

Crystals of native and selenomethionine-labelled STING138–344 protein from M. musculus diffracted to resolutions of 2.39 and 2.2 Å, respectively
Keywords: STING, c-di-GMP, innate immune response, pathogens
Abstract
The innate immune response is the first defence system against pathogenic microorganisms, and cytosolic detection of pathogen-derived DNA is believed to be one of the major mechanisms of interferon production. Recently, the mammalian ER membrane protein STING (stimulator of IFN genes; also known as MITA, ERIS, MPYS and TMEM173) has been found to be the master regulator linking the detection of cytosolic DNA to TANK-binding kinase 1 (TBK1) and its downstream transcription factor IFN regulatory factor 3 (IRF3). In addition, STING itself was soon discovered to be a direct sensor of bacterial cyclic dinucleotides such as c-di-GMP or c-di-AMP. However, structural studies of apo STING and its complexes with these cyclic dinucleotides and with other cognate binding proteins are essential in order to fully understand the roles played by STING in these crucial signalling pathways. In this manuscript, the successful crystallization of the C-terminal domain of murine STING (STING-CTD; residues 138–344) is reported. Native and SeMet-labelled crystals were obtained and diffracted to moderate resolutions of 2.39 and 2.2 Å, respectively.
1. Introduction
Cyclic di-GMP (c-di-GMP) is a unique secondary messenger that controls a plethora of cellular activities such as biofilm formation, biogenesis of flagella and pili, secretion of pathogenic factors etc. in diverse bacteria (Römling et al., 2005 ▶; Jenal & Malone, 2006 ▶; Römling & Amikam, 2006 ▶; Hengge, 2009 ▶; Schirmer & Jenal, 2009 ▶). Its synthesis via GGDEF-domain-containing diguanlyate cyclases (DGCs) and degradation via EAL-domain-containing (Tal et al., 1998 ▶; Simm et al., 2004 ▶; Tischler & Camilli, 2004 ▶; Römling et al., 2005 ▶) or HD-GYP-domain-containing (Slater et al., 2000 ▶; Ryan et al., 2006 ▶) phosphodiesterases (PDEs) has been well investigated in recent years. However, it remains unclear how many distinct c-di-GMP receptors are available and how these receptors execute their functions upon c-di-GMP binding in the cell, although a wide variety of different protein-based or RNA-based recognition motifs for c-di-GMP have been discovered, including those from the transcriptional factors Clp (Leduc & Roberts, 2009 ▶; Chin et al., 2010 ▶; Tao et al., 2010 ▶), FleQ (Hickman & Harwood, 2008 ▶) and VspT (Krasteva et al., 2010 ▶), from RNA-processing polynucleotide phosphorylase (PNPase; Tuckerman et al., 2011 ▶), from degenerate GGDEF or EAL domains (Navarro et al., 2009 ▶, 2011 ▶), from PilZ-domain proteins (Amikam & Galperin, 2006 ▶; Benach et al., 2007 ▶; Li et al., 2009 ▶; Ko et al., 2010 ▶; Habazettl et al., 2011 ▶; Li et al., 2011 ▶; Liao et al., 2012 ▶) and from riboswitches (Kulshina et al., 2009 ▶; Smith et al., 2009 ▶, 2011 ▶). The search for novel c-di-GMP receptors is still ongoing (Römling, 2011 ▶; Sondermann et al., 2011 ▶; Ryan et al., 2012 ▶).
Another unique cyclic dinucleotide, c-di-AMP, has recently been discovered and found to play roles in regulating cell-cycle progression (Römling, 2008 ▶; Witte et al., 2008 ▶; Corrigan et al., 2011 ▶; Oppenheimer-Shaanan et al., 2011 ▶) as well as controlling cell size and envelope stress (Corrigan et al., 2011 ▶). Interestingly, both c-di-AMP and c-di-GMP have been found to activate a host type I interferon response (Karaolis et al., 2007 ▶; McWhirter et al., 2009 ▶; Woodward et al., 2010 ▶; Jin et al., 2011 ▶; Sauer et al., 2011 ▶) and the C-terminal domain of the STING protein (STING-CTD) has been identified as the direct innate immune sensor of c-di-GMP (Burdette et al., 2011 ▶), providing a scaffold to specify and promote phosphorylation of IFN regulatory factor 3 (IRF3) by TANK-binding kinase 1 (TBK1) (Tanaka & Chen, 2012 ▶). The phosphorylated IRF3 then dimerizes and translocates into the nucleus to bind at the IFNB promoter to induce interferon expression (Bowie, 2012 ▶). Structural studies are required to better characterize the interactions between STING and cyclic dinucleotides or other cognate binding proteins, which will allow a more detailed understanding of the roles played by STING in these crucial self-defence signalling pathways in eukaryotic cells.
