression protocol.
Our study demonstrated high level AAV vector-mediated c-µdys expression, which contrasts with undetectable or low levels of transgene expression reported in the majority of the studies that did not use immunosuppression during and after vector delivery.6,7,8,12,22,30 For example, a phase I clinical trial of limb-girdle muscular dystrophy (LGMD-2D)12 reported humoral and T-cell responses to AAV1, in one patient who failed to show transgene expression. In a phase I human trial involving DMD patients by Mendell's group,7 none of the patients displayed mini-dystrophin expressing myofibers, with four of six enrolled patients having detectable dystrophin-specific T cells in peripheral blood, and two of them displayed pre-existing T cells specific to dystrophin, raising the concern that potential transgene expressing fibers might have been rejected. These results suggest that exogenously expressed dystrophin in muscles of DMD patients might be as immunogenic, if not more so, than the viral capsid, especially when expressed by a ubiquitously active promoter such as CMV. It is also a possibility that novel epitopes in the shortened µdys protein could also elicit a cytotoxic T lymphocyte response. However, at present there is no direct evidence that the immune response seen against dystrophin in the clinical trial was the reason that no vector-encoded dystrophin expression was observed in AAV-injected muscles. A recent study by Bowles et al. on the same set of patients reported that antibodies to the chimeric AAV2.5 capsid were detected following the treatment in all six enrolled patients, and two patients had pre-existing neutralizing antibodies to the chimeric capsid.31 T-cell responses to the AAV capsid were detected at substantial levels in one patient and weakly in three patients, which was not observed in the earlier paper.7 Furthermore, T-cell responses to dystrophin epitopes were observed in three patients while two of them had pre-existing dystrophin-specific T cells. The authors confirmed that no dystrophin expression was detected in the majority of the treated patients other than two patients who had a few dystrophin-positive fibers31.
Similarly, Herson and colleagues30 reported modest cellular infiltration 4 weeks after injection of an AAV1 vector into muscles of LGMD-2C patients with transgene expression being detected only in a limited muscle of fibers (4.7–10.5%). T-cell responses to AAV1 capsid proteins were also observed in a clinical trial for lipoprotein lipase deficiency.11 Specifically, AAV1 capsid-specific CD8+ T cells were detected at 4 weeks in all patients after i.m. injection with kinetics that was dose-dependent. In dog studies, while marked inflammation was observed following either i.m. or intravenous injection of AAV vectors into neonatal or adult dogs without immunosuppression,21,32 the use of transient immunosuppression enhanced transgene expression in muscles,22,24 as well as in lung tissues.33 By using a mRNA sequence-optimized µdys with a muscle specific promoter, Koo et al. reported transgene expression from AAV8 for 2 months in one DMD-affected dog without the use of immunosuppression.34 The data are encouraging, but need to be interpreted cautiously and confirmed in additional animals. Immunity to AAV capsid and neo-epitopes in transgene products, both before and after treatment should be analyzed, and it is unlikely that repeated administration would be possible without the use of immunosuppression. Our previous studies in normal dogs24 and the current study in cxmd dogs with sustained, widespread c-µdys expression for at least 2 years demonstrate that immune modulation might be required to prevent host immune responses following AAV administration and thereby promote long-term transgene expression following discontinuation of immunosuppression.
Despite the use of immunosuppression, neutralizing antibody responses against AAV developed in both dogs in the current study. Two recent studies reported pre-existing neutralizing antibodies to AAV6 in dogs including neonatal pups,35,36 however, a few other studies did not.33,37 In the present study, dog H222, but not G844, showed detectable anti-AAV antibodies before treatment. Interestingly, the expression of µdys was not as uniform and at a lower level in H222 compared to that in G844, which corresponded to the detection of pre-existing anti-AAV antibodies in H222. However, it is notable that this pre-existing neutralizing activity (i) did not prevent AAV6 vector transduction, and (ii) did not affect the persistence of canine dystrophin expression. On the other hand, antibodies against the c-µdys protein either before or after c-µdys vector administration were not observed, at least not at a detectable level.
