

The atomic structure of a new crystal form of the human histidine triad nucleotide-binding protein 1 (hHINT1)–AMP complex is reported at 1.38 Å resolution.
Keywords: HINT, histidine triad nucleotide-binding proteins
Abstract
Histidine triad nucleotide-binding protein 1 (HINT1) represents the most ancient and widespread branch of the histidine triad protein superfamily. HINT1 plays an important role in various biological processes and has been found in many species. Here, the structure of the human HINT1–adenosine 5′-monophosphate (AMP) complex at 1.38 Å resolution obtained from a new monoclinic crystal form is reported. The final structure has R cryst = 0.1207 (R free = 0.1615) and the model exhibits good stereochemical quality. Detailed analysis of the high-resolution data allowed the details of the protein structure to be updated in comparison to the previously published data.
1. Introduction
Histidine triad nucleotide-binding protein 1 (HINT1) belongs to a branch of the histidine triad (HIT) protein superfamily that consists primarily of mononucleotide and dinucleotide hydrolases and nucleotide transferases that have a characteristic C-terminal active-site motif HXHXHXX, where X is a hydrophobic residue (Brenner, 2002 ▶). HINT proteins are the most conserved members of the HIT superfamily with nucleotide hydrolase activity. HINT protein homologues are present in a wide variety of organisms, including the metazoan (Bieganowski et al., 2002 ▶; Parks et al., 2004 ▶; Wu et al., 2010 ▶), plant (Guranowski et al., 2011 ▶), fungal (Korsisaari et al., 2003 ▶) and bacterial (Chou et al., 2005 ▶; Bardaweel et al., 2010 ▶) kingdoms. The strongly conserved sequences of the HINT proteins suggest their fundamental role in cell metabolism. HINT1 is expressed in multiple tissues and is present both in the nucleus and cytoplasm, but its exact cellular function has still not been precisely defined.
In vitro studies indicate that human HINT1 is able to bind various nucleotides, e.g. adenosine 5′-monophosphate (AMP), adenosine 5′-diphosphate (ADP) and the diadenosine polyphosphates 5′,5′′-diadenosine triphosphate (Ap3A) and 5′,5′′-diadenosine tetraphosphate (Ap4A) (Lima et al., 1996 ▶). Rabbit HINT1 (rHINT1) also binds certain purine nucleosides and nucleotides (Brenner et al., 1997 ▶). HINT proteins demonstrate phosphoramidase activity towards adenosine 5′-O-phosphoramidate (AMP-NH2; Bieganowski et al., 2002 ▶). Lysyl-adenylate (generated by lysyl-tRNA synthetase; LysRS) has also been suggested to be a substrate of the HINT1 hydrolase (Chou & Wagner, 2007 ▶; Chou et al., 2007 ▶). The ability to hydrolyse the P—N bond in nucleoside phosphoramidates has been demonstrated in studies on the conversion of PSI-7851 (2′-deoxy-2′-fluoro-2′-C-methyluridine-5′-monophosphate), a potent anti-HCV prodrug with in vitro and in vivo activity (Murakami et al., 2010 ▶). Recently conducted research has confirmed that rHINT1 is able to desulfurate 5′-O-phosphorothioylated nucleosides (Krakowiak et al., 2007 ▶; Ozga, 2010 ▶).
HINT1 is involved in transcription regulation (Lee et al., 2004 ▶; Lee & Razin, 2005 ▶; Weiske & Huber, 2006 ▶; Cen et al., 2009 ▶; Wang et al., 2009 ▶) and cell-cycle control (Weiske & Huber, 2005 ▶). HINT1 plays a role in psychiatric disorders such as schizophrenia, bipolar disorder, major depression and anxiety (Guang et al., 2004 ▶; Vawter et al., 2004 ▶; Barbier et al., 2007 ▶; Barbier & Wang, 2009 ▶; Elashoff et al., 2007 ▶; Chen et al., 2008 ▶; Liu et al., 2008 ▶; Varadarajulu et al., 2011 ▶). Recent studies suggest that mammalian HINT1 modulates apoptosis in cancer cells and is a potential tumour suppressor (Su et al., 2003 ▶; Li et al., 2006 ▶; Weiske & Huber, 2006 ▶; Wang et al., 2007 ▶, 2009 ▶; Hsieh et al., 2009 ▶; Zhang et al., 2009 ▶; Zambelli et al., 2010 ▶).