To date, however, no such information about STING and/or its complexes is available. In this manuscript, we report the first successful crystallization of the murine STING138–344 domain. Native and SeMet-labelled crystals have been obtained and diffracted to resolutions of 2.39 and 2.2 Å, respectively.
2. Materials and methods
2.1. Reagents
c-di-GMP was produced by an enzymatic method using an altered thermophilic DGC enzyme as described previously (Rao et al., 2009 ▶).
2.2. Cloning and purification
The whole Mus musculus (murine) STING gene was synthesized using a cost-effective PCR-based two-step DNA-synthesis method (Xiong et al., 2008 ▶). The codons were optimized and designed using the DNAWorks software (Hoover & Lubkowski, 2002 ▶) to achieve a higher level of expression in Escherichia coli. The optimized oligomer and primer sequences used for STING gene assembly are listed in Table 1 ▶.
Table 1. List of the oligomers/primers used in assembling the M. musculus (murine) STING gene.
| Primer No. | Oligomer sequence 5′–3′ |
|---|---|
| 1 | TACTTCCAATCCAATGCTCTGACACCTGC |
| 2 | CCACATTCAGTTTCTTTTCTTCGCACACCGCGCTCACTTCCGCAGGTGTCAGAGCATTGG |
| 3 | CGAAGAAAAGAAACTGAATGTGGCGCATGGCCTGGCGTGGAGCTACTATATTGGCTATCT |
| 4 | CGAATCCGCGCTTGCAGCCCAGGCAGAATCAGCCGCAGATAGCCAATATAGTAGCTCCAC |
| 5 | CTGCAAGCGCGGATTCGTATGTTTAATCAGCTGCATAACAACATGCTGAGCGGTGCCGGC |
| 6 | CTGGCACGCCGCAATCCAGCGGAAACAGGATATACAGACGACGGGAGCCGGCACCGCTCA |
| 7 | GATTGCGGCGTGCCAGATAATCTGAGCGTGGTGGATCCGAACATTCGTTTTCGTGATATG |
| 8 | TCTTAATGCCCGCACGATCAATATTCTGCTGCGGCAGCATATCACGAAAACGAATGTTCG |
| 9 | GATCGTGCGGGCATTAAGAATCGTGTGTATAGCAATAGCGTGTATGAAATCCTGGAAAAT |
| 10 | TCGCATATTCCAGAATGCACACTCCAGCCGGTTGGCCATTTTCCAGGATTTCATACACGC |
| 11 | TGTGCATTCTGGAATATGCGACCCCGCTGCAGACCCTGTTTGCAATGTCACAGGACGCGA |
| 12 | GTTTGGCCTGTTCTAAACGATCTTCACGGCTAAAGCCCGCTTTCGCGTCCTGTGACATTG |
| 13 | ATCGTTTAGAACAGGCCAAACTGTTTTGCCGTACCCTGGAAGAAATTTTGGAAGACGTGC |
| 14 | TTGATACACTATCAGACGGCAATTGTTACGGCTTTCCGGCACGTCTTCCAAAATTTCTTC |
| 15 | TGCCGTCTGATAGTGTATCAAGAACCGACCGATGGCAATTCATTTTCACTGTCGCAGGAA |
| 16 | CACTTCTTCTTTCTCTTCCTGACGAATATGACGCAGCACTTCCTGCGACAGTGAAAATGA |
| 17 | GTCAGGAAGAGAAAGAAGAAGTGACCATGAATGCGCCGATGACCAGCGTGGCGCCTCCGC |
| 18 | TCCATACCGCTTATCAGCAGACGCGGCTCCTGGCTCAACACGCTCGGCGGAGGCGCCACG |
| 19 | CTGCTGATAAGCGGTATGGACCAGCCGCTGCCCCTGCGTACGGATCTGATTTGACATTGG |
| 20 | TTATCCACTTCCAATGTCAAATCAGATCCGTAC |
In order to obtain STING protein with improved solubility, we constructed a series of STING gene fragments of different lengths (Fig. 1 ▶ a). The STING gene fragments STING138–333, STING138–344, STING138–378, STING179–344 and STING179–333 were chosen based on their hydropathic profiles. The STING138–333, STING138–344 and STING138–378 truncations were PCR-amplified directly from the whole synthesized gene template by using the same forward primer 5′-TACTTCCAATCCAATGCTCTGACACCTGCGGAAGTGAG-3′ with different reverse primers 5′-TATCCACTTCCAATGTCAACGAATATGACGCAGCACTTC-3′ for the STING138–333 domain, 5′-TATCCACTTCCAATGTCACGCATTCATGGTCACTTCTTC-3′ for the STING138–344 domain and 5′-TTATCCACTTCCAATGTCAAATCAGATCCGTAC-3′ for the STING138–378 domain, while the STING179–344 and STING179–333 truncations were synthesized using the same forward primer 5′-TACTTCCAATCCAATGCTCGTATGTTTAATCAGCTGCATAAC-3′ with different reverse primers 5′-TATCCACTTCCAATGTCAACGAATATGACGCAGCACTTC-3′ for the STING179–333 domain and 5′-TATCCACTTCCAATGTCACGCATTCATGGTCACTTCTTC-3′ for the STING179–344 domain.
