

Abstract
To meet the challenge of antibiotic resistance worldwide, a new generation of antimicrobials must be developed.1 This communication demonstrates ab initio design of potent peptides against methicillin-resistant Staphylococcus aureus (MRSA). Our idea is that the peptide is very likely to be active when most probable parameters are utilized in each step of the design. We derived the most probable parameters (e.g. amino acid composition, peptide hydrophobic content, and net charge) from the antimicrobial peptide database2 by developing a database filtering technology (DFT). Different from classic cationic antimicrobial peptides usually with high cationicity, DFTamP1, the first anti-MRSA peptide designed using this technology, is a short peptide with high hydrophobicity but low cationicity. Such a molecular design made the peptide highly potent. Indeed, the peptide caused bacterial surface damage and killed community-associated MRSA USA300 in 60 minutes. Structural determination of DFTamP1 by NMR spectroscopy revealed a broad hydrophobic surface, providing a basis for its potency against MRSA known to deploy positively charged moieties on the surface as a mechanism for resistance. A combination of our ab initio design with database screening3 led to yet another peptide with enhanced potency. Because of simple composition, short length, stability to proteases, and membrane targeting, the designed peptides are attractive leads for developing novel anti-MRSA therapeutics. Our database-derived design concept can be applied to the design of peptide mimicries to combat MRSA as well.
Natural antimicrobial peptides (AMPs) are essential host defense molecules discovered in bacteria, plants, and animals.4 These fascinating polypeptides can adopt a variety of three-dimensional structures and constitute excellent templates for engineering a new generation of antimicrobials. Numerous methods have been developed for peptide design.1 The first method is natural template optimization via truncation or substitution.5 The second method is sequence shuffling, i.e. changing the positions of amino acids in the sequence. This spans from sequence reversal to combinatorial library, where all the residues may be optimized.6 The third method is motif hybridization. Examples include early cecropin-melittin hybrids to recent “Grammar”-generated peptides.6b,7 Finally, novel AMPs can also be de novo or rationally designed from a few amino acids (e.g. KL peptides).8
This communication describes a novel database-derived technology, which differs from all the above methods in several aspects. First, the antimicrobial peptide database (APD) enabled us to design potent antimicrobials from the beginning. Because no assumption was taken in our design, we referred to this innovative approach as ab initio design to distinguish it from other de novo approaches.8 Second, a clear advantage of this ab initio approach is that the designed peptide has a good chance of being antimicrobial because it utilized most probable parameters derived from a set of natural AMPs with the desired activity. Because only a few peptides were synthesized, our method is cost effective compared to sequencing shuffling or library screening. Third, we were able to derive the most critical parameters from our design. The combination of the two most critical parameters led to a new design concept that guides the synthesis of other types of potent anti-MRSA compounds. Our design is possible due to our continued effort in developing and updating the comprehensive APD in the past several years.2 In our database, the peptide information is so well registered that it allows users to effectively retrieve peptides with defined properties. Furthermore, peptides can be sorted according to key parameters such as length, charge and hydrophobicity. This feature makes it possible to locate the maximum in each case (Figure 1, filled columns). In our opinion, these maxima represent the most probable parameters that determine the properties of natural AMPs and can be merged for designing novel peptides.
Figure 1.
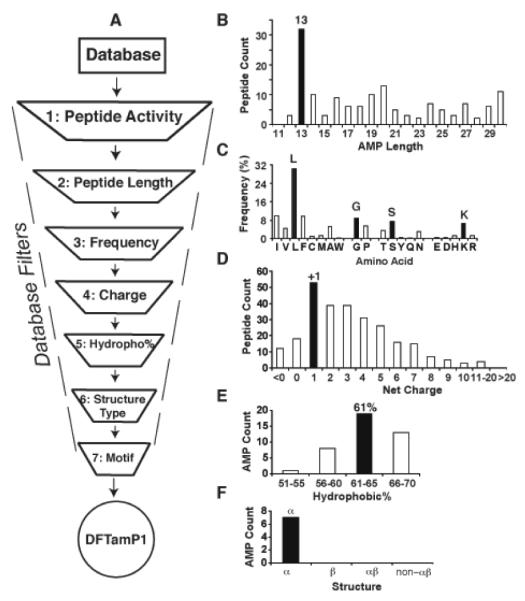
The database filtering technology (A) and the determination of peptide length (B), amino acids (C), charge (D), hydrophobic content (E), and structure type (F). For additional description of each filter, see the text. The designed peptide is called DFTamP1.