The first HINT1 crystal structure was published in 1996 (Lima et al., 1996 ▶) and the protein was initially described as an inhibitor of protein kinase C (Pearson et al., 1990 ▶) and further as PKCI (protein kinase C-interacting; Klein et al., 1998 ▶). Finally, the ‘HINT’ name was coined after structural analysis performed by Brenner et al. (1997 ▶). At present, 14 HINT1 structures are available in the PDB: six human (Lima et al., 1996 ▶, 1997 ▶), six rabbit (Brenner et al., 1997 ▶; Krakowiak et al., 2004 ▶; Dolot et al., 2011 ▶) and two from Escherichia coli (Bardaweel et al., 2010 ▶). The deposited structures display HINT in the apo state (Lima et al., 1996 ▶) or in complex with selected ligands: substrate analogues or products of their hydrolysis (Lima et al., 1997 ▶; Krakowiak et al., 2004 ▶; Bardaweel et al., 2010 ▶). To date, the highest registered data resolution for hHINT1 was 1.50 Å (PDB entry 1kpf; Lima et al., 1997 ▶).
The structure of hHINT1 is characterized as a general α+β fold type and is further subclassed as an α+β meander fold. The monomer consists of a five-stranded antiparallel β-sheet and two α-helices: a short α1 helix localized close to the N-terminus and a long α2 helix localized between the β3 and β5 strands. Helices α1 (residues 18–24) and α2 (residues 68–86) pack on opposite sides of the β-sheet, which is wrapped around the longest helix. X-ray crystal structure and size-exclusion chromatography analyses have revealed that hHINT1 exists as a homodimer (Chou et al., 2005 ▶; Lima et al., 1996 ▶, 1997 ▶). The dimer consists of two monomers which form a ten-stranded antiparallel β-sheet, while the α2 helix from one monomer is localized opposite the α2′ helix from the other monomer (Fig. 1 ▶). Each of the constituent monomers contains a well separated active site outside the dimer interface (Boulanger & Kantrowitz, 2003 ▶).
Figure 1.

(a) Cartoon representation of the hHINT1 dimer (based on PDB entry 3tw2). Structure elements are indicated in orange (α-helices) and blue (β-strands). Each monomer is shown in a darker (chain A) and a lighter (chain B) colour scheme. The location of the active site is indicated by the bound AMP molecule (shown in stick representation). Secondary-structure elements were assigned with DSSP (Kabsch & Sander, 1983 ▶). The same dimer structure after a 90° rotation is shown below. (b) B-factor analysis. The structures of the A and B chains are drawn in cartoon putty representation, in which colour is ramped by residue from blue for the lowest B-factor value to red for the highest B-factor value. In addition, the size of the tube also reflects the value of the B factor, where the larger the B factor the thicker the tube.
The nucleotide-binding pocket is formed by loops L1, L3 and L6, helix α1 and a fragment of β-sheet. The purine base-binding site is localized between the helix and the L1 loop and mostly consists of most conserved hydrophobic residues. Loop L3 binds the ribose residue. Loop L6 is in contact with the sugar and phosphate moieties. Conserved polar residues, including His112 and His114 from the HIT motif, are involved in binding the α-phosphate moiety (Lima et al., 1996 ▶, 1997 ▶). The cleft is localized away from the dimer contact interface and each of the monomeric components can independently bind one nucleoside molecule. Nevertheless, the C-terminus of each monomer makes a range of contacts with the adjacent monomer in close proximity to the active site, including hydrogen-bond interactions between Gly126 and Arg119′ and Asn122′ from the second chain. In addition, the C-terminal fragment of one monomer is involved in closing the binding pocket of the second monomer using the Trp123 residue, which is situated at the opposite position to helix α1, but these residues do not interact with the nucleoside molecule.
Because the C-terminus of HINT1 (which exerts hydrolytic activity and mediates in homodimer formation and nucleotide binding) does not interact with other proteins, especially with transcription factors, it has been suggested that only the N-terminus of HINT1 or some structural motifs from the N-terminal part may be responsible for the formation of contacts with its binding partners (Cen et al., 2009 ▶).