Figure 1.

(a) The domain architecture and constructs used in these studies. Strongly predicted transmembrane segments are shown as boxes containing solid lines, while weakly predicted transmembrane segments are shown as boxes containing dotted lines. The constructs are indicated by white boxes; the starting and ending positions are indicated by arrows. (b) 13% SDS–PAGE monitoring of the overexpression and purification of the STING138–378 (lanes 1, 2, 3 and 4), STING138–344 (lanes 5, 6, 7 and 8) and STING138–333 (lanes 9, 10, 11 and 12) domains. Lane M, protein marker (labelled in kDa); lanes 1, 5 and 9, whole cell lysate before IPTG induction; lanes 2, 6 and 10, whole cell lysate after IPTG induction; lanes 3, 7 and 11, nickel-column-purified domains after IPTG induction; lanes 4, 8 and 12, nickel-column-purified domains after TEV cleavage.
The obtained PCR fragment exhibited the correct size in an agarose-gel electrophoresis experiment and was confirmed by DNA sequencing. A ligation-independent cloning (LIC) approach (Aslanidis & de Jong, 1990 ▶; Stols et al., 2001 ▶; Wu et al., 2005 ▶) was then used to obtain the desired constructs. The final constructs code for an N-terminal His6 tag, a 17-amino-acid linker and the STING138–333, STING138–344, STING138–378, STING179–333 and STING179–344 truncations under the control of a T7 promoter. Overexpression of the His6-tagged proteins was induced by the addition of 800 µl 500 mM IPTG to the medium (to give a final IPTG concentration of 0.5 mM) at 293 K for 18 h. The cells were harvested, resuspended in lysis buffer (20 mM Tris–HCl pH 8.0, 80 mM NaCl) and ruptured using a microfluidizer (Microfluidics). Most of the target protein was found to be present in the soluble fraction after centrifugation. Surprisingly, exclusion of the last putative transmembrane-containing region (residues 138–179; Burdette et al., 2011 ▶) only gave inclusion bodies; only truncations starting from residue 138 delivered soluble proteins. The three soluble truncated proteins were purified by immobilized metal-affinity chromatography (IMAC) on a nickel column (Sigma) equilibrated with a buffer consisting of 20 mM Tris–HCl pH 8.0, 80 mM NaCl and eluted with a gradient of 50–300 mM imidazole in the same buffer. The fractions containing the STING138–333, STING138–344 and STING138–378 domains were monitored using 13% SDS–PAGE and recombined. The His6 tag and linker were further cleaved from the STING138–333, STING138–344 and STING138–378 domains using tobacco etch virus (TEV) protease at 289 K for 16 h (Fig. 1 ▶ b). The final product contained an extra tripeptide SNA at the N-terminal end after cleavage of the His6-tag and linker sequence (MHHHHHHSTSVDLGTENLYFQ) from the ligation vector. For crystallization, the STING138–333, STING138–344 and STING138–378 domains were further purified on a Sephadex gel-filtration column (ÄKTA; Pharmacia Inc.) using the lysis buffer. SeMet-labelled