Figure 1A depicts a pipeline of database filters, each defining one peptide parameter. For short, we call this pipeline database filtering technology (DFT). In the following, we design an anti-MRSA peptide to illustrate this technology. Because S. aureus is Gram-positive (G+) bacterium, we selected all the 268 AMPs with anti-G+ activity from the APD at the time of our design (filter 1). This set of AMPs was utilized as models to derive most probable parameters for peptide design. To determine the peptide length, the number of peptides in a defined length group (step size 5) was plotted. The anti-G+ peptides with 11-15 residues were found to be dominant (not shown). A further analysis revealed that the majority of these peptides had a peptide length of 13 (Figure 1B), which was thus determined as the most probable peptide length for our design (filter 2). A third filter was used to determine the types of amino acids. To simplify our design, we selected four most frequently occurring amino acids: leucines (L), glycines (G), serines (S) and lysines (K), one from each of the four amino acid groups (Figure 1C). The general importance of G, S, and K can be appreciated from their high frequencies in the aver-aged amino acid profiles of 44 bacterial lantibiotics, 131 plant cyclotides, and 75 amphibian temporins (see Figure S1 in the Supporting Information).
Likewise, a fourth filter was introduced to determine the number of charged residues. A plot of peptide entries as a function of net charge showed that the largest anti-G+ group had a net charge of +1 (Figure 1D), leading to the choice of one lysine. In addition to positive charge, hydrophobicity is another essential element in natural AMPs.1 Therefore, we subsequently determined the hydrophobic content for the peptide. In the APD, the hydrophobic content is represented as a ratio between the total hydrophobic residues and the total amino acids in a peptide.1 Figure 1E showed that most peptides possessed hydrophobic contents in the 61-65% range, which corresponded to eight leucines for a 13-residue peptide. With one K and eight L amino acids determined, there were only four additional amino acids (G or S) to be assigned. We assigned two G and two S amino acids owing to their similar frequencies in Figure 1C. Taken together, this 13-residue peptide designed here comprised 1K, 2G, 2S, and 8L.
There are numerous ways of combining this set of residues into a unique peptide sequence. To reduce the possibilities, we utilized additional filters. Figure 1F presented structure distribution of the anti-G+ peptides with 11-15 residues. All the known structures were helical. Therefore, the sixth filter suggested a helical conformation for the designed peptide. In general, there are two models for building an amphipathic helix.1 In the first model, all hydrophobic residues are clustered at one end and all hydrophilic residues at the opposite side. In the second model, hydrophobic residues are interspersed with hydrophilic residues at every 2-3 residues. We introduced the seventh motif filter in the APD2,3 to help to decide on the helix model. Motifs are a cluster of amino acid residues that occur frequently in natural AMPs. Because there was no peptide in this database that contained four or more leucines in a row, it is unlikely that the eight leucines could all appear at one end of the peptide sequence. This allowed us to reject the first model and to design the peptide according to the second model.
As the combinations LL, LLL, and LLLL occurred in 449, 11, and 0 AMP sequences in the APD, respectively, it is best to split the eight leucines into four pairs (i.e. LL-LL-LL-LL). To place G, S, and K into this leucine pattern, we compared three residue motifs and found that GLL, KLL, and SLL are all well represented in the AMP database (GLL, 147; KLL, 69; and SLL, 58). We rejected alternatives such as LLG, LLK, and LLS to avoid the generation of a string of LLLL, which does not occur in the APD. If three 3-residue motifs (GLL, KLL, and SLL) are chosen, a 4-residue motif is required to reach the 13-residue limit. We compared the frequencies of possible 4-residue motifs and GKLL was found to have the highest occurrence in the database. This led to four highly occurring motifs GLL, SLL, SLL, and GKLL that exactly match the amino acid composition of the designed peptide. Logically, two peptide sequences are possible depending on whether the four motifs are connected in ascending (increase in frequency) or descending (decrease in frequency) order. Sequence 1 was arranged in the ascending order GKLL(41)-SLL(58)-SLL(58)-GLL(147), whereas sequence 2 was assembled in the descending order GLL(147)-SLL(58)-SLL(58)-GKLL(41) (frequency in parenthesis). We rejected random combinations because they are in conflict with the spirit of our “rational design”. We also investigated additional motifs that define the optimal connections between the above four motifs. There are three possible joints between the four motifs in each sequence. Two joints are identical (LL-S or L-SL) and only the C-terminal joints differ (LL-G or L-GL for sequence 1 and LL-G or L-GK for sequence 2). Because the linking motif LGK appeared more frequently in the database (89 counts) than LGL (31 counts), sequence 2 that contains LGK was thus established as the final sequence. Therefore, we obtained a novel peptide sequence GLLSLLSLLGKLL by applying the most probable AMP parameters to each step of our design. Since this is the first anti-MRSA peptide designed using DFT, it is hereinafter referred to as DFTamP1 (Figure 1A). This peptide is designed to target Gram-positive bacteria by adopting a helical conformation. In the following, we provide proof for both structure and activity of DFTamP1.