AAV-mediated gene therapy for muscular dystrophies is a promising yet complex therapeutic approach that has been under development for the past 15 years. The fact that persistent, widespread transgene expression and molecular reconstitution can be achieved as shown in the current preclinical studies in DMD-affected dogs is encouraging. Intravascular gene delivery is an alternative for widespread vector delivery. However, at this time, systemic intravascular injection remains orders of magnitude less efficient than direct i.m. injection and also delivers the majority of the virus to liver, lung, and other non-muscle tissues. Until a reliable systemic delivery method is developed and evaluated, multiple-injection i.m. gene delivery may be an option for treating one or a group of muscles that are important for activities of daily living and improving quality of life. Whether the molecular reconstitution lead to functional improvement of the treated dystrophic muscle remains to be analyzed. CSP, MMF and ATG are standard immunosuppressants commonly used in bone marrow and solid organ transplantation in humans.38,39,40,41,42,43,44 The feasibility of combining large-scale regional AAV-mediated gene delivery with a clinically acceptable immunosuppression demonstrated in a clinically relevant animal model is a critical step in translating the approach to humans.
Materials and Methods
Animals. Research was performed according to the principles outlined in the Guide for Laboratory Animal Facilities and Care prepared by the National Academy of Sciences, Washington, DC; National Research Council, Washington, DC. Dogs were housed in kennels certified by the American Association for Accreditation of Laboratory Animal Care, Washington, DC. This study was approved by the Institutional Animal Care and Use Committee of the Fred Hutchinson Cancer Research Center (Seattle, WA). All dogs were immunized for leptospirosis, distemper, hepatitis, papillomavirus and parvovirus, dewormed, and observed for disease for at least 2 months before being entered in the study. Cxmd dogs were diagnosed by genotyping as described.45 Serum creatine kinase levels were determined on all dogs within 24 hours of birth and at various time points after transplantation as clinically indicated. Progressive weakness, excessive salivation, progressive enlargement of the base of the tongue, inability to fully open the jaw, dysphagia, signs of pharyngeal and esophageal dysfunction, marked muscle atrophy of truncal and limb muscles accompanied by fibrosis and contractures, decreased growth rate, plantigrade stance, an abnormal stiff-limbed, short-strided, and shuffling gait, and exercise intolerance were apparent in all cxmd dogs. Both G844 and H222 were 5 months old, and weighed 7.2 kg and 7.4 kg at the start of the study, respectively.
AAV vector construction, production and quantitation. The AAV6-CMV-c-µdys vector (previously called AAV6-CMV-k9-µ-dysΔR4-R23/ΔCT) was produced as described previously.5,18,46 Briefly, the vector plasmid with recombinant AAV genomes and a plasmid (pDGM6) encoding AAV Rep and Cap proteins, and adenovirus proteins required for AAV replication, were co-transfected into human embryonic kidney 293 cells by CaPO4 co-precipitation. The cells and culture medium were collected and processed through a microfluidizer, sterile filtered, and affinity purified with a HiTrap heparin column (Amersham, Piscataway, NJ). The peak AAV6 fraction was then layered onto a 40% sucrose solution and pelleted at 131,000g for 16 hours at 6 °C, then resuspended in Hanks' balanced salt solution (Invitrogen, Carlsbad, CA). Vector genome (vg) concentrations were determined relative to a plasmid standard by Southern blot hybridization.
Immunosuppression. Immunosuppression consisted of oral CSP (Teva Pharmaceuticals USA, Sellersville, PA), 5 mg/kg twice daily, on days −2 to 24 weeks postinjection, subcutaneous MMF (Roche, Nutley, NJ), 7.5 mg/kg twice daily subcutaneously on days 0 to 24 weeks after injection, and subcutaneous ATG (produced in collaboration with SangStat), 1 mg/kg from day −2 to day 2. Blood CSP levels were measured using the CSP Monoclonal Whole Blood Assay (Abbott Laboratories, Abbott Park, IL) on days 3, 7 and weekly thereafter to guide CSP dosing in recipients. Complete blood counts were monitored before the initiation of the study, on days 0, 3 and 7 after injection, and then once weekly thereafter. Serum chemistry levels were obtained once weekly to monitor kidney and liver changes.