STING138–344 was further prepared in order to solve the phase problem. The labelled domain was generated in a similar way except that it was produced using the non-auxotrophic E. coli strain Rosetta (DE3) as the host in the absence of methionine but with ample amounts of SeMet (100 mg l−1). The M9 medium consisted of 1 g NH4Cl, 3 g KH2PO4 and 6 g Na2HPO4 supplemented with 20%(w/v) glucose, 0.3%(w/v) MgSO4 and 10 mg FeSO4 in 1 l double-distilled water. Induction was performed at 293 K for 18 h by the addition of IPTG to 450 ml M9 medium (to give a final IPTG concentration of 0.5 mM). Purification of the SeMet-labelled STING138–344 protein was performed using the same procedure as used for the native protein.
2.3. Crystallization
For crystallization, native STING138–333, STING138–344 and STING138–378 domains and SeMet-labelled STING138–344 domain were concentrated to approximately 6.5 mg ml−1 in 20 mM Tris–HCl pH 8.0, 80 mM sodium chloride using an Amicon Ultra-10 (Millipore). Appropriate volumes of 0.5 mM c-di-GMP were also added to the solutions of the native and SeMet-labelled STING138–333, STING138–344 and STING138–378 domains to prepare samples for STING–c-di-GMP cocrystallization at a 2:1 ligand:protein ratio. Screening for crystallization conditions of the SeMet-labelled protein were performed using sitting-drop vapour diffusion in 96-well plates (Hampton Research) at 277 K by mixing 0.5 µl protein solution with 0.5 µl reservoir solution and equilibrating against 50 µl reservoir solution. Initial screens, including the Crystal Screen and Crystal Screen 2 sparse-matrix screens (Hampton Research), a systematic PEG–pH screen and the PEG/Ion screen (Hampton Research), were performed using a Gilson C240 crystallization workstation. Samples of the native STING138–333, STING138–344 and STING138–378 domains and their ligand-bound complexes were screened under similar conditions. Pyrimid-shaped crystals of the STING138–344 domain appeared in 7 d from drops equilibrated against 50 µl reservoir solution comprising 2% PEG 400, 1.6 M ammonium sulfate, 0.1 M MES monohydrate, while pyrimid-shaped crystals of SeMet-labelled STING138–344 appeared in 7 d from drops equilibrated against 50 µl reservoir solution comprising 1.6 M potassium/sodium phosphate, 0.1 M Na HEPES pH 7.5 (Fig. 2 ▶). Crystals of both proteins suitable for diffraction experiments were grown from drops by mixing 1.5 µl protein solution with 1.5 µl reservoir solution and equilibrating against 500 µl reservoir solution at 277 K. Crystals of STING138–344 reached approximate dimensions of 0.05 × 0.05 × 0.1 mm and those of SeMet-STING138–344 reached approximate dimensions of 0.1 × 0.1 × 0.15 mm after one week.
Figure 2.

Crystals of the STING138–344 and SeMet-labelled STING138–344 domains. (a) STING138–344 crystals grown in 2% PEG 400, 1.6 M ammonium sulfate, 0.1 M MES monohydrate using the hanging-drop vapour-diffusion method at 298 K. These crystals reached average dimensions of 0.05 × 0.05 × 0.1 mm after one week. (b) SeMet-labelled STING138–344 crystals grown in 1.6 M potassium/sodium phosphate, 0.1 M Na HEPES pH 7.5. These crystals reached average dimensions of 0.1 × 0.1 × 0.15 mm after one week.