In this manuscript, we report a detailed 1.38 Å resolution structure of hHINT1 complexed with AMP obtained from a new monoclinic crystal form.
2. Materials and methods
2.1. Cloning, expression, purification and crystallization of hHINT1
Human HINT1 protein was expressed and purified in E. coli BL21 strain using the pSGA02-hHINT1 plasmid as previously described for rabbit HINT1 (Gilmour et al., 1997 ▶). The single-step purification was performed using AMP–agarose (Sigma) affinity chromatography. The homogenous protein preparation was dialyzed against a buffer consisting of 20 mM Tris–HCl pH 7.5, 150 mM NaCl, concentrated to a protein concentration of 10 mg ml−1, frozen in liquid nitrogen and stored at 193 K.
The hHINT1–AMP complex was crystallized by the vapour-diffusion method in the standard hanging-drop format using a mixture of 10 mg ml−1 protein solution and a sixfold molar excess of AMP. Initial crystallization conditions were similar to those previously used for rHINT1 and were used as a starting point for optimization. The final precipitant solution consisted of 100 µM sodium cacodylate buffer pH 5.5, 6.0, 6.5 or 7.0 and 15, 17, 19, 21, 23 or 25%(w/v) PEG 8000 with or without the addition of 100 µM sodium acetate at two temperatures (ambient and 278 K). Crystallization drops were set up by mixing 2 µl protein solution with 2 µl precipitant solution and were suspended over 1 ml precipitant solution. At ambient temperature, most crystallization conditions produced needles. After 1–2 months, the growth of additional crystals with a tetragonal bipyramid shape, identical to previously obtained rHINT1 crystals (Dolot et al., 2011 ▶), was observed in the same drops. Crystallization at lower temperature using conditions consisting of 19%(w/v) PEG 8000, 100 µM sodium cacodylate pH 5.5 allowed crystals of a new crystal form with a plate shape and typical dimensions of 0.8 × 0.4 × 0.1 mm to be obtained. These crystals appeared after 48–72 h.
2.2. X-ray data collection and structure determination
Crystals of hHINT1 were flash-cooled by transfer into a mixture of PEG 400 and mother liquor [1:3(v:v)] as a cryoprotectant, followed by cooling in an N2 stream at 100 K (Teng, 1990 ▶). Diffraction data were collected using synchrotron radiation with a MAR CCD 165 mm detector on the EMBL X13 beamline at the DESY sychrotron, Hamburg, Germany. The data were processed, integrated and scaled with iMOSFLM (Battye et al., 2011 ▶) and SCALA from the CCP4 suite (Winn et al., 2011 ▶). The data-collection and processing statistics are given in Table 1 ▶. Assuming the presence of two molecules in the asymmetric unit, the Matthews coefficient V M was 2.09 Å3 Da−1, corresponding to a solvent content of approximately 40%.
Table 1. Data-collection and final model-refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | C2 |
| Wavelength (Å) | 0.8123 |
| Unit-cell parameters | |
| a (Å) | 77.42 |
| b (Å) | 46.45 |
| c (Å) | 64.03 |
| β (°) | 94.42 |
| Total No. of reflections | 201262 |
| Unique reflections | 45932 |
| Completeness (%) | 97.5 (84.0) |
| Resolution (Å) | 25.58–1.38 (1.45–1.38) |
| R merge † | 0.122 (0.291) |
| Multiplicity | 4.4 (2.8) |
| Mosaicity (°) | 0.47 |
| Wilson B factor (Å2) | 11.17 |
| 〈I/σ(I)〉 | 13.1 (3.4) |
| Refinement | |
| No. of reflections used in refinement | 43580 |
| No. of reflections used for R free | 2320 |
| R cryst/R free ‡ | 0.1207/0.1615 |
| No. of non-H atoms | |
| Protein | 2049 |
| Solvent | 410 |
| Ligand | 23 |
| R.m.s.d.s from ideal values | |
| Bond lengths (Å) | 0.012 |
| Bond angles (°) | 1.479 |
| Ramachandran plot§ | |
| Most favoured (%) | 89.9 |
| Additional allowed (%) | 10.1 |
| Mean B values¶ (Å2) | |
| Protein | 13.87 |
| AMP | 14.66 |
| Water | 37.12 |
R
merge = 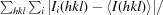
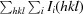 , where Ii(hkl) is the intensity of an individual measurement of reflection hkl and 〈I(hkl)〉 is the mean intensity of the reflection.
