

Abstract
Activation of the Sonic Hedgehog (Shh) pathway and increased expression of Gli1 play an important role in proliferation and transformation of granule cell progenitors (GCPs) in the developing cerebellum. Medulloblastomas arising from cerebellar GCPs are frequently driven by Shh pathway-activating mutations, however molecular mechanisms of Shh pathway dysregulation and transformation of neural progenitors remain poorly defined. We report that the transcription factor and oncogene Snail1 (Sna1) is directly induced by Shh pathway activity in GCPs, murine medulloblastomas, and human medulloblastoma cells. Enforced expression of Sna1 was sufficient to induce GCPs and medulloblastoma cell proliferation in the absence of Shh/Gli1 exposure. Additionally, enforced expression of Sna1 increased transformation of medulloblastoma cells in vitro and in vivo. Analysis of potential Sna1 targets in neural cells revealed a novel Sna1 target, N-Myc, a transcription factor known to play a role in Shh-mediated GCP proliferation and medulloblastoma formation. We found that Sna1 directly induced transcription of N-Myc in human medulloblastoma cells, and that depletion of N-Myc ablated the Sna1-induced proliferation and transformation. Taken together, these results provide further insight into the mechanism of Shh-induced transformation of neural progenitor cells and suggest that induction of Sna1 may serve to amplify the oncogenic potential of Shh pathway activation through N-Myc induction.
Keywords: Snail, Shh, Medulloblastoma, N-Myc, Granule Cell Progenitors
INTRODUCTION
Sonic Hedgehog (Shh) is a secreted morphogen that plays a major role in patterning, proliferation and cell fate determination during embryonic development(1). Within the central nervous system (CNS), the concentration and duration of Shh exposure confers positional information, influences progenitor cell fate along the dorsal-ventral axis of the developing neural tube, and mediates axonal pathfinding(2). Postnatally, Shh promotes the dramatic expansion of cerebellar granule cell progenitors (GCPs) (3, 4) and maintains the neural stem cell niche in the mature nervous system(5). Binding of Shh to the inhibitory receptor, Patched1 (Ptc1), results in activation of Smoothened (Smo) and subsequent activation of Gli transcription factors(2). In addition to its role in mediating interactions among normal cells, aberrant activation of Shh signaling contributes to unrestrained proliferation and tumorigenesis in a number of tissues including the digestive tract, skin and CNS(6–11).
Medulloblastomas comprise a heterogeneous group of primitive neuroectodermal tumors thought to derive from neural progenitors within the developing cerebellum(12). Activating mutations in Shh pathway components have been detected in a subset of syndromic and sporadic medulloblastoma(12, 13). Moreover, mice harboring heterozygous inactivation of Ptc1 or activating mutation in Smo develop spontaneous medulloblastomas(7, 10, 14, 15).Gli1 is sufficient to induce neural progenitor cell proliferation in vitro and in vivo, and is highly expressed in both human and murine medulloblastomas, the latter of which fail to develop in the absence of Gli1(10, 13, 15–17). Characterization of genes promoting Shh/Gli1-mediated malignant transformation is of significant interest in understanding the biology of medulloblastoma and identifying potential targets for therapeutic intervention.
Using a combination of in vivo and in vitro systems, we identified Sna1 as a mediator of Shh-induced proliferation in untransformed and transformed cells of the CNS. Sna1 is a zinc-finger transcription factor that plays a prominent role in epithelial-mesenchymal transition (EMT), cellular transformation, and enhancing Gli1-induced epithelial transformation (18–20). A possible role for Sna1 in malignant proliferation within the postnatal CNS has not previously been explored. We found that Sna1 contributes to proliferation of transformed and untransformed primary neural cells. Specifically, we report that Sna1 is induced in GCPs and medulloblastoma cells in a Gli1-responsive manner. Enforced expression of Sna1 is sufficient to increase proliferation of both cell types through induction of its novel target, N-Myc. Our data indicate a role for Sna1 in the normal and neoplastic proliferation of neural progenitor cells and suggest additional avenues for therapeutic targeting of oncogenic Shh signaling.
MATERIALS AND METHODS
Mice
Experimental protocols were approved by the Institutional Animal Care and Use Committee of Mayo Clinic. Mice carrying a deletion in Patched1 (Ptc+/−) (7, 10) were maintained on a C57Bl6/129SvJ background. Gli1−/− (21) and ND2:SmoA1 mice (15) were maintained on a C57BL/6 background. Ptc+/− and ND2:SmoA1 mice were monitored for signs of increased intracranial pressure; when symptomatic, the animal was sacrificed and the tumor was separated from adjacent cerebellum for analysis as described (10).
Xenografts were generated by injecting 2.5×106 Daoy cells expressing Sna1 or control lentiviral vector subcutaneously into the flank of six athymic nu/nu mice per condition. After two weeks, the tumors were resected and their volume was measured and averaged.
Plasmid Constructs and Viral Production
Sequences encoding murine and human Sna1 were amplified by PCR and cloned into pAAV SIRES eGFP and pSIN-Luc-UbEm. Gli1 and Gli1ZFD expressing constructs were previously described(22). Oligonucleotides corresponding to Sna1 and N-Myc sequences were synthesized (IDT, Coralville, IA), annealed and ligated into the appropriate vector to generate shRNA constructs (Supplemental Table 1). pAAV H1 siRNA eGFP vector was used for shSna1. Daoy studies used N-Myc shRNA in the pFRT vector, which allows for hygromycin selection of positive transfection. ONS76 studies used N-Myc shRNA in pKLO.1 (Open Biosystems, Huntsville, AL), as ONS76 cells were unresponsive to hygromycin.
For virus production, pSIRES or pSIN-Luc-UbEm expression vectors were transfected into HEK293T cells (ATCC, Manassas, VA). Lentiviral supernatants were filtered and virus was concentrated via ultracentrifugation; adenovirus was concentrated from cell lysate on a deoxycholate gradient(23).
For luciferase assays human Sna1 and N-Myc promoter segments were amplified by PCR and cloned into the pGL4.17 firefly luciferase expression vector (Promega, Madison, WI) (Supplemental Table 3). Site-directed mutagenesis of putative Gli binding elements (GBEs) and E-Box sequences in the Sna1 and N-Myc promoters(24), respectively, was performed using the Quickchange II XL Kit (Stratagene, Santa Clara, CA) according to manufacturer instructions (Supplemental Table 2).
Cell Culture and Treatment
HEK293T and medulloblastoma cell lines Daoy (ATCC, Manassas, VA) and ONS76 (a generous gift from Peter Forsythe, Calgary, Ontario) were grown in DMEM containing 10% FBS, and transfected using poly(ethylenimine) (PEI, Polysciences, Warrington, PA)(25) or Lipofectamine (Invitrogen, Carlsbad, CA).