2.4. Data collection and processing
Crystals of both proteins/complexes were flash-cooled at 100 K under a stream of cold nitrogen gas using the reservoir solution with 10% glycerol as cryoprotectant. Prior to data collection, the crystals were scanned for Se absorption and 0.97622 Å was found to be the peak wavelength of the selenium anomalous signal. X-ray diffraction data were collected from native STING138–344 and SeMet-labelled STING138–344 crystals on beamlines BL13C1 and BL13B1, respectively, at the National Synchrotron Radiation Research Center (NSRRC) in Taiwan and reached resolutions of 2.39 and 2.2 Å, respectively (Fig. 3 ▶). The native and SeMet-labelled STING138–344 data were indexed and integrated using the HKL-2000 processing software (Otwinowski & Minor, 1997 ▶), generating data sets that were 100% complete. The crystals of STING138–344 and SeMet-labelled STING138–344 were found to belong to space group P31 or P32. Detailed statistics for the quality of the collected data are listed in Table 2 ▶. The Matthews coefficient and solvent content were 1.91 Å3 Da−1 and 35.59%, respectively, for native STING138–344 and were 1.90 Å3 Da−1 and 35.37%, respectively, for SeMet-labelled STING138–344.
Figure 3.

Diffraction pattern of SeMet-labelled STING138–344 protein collected using a MAR CCD detector on the BL13B1 beamline at the National Synchrotron Radiation Research Center (NSRRC) in Taiwan. The exposure time was 8 s, the oscillation range was 1° per frame and the crystal-to-detector distance was 300 mm. The edge of the detector corresponds to a resolution of 2.2 Å.
Table 2. Summary of the crystallographic data for the native STING138–344 and SeMet-labelled STING138–344 targets.
Values in parentheses are for the outermost resolution shell.
| STING138–344 | SeMet STING138–344, peak | |
|---|---|---|
| Beamline | BL13C1, NSRRC | BL13B1, NSRRC |
| Wavelength (Å) | 0.98922 | 0.97622 |
| Space group | P31 or P32 | P31 or P32 |
| Unit-cell parameters (Å, °) | a = b = 78.619, c = 50.418, α = β = 90, γ = 120 | a = b = 78.493, c = 50.409, α = β = 90, γ = 120 |
| Resolution range (Å) | 30–2.39 (2.48–2.39) | 30–2.20 (2.28–2.20) |
| Total reflections | 44542 (4387) | 125788 (12546) |
| Unique reflections | 13794 (1371) | 17676 (1767) |
| Multiplicity | 3.2 (3.2) | 7.1 (7.1) |
| Completeness (%) | 100 (100) | 100 (100) |
| R merge † (%) | 6.1 (59.5) | 6.5 (44.7) |
| 〈I/σ(I)〉 | 19.4 (2.3) | 23.8 (4.9) |
| Matthews coefficient (Å3 Da−1) | 1.91 | 1.90 |
| Solvent content (%) | 35.59 | 35.37 |
R
merge = 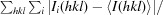
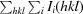 , where I
i(hkl) is the ith intensity measurement of reflection hkl, including symmetry-related reflections, and 〈I(hkl)〉 is its average.
, where I
i(hkl) is the ith intensity measurement of reflection hkl, including symmetry-related reflections, and 〈I(hkl)〉 is its average.
3. Results and discussion
In this manuscript, we report the successful cloning, protein expression and purification of the STING138–344 protein and the crystal screening and preliminary X-ray data analyses of native and SeMet-labelled STING138–344 proteins. Initially, we constructed a series of STING clones with different N-terminal and C-terminal sequences (Fig. 1 ▶ a). Unexpectedly, constructs starting from residue 179 that lacked a putative transmembrane segment gave proteins in inclusion bodies, and only constructs starting from residue 138 (STING138–333, STING138–344 and STING138–378) that contained a putative transmembrane segment gave soluble protein. As shown in Fig. 1 ▶(b), the His6 tag and linker of the STING138–333, STING138–344 and STING138–378 domains could be successfully cleaved by TEV protease at 289 K for 16 h to obtain the domains with a purity of >95%. The domains contained an extra tripeptide SNA at the N-terminal end after cleavage of the His6-tag and linker sequence (MHHHHHHSTSVDLGTENLYFQ) from the ligation vector. These domains were further purified by gel-filtration chromatography. However, no crystal formation was observed for the STING138–333 and STING138–378 domains. Hence, only the STING138–344 domain was further studied.