Large-scale i.m. injection and muscle biopsy. The animals were anesthetized with isoflurane and placed in a lateral decubitus position. The skin of the left hind limb was opened to expose GAS, and TC and extensor digitorum longus muscles, and 250 µl of Ringer's solution containing the appropriate amount of vector was injected into the muscle belly 4 mm beneath the aponeurosis using 31-gauge syringes (Becton–Dickinson, Franklin Lakes, NJ). The skin was closed with 4–0 Maxon, and all dogs were monitored daily for recovery. Muscle biopsy samples were obtained 2, 4 and 12 weeks after viral injection, embedded in optimal cutting temperature medium (Tissue-Tek, Hatfield, PA), and frozen in liquid nitrogen-cooled isopentane, and stored at −80 °C until use. At necropsy, the muscles were dissected into 1 cm3 pieces, embedded in OCT medium, and subjected for further cutting into 10 µm sections for analysis.
Histological analysis and immunofluorescence staining. Histological analysis and immunofluorescence staining were performed as described.23,24 Briefly, 6 µm cryostat sections were cut for hematoxylin and eosin-phloxine staining for basic histological analysis. For immunofluorescence, rabbit anti-mouse dystrophin (N-terminus) polyclonal antibody, an affinity purified rabbit polyclonal antibody raised against the N-terminal 246 amino acids of mouse dystrophin;47 mouse anti-human dystrophin (C-terminus) monoclonal antibody (VP-D505, Vector laboratories, Burlingame, CA); mouse anti-human β-sarcoglycan (Novacastra, Newcastle upon Tyne, UK) and rabbit anti-human alpha-dystrobrevin-2 (provided by Dr Froehner, University of Washington, Seattle, WA); and mouse anti-canine CD8 monoclonal antibody (JD3 1.2 mg/ml, FHCRC Biologics Production Facility, Seattle, WA). Sections were counterstained with 4′, 6-diamidino-2-phenylindole (DAPI, Sigma, St Louis, MO), then rinsed in phosphate-buffered saline, and mounted in Vectashield (Vector Laboratories). Staining was examined and photographed using a Nikon Eclipse 800 fluorescent microscope.
Electron microscopy and fluorescence microscopy. Electron microscopy was performed as previously described.48 Briefly, small (~2 mm3) regions of the wild-type, cxmd and treated cxmd Achilles myotendinous junction were dissected and fixed in half strength Karnovskys fixative for at least 24 hours at 4 °C. After fixation, muscles were washed in 0.1 mol/l cacodylate buffer, post-fixed in 1% osmium tetroxide/cacodylate buffer for 2–3 hours, washed in 0.1 mol/l cacodylate buffer, dehydrated through ethanol into Epon, embedded, and polymerized at 60 °C overnight. Thick 1 m sections were cut and stained with toluidine blue. Ultra thin sections were cut between 70 and 100 nm and stained with saturated aqueous uranyl acetate and Reynolds lead citrate and viewed with a JEOL 1010 Transmission Electron Microscope (JEOL USA, Peabody, MA). Images were photographed with a wide-angle 1024 × 1024 Gatan 792 Multiscan 600W CCD camera (Gatan, Pleasanton, CA).
Cell culture. Human embryonic kidney 293 cells (ATCC CRL 1573) and HTX cells, an approximately diploid subclone of human HT-1080 fibrosarcoma cells (ATCC CCL 121), were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum.
Western blot. Canine dystrophin and µdys proteins were visualized by western blot analysis using standard procedures. Thirty micrograms each of dog muscle tissue lysates and 5 µg each of 293 cell lysates were loaded in designated lanes. Mouse monoclonal manex1011b (Developmental Studies Hybridoma Bank) was used as the primary antibody, and goat anti-mouse conjugated with horseradish peroxidase (Sigma) was used as the secondary antibody. A rabbit polyclonal antibody to actin (Sigma) was used as a control for the amount of protein loaded in lanes. A goat anti-rabbit conjugated to horseradish peroxidase was used as the secondary antibody to detect the rabbit antibody. Western blots were developed using ECL Plus Western Blotting Detection System (GE Healthcare, Pittsburgh, PA).
Enzyme-linked immunosorbent assay to detect serum antibodies to c-µdys. A standard sandwich enzyme-linked immunosorbent assay similar to a previously described protocol33 was employed to detect serum antibodies to c-µdys. Briefly, a rabbit polyclonal anti-dystrophin antibody was used47 to coat wells of 96-well plates. Then cell lysate from 293 cells or 293 cells transfected with the plasmid pACMV-c-µdys (200 µg protein per ml) was added to the wells and unbound proteins were removed one day later by washing. Next, dog serum samples diluted 1:20 were added and binding was done at 4 °C overnight. Afterwards, rabbit anti-canine IgG conjugated to peroxidase (Sigma) diluted 1:500 or 1:1,000 was used to detect bound canine anti-c-µdys antibodies. Lastly, a peroxidase substrate was added and absorbance was read at 405 nm after product development (30 minutes to 1 hour).