Primary cerebellar GCPs were isolated from postnatal day 5/6 (P5/6) mice and grown on poly-D-lysine-coated plates in Neurobasal medium (Invitrogen, Carlsbad, CA) supplemented with B27 and EGF(3). Cerebellar stem cells (CBSCs) were generated from cerebella from P10 mice and cultured under the same conditions. Hippocampal neural stem cells (NSCs) were derived from P5/6 WT mice and cultured as neurospheres(26). Ptc+/− stem-like tumor cells were isolated from freshly dissected medulloblastomas, dissociated, filtered, triturated and cultured in NSC culture medium supplemented with EGF and bFGF(22).
Where indicated, media was supplemented with 2µg/mL (GCPs) or 1µg/mL (Daoy and ONS76) N-Shh(26), 10µg/mL cycloheximide for 8 hours,50µg/mL MG132 for 1 hour (both from Sigma-Aldrich, St. Louis, MO), 10µg/mL cyclopamine for 24 hours (TRC, Ontario, Canada), or appropriate vehicle.
For clonogenic growth assays, Daoy cells were transfected and allowed to recover for 48 hours. Cells expressing pFRT selection vector were treated with hygromycin for 36 hours before plating. 500 cells were plated per dish in triplicate per condition. Colonies grew for 2 weeks, then were fixed and stained with 1% crystal violet.
Fluorescent Staining of GCPs
GCPs were cultured on glass slides and incubated with adenovirus for 48 hours. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton-X-100, then incubated with antibodies to Sna1 (Abcam, Cambridge, MA), Ki67 (Santa Cruz Biotechnologies, Santa Cruz, CA) or phosphorylated Histone H3Ser10 (Cell Signaling Technology, Boston, MA) and counterstained with DAPI (Sigma-Aldrich, St. Louis, MO). For quantification of labeled cells, micrographs were taken of 10 random fields per condition at 40X magnification. The number of positive cells was counted, averaged and normalized to the uninfected control.
mRNA Expression Analyses
After NSC nucleofection, cells were sorted by FACS to isolate GFP-expressing as described(22). RNA was extracted from tissue samples with TRIzol® (Invitrogen, Carlsbad, CA) and from cultured cells with Absolutely RNA® (Stratagene, Santa Clara, CA). cDNA was generated using Sprint™ RT Complete – Random Hexamer (Clontech, Mountain View, CA). Quantitative RT-PCR (qRT-PCR) was performed in triplicate using Stratagene's MX3000p with Brilliant SYBR Green (Stratagene, Santa Clara, CA). Amplification efficiencies were optimized for all gene specific primer pairs (Supplemental Table 3). PCR product expression was normalized to Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) on the same plate and calibrated to controls. qRT-PCR data are portrayed with the calibrator cDNA normalized to one copy and the experimental samples showing fold induction.
Luciferase assays were performed on 10µg protein extracted from Daoy cells transfected with pGL4.17 firefly promoter and pGL4.74 Renilla luciferase expression vectors using the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI). Relative luciferase activity is expressed as firefly luciferase activity divided by Renilla luciferase activity, normalized to the control.
Western Blot Analysis
Cell pellets and resected murine tissues were triturated in lysis buffer(26) followed by removal of cellular debris by centrifugation. Protein concentrations were determined through Bradford analysis (Bio-Rad, Hercules, CA) and 5–10µg of protein was separated by SDS-PAGE using 10% Tris–HCl poly-acrylamide gels. Antibodies used in this study include: Snail1 (Abcam, Cambridge, MA), GAPDH (Millipore, Billerica, MA), N-Myc (Santa Cruz Biotechnologies, Santa Cruz, CA), β-catenin (Millipore, Billerica, MA) and CyclinD1 (Sigma-Aldrich, St. Louis, MO). Bands were detected by application of peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence (Pierce, Rockford, IL) and visualized using EpiChemi3 Darkroom (UVP BioImaging Systems, Upland, CA).
Cell Proliferation Analysis
36 hours after transfection, Daoy and ONS76 cells were pulsed for two hours with 5µM of 5-ethynyl-2'-deoxyuridine (EdU), then fluorescently labeled using the Click-iT® EdU Alexa Fluor® 647 Flow Cytommetry Assay Kit (Invitrogen, Carlsbad, CA). Transfected cells were detected with Alexa Fluor® 488-conjugated anti-GFP (Invitrogen, Carlsbad, CA), as copper sulfate in the Click-iT® reaction quenches eGFP fluorescence. The proportion of proliferating transfected cells was determined by dividing the number of transfected cells positive for EdU by the number of transfected cells, as detected by flow cytommetry and normalized to the control.
Chromatin Immunoprecipitation Assay
24 to 36 hours after cyclopamine treatment or transfection, respectively, Daoy cell DNA and associated proteins were crosslinked with 1% formaldehyde followed by cell lysis and DNA shearing via fifteen minutes of repeated cycles of 30 seconds of sonication followed by 10 seconds of rest. Chromatin immunoprecipitation (ChIP) was performed with EZ-Magna ChIP™ G (Millipore, Billerica, MA) using 1µg antibody. Antibodies used include Gli1 (R&D Systems, Minneapolis, MN), Sna1 (Abcam, Cambridge, MA), Goat IgG (Santa Cruz Biotechnologies, Santa Cruz, CA) and Rabbit IgG (Jackson Immunoresearch). PCR primers were designed to amplify regions spanning putative E-Boxes and GBEs identified within the human Sna1 and N-Myc promoters up to two kilobases upstream of transcription start, respectively(24, 27, 28). (Supplemental Table 4). After ChIP, PCR was performed on the DNA that co-immunoprecipitated with the indicated antibody as compared to total cell lysate.
Data Analysis
Results were graphed to demonstrate mean ± SEM and normalized to controls for a single experiment containing at least three replicates. After an f test to ensure equal variance, data were compared using a t test in GraphPad Prism Software (La Jolla, CA); a p value of p<0.01 was considered significant.
RESULTS
Shh pathway activation induces Sna1 expression in neural progenitor cells in vivo and in vitro
Medulloblastoma is a heterogeneous tumor that can arise from cerebellar granule cell progenitors (GCPs) or neural stem cells (NSCs)(29, 30). The present study was devised to determine the involvement of Sna1 in Shh-mediated neural tumorigenesis through analyses of neural progenitor and medulloblastoma cells. We found that Sna1 is present in cerebellum during a period of maximal proliferation (P5/6); its mRNA level is diminished once proliferation of GCPs has ceased and the cerebellum had matured (Figure 1A, Supplemental Figure 1). Mice harboring genetic activation of the Shh pathway through haploinsufficiency of Ptc1 express 6.7-fold higher levels of Sna1 mRNA than WT littermates at P6 (Figure 1A). In adults, Sna1 mRNA can be seen at trace amounts in the cerebella of non-tumor bearing Ptc+/− mice (Figure 1A).
Figure 1. Shh pathway activation induces Sna1 expression in neural progenitor cells.