Since STING does not seem to share homology with any known immunosensors and may represent a novel category of microbial detector (Burdette et al., 2011 ▶), solution of its structure using a molecular-replacement approach is unlikely. Therefore, in order to obtain the essential phase information, we further screened the SeMet-labelled STING138–344 domain for crystallization. Luckily, crystals of the SeMet-labelled STING138–344 domain were obtained after one week. Further refinements of the STING138–344 protein/complex are now in progress.
Acknowledgments
This work was supported in part by the Ministry of Education, Taiwan, Republic of China under the ATU plan and by the National Science Council, Taiwan, Republic of China (grant 97-2113-M005-005-MY3 to S-HC). We appreciate the Structural Genomics Databases service provided by the GMBD Bioinformatics Core (http://www.tbi.org.tw), NRPGM, Taiwan, Republic of China. We would also like to thank the Core Facilities for Protein X-ray Crystallography in the Academia Sinica, Taiwan, Republic of China for help in crystal screening, the National Synchrotron Radiation Research Center (NSRRC) in Taiwan and the SPring-8 Synchrotron facility in Japan for assistance in X-ray data collection. The National Synchrotron Radiation Research Center is a user facility supported by the National Science Council, Taiwan, Republic of China and the Protein Crystallography Facility is supported by the National Research Program for Genomic Medicine, Taiwan, Republic of China.
References
- Amikam, D. & Galperin, M. Y. (2006). Bioinformatics, 22, 3–6. [DOI] [PubMed]
- Aslanidis, C. & de Jong, P. J. (1990). Nucleic Acids Res. 18, 6069–6074. [DOI] [PMC free article] [PubMed]
- Benach, J., Swaminathan, S. S., Tamayo, R., Handelman, S. K., Folta-Stogniew, E., Ramos, J. E., Forouhar, F., Neely, H., Seetharaman, J., Camilli, A. & Hunt, J. F. (2007). EMBO J. 26, 5153–5166. [DOI] [PMC free article] [PubMed]
- Bowie, A. (2012). Sci. Signal. 5, pe9. [DOI] [PubMed]
- Burdette, D. L., Monroe, K. M., Sotelo-Troha, K., Iwig, J. S., Eckert, B., Hyodo, M., Hayakawa, Y. & Vance, R. E. (2011). Nature (London), 478, 515–518. [DOI] [PMC free article] [PubMed]
- Chin, K.-H., Lee, Y.-C., Tu, Z.-L., Chen, C.-H., Tseng, Y.-H., Yang, J.-M., Ryan, R. P., McCarthy, Y., Dow, J. M., Wang, A. H.-J. & Chou, S.-H. (2010). J. Mol. Biol. 396, 646–662. [DOI] [PubMed]
- Corrigan, R. M., Abbott, J. C., Burhenne, H., Kaever, V. & Gründling, A. (2011). PLoS Pathog. 7, e1002217. [DOI] [PMC free article] [PubMed]
- Habazettl, J., Allan, M. G., Jenal, U. & Grzesiek, S. (2011). J. Biol. Chem. 286, 14304–14314. [DOI] [PMC free article] [PubMed]
- Hengge, R. (2009). Nature Rev. Microbiol. 7, 263–273. [DOI] [PubMed]
- Hickman, J. W. & Harwood, C. S. (2008). Mol. Microbiol. 69, 376–389. [DOI] [PMC free article] [PubMed]