, where Ii(hkl) is the intensity of an individual measurement of reflection hkl and 〈I(hkl)〉 is the mean intensity of the reflection.
R
cryst = 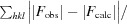
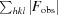 for all reflections, where F
obs and F
calc are observed and calculated structure factors, respectively. R
free is calculated analogously for the test reflections, which were randomly selected and excluded from the refinement.
for all reflections, where F
obs and F
calc are observed and calculated structure factors, respectively. R
free is calculated analogously for the test reflections, which were randomly selected and excluded from the refinement.
Calculated using PROCHECK.
Calculated using BAVERAGE.
The structure was solved by molecular replacement with MOLREP (Vagin & Teplyakov, 2010 ▶) using the hHINT1 crystal structure (PDB entry 1kpf; Lima et al., 1997 ▶) as a search model. Refinement was carried out with REFMAC5 (Murshudov et al., 2011 ▶). After the calculation of an initial electron-density map, the structure was completed using alternate cycles of manual building, including main-chain and side-chain rebuilding and the addition of alternative residue conformations, loop fragments and solvent molecules, in the Coot program (Emsley et al., 2010 ▶). All refinement steps were monitored using R cryst and R free values. After a few rounds of refinement, the non-H atoms were refined anisotropically in all subsequent refinement cycles. During the first round of anisotropic refinement, the R cryst and R free values dropped considerably (by approximately 2.5%) in comparison with isotropic refinement. The stereochemical quality of the resulting model was judged using the PROCHECK program (Laskowski et al., 1993 ▶) and the validation tools implemented in Coot. The mean temperature factors for protein main chains and side chains and water molecules were calculated using the BAVERAGE program from the CCP4 program suite. Protein-structure superpositions were performed using the program LSQKAB (Kabsch, 1976 ▶). Graphic representations of superimposed structures were visually inspected. Interface analysis was performed using the EBI PISA server (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html; Krissinel & Henrick, 2007 ▶). Contacts were measured using the CONTACT program from the CCP4 suite. The refinement statistics of the described structure are given in Table 1 ▶. All figures were generated using PyMOL v.0.99 (DeLano, 2002 ▶).
3. Results and discussion
3.1. Overall structure
The crystal structure of hHINT1 (space group C2) contained two independent protein molecules, an AMP molecule and 410 well defined water molecules in the asymmetric unit and had an R factor of 0.1207 (R free = 0.1615). In each subunit, there were no residues in disallowed or generously allowed Ramachandran conformations. The pair of molecules around the noncrystallographic twofold-symmetry axis, which is parallel to the crystal y axis, formed a homodimer. Each subunit exhibited the typical HINT1 fold and was composed of five β-strands and two α-helices. Because of a lack of well defined electron density in 2|F o − F c| and |F o − F c| maps for the N-terminal regions of both protein chains, as in other previously deposited mammalian HINT1 structures, residues 1–10 of chain A and 1–14 of chain B were not included in the protein model. The dimerization of the subunits creates an extensive interface region, with a large number of polar interactions as well as salt bridges between the chains. A total of 20% or 1930 Å2 of the accessible surface area of the subunits (9660 Å) is buried in the interface. The main structural elements that participate in dimerization are the β4 strand, the middle part of the α2 helix and the C-terminal fragment of the protein chain, which is very important for dimer formation. The high-resolution data allowed the previously deposited model of the hHINT1–AMP complex (PDB entry 1kpf) to be updated and a more detailed hHINT1 structure model with higher precision, including side-chain rotamers, to be built. The side chains of 16 residues of chain A and 14 residues of chain B exhibit double or multiple possible conformations.
3.2. Crystal packing and crystal contacts
The hHINT1–AMP complex crystal that we describe here belongs to space group C2, which has not previously been observed for hHINT1. However, it contained two protein molecules which form a biologically active dimer in the asymmetric unit, as in orthorhombic (space group P212121) crystals which account for the majority of deposited hHINT1 structures (PDB entries 1av5, 1kpa, 1kpb and 1kpe; Lima et al., 1996 ▶, 1997 ▶). The other known monoclinic crystal form (PDB entry 1kpc; space group P21; Lima et al., 1996 ▶) contains four hHINT1 molecules arranged into two dimers in the asymmetric unit. hHINT1 is also able to crystallize in a tetragonal form (PDB entry 1kpf; space group P43212; Lima et al., 1997 ▶), as in the majority of deposited rabbit HINT1 structures, with one protein molecule in the asymmetric unit; this is half of the biological dimer, which can be reproduced by a symmetry operation.