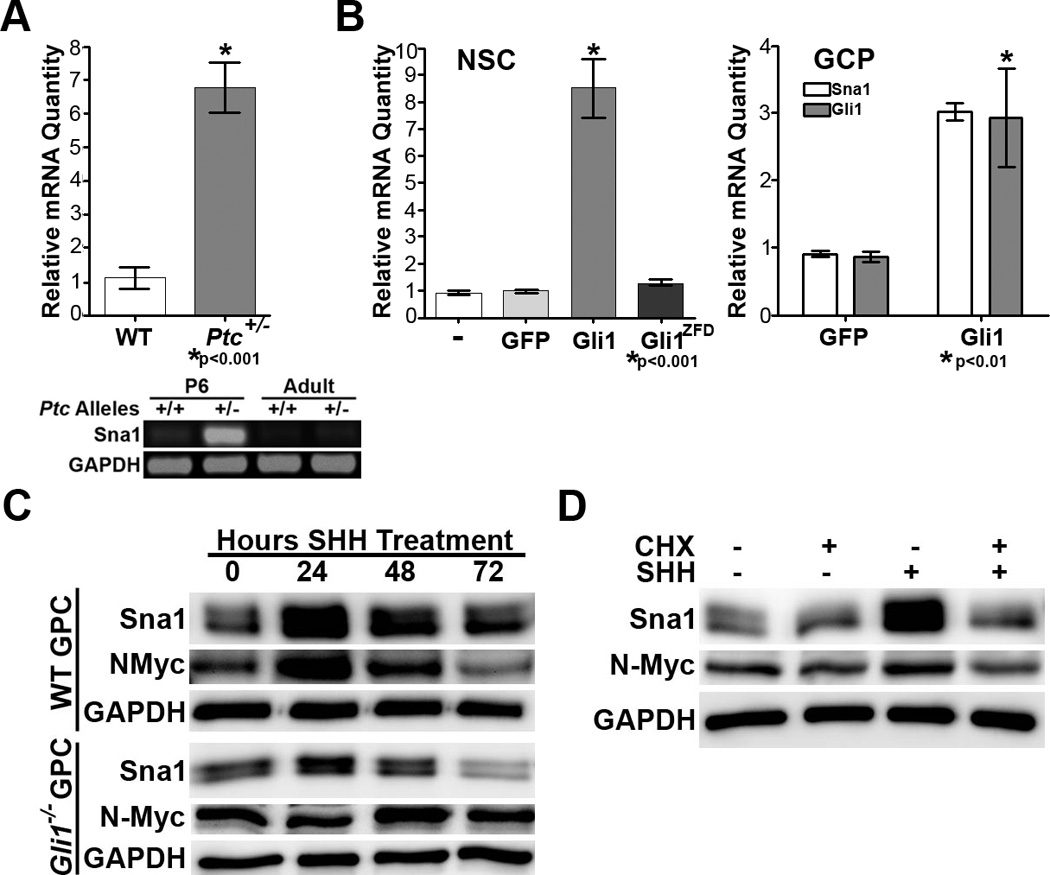
A. Top, qRT-PCR shows Sna1 mRNA is increased 6.7-fold in the cerebella of P6 Ptc+/− mice versus WT siblings. Bottom, RT-PCR shows Sna1 mRNA is increased in the cerebella of P6 and adult Ptc+/− mice versus WT siblings. B. Left, Sna1 mRNA was induced 8.51-fold in hippocampal NSCs in the presence of elevated Gli1, but not Gli1ZFD. Right, Sna1 mRNA was induced in GCPs expressing Gli1. C. Western blot analyses shows Sna1 protein was increased in both WT and Gli1 null cells exposed to Shh ligand for 24 hours. D. Western blot analysis shows that Sna1 and N-Myc protein were increased in GCPs exposed to Shh ligand alone, but not in the presence of cycloheximide (CHX).
To determine whether Sna1 mRNA is expressed in response to Shh pathway activation in neural tissue, we performed expression analyses on mRNA isolated from neural precursor cells transfected with Gli1 or control vectors (Figure 1B). We found that Sna1 mRNA was induced in multipotent hippocampal NSCs expressing Gli1 compared to NSCs expressing GFP or Gli1ZFD, which lacks the DNA binding domain and is transcriptionally silent (Figure 1B). We similarly found that Sna1 mRNA is induced in lineage-restricted GCPs expressing Gli1 (Figure 1B). Furthermore, treatment of WT GCPs with exogenous Shh ligand induced Sna1 protein, indicating that Sna1 is induced in response to ligand activation of the Shh pathway in neural progenitors (Figure 1C).
Given our result that enforced expression of Gli1 was sufficient to induce Sna1 in NSCs and GCPs, we sought to test whether Gli1 was required for Sna1 induction. We found that Sna1 was induced to a lesser extent in GCPs isolated from Gli1−/− mice than WT mice (Figure 1C). This suggests that Sna1 induction by Shh is partially dependent on the presence of Gli1. This stands in contrast to the well-established Gli1 target, N-Myc(31), which was not induced in Gli1−/− GCPs after exposure to Shh (Figure 1C). Furthermore, WT GCPs treated with the protein synthesis inhibitor cycloheximide failed to show an increase in Sna1 or N-Myc protein when exposed to Shh (Figure 1D). GCPs coincubated with Shh and proteosome inhibitor MG132 failed to show further stabilization of Sna1 than GCPs treated with Shh alone (Supplemental Figure 2). Together these data indicate that Sna1 is present in GCPs during the period of maximal proliferative expansion in vitro and in vivo, that and that Sna1 is induced in GCPs by genetic and biochemical Shh pathway activation in a manner dependent upon protein synthesis, but not protein stabilization.
Sna1 is increased in medulloblastoma tumors and cells in response to elevated Shh pathway activity
Since aberrant activation of the Shh pathway is associated with medulloblastoma formation, we hypothesized that Sna1 would also be induced by elevated Shh pathway activity in medulloblastoma lesions. To explore this question we used ND2:SmoA1 and Ptc+/− murine models of medulloblastoma, which harbor Shh pathway-activating mutations. ND2:SmoA1 mice carry a mutation conferring constitutive activation of Smo in GCPs and develop medulloblastoma between 3 and 4 months of age(15); Ptc+/− mice harbor one inactive allele of Ptc1, developing tumors at a lower incidence and longer latency than ND2:SmoA1 mice(7, 10). We found that tumors collected from ND2:SmoA1 mice express higher levels of Sna1 protein than the surrounding normal cerebellum (Figure 2A). Of these tumors, a subset was chosen for mRNA expression analysis (Figure 2B). Tumors collected from ND2:SmoA1 mice and TSCs generated from Ptc+/− mice with elevated Shh pathway activation, as measured by Gli1 expression, had increased Sna1 mRNA as compared with normal cerebellar stem cells. However, not all tumors or TSCs demonstrated increased elevated Shh pathway activity; those lacking elevation of Gli1 also lacked elevation of Sna1 (Figure 2B and 2C). Interestingly, tumors showing low Gli1 induction did not show appreciable Sna1 mRNA induction, indicating that Sna1 mRNA induction in Shh pathway-driven medulloblastoma lesions is associated with significant Shh pathway activation (Figure 2B).