The in vitro activity of DFTamP1 was assayed using the standard microdilution method.9 The peptide was found to be active against S. aureus USA300 with a minimal inhibitory concentration (MIC) of 3.1 μM. However, it did not inhibit the growth of B. subtilis, E. coli or P. aeruginosa until 120 μM (Table 1). In the presence of DFTamP1, the continuous and round surface of S. aureus USA300 (Figure 2A) was damaged and cell leakage (blebs) was detected by transmission electron microscopy (Figure 2B, arrow). This cell leakage agreed with the results of flow cytometry (Figure 2C), because surface damage by the peptide enabled the entrance of the DNA-binding dye into bacteria, leading to a steep fluorescence buildup in 30 min. Rapid killing of MRSA USA300 is also supported by killing kinetics experiments (Figure S2). These results indicate that the peptide acted on bacterial membranes.10
Table 1.
Bacterial minimal inhibitory concentration (MIC), selectivity index and hydrophobicity of database-derived anti-MRSA peptides
| Peptide | Sequencea | MIC (μM) b | CSc | tRP (min)d |
|||
|---|---|---|---|---|---|---|---|
| SA | BS | EC | PA | ||||
| DFTamP1 | GLLSLLSL LGKLL |
3.12 | >120 | >120 | >120 | 3.3 | 19.79 |
| DFTamP1-p | GLLPLLSL LGKLL |
1.56 | 3.12 | 25 | >100 | 1.3 | 18.72 |
| DFTamP1-pi | GIIPIISIIG KII |
>34 | >30 | >34 | >30 | NAe | 17.57 |
| DFTamP1-pv | GVVPVVS VVGKVV |
>100 | >100 | >100 | >100 | NA | 12.36 |
| Temporin- PTa |
FFGSVLK LIPKIL |
3.12 | 6.25 | 25 | >100 | 40 | 14.50 |
| Temporin- PTa (D-form) |
FFGSVLK LIPKIL |
3.12 | 6.25 | 25 | >100 | NA | 14.48 |
| Temporin- PTa6L |
FFGSLLKL LPKLL |
0.78- 1.56 |
3.12 | 12.5 | 50- 100 |
32 | 15.39 |
| Temporin- PTa8L |
LLGSLLK LLPKLL |
0.78- 1.56 |
3.12 | 12.5 | >100 | 32 | 15.43 |
| DASamP1f | FFGKVLK LIRKIF |
3.12 | >100 | >100 | >100 | 8 | 14.92 |
The C-termini of the peptides are amidated.
The bacteria are SA, S. aureus USA300; BS, B. subtilis; EC, E. coli K12; PA, P. aeruginosa.
Cell selectivity index of the peptide.
The HPLC retention time of the peptide (see Methods).
Not available.
Database screening obtained antimicrobial peptide 1, which is a mutant of temporin-PTa.14
Figure 2.