Among all of the crystal forms listed, the structure described here (PDB entry 3tw2) displays crystal packing at a similar level to that in the 1kpc and 1kpf crystals, as indicated by the Matthews coefficients (V M; Matthews, 1968 ▶) of 2.08, 2.10 and 2.12 Å3 Da−1, respectively, in contrast to the loosely packed orthorhombic structure (e.g. 1av5, with a V M value of 2.65 Å3 Da−1). Similar results were obtained after calculation of the packing coefficient k (Andersson & Hovmöller, 2000 ▶), with values of 0.694, 0.685, 0.691 and 0.549, respectively. However, the similarity of the packing level is not connected to the observed arrangement of the protein molecules inside the crystal structure, which is completely different in each of the cases discussed (Fig. 2 ▶). It should be pointed out that the crystal described here (PDB entry 3tw2) contains small (10 Å wide) but clearly defined solvent channels that are parallel to the crystallographic a axis. A comparative analysis of the contacts between symmetry-related molecules in the crystal forms discussed provided some interesting results. The largest number of contacts is observed for the 3tw2 structure, in which ten contacts are realised via hydrogen bonds with three additional salt bridges, in contrast to the remaining crystal forms, in which the numbers of contacts are much lower.
Figure 2.

Crystal-packing diagrams of known hHINT1 crystal forms. (a) hHINT1 crystallized in space group C2 with a dimer per asymmetric unit (based on PDB entry 3tw2). Views along the a, b and c axes are shown. (b) hHINT1 crystallized in space group P212121 with a dimer per asymmetric unit (based on PDB entry 1av5). Views along the a, b and c axes are shown. (c) hHINT1 crystallized in space group P21 with a dimer of dimers per asymmetric unit (based on PDB entry 1kpc). Views along the a, b and c axes are shown. (d) hHINT1 crystallized in space group P43212 with a monomer per asymmetric unit (based on PDB entry 1kpf). Views along the a and c axes are shown.
3.3. Nucleotide-binding site
Previously published ligand-binding studies have shown that HINT proteins are able to bind purine nucleosides and nucleotides, despite the fact that the HINT nucleotide-binding motif possesses a different sequence compared with other nucleotide-binding proteins (Gilmour et al., 1997 ▶). As shown in Fig. 3 ▶, the AMP molecule is bound in a cleft of the enzyme commonly described as the active site (Brenner et al., 1997 ▶; Lima et al., 1997 ▶). The location of AMP was clearly identified in the |F o − F c| electron-density map and the ligand model was easily fitted. The active-site cleft has traditionally been divided into the mostly hydrophobic purine-binding pocket (isoleucines Ile18, Ile22, Ile27 and Ile44 and phenylalanines Phe19 and Phe41) and the sugar-binding pocket, in which the Asp43 residue makes hydrogen bonds to the O2′ and O3′ hydroxyl O atoms of the ribose ring. The phosphate-group O atoms also play important roles in the ligand–protein interactions. The O2P atom makes a strong hydrogen bond to His112 and His114, O3P interacts directly with the main-chain N atoms of Ser107 and Val108, and an additional hydrogen bond is generated between the O1P atom and the ND2 atom of Asn99. In this data set, one AMP molecule is observed in the binding cleft of the A chain and there was nothing exceptional in this canonical mode of product binding, while the binding cleft of chain B is filled by structural water molecules only. AMP is bound, as expected, in the purine-binding site and it is also in close hydrophobic interaction with another symmetry-related hHINT1 molecule in the lattice. One of the possible reasons for the observed single ligand molecule binding per HINT1 dimer is that the active site of chain B is blocked by the lattice contacts and access of AMP is difficult, while the active site of chain A is oriented towards the internal water channels. An almost identical assembly was observed in the previously published hHINT1–ligand complex structure with a dimer in the asymmetric unit (PDB entry 1kpe), as well as in recently published structures of hHINT1 complexed with aminoacyl adenylates (PDB entries 4eqe, 4eqg and 4eqh; Wang et al., 2012 ▶).