Figure 2. Shh pathway activation induces Sna1 in medulloblastoma.

Protein (A) and cDNA (B) samples were prepared from the tumors and contiguous normal cerebella of four tumor-bearing ND2:SmoA1 mice. A. Western blot analysis shows that three tumors (T) show increased Sna1 expression as compared to contiguous cerebellum (C). B. qRT-PCR analyses show that the tumor with highly elevated Gli1 also shows an increase in Sna1 mRNA, however Sna1 mRNA is not increased when Gli is only slightly induced or not induced. C. qRT-PCR analyses of mRNA expression in tumor stem-like cell lines developed from Ptc+/− medulloblastomas show that the cell line with elevated Gli1 expression (5808) also has high Sna1 mRNA expression. Sna1 is not detected in the cell line with low Gli1 expression (4895) or in WT CBSCs.
We extended our study to include human medulloblastoma-derived cell lines Daoy and ONS76. In Daoy cells, Sna1 mRNA induction follows a similar timecourse to Gli1 mRNA induction as it is moderately induced after 4 hours and reaches maximal induction at 8 hours of Shh treatment (Figure 3A). In ONS76 cells, there is a slight delay in Sna1 induction relative to Gli1: Gli1 is moderately induced at 2 hours and maximally induced after 6 hours of Shh treatment, while Sna1 is moderately induced after 4 hours and maximally induced after 8 hours of Shh treatment (Figure 3B). While activation of Sna1 and Gli1 in response to Shh treatment show different relational kinetics between the two cell lines, Sna1 mRNA peaks with or slightly after Gli1 mRNA expression in both, indicating that it is increased in response to activation of the Shh pathway. In support of this hypothesis, treatment of both cell lines with cyclopamine reduced Sna1 mRNA (Supplemental Figure 3).
Figure 3. Shh pathway activation induces Sna1 in human medulloblastoma cells.

A and B. qRT-PCR analysis shows that Sna1 mRNA expression is robustly increased by exposure to Shh ligand in ONS76 (A) and Daoy cells (B) after 8 hours as compared toGli1 mRNA expression. C. A ChIP assay performed on Daoy cells shows that Gli1 can bind to the promoter of Sna1 between 1217 and 1709 bases upstream of transcription start. D. ChIP performed on cells treated with cyclopamine shows a reduction of Gli1 occupancy on the Sna1 promoter as compared to vehicle control. E. Daoy cells transfected with the wild-type Sna1 promoter (WT) driving luciferase expression show a 1383% increase in promoter induction in by Gli1 as compared to control cells. Mutation of putative GBEs in the region of Gli11 binding prevents (sites −1685, −1449 and −1333) or reduces (site −1297) Sna1 promoter induction in the presence of Gli1.
To investigate whether Gli1 could directly induce the Sna1 promoter, chromatin immunoprecipitation (ChIP) was performed in untransfected Daoy cells using antisera against Gli1 to analyze four amplicons spanning from positions −1709 to −50, encompassing putative Gli1 binding elements upstream of the start of Sna1 transcription, finding that Gli1 binds to the region between −1709 and −1217 (Figure 3C), and treatment of Daoy cells with cyclopamine reduced Gli1 occupancy of this region of the Sna1 promoter (Figure 3D), indicating that Gli1 is capable of binding to the Sna1 promoter in a Shh-dependent manner. To analyze the ability of Gli1 to induce the Sna1 promoter via putative GBEs(32), the −1709 to −1217 region of the Sna1 promoter was used to drive luciferase expression in the presence of enforced Gli1 expression (Figure 3E). Gli1 was able to potently induce the wild-type Sna1 promoter. Mutation of GBEs at positions −1685, −1449 and −1333 abolished activation of the Sna1 promoter by Gli1, indicating these sites to be essential in Gli1-mediated Sna1 induction. Mutation of the GBE at position −1297 reduced but did not abolish the ability of Gli1 to induce the Sna1 promoter, indicating that this site plays a positive but nonessential role in Sna1 induction. As we found in normal GCPs, Sna1 is induced in response to genetic and biochemical Shh pathway activation in primary murine medulloblastomas and human medulloblastoma cells in vitro, and that Sna1 is a direct transcriptional target of Gli1.
Sna1 promotes proliferation of GCPs and medulloblastoma cells
As Shh is known to promote GCP and medulloblastoma cell proliferation, we hypothesized that Sna1 may promote proliferation of these neural cells. Phosphorylated histone H3 (pH3) and Ki67 immunodetection to indicate proliferation revealed increased pH3- or Ki67-positive staining in GCPs infected with adenovirus endocing Sna1 than controls (Figure 4A and 4B). This increase in proliferation was slightly lower than that of GCPs treated with Shh (Figure 4C). We next analyzed the ability of Sna1 to promote proliferation of human medulloblastoma cells by measuring incorporation of EdU during DNA synthesis via flow cytommetry. Medulloblastoma cells transfected with Sna1 showed a dramatic increase in EdU incorporation over control cells (Figure 5A). Furthermore, Daoy cells transfected with shRNA against Sna1 (Supplemental Figure 4) showed a decrease in proliferation versus cells transfected with scrambled shRNA; this was rescued by re-expression of Sna1 (Figure 5B). Together these data indicate that Sna1 is sufficient to induce proliferation of both primary GCPs and medulloblastoma cells in vitro in the absence of exogenous Shh.
Figure 4. Sna1 promotes GCP proliferation.
A. Phosphorylated histone H3 (pH3) and (B) Ki67 immunoreactivity is increased approximately 200% in GCPs infected with adenovirus expressing Sna1 versus uninfected controls. C. Ki67 immunoreactivity is increased approximately 276% in GCPs treated with Shh versus vehicle control.
Figure 5. Sna1 promotes medulloblastoma cell proliferation.
A. EdU incorporation by ONS76 and Daoy cells increased by 231% and 41%, respectively in Sna1-transfected cells versus controls. B. Proliferation of Daoy cells decreased by 15% in cells transfected with shRNA against Sna1 (shSna1) versus cells transfected with scrambled shRNA (Scr.; p=0.0027) as quantified by EdU incorporation; this decrease is rescued by re-expression of Sna1 (p=0.0002).