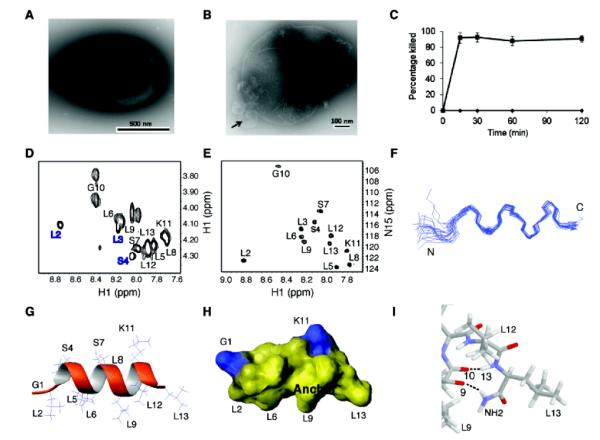
Structure and activity of DFTamP1. Shown are transmission electron microscopy images of MRSA USA300 before (A) and after peptide treatment (B), rapid killing of the 108 colony-forming units of S. aureus USA300 by 2× MIC followed by flow cytometry (C), and NMR studies (D and E) that enabled us to view the active conformation of the peptide (F-I). DFTamP1 has the highest content of leucines (61.5%) compared to natural temporins (Table S3 in the Supporting Information). Despite this difficulty, the cross peaks of these leucines are resolved in the natural abundance 2D 1H and 15N correlated NMR spectrum of DFTamP1 in complex with 40-fold deuterated sodium dodecyl sulfate at 25°C and pH 5.4 (E). Displayed structural views of membrane-bound DFTamP1 are superimposed 20 backbone structures (F), a ribbon diagram indicating a clear segregation of hydrophilic (residues S4, S7, and K11) and hydrophobic leucine side chains (G), potential surface (H), and local structure at the C-terminus indicating hydrogen bonding between the C-terminal amide NH2 and the carbonyl atom of residue L9 of the peptide (I).
To provide insight into the potency of DFTamP1, we determined its 3D structure in membrane-mimetic micelles.11 Homonuclear and heteronuclear 2D NMR spectra are shown in Figure 2, panels D and E, respectively. The natural abundance 1H and 15N correlated spectrum was very helpful in this case because of the highest content of leucines (61.5%) in DFTamP1 of the known natural AMPs. The data enabled not only the substantiation of spectral assignments but also structural refinement.9 DFTamP1 was found to adopt a helical structure (Figure 2F), where hydrophilic and hydrophobic side chains were clustered on opposite faces (Figure 2G,H). The C-terminal amide protons formed hydrogen bonds with the carbonyl oxygen of residue L9 (Figure 2I). Statistics of the structure is summarized in Supporting Information Table S2. The fraying N-terminus of DFTamP1 (Figure 2F) is consistent with sharp spectral lines for residues L2 to S4 (Figure 2D). These dynamic data implied that residues L5-L13 of the peptide were more important for membrane binding. We propose that the broad hydrophobic surface of DFTamP1 (Fig. 2H) is essential for its potency against MRSA that deploys positively charged moieties on cellular surface as a mechanism to reduce the effect of classical cationic AMPs usually rich in cationic residues.12 According to our membrane perturbation potential model,9 the broad surface of DFTamP1 allowed the peptide to penetrate deeper into bacterial membranes to exert its damaging effect (Fig. 2B).
DFTamP1 was designed based on the most probable parameters from a set of peptides that are active against Gram-positive bacteria. It is likely that this peptide shares some features with candidates in the APD. Database analysis revealed that the amino acid sequence of DFTamP1 resembles amphibian temporins8d, where position 3 is usually a proline residue (Table S3). When a similar amino acid S4 of DFTamP1 was changed to a proline (i.e. DFTamP1-p), the peptide gained activity against B. subtilis and E. coli, underscoring the important role of this proline in determining the activity spectrum of the peptide. To confirm the importance of the leucine surface of DFTamP1 (Figure 2H) for bacterial targeting, we substituted the eight leucines with isoleucines or valines (Table 1). The isoleucine analog (DFTamP1-pi) was poorly soluble and showed no activity at the maximal concentration achieved. The valine analog (DFTamP1-pv) was also inactive. Our results from real time bacterial growth and killing confirmed that the valine analog was unable to kill MRSA even at a concentration 32-fold of the MIC of DFTamP1, while DFTamP1 showed killing at its MIC and bacteria were lysed and inhibited at 8-fold the MIC (Figure S3). Thus, the leucine surface of DFTamP1 is critical for membrane targeting (Figure 2H) and MRSA killing. The reduced activity of these peptide mutants could be attributed to a decrease in hydrophobicity based on HPLC retention time measurements (Table 1)5c,8c as well as the higher helix-forming tendency of leucines than either isoleucines or valines.13
Interestingly, our recent work found that DASamP1, a temporin-PTa analog with increased cationic residues (Table 1), showed anti-MRSA activity both in vitro and in vivo.3b The antimicrobial activity of temporin-PTa was not reported originally due to a limited amount of material.14 Here we found that synthetic temporin-PTa displayed an antibacterial spectrum comparable to DFTamP1-p. Furthermore, a D-form of temporin-PTa, consisting of all D-amino acids, was equally potent against MRSA, E. coli, and B. subtilis, substantiating membrane targeting.6b To further test the importance of leucines in DFTamP1 (Figure 2H), we also incorporated additional leucines to temporin-PTa to obtain the 6L and 8L analogs (Table 1). These two leucine-rich analogs displayed improved antibacterial activity and retained a cell selectivity index of 32. In contrast, increasing positively charged residues did not improve peptide potency but reduced cell selectivity of DASamP1 (Table 1).3b Hence, mimicking the leucine anchor of DFTamP1 offers a useful strategy to enhance anti-MRSA potency, whereas increasing the number of cationic residues in DASamP1 was not helpful (Table 1). These results indicate that high hydrophobicity and low cationicity are two critical parameters for designing anti-MRSA peptides.