Figure 3.

The nucleotide-binding site of the hHINT1–AMP complex. Hydrogen-bonding interactions between AMP atoms and nearby side chains or water molecules are shown as dashed lines. The OMIT F o − F c electron-density map of AMP is contoured at the 3.0σ level based on 1.38 Å resolution data (PDB entry 3tw2).
3.4. Structural comparison
The overall shape of the obtained hHINT1 structure model at 1.38 Å resolution is almost identical to the model used for molecular replacement (PDB entry 1kpf; Lima et al., 1997 ▶) and its fold is in agreement with those of other hHINT1 structures deposited in the PDB. The hHINT1 structure described here contains both the bound and the unbound states. Least-squares superposition of the Cα coordinates of the A (bound) and B (unbound) chains, with an r.m.s.d. value of 0.244 Å, did not exhibit significant structural differences caused by ligand binding, except in the regions of loops L1 and L6 that showed small changes in position. The major differences in the Cα trace can be found in the N-terminal region, together with a higher averaged temperature B factor (Fig. 1 ▶ b), which is an effect of the high flexibility of this part of the protein chain. In addition, a higher level of thermal vibration is observed in almost all loop regions of chain B, with the exception of the L2 loop. The remaining parts of the model, including the core and the C-terminal fragment, exhibit comparable low B-factor values, as is reflected in the quality of the obtained electron-density maps, which did not cause any difficulties in interpretation during fitting and rebuilding of the model.
Comparison of the known E. coli, rabbit and human HINT1 structures indicates some level of flexibility of the L1, L3 and L6 loop regions which are involved in building the binding site (Fig. 4 ▶). In addition, the bacterial and mammalian HINTs also show significant differences in the N-terminal and C-terminal structure fragments. Mammalians HINTs have a longer polypeptide chain, and sequence comparison with the E. coli homologue indicates that this difference relates to the N-terminal part, which is involved in direct interaction with the pontin and reptin proteins (Weiske & Huber, 2006 ▶), while the bacterial homologue possesses a short extension of the C-terminal part which apparently does not prevent dimer formation. However, analysis of superposed HINT1 protein structures did not exhibit significant differences in the placement of ligand molecules inside the binding pocket.
Figure 4.

Superposition of the Cα atoms of all E. coli (blue), human (red) and rabbit (green) HINT1 protein structures deposited in the PDB.
4. Conclusion
The crystal structure of human HINT1 in complex with AMP was determined at 1.38 Å resolution, the highest resolution yet recorded for a deposited hHINT structure, and is the first structure determined in a new monoclinic crystal form. Anisotropic refinement of the non-H atoms resulted in a final structure with good crystallographic statistics and stereochemical quality. The presented hHINT1 structure shows that binding of AMP in the binding cleft does not lead to significant conformational changes. Binding of one ligand molecule per HINT1 dimer is an additional interesting observation. However, these excellent resolution data did not allow definition of the structure of the N-terminal fragment of the hHINT1 chain, similarly to previously reported data. The influence of crystallization conditions on the crystal forms obtained and on the packing of hHINT1 crystals needs to be investigated.
Supplementary Material
PDB reference: hHINT1–AMP complex, 3tw2
Acknowledgments
This research was financially assisted by Polish Ministry of Science and Higher Education grant NN204516139 (to RD) for the years 2010–2013.