N-Myc is novel direct target of Sna1 in neural cells
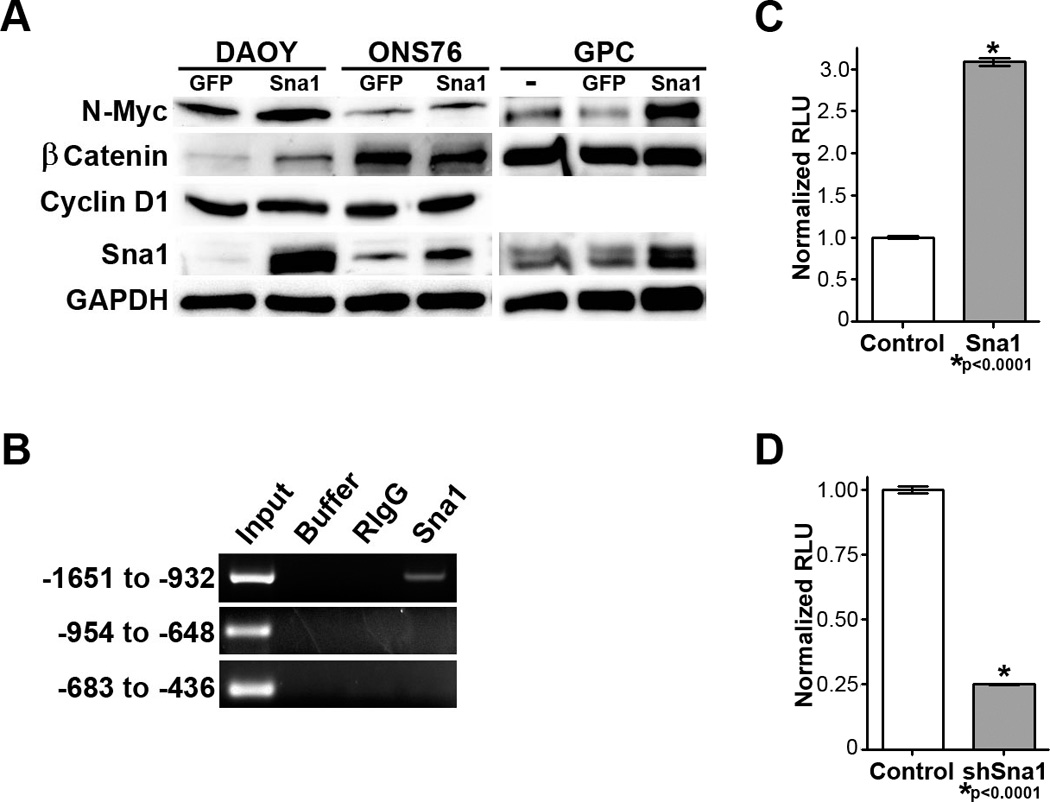
Extending our observation that Sna1 induces proliferation of GCPs, we sought to determine whether Sna1 induced expression of proteins known to promote GCP proliferation. We interrogated a number of targets, finding that N-Myc protein levels were increased in GCPs and human medulloblastoma cells in the presence of enforced Sna1 expression (Figure 6A). This is particularly intriguing because N-Myc is known to play an essential role in Shh-mediated proliferation and transformation of GCPs(31, 33). Although we found no other targets consistently induced by Sna1, β-catenin protein is induced by Sna1 expression in Daoy cells, which harbor a Trp53-inactivating mutation, however β-catenin levels were unaffected by Sna1 expression in GCPs and ONS76 cells, which express WT Trp53 (Figure 6A). These data demonstrate that Sna1 has cell-type specific targets and that N-Myc is a consistent target of Sna1 across neural progenitor and medulloblastoma cells.
Figure 6. Sna1 induces N-Myc in neural cells.

A. Western blot analyses revealed increased expression of N-Myc in medulloblastoma cells and GCPs expressing Sna1. β-catenin is induced by Sna1 only in Daoy cells. CyclinD1 was unaffected by Sna1. B. RT-PCR shows increased expression of N-Myc in medulloblastoma cells expressing Sna1. C. A ChIP assay revealed that Sna1 binds to the N-Myc promoter between positions −1651 and −932. D. Daoy cells transfected with the wild-type N-Myc promoter (WT) driving luciferase expression show a 258% increase in promoter induction by Sna1 as compared to the control. Mutation of E-box sequences in the region of Sna1 binding prevents N-Myc promoter by Sna1. E. Daoy cells transfected with shRNA against Sna1 show a 75% reduction in N-Myc promoter activity compared to scrambled shRNA.
As Sna1 is a transcription factor which binds to E-Box elements(28), we next sought to determine whether Sna1 directly induces N-Myc RNA expression. Comparative qRT-PCR performed on Daoy and ONS76 cells transduced with Sna1 showed an increase in N-Myc mRNA (Figure 6B). ChIP performed using antisera against Sna1 to analyze three N-Myc promoter amplicons - encompassing putative E-box sequences up to 2000 bases upstream of transcription start- shows that Sna1 binds to the region −1651 to −932 (Figure 6C). When this region was used to drive luciferase expression in Daoy, the N-Myc promoter was induced in the presence of Sna1; mutation of putative E-box sequences in this region demonstrates that loss of any E-Box ameliorates the ability of Sna1 to induce the N-Myc promoter (Figure 6D). Likewise, expression of shRNA against Sna1 dramatically reduced N-Myc promoter induction (Figure 6E), arguing that Sna1 is important in direct N-Myc promoter induction in medulloblastoma cells.
N-Myc is required for Sna1-mediated neural growth and transformation
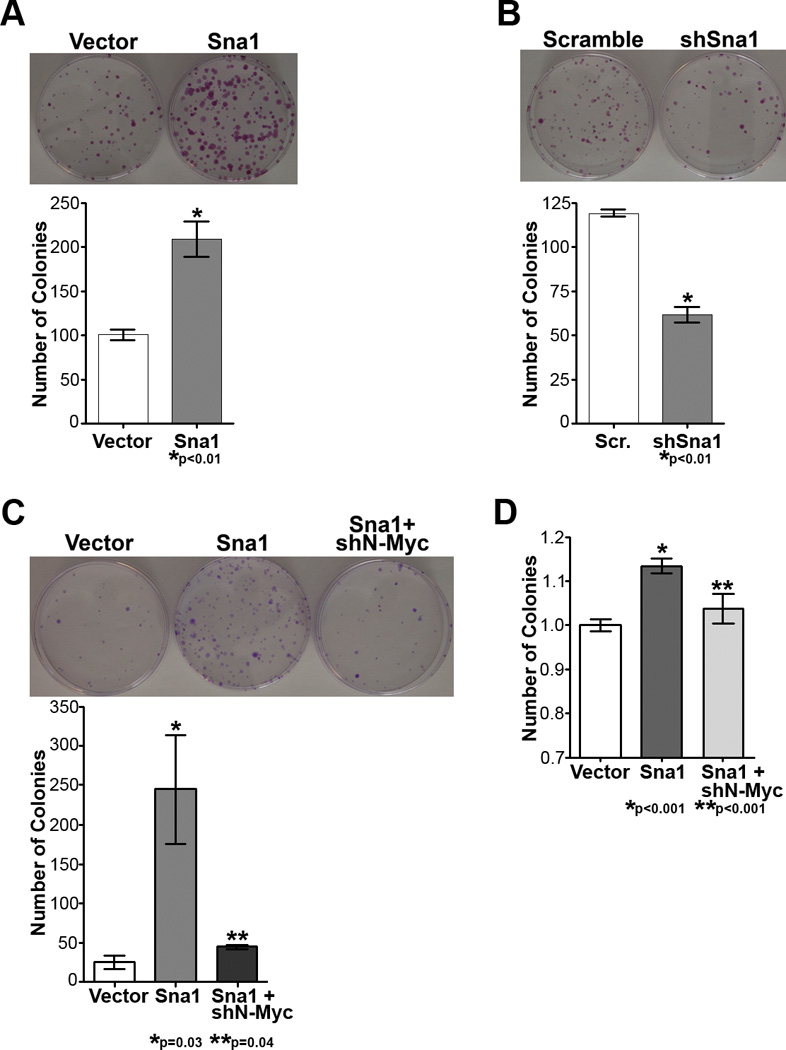
As Sna1 is sufficient to transform MEFs and MDCK cells(34, 35) and we found that Sna1 increased proliferation of medulloblastoma cells, we hypothesized that Sna1 might also promote transformation of neural lineage cells. Colony formation assays to measure loss of contact inhibition were used to indicate in vitro transformation. Daoy cells overexpressing Sna1 formed nearly twice as many colonies as control cells (Figure 7A) and colony formation was reduced in Daoy cells expressing shRNA against Sna1 (Figure 7B), indicating that Sna1 contributes to medulloblastoma cell transformation in vitro. To determine the ability of Sna1 to transform medulloblastoma cells in vivo, Daoy cells were infected with control (GFP) or Sna1-expressing lentivirus and injected subcutaneously into athymic mice. Upon resection, the resultant Sna1-expressing tumors were found to be larger than control tumors, indicating that Sna1 drives medulloblastoma tumor growth in vivo (Figure 7C).