In conclusion, it is feasible to retrieve all the parameters from our database to design potent anti-MRSA peptides from the beginning (i.e. ab initio design). In the future, the DFT may be further optimized and incorporated into the database for peptide design. In addition, we also succeeded in identifying potent anti-MRSA peptides based on database screening. By combining these two database approaches, we succeeded in obtaining temporin-PTa8L with higher activity and good cell selectivity (Table 1). Our practices not only uncovered the important parameters for peptide engineering but also led to the discovery of new anti-MRSA candidates. Such peptides possess several attractive features. First, they have short sequences and simple amino acid compositions, allowing for cost-effective chemical synthesis. Second, membrane targeting of these peptides enabled us to synthesize an all D-amino acid analog, which showed equal antibacterial activities (Table 1) but superior stability to proteases (Figure S4). Third, we already demonstrated the in vivo efficacy of DASamP1 in preventing biofilm formation in a mouse model.3b Finally and most importantly, a combination of high hydrophobicity and low cationicity leads to a useful concept for designing anti-MRSA peptides. To better appreciate this design concept, we have summarized the three scenarios in the graphics for the Table of Contents. A classic cationic AMP with 3-4 positive charges on average1,2 is ideal for targeting susceptible bacteria with a negatively charged surface (A). Such a highly charged peptide, however, could be repelled from resistant bacteria due to the bacterial deployment of positively charged groups on the surface (B). Our ab initio designed peptides are effective against resistant bacteria, especially MRSA, because cationic amino acids in these peptides have been minimized (C). In particular, DFTamP1 and temporin-PTa8L are promising templates for our next round of peptide engineering with a goal of optimizing cell selectivity by fine-tuning the hydrophobic surface (Figure 2H). Our database-derived molecular design concept can guide the construction of other anti-MRSA compounds, including peptide mimicries.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIAID/NIH grant R56AI81975 to GW. We thank Tom Bargar for conducting Transmission Electron Microscopy, Vinai Chittezham Thomas for assistance with Flow Cytometry, and Edward Ezell for maintaining the 600-MHz NMR spectrometer.
ABBREVIATIONS
- AMPs
antimicrobial peptides
- DASamP1
database screening obtained anti-MRSA peptide 1
- DFT
database filtering technology
- DFTamP1
DFT-designed anti-MRSA peptide 1
- MIC
minimal inhibitory concentration
- MRSA
methicillin-resistant Staphylococcus aureus
- NMR
nuclear magnetic resonance.
Footnotes
ASSOCIATED CONTENT Further experimental details are provided as supporting information, together with supporting figures and tables.