Neutralization assay. Detection of neutralizing antibodies against AAV6 vectors was done as described.49 Briefly, 108 vg aliquots of an AAV6 vector that encodes human placental alkaline phosphatase were incubated with dog serum diluted to final concentrations of from 1:100 to 1:12,800 in twofold intervals, or with no serum as a negative control, for 1 hour at 37 °C in a total volume of 400 µl Dulbecco's modified Eagle medium. Next, duplicate 80, 10 and 2 µl samples of the mixtures were added to HTX cells seeded the day before at 2 × 104 cells per well in 12-well plates. The HTX cells were grown for 2–3 days, were fixed and stained for alkaline phosphatase expression, and alkaline phosphatase-positive foci were counted to determine the transduction rate. The reciprocal of the highest dilution of serum that inhibited AAV vector transduction by ≥50% compared to vector incubated without serum was defined as the neutralizing titer.
Acknowledgments
We thank E. Zellmer, and Patrice Stroup for technical assistance, A. Joslyn, Jennifer Duncan, DVM, M. Spector, DVM and their team for their care of the dogs. We further thank S. Carbonneau, H. Crawford, B. Larson, K. Carbonneau, J. Vermeulen, and D. Gayle for administrative assistance and manuscript preparation. The authors are grateful for research funding from the National Institutes of Health, Bethesda, MD grants U54HD047175 (J.S.C. and S.J.T.); R01AR056949 (S.J.T.); and R37-AR40864 (J.S.C.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health nor its subsidiary Institutes and Centers. In addition, this work was supported by Development Grant (to Z.W.) from the Muscular Dystrophy Association (MDA 114979). The authors declare no conflicts of interest.
REFERENCES
- Muntoni F, Torelli S., and, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- Ervasti JM.2006Structure and function of the dystrophin-glycoprotein complexIn Winder, SJ (ed). Molecular Mechanisms of Muscular DystrophiesLandes Bioscience: Georgetown, TX, 1–13.
- Ahn AH., and, Kunkel LM. The structural and functional diversity of dystrophin. Nat Genet. 1993;3:283–291. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- Wang Z, Chamberlain JS, Tapscott SJ., and, Storb R. Gene therapy in large animal models of muscular dystrophy. ILAR J. 2009;50:187–198. doi: 10.1093/ilar.50.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG.et al. (2004Systemic delivery of genes to striated muscles using adeno-associated viral vectors Nat Med 10828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Rodino-Klapac LR, Rosales-Quintero X, Kota J, Coley BD, Galloway G.et al. (2009Limb-girdle muscular dystrophy type 2D gene therapy restores alpha-sarcoglycan and associated proteins Ann Neurol 66290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S.et al. (2010Dystrophin immunity in Duchenne's muscular dystrophy N Engl J Med 3631429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Tapscott SJ, Chamberlain JS., and, Storb R. Immunity and AAV-Mediated Gene Therapy for Muscular Dystrophies in Large Animal Models and Human Trials. Front Microbiol. 2011;2:201. doi: 10.3389/fmicb.2011.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ.et al. (2006Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune responseerratum appears in Nat Med. 2006 May;12(5):592]. Nat Med 12342–347. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE.et al. (2007CD8(+) T-cell responses to adeno-associated virus capsid in humans Nat Med 13419–422. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, Meulenberg JJ, Hui DJ, Basner-Tschakarjan E, Hasbrouck NC, Edmonson SA.et al. (2009AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells Blood 1142077–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Rodino-Klapac LR, Rosales XQ, Coley BD, Galloway G, Lewis S.et al. (2010Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D Ann Neurol 68629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EP, Bronson A, Levin AA, Takeda S, Yokota T, Baudy AR.et al. (2011Restoring dystrophin expression in duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through Am J Pathol 17912–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus A, den Dunnen JT., and, van Ommen GJ. New insights in gene-derived therapy: the example of Duchenne muscular dystrophy. Ann N Y Acad Sci. 2010;1214:199–212. doi: 10.1111/j.1749-6632.2010.05836.x. [DOI] [PubMed] [Google Scholar]