Figure 7. Sna1 increases transformation of medulloblastoma cells, which is abolished by N-Myc depletion.

A. Daoy cells expressing Sna1 formed twice the number of colonies than did cells expressing vector alone. B. Daoy colony formation was reduced with depletion of Sna1 with Sna1-specific shRNA (shSna1). C. Subcutaneous xenograft tumors derived from Sna1-expressing Daoy cells were larger upon resection than control (GFP) tumors. D. EdU incorporation by ONS76 cells shows increased proliferation in cells expressing Sna1 (p<0.0001) was abolished in cells also transfected with shN-Myc (p=0.002); this is rescued by re-expression of N-Myc (p=0.0009). E. The contact-independent growth advantage of Sna1-expressing Daoy (p=0.03) and ONS76 cells (p=0.001) was abolished with N-Myc shRNA (shN-Myc; p=0.04 and p=0.014, respectively).
Finally, we sought to determine if induction of N-Myc by Sna1 resulted in a biologically notable response and whether depletion of N-Myc would affect the proliferative and transforming effects of Sna1 in medulloblastoma cells. We found that ONS76 cells expressing Sna1 and shRNA against N-Myc show reduced proliferation versus cells expressing Sna1 alone; this was rescued by re-expression of N-Myc (Figure 7D). Likewise, Daoy and ONS76 cells expressing shRNA against N-Myc in conjunction with Sna1 overexpression showed a reduction in the number of colonies formed by Sna1 overexpression alone (Figure 7E). These data show that reduction of N-Myc can modulate the ability of Sna1 to induce medulloblastoma cell transformation and argue that the proliferative and transformative effects of Sna1 are mediated through N-Myc. Collectively, our data suggest that Shh pathway–induced Sna1 may amplify aberrant Shh signaling through the induction of N-Myc in GCPs and contribute to malignant transformation within the cerebellum.
DISCUSSION
Our results demonstrate that not only is Sna1 is present in the postembryonic mammalian CNS, but it is also induced by Shh pathway activation in neural progenitor cells. While it is known that Sna1 is regulated by Shh pathway activity in epithelial cells(18–20, 36, 37), we present the first data that this pathway is conserved in vertebrate neural cells and contributes to the normal and neoplastic proliferation thereof. Surprisingly, we found that Shh stimulation of GCPs results in induction of Sna1 even in the absence of Gli1, suggesting that Gli1 is sufficient but not necessary for Sna1 induction. These data are consistent with prior work demonstrating activation of Shh target genes and normal development of Gli1 null mice(21).
Building upon our data in GCPs, we found that Sna1 is induced in conjunction with Shh pathway activation in primary murine tumors and human medulloblastoma lines. This is consistent with prior studies demonstrating that Sna1 is induced by Gli1 and enhances Gli1-mediated transformation(18–20, 37). Given that Sna1 is induced in response to biochemical and genetic activation of Shh pathway in GCPs and medulloblastomas of human and murine origin, we propose that Sna1 induction is a feature of Shh pathway activity in the CNS and may serve to amplify the oncogenic Shh signal.
Interestingly, application of Shh ligand to human medulloblastoma cells results in a small increase in Sna1 mRNA at four hours followed by a robust increase at 8 hours, suggesting a bimodal induction of Sna1 mRNA with respect to time of Shh ligand stimulation. Gli1 is regulated by a positive feedback loop, as its mRNA is induced by Shh signaling(38, 39); it is possible that Sna1 mRNA is initially transcribed by cytoplasmic Gli protein reserves already present in the medulloblastoma cell at 4 hours, however robust activation only occurs at later timepoints after Gli1 is itself produced in response to Shh. In support of this hypothesis, we found that increases (and decreases) in Gli1 mRNA in both cell lines precede the increases (and decreases) in Sna1 mRNA. Combined with our finding that Gli1 was able to bind and directly induce the Sna1 promoter, the magnitudinal and temporal correlation between Sna1 and Gli1 mRNA induction by Shh ligand indicates that Shh pathway activation leads to Sna1 induction in medulloblastoma cells.
Our data show that Sna1 promotes GCP and medulloblastoma cell proliferation and, transformation in vitro and in vivo. Sna1 is traditionally thought to favor EMT versus proliferation in the context of tumorigenesis (28, 34, 40, 41) and to impair cell division(42). Conversely, Sna1 expression correlates with increased proliferation in several systems including during hair bud formation(43), at the tumor-stroma interface of sarcomas(44), and in Gli1-transformed cells(20); loss of Sna1 was found to decrease tumor growth of mouse skin carcinoma and human breast cancer cell xenografts(45, 46). Furthermore, Sna1 was found to be induced by Gli1 and to enhance Gli1-mediated transformation of RK3E epithelial cells through enhancing nuclear β-catenin activity, although Sna1 alone was insufficient to transform these cells in vitro(19, 20). Combined with our data, these findings suggest that the effect of Sna1 on cell proliferation is context-specific, depending on the cell type in which Sna1 is expressed as well as the presence of molecular factors such as activated Shh pathway signaling. In support of this hypothesis, we also found that cyclinD1 – previously reported to be repressed by Sna1(42) – was unaffected by Sna1 expression in medulloblastoma cells. Additionally, increased expression of both Sna1 and Gli1 was noted in a subset of medulloblastomas(47, 48).