References
- Andersson, K. M. & Hovmöller, S. (2000). Acta Cryst. D56, 789–790. [DOI] [PubMed]
- Barbier, E. & Wang, J. B. (2009). BMC Neurosci. 10, 132. [DOI] [PMC free article] [PubMed]
- Barbier, E., Zapata, A., Oh, E., Liu, Q., Zhu, F., Undie, A., Shippenberg, T. & Wang, J. B. (2007). Neuropsychopharmacology, 32, 1774–1782. [DOI] [PubMed]
- Bardaweel, S., Pace, J., Chou, T.-F., Cody, V. & Wagner, C. R. (2010). J. Mol. Biol. 404, 627–638. [DOI] [PubMed]
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Bieganowski, P., Garrison, P. N., Hodawadekar, S. C., Faye, G., Barnes, L. D. & Brenner, C. (2002). J. Biol. Chem. 277, 10852–10860. [DOI] [PMC free article] [PubMed]
- Boulanger, R. R. & Kantrowitz, E. R. (2003). J. Biol. Chem. 278, 23497–23501. [DOI] [PubMed]
- Brenner, C. (2002). Biochemistry, 41, 9003–9014. [DOI] [PMC free article] [PubMed]
- Brenner, C., Garrison, P., Gilmour, J., Peisach, D., Ringe, D., Petsko, G. A. & Lowenstein, J. M. (1997). Nature Struct. Biol. 4, 231–238. [DOI] [PMC free article] [PubMed]
- Cen, B., Li, H. & Weinstein, I. B. (2009). J. Biol. Chem. 284, 5265–5276. [DOI] [PubMed]
- Chen, Q., Wang, X., O’Neill, F. A., Walsh, D., Kendler, K. S. & Chen, X. (2008). Schizophr. Res. 106, 200–207. [DOI] [PMC free article] [PubMed]
- Chou, T.-F., Bieganowski, P., Shilinski, K., Cheng, J., Brenner, C. & Wagner, C. R. (2005). J. Biol. Chem. 280, 15356–15361. [DOI] [PMC free article] [PubMed]
- Chou, T.-F., Tikh, I. B., Horta, B. A., Ghosh, B., De Alencastro, R. B. & Wagner, C. R. (2007). J. Biol. Chem. 282, 15137–15147. [DOI] [PubMed]
- Chou, T.-F. & Wagner, C. R. (2007). J. Biol. Chem. 282, 4719–4727. [DOI] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Dolot, R., Ozga, M., Krakowiak, A. & Nawrot, B. (2011). Acta Cryst. D67, 601–607. [DOI] [PubMed]
- Elashoff, M., Higgs, B. W., Yolken, R. H., Knable, M. B., Weis, S., Webster, M. J., Barci, B. M. & Torrey, E. F. (2007). J. Mol. Neurosci. 31, 221–243. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Gilmour, J., Liang, N. & Lowenstein, J. M. (1997). Biochem. J. 326, 471–477. [DOI] [PMC free article] [PubMed]
- Guang, W., Wang, H., Su, T., Weinstein, I. B. & Wang, J. B. (2004). Mol. Pharmacol. 66, 1285–1292. [DOI] [PubMed]
- Guranowski, A., Wojdyła, A. M., Rydzik, A. M., Stepiński, J. & Jemielity, J. (2011). Acta Biochim. Pol. 58, 131–136. [PubMed]
- Hsieh, S.-Y., Hsu, C.-Y., He, J.-R., Liu, C.-L., Lo, S.-J., Chen, Y.-C. & Huang, H.-Y. (2009). J. Proteome Res. 8, 3977–3986. [DOI] [PubMed]
- Kabsch, W. & Sander, C. (1983). Biopolymers, 22, 2577–2637. [DOI] [PubMed]
- Klein, M. G., Yao, Y., Slosberg, E. D., Lima, C. D., Doki, Y. & Weinstein, I. B. (1998). Exp. Cell Res. 244, 26–32. [DOI] [PubMed]
- Korsisaari, N., Rossi, D. J., Luukko, K., Huebner, K., Henkemeyer, M. & Mäkelä, T. P. (2003). Mol. Cell. Biol. 23, 3929–3935. [DOI] [PMC free article] [PubMed]
- Krakowiak, A., Kaczmarek, R., Baraniak, J., Wieczorek, M. & Stec, W. J. (2007). Chem. Commun., pp. 2163–2165. [DOI] [PubMed]
- Krakowiak, A., Pace, H. C., Blackburn, G. M., Adams, M., Mekhalfia, A., Kaczmarek, R., Baraniak, J., Stec, W. J. & Brenner, C. (2004). J. Biol. Chem. 279, 18711–18716. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lee, Y.-N., Nechushtan, H., Figov, N. & Razin, E. (2004). Immunity, 20, 145–151. [DOI] [PubMed]