- Hoover, D. M. & Lubkowski, J. (2002). Nucleic Acids Res. 30, e43. [DOI] [PMC free article] [PubMed]
- Jenal, U. & Malone, J. (2006). Annu. Rev. Genet. 40, 385–407. [DOI] [PubMed]
- Jin, L., Hill, K. K., Filak, H., Mogan, J., Knowles, H., Zhang, B., Perraud, A.-L., Cambier, J. C. & Lenz, L. L. (2011). J. Immunol. 187, 2595–2601. [DOI] [PMC free article] [PubMed]
- Karaolis, D. K., Means, T. K., Yang, D., Takahashi, M., Yoshimura, T., Muraille, E., Philpott, D., Schroeder, J. T., Hyodo, M., Hayakawa, Y., Talbot, B. G., Brouillette, E. & Malouin, F. (2007). J. Immunol. 178, 2171–2181. [DOI] [PubMed]
- Ko, J., Ryu, K.-S., Kim, H., Shin, J.-S., Lee, J.-O., Cheong, C. & Choi, B.-S. (2010). J. Mol. Biol. 398, 97–110. [DOI] [PubMed]
- Krasteva, P. V., Fong, J. C., Shikuma, N. J., Beyhan, S., Navarro, M. V., Yildiz, F. H. & Sondermann, H. (2010). Science, 327, 866–868. [DOI] [PMC free article] [PubMed]
- Kulshina, N., Baird, N. J. & Ferré-D’Amaré, A. R. (2009). Nature Struct. Mol. Biol. 16, 1212–1217. [DOI] [PMC free article] [PubMed]
- Leduc, J. L. & Roberts, G. P. (2009). J. Bacteriol. 191, 7121–7122. [DOI] [PMC free article] [PubMed]
- Li, T.-N., Chin, K.-H., Fung, K.-M., Yang, M.-T., Wang, A. H.-J. & Chou, S.-H. (2011). PLoS One, 6, e22036. [DOI] [PMC free article] [PubMed]
- Li, T.-N., Chin, K.-H., Liu, J.-H., Wang, A. H.-J. & Chou, S.-H. (2009). Proteins, 75, 282–288. [DOI] [PubMed]
- Liao, Y.-T., Chin, K.-H., Kuo, W.-T., Chuah, M. L.-C., Liang, Z.-X. & Chou, S.-H. (2012). Acta Cryst. F68, 301–305. [DOI] [PMC free article] [PubMed]
- McWhirter, S. M., Barbalat, R., Monroe, K. M., Fontana, M. F., Hyodo, M., Joncker, N. T., Ishii, K. J., Akira, S., Colonna, M., Chen, Z. J., Fitzgerald, K. A., Hayakawa, Y. & Vance, R. E. (2009). J. Exp. Med. 206, 1899–1911. [DOI] [PMC free article] [PubMed]
- Navarro, M. V., De, N., Bae, N., Wang, Q. & Sondermann, H. (2009). Structure, 17, 1104–1116. [DOI] [PMC free article] [PubMed]
- Navarro, M. V., Newell, P. D., Krasteva, P. V., Chatterjee, D., Madden, D. R., O’Toole, G. A. & Sondermann, H. (2011). PLoS Biol. 9, e1000588. [DOI] [PMC free article] [PubMed]
- Oppenheimer-Shaanan, Y., Wexselblatt, E., Katzhendler, J., Yavin, E. & Ben-Yehuda, S. (2011). EMBO Rep. 12, 594–601. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Rao, F., Pasunooti, S., Ng, Y., Zhuo, W., Lim, L., Liu, A. W. & Liang, Z.-X. (2009). Anal. Biochem. 389, 138–142. [DOI] [PubMed]
- Römling, U. (2008). Sci. Signal. 1, pe39. [DOI] [PubMed]
- Römling, U. (2011). Environ. Microbiol., doi:10.1111/j.1462-2920.2011.02617.x.
- Römling, U. & Amikam, D. (2006). Curr. Opin. Microbiol. 9, 218–228. [DOI] [PubMed]
- Römling, U., Gomelsky, M. & Galperin, M. Y. (2005). Mol. Microbiol. 57, 629–639. [DOI] [PubMed]
- Ryan, R. P., Fouhy, Y., Lucey, J. F., Crossman, L. C., Spiro, S., He, Y.-W., Zhang, L.-H., Heeb, S., Cámara, M., Williams, P. & Dow, J