N-Myc is transcribed in response to Shh signaling and plays an important role in both Shh-mediated GCP proliferation and medulloblastoma formation(49, 50). Our data show that Sna1 directly binds and induces the N-Myc promoter. Furthermore, N-Myc modulates Sna1-mediated medulloblastoma cell proliferation and transformation, indicating that the action of Sna1 on cerebellar cell growth and transformation is mediated through N-Myc. Previous work demonstrates that N-Myc is an intermediate early gene (IEG) directly induced within 3 hours of Shh ligand exposure(31). As we did not detect appreciable induction of Sna1 mRNA until 8 hours Shh ligand stimulation, Sna1 cannot be the only mechanism by which N-Myc transcription responds to Shh activity. Rather, our findings complement this research, showing that N-Myc protein expression can be induced by prolonged Shh activity and by Sna1 overexpression. Sna1 induction by Shh may be a mechanism by which N-Myc levels are sustained in response to Shh, amplifying the mitogenic signal.
This study represents the first demonstration of direct transcriptional activation of a promoter region by Sna1, which has been traditionally thought of as a transcriptional repressor(28, 34, 51). Conversely, several studies also show increases in RNA or protein in response to Snail activity: mesenchymal markers such as vimentin and LEF-1 are upregulated in epithelial cells upon Snail expression(34, 52). cDNA and protein array studies reveal a subset of targets elevated in the presence of Sna1, including Id3, Wnt5a, and β-catenin(52, 53). Recent studies have shown that Sna1 can act as a coactivator for β-Catenin and NFκB(54, 55). Notably, β-catenin was previously found to activate the N-Myc promoter(56); it is possible that Sna1 and β-catenin act cooperatively to promote N-Myc transcription. Our finding that Sna1 directly induces the N-Myc promoter, combined with previous studies, supports a role for Sna1 as a context-specific transcriptional activator in addition to its canonical role in transcriptional repression.
It is essential to understand the biological function of factors downstream of Gli proteins in the cerebellum, given the critical role of Shh pathway activity in GCP development and transformation. To this end, we have found that Sna1 is induced in GCPs and medulloblastoma cells in response to Shh pathway activation, and increases proliferation and transformation of these cells. We have described the direct induction of the novel Sna1 target, N-Myc and demonstrate its ability to modulate Sna1- induced medulloblastoma cell proliferation and transformation. This novel interaction gives further insight into the involvement of Sna1 in Shh signaling in neural progenitors and in the proliferation and transformation of neural tissue, suggesting that sequential activation of the Sna1 and N-Myc transcription factors plays a role in Shh-pathway mediated neural tumorigenesis.
Supplementary Material
Acknowledgments
Grant Support: American Cancer Society RSG116446; NCI Brain SPORE CA108961-04; Watermann Family Foundation for Cancer Genetics; MEFZ was supported by the Mayo Clinic Cancer Center, Mayo Clinic Pancreatic SPORE P50 CA102701, and Mayo Clinic Center for Cell Signaling in Gastroenterology P30 DK084567
Footnotes
Conflicts of Interest: The authors report no potential conflicts of interest.
Works Cited
- 1.Fuccillo M, Joyner AL, Fishell G. Morphogen to mitogen: the multiple roles of hedgehog signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7:772–783. doi: 10.1038/nrn1990. [DOI] [PubMed] [Google Scholar]
- 2.Charron F, Tessier-Lavigne M. Novel brain wiring functions for classical morphogens: a role as graded positional cues in axon guidance. Development (Cambridge, England) 2005;132:2251–2262. doi: 10.1242/dev.01830. [DOI] [PubMed] [Google Scholar]
- 3.Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Molecular and cellular biology. 2000;20:9055–9067. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 5.Lai K, Kaspar BK, Gage FH, Schaffer DV. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nature neuroscience. 2003;6:21–27. doi: 10.1038/nn983. [DOI] [PubMed] [Google Scholar]
- 6.Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development (Cambridge, England) 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 7.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science (New York, NY. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 8.Ruiz i Altaba A, Sanchez P, Dahmane N. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nature reviews. 2002;2:361–372. doi: 10.1038/nrc796. [DOI] [PubMed] [Google Scholar]
- 9.Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, et al. Mutations in SUFU predispose to medulloblastoma. Nature genetics. 2002;31:306–310. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- 10.Wetmore C, Eberhart DE, Curran T. The normal patched allele is expressed in medulloblastomas from mice with heterozygous germ-line mutation of patched. Cancer research. 2000;60:2239–2246. [PubMed] [Google Scholar]
- 11.Xie K, Abbruzzese JL. Developmental biology informs cancer: the emerging role of the hedgehog signaling pathway in upper gastrointestinal cancers. Cancer cell. 2003;4:245–247. doi: 10.1016/s1535-6108(03)00246-0. [DOI] [PubMed] [Google Scholar]
- 12.Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 13.Wetmore C. Sonic hedgehog in normal and neoplastic proliferation: insight gained from human tumors and animal models. Current opinion in genetics & development. 2003;13:34–42. doi: 10.1016/s0959-437x(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 14.Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B, et al. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer research. 2008;68:1768–1776. doi: 10.1158/0008-5472.CAN-07-5092. [DOI] [PubMed] [Google Scholar]
- 15.Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer research. 2004;64:7794–7800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 16.Kimura H, Stephen D, Joyner A, Curran T. Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene. 2005;24:4026–4036. doi: 10.1038/sj.onc.1208567. [DOI] [PubMed] [Google Scholar]
- 17.Weiner HL, Bakst R, Hurlbert MS, Ruggiero J, Ahn E, Lee WS, et al. Induction of medulloblastomas in mice by sonic hedgehog, independent of Gli1. Cancer research. 2002;62:6385–6389. [PubMed] [Google Scholar]
- 18.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Deng W, Lobo-Ruppert SM, Ruppert JM. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by beta-catenin. Oncogene. 2007;26:4489–4498. doi: 10.1038/sj.onc.1210241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Deng W, Nail CD, Bailey SK, Kraus MH, Ruppert JM, et al. Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene. 2006;25:609–621. doi: 10.1038/sj.onc.1209077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development (Cambridge, England) 2002;129:4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- 22.Galvin KE, Ye H, Erstad DJ, Feddersen R, Wetmore C. Gli1 induces G2/M arrest and apoptosis in hippocampal but not tumor-derived neural stem cells. Stem cells (Dayton, Ohio) 2008;26:1027–1036. doi: 10.1634/stemcells.2007-0879. [DOI] [PubMed] [Google Scholar]