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Wang G. Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies. CABI; England: 2010. [Google Scholar]
- (2)(a).Wang Z, Wang G. Nucleic Acid Res. 2004;32:D590–D592. doi: 10.1093/nar/gkh025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang G, Li X, Wang Z. Nucleic Acids Res. 2009;37:D933–D937. doi: 10.1093/nar/gkn823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3)(a).Wang G, Watson KM, Peterkofsky A, Buckheit RW., Jr. Antimicrob. Agents Chemother. 2010;54:1343–1346. doi: 10.1128/AAC.01448-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Menousek J, Mishra B, Hanke ML, Heim CE, Kielian T, Wang G. Int. J. Antimicrob. Agents. 2012;39:402–406. doi: 10.1016/j.ijantimicag.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4)(a).Zasloff M. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]; (b) Hancock RE, Sahl HG. Nat. Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]; (c) Brogden KA. Nat. Rev. Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- (5)(a).Wang G. J. Biol. Chem. 2008;283:32637–32643. doi: 10.1074/jbc.M805533200. [DOI] [PubMed] [Google Scholar]; (b) Monincová L, Budesínský M, Slaninová J, Hovorka O, Cvacka J, Voburka Z, Fucík V, Borovicková L, Bednárová L, Straka J, Cerovský V. Amino Acids. 2010;39:763–775. doi: 10.1007/s00726-010-0519-1. [DOI] [PubMed] [Google Scholar]; (c) Li X, Li Y, Han H, Miller DW, Wang G. J. Am. Chem. Soc. 2006;128:5776–5785. doi: 10.1021/ja0584875. [DOI] [PubMed] [Google Scholar]
- (6)(a).Cherkasov A, Hilpert K, Jenssen H, Fjell CD, Waldbrook M, Mullaly SC, Volkmer R, Hancock RE. ACS Chem. Biol. 2009;4:65–74. doi: 10.1021/cb800240j. [DOI] [PubMed] [Google Scholar]; (b) Merrifield RB, Juvvadi P, Andreu D, Ubach J, Boman A, Boman HG. Proc. Natl. Acad. Sci. USA. 1995;92:3449–3453. doi: 10.1073/pnas.92.8.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li X, Li Y, Peterkofsky A, Wang G. Biochim. Biophys. Acta. 2006;1758:1203–1214. doi: 10.1016/j.bbamem.2006.03.032. [DOI] [PubMed] [Google Scholar]; (d) Monroc S, Badosa E, Besalú E, Planas M, Bardají E, Montesinos E, Feliu L. Peptides. 2006;27:2575–2584. doi: 10.1016/j.peptides.2006.05.001. [DOI] [PubMed] [Google Scholar]
- (7)(a).Haney EF, Nazmi K, Bolscher JG, Vogel HJ. Biochim. Biophys. Acta. 2012;1818:762–775. doi: 10.1016/j.bbamem.2011.11.023. [DOI] [PubMed] [Google Scholar]; (b) Loose C, Jensen K, Rigoutsos I, Stephanopoulos G. Nature. 2006;443:867–869. doi: 10.1038/nature05233. [DOI] [PubMed] [Google Scholar]
- (8)(a).Blondelle SE, Houghten RA. Biochemistry. 1992;31:12688–12694. doi: 10.1021/bi00165a020. [DOI] [PubMed] [Google Scholar]; (b) Kang SJ, Won HS, Choi WS, Lee BJ. J. Pept. Sci. 2009;15:583–588. doi: 10.1002/psc.1149. [DOI] [PubMed] [Google Scholar]; (c) Chen Y, Mant CT, Farmer SW, Hancock RE, Vasil ML, Hodges RS. J. Biol. Chem. 2005;280:12316–12329. doi: 10.1074/jbc.M413406200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mangoni ML, Shai Y. Cell. Mol. Life Sci. 2011;68:2267–2280. doi: 10.1007/s00018-011-0718-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wang G, Li Y, Li X. J. Biol. Chem. 2005;280:5803–5811. doi: 10.1074/jbc.M410116200. [DOI] [PubMed] [Google Scholar]
- (10).Wang G, Epand RF, Mishra B, Lushnikova T, Thomas VC, Bayles KW, Epand RM. Antimicrob. Agents Chemother. 2012;56:845–856. doi: 10.1128/AAC.05637-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wüthrich K. NMR of proteins and nucleic acids. Wiley; New York, NY: 1986. [Google Scholar]
- (12).Andra J, Goldmann T, Ernst CM, Peschel A, Gutsmann T. J. Biol. Chem. 2011;286:18692–18700. doi: 10.1074/jbc.M111.226886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pace CN, Scholtz JM. Biophys. J. 1998;75:422–427. doi: 10.1016/s0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Conlon JM, Kolodziejek J, Nowotny N, Leprince J, Vaudry H, Coquet L, Jouenne T, King JD. Toxicon. 2008;52:465–473. doi: 10.1016/j.toxicon.2008.06.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.