- Lee, Y.-N. & Razin, E. (2005). Mol. Cell. Biol. 25, 8904–8912. [DOI] [PMC free article] [PubMed]
- Li, H., Zhang, Y., Su, T., Santella, R. M. & Weinstein, I. B. (2006). Oncogene, 25, 713–721. [DOI] [PubMed]
- Lima, C. D., Klein, M. G. & Hendrickson, W. A. (1997). Science, 278, 286–290. [DOI] [PubMed]
- Lima, C. D., Klein, M. G., Weinstein, I. B. & Hendrickson, W. A. (1996). Proc. Natl Acad. Sci. USA, 93, 5357–5362. [DOI] [PMC free article] [PubMed]
- Liu, Q., Puche, A. C. & Wang, J. B. (2008). Neurochem. Res. 33, 1263–1276. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Murakami, E., Tolstykh, T., Bao, H., Niu, C., Steuer, H. M., Bao, D., Chang, W., Espiritu, C., Bansal, S., Lam, A. M., Otto, M. J., Sofia, M. J. & Furman, P. A. (2010). J. Biol. Chem. 285, 34337–34347. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Ozga, M. (2010). Postepy Biochem. 56, 55–60. [PubMed]
- Parks, K. P., Seidle, H., Wright, N., Sperry, J. B., Bieganowski, P., Howitz, K., Wright, D. L. & Brenner, C. (2004). Physiol. Genomics, 20, 12–14. [DOI] [PMC free article] [PubMed]
- Pearson, J. D., DeWald, D. B., Mathews, W. R., Mozier, N. M., Zürcher-Neely, H. A., Heinrikson, R. L., Morris, M. A., McCubbin, W. D., McDonald, J. R., Fraser, E. D., Vogel, H. J., Kay, C. M. & Walsh, M. P. (1990). J. Biol. Chem. 265, 4583–4591. [PubMed]
- Su, T., Suzui, M., Wang, L., Lin, C.-S., Xing, W.-Q. & Weinstein, I. B. (2003). Proc. Natl Acad. Sci. USA, 100, 7824–7829. [DOI] [PMC free article] [PubMed]
- Teng, T.-Y. (1990). J. Appl. Cryst. 23, 387–391.
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Varadarajulu, J., Lebar, M., Krishnamoorthy, G., Habelt, S., Lu, J., Weinstein, I. B., Li, H., Holsboer, F., Turck, C. W. & Touma, C. (2011). Behav. Brain Res. 220, 305–311. [DOI] [PubMed]
- Vawter, M. P., Weickert, C. S., Ferran, E., Matsumoto, M., Overman, K., Hyde, T. M., Weinberger, D. R., Bunney, W. E. & Kleinman, J. E. (2004). Neurochem. Res. 29, 1245–1255. [DOI] [PubMed]
- Wang, J., Fang, P., Schimmel, P. & Guo, M. (2012). J. Phys. Chem. B, 116, 6798–6805. [DOI] [PMC free article] [PubMed]
- Wang, L., Li, H., Zhang, Y., Santella, R. M. & Weinstein, I. B. (2009). Int. J. Cancer, 124, 1526–1534. [DOI] [PMC free article] [PubMed]
- Wang, L., Zhang, Y., Li, H., Xu, Z., Santella, R. M. & Weinstein, I. B. (2007). Cancer Res. 67, 4700–4708. [DOI] [PubMed]
- Weiske, J. & Huber, O. (2005). J. Cell Sci. 118, 3117–3129. [DOI] [PubMed]
- Weiske, J. & Huber, O. (2006). J. Biol. Chem. 281, 27356–27366. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wu, L., Wu, X., Deng, H. & Huang, Y. (2010). Dev. Comp. Immunol. 34, 76–83. [DOI] [PubMed]
- Zambelli, D., Zuntini, M., Nardi, F., Manara, M. C., Serra, M., Landuzzi, L., Lollini, P.-L., Ferrari, S., Alberghini, M., Llombart-Bosch, A., Piccolo, E., Iacobelli, S., Picci, P. & Scotlandi, K. (2010). Int. J. Cancer, 126, 41–52. [DOI] [PubMed]
- Zhang, Y.-J., Li, H., Wu, H.-C., Shen, J., Wang, L., Yu, M.-W., Lee, P.-H., Weinstein, I. B. & Santella, R. M. (2009). Cancer Lett. 275, 277–284. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: hHINT1–AMP complex, 3tw2