- 23.Kim J, Miller VM, Levites Y, West KJ, Zwizinski CW, Moore BD, et al. BRI2 (ITM2b) inhibits Abeta deposition in vivo. J Neurosci. 2008;28:6030–6036. doi: 10.1523/JNEUROSCI.0891-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.ConTra [Internet] [cited 2009 July 9]; Available from: http://bioit.dmbr.ugent.be/ConTra/indexphp. [Google Scholar]
- 25.Godbey WT, Wu KK, Hirasaki GJ, Mikos AG. Improved packing of poly(ethylenimine)/DNA complexes increases transfection efficiency. Gene therapy. 1999;6:1380–1388. doi: 10.1038/sj.gt.3300976. [DOI] [PubMed] [Google Scholar]
- 26.Galvin KE, Ye H, Wetmore C. Differential gene induction by genetic and ligand-mediated activation of the Sonic hedgehog pathway in neural stem cells. Developmental biology. 2007;308:331–342. doi: 10.1016/j.ydbio.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 27.Hooghe B, Hulpiau P, van Roy F, De Bleser P. ConTra: a promoter alignment analysis tool for identification of transcription factor binding sites across species. Nucleic acids research. 2008;36:W128–W132. doi: 10.1093/nar/gkn195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nature cell biology. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 29.Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer cell. 2008;14:135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development (Cambridge, England) 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- 32.Ruppert JM, Vogelstein B, Arheden K, Kinzler KW. GLI3 encodes a 190-kilodalton protein with multiple regions of GLI similarity. Molecular and cellular biology. 1990;10:5408–5415. doi: 10.1128/mcb.10.10.5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hatton BA, Knoepfler PS, Kenney AM, Rowitch DH, de Alboran IM, Olson JM, et al. N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer research. 2006;66:8655–8661. doi: 10.1158/0008-5472.CAN-06-1621. [DOI] [PubMed] [Google Scholar]
- 34.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nature cell biology. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 35.Perez-Mancera PA, Perez-Caro M, Gonzalez-Herrero I, Flores T, Orfao A, de Herreros AG, et al. Cancer development induced by graded expression of Snail in mice. Human molecular genetics. 2005;14:3449–3461. doi: 10.1093/hmg/ddi373. [DOI] [PubMed] [Google Scholar]
- 36.Fiaschi M, Rozell B, Bergstrom A, Toftgard R, Kleman MI. Targeted expression of GLI1 in the mammary gland disrupts pregnancy-induced maturation and causes lactation failure. The Journal of biological chemistry. 2007;282:36090–36101. doi: 10.1074/jbc.M704280200. [DOI] [PubMed] [Google Scholar]
- 37.Louro ID, Bailey EC, Li X, South LS, McKie-Bell PR, Yoder BK, et al. Comparative gene expression profile analysis of GLI and c-MYC in an epithelial model of malignant transformation. Cancer research. 2002;62:5867–5873. [PubMed] [Google Scholar]
- 38.Hynes M, Stone DM, Dowd M, Pitts-Meek S, Goddard A, Gurney A, et al. Control of cell pattern in the neural tube by the zinc finger transcription factor and oncogene Gli-1. Neuron. 1997;19:15–26. doi: 10.1016/s0896-6273(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 39.Lee J, Platt KA, Censullo P, Ruiz i Altaba A. Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development (Cambridge, England) 1997;124:2537–2552. doi: 10.1242/dev.124.13.2537. [DOI] [PubMed] [Google Scholar]
- 40.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development (Cambridge, England) 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 41.Hemavathy K, Ashraf SI, Ip YT. Snail/slug family of repressors: slowly going into the fast lane of development and cancer. Gene. 2000;257:1–12. doi: 10.1016/s0378-1119(00)00371-1. [DOI] [PubMed] [Google Scholar]
- 42.Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes & development. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jamora C, Lee P, Kocieniewski P, Azhar M, Hosokawa R, Chai Y, et al. A signaling pathway involving TGF-beta2 and snail in hair follicle morphogenesis. PLoS biology. 2005;3:e11. doi: 10.1371/journal.pbio.0030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Franci C, Takkunen M, Dave N, Alameda F, Gomez S, Rodriguez R, et al. Expression of Snail protein in tumor-stroma interface. Oncogene. 2006;25:5134–5144. doi: 10.1038/sj.onc.1209519. [DOI] [PubMed] [Google Scholar]
- 45.Olmeda D, Montes A, Moreno-Bueno G, Flores JM, Portillo F, Cano A. Snai1 and Snai2 collaborate on tumor growth and metastasis properties of mouse skin carcinoma cell lines. Oncogene. 2008;27:4690–4701. doi: 10.1038/onc.2008.118. [DOI] [PubMed] [Google Scholar]
- 46.Olmeda D, Moreno-Bueno G, Flores JM, Fabra A, Portillo F, Cano A. SNAI1 is required for tumor growth and lymph node metastasis of human breast carcinoma MDA-MB-231 cells. Cancer research. 2007;67:11721–11731. doi: 10.1158/0008-5472.CAN-07-2318. [DOI] [PubMed] [Google Scholar]
- 47.Oncomine 4.4 [Internet] [cited 2010 Nov 5];2010 Oct 6; Available from: www.oncomine.org. [Google Scholar]
- 48.Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nature genetics. 2009;41:465–472. doi: 10.1038/ng.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development (Cambridge, England) 2004;131:217–228. doi: 10.1242/dev.00891. [DOI] [PubMed] [Google Scholar]
- 50.Thomas WD, Chen J, Gao YR, Cheung B, Koach J, Sekyere E, et al. Patched1 deletion increases N-Myc protein stability as a mechanism of medulloblastoma initiation and progression. Oncogene. 2009;28:1605–1615. doi: 10.1038/onc.2009.3. [DOI] [PubMed] [Google Scholar]
- 51.Nakayama H, Scott IC, Cross JC. The transition to endoreduplication in trophoblast giant cells is regulated by the mSNA zinc finger transcription factor. Developmental biology. 1998;199:150–163. doi: 10.1006/dbio.1998.8914. [DOI] [PubMed] [Google Scholar]
- 52.Larriba MJ, Casado-Vela J, Pendas-Franco N, Pena R, Garcia de Herreros A, Berciano MT, et al. Novel snail1 target proteins in human colon cancer identified by proteomic analysis. PloS one. 2010;5:e10221. doi: 10.1371/journal.pone.0010221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moreno-Bueno G, Cubillo E, Sarrio D, Peinado H, Rodriguez-Pinilla SM, Villa S, et al. Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer research. 2006;66:9543–9556. doi: 10.1158/0008-5472.CAN-06-0479. [DOI] [PubMed] [Google Scholar]
- 54.Solanas G, Porta-de-la-Riva M, Agusti C, Casagolda D, Sanchez-Aguilera F, Larriba MJ, et al. E-cadherin controls beta-catenin and NF-kappaB transcriptional activity in mesenchymal gene expression. Journal of cell science. 2008;121:2224–2234. doi: 10.1242/jcs.021667. [DOI] [PubMed] [Google Scholar]
- 55.Stemmer V, de Craene B, Berx G, Behrens J. Snail promotes Wnt target gene expression and interacts with beta-catenin. Oncogene. 2008;27:5075–5080. doi: 10.1038/onc.2008.140. [DOI] [PubMed] [Google Scholar]
- 56.Shu W, Guttentag S, Wang Z, Andl T, Ballard P, Lu MM, et al. Wnt/beta-catenin signaling acts upstream of N-myc, BMP4, and FGF signaling to regulate proximal-distal patterning in the lung. Developmental biology. 2005;283:226–239. doi: 10.1016/j.ydbio.2005.04.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.