- Vilquin JT, Catelain C., and, Vauchez K. Cell therapy for muscular dystrophies: advances and challenges. Curr Opin Organ Transplant. 2011;16:640–649. doi: 10.1097/MOT.0b013e32834cfb70. [DOI] [PubMed] [Google Scholar]
- Negroni E, Vallese D, Vilquin JT, Butler-Browne G, Mouly V., and, Trollet C. Current advances in cell therapy strategies for muscular dystrophies. Expert Opin Biol Ther. 2011;11:157–176. doi: 10.1517/14712598.2011.542748. [DOI] [PubMed] [Google Scholar]
- Palmieri B, Tremblay JP., and, Daniele L. Past, present and future of myoblast transplantation in the treatment of Duchenne muscular dystrophy. Pediatr Transplant. 2010;14:813–819. doi: 10.1111/j.1399-3046.2010.01377.x. [DOI] [PubMed] [Google Scholar]
- Wang Z, Allen JM, Riddell SR, Gregorevic P, Storb R, Tapscott SJ.et al. (2007Immunity to adeno-associated virus-mediated gene transfer in a random-bred canine model of Duchenne muscular dystrophy Hum Gene Ther 1818–26. [DOI] [PubMed] [Google Scholar]
- Yue Y, Ghosh A, Long C, Bostick B, Smith BF, Kornegay JN.et al. (2008A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs Mol Ther 161944–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa K, Yoshimura M, Urasawa N, Ohshima S, Howell JM, Nakamura A.et al. (2007Injection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene products Gene Ther 141249–1260. [DOI] [PubMed] [Google Scholar]
- Ohshima S, Shin JH, Yuasa K, Nishiyama A, Kira J, Okada T.et al. (2009Transduction efficiency and immune response associated with the administration of AAV8 vector into dog skeletal muscle Mol Ther 1773–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic P, Schultz BR, Allen JM, Halldorson JB, Blankinship MJ, Meznarich NA.et al. (2009Evaluation of vascular delivery methodologies to enhance rAAV6-mediated gene transfer to canine striated musculature Mol Ther 171427–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Storb R, Lee D, Kushmerick MJ, Chu B, Berger C.et al. (2010Immune responses to AAV in canine muscle monitored by cellular assays and noninvasive imaging Mol Ther 18617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kuhr CS, Allen JM, Blankinship M, Gregorevic P, Chamberlain JS.et al. (2007Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression Mol Ther 151160–1166. [DOI] [PubMed] [Google Scholar]
- Lapidos KA, Kakkar R., and, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res. 2004;94:1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- Ervasti JM., and, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Tidball JG. Force transmission across muscle cell membranes. J Biomech. 1991;24 (Suppl. 1):43–52. doi: 10.1016/0021-9290(91)90376-x. [DOI] [PubMed] [Google Scholar]
- Tidball JG., and, Law DJ. Dystrophin is required for normal thin filament-membrane associations at myotendinous junctions. Am J Pathol. 1991;138:17–21. [PMC free article] [PubMed] [Google Scholar]
- Banks GB, Judge LM, Allen JM., and, Chamberlain JS. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet. 2010;6:e1000958. doi: 10.1371/journal.pgen.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herson S, Hentati F, Rigolet A, Romero NB, Behin A, Leturcq F.et al. (2011A phase I dose-escalating study of AAV1–γ-sarcoglycan gene therapy for limb girdle muscular dystrophy type 2C Mol Ther 19 (Suppl. 1)S20–S21. [Google Scholar]
- Bowles DE, McPhee SW, Li C, Gray SJ, Samulski JJ, Camp AS.et al. (2012Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector Mol Ther 20443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornegay JN, Li J, Bogan JR, Bogan DJ, Chen C, Zheng H.et al. (2010Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs Mol Ther 181501–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert CL, Madtes DK, Vaughan AE, Wang Z, Storb R, Tapscott SJ.et al. (2010Expression of human alpha1-antitrypsin in mice and dogs following AAV6 vector-mediated gene transfer to the lungs Mol Ther 181165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo T, Okada T, Athanasopoulos T, Foster H, Takeda S., and, Dickson G. Long-term functional adeno-associated virus-microdystrophin expression in the dystrophic CXMDj dog. J Gene Med. 2011;13:497–506. doi: 10.1002/jgm.1602. [DOI] [PubMed] [Google Scholar]
- Shin JH, Yue Y, Smith B., and, Duan D. Humoral immunity to AAV-6, 8, and 9 in normal and dystrophic dogs. Hum Gene Ther. 2012;23:287–289. doi: 10.1089/hum.2011.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapti K, Louis-Jeune V, Kohlbrenner E, Ishikawa K, Ladage D, Zolotukhin S.et al. (2012Neutralizing antibodies against AAV serotypes 1, 2, 6, and 9 in sera of commonly used animal models Mol Ther 2073–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett AL, Garikipati D, Wang Z, Tapscott S., and, Chamberlain JS. Immune Responses to rAAV6: The Influence of Canine Parvovirus Vaccination and Neonatal Administration of Viral Vector. Front Microbiol. 2011;2:220. doi: 10.3389/fmicb.2011.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storb R, Yu C, Wagner JL, Deeg HJ, Nash RA, Kiem HP.et al. (1997Stable mixed hematopoietic chimerism in DLA-identical littermate dogs given sublethal total body irradiation before and pharmacological immunosuppression after marrow transplantation Blood 893048–3054. [PubMed] [Google Scholar]
- Yu C, Seidel K, Nash RA, Deeg HJ, Sandmaier BM, Barsoukov A.et al. (1998Synergism between mycophenolate mofetil and cyclosporine in preventing graft-versus-host disease among lethally irradiated dogs given DLA-nonidentical unrelated marrow grafts Blood 912581–2587. [PubMed] [Google Scholar]
- Storb R, Kolb HJ, Graham TC, Kolb H, Weiden PL., and, Thomas ED. Treatment of established graft-versus-host disease in dogs by antithymocyte serum or prednisone. Blood. 1973;42:601–609. [PubMed] [Google Scholar]
- Storb R, Floersheim GL, Weiden PL, Graham TC, Kolb HJ, Lerner KG.et al. (1974Effect of prior blood transfusions on marrow grafts: abrogation of sensitization by procarbazine and antithymocyte serum J Immunol 1121508–1516. [PubMed] [Google Scholar]
- Storb R, Gluckman E, Thomas ED, Buckner CD, Clift RA, Fefer A.et al. (1974Treatment of established human graft-versus-host disease by antithymocyte globulin Blood 4456–75. [PubMed] [Google Scholar]
- Storb R, Etzioni R, Anasetti C, Appelbaum FR, Buckner CD, Bensinger W.et al. (1994Cyclophosphamide combined with antithymocyte globulin in preparation for allogeneic marrow transplants in patients with aplastic anemia Blood 84941–949. [PubMed] [Google Scholar]
- Kahl C, Leisenring W, Deeg HJ, Chauncey TR, Flowers ME, Martin PJ.et al. (2005Cyclophosphamide and antithymocyte globulin as a conditioning regimen for allogeneic marrow transplantation in patients with aplastic anaemia: a long-term follow-up Br J Haematol 130747–751. [DOI] [PubMed] [Google Scholar]
- Dell'Agnola C, Wang Z, Storb R, Tapscott SJ, Kuhr CS, Hauschka SD.et al. (2004Hematopoietic stem cell transplantation does not restore dystrophin expression in Duchenne muscular dystrophy dogs Blood 1044311–4318. [DOI] [PubMed] [Google Scholar]
- Blankinship MJ, Gregorevic P, Allen JM, Harper SQ, Harper H, Halbert CL.et al. (2004Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6 Mol Ther 10671–678. [DOI] [PubMed] [Google Scholar]
- Rafael JA, Cox GA, Corrado K, Jung D, Campbell KP., and, Chamberlain JS. Forced expression of dystrophin deletion constructs reveals structure-function correlations. J Cell Biol. 1996;134:93–102. doi: 10.1083/jcb.134.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks GB, Combs AC, Chamberlain JR., and, Chamberlain JS. Molecular and cellular adaptations to chronic myotendinous strain injury in mdx mice expressing a truncated dystrophin. Hum Mol Genet. 2008;17:3975–3986. doi: 10.1093/hmg/ddn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert CL, Miller AD, McNamara S, Emerson J, Gibson RL, Ramsey B.et al. (2006Prevalence of neutralizing antibodies against adeno-associated virus (AAV) types 2, 5, and 6 in cystic fibrosis and normal populations: Implications for gene therapy using AAV vectors Hum Gene Ther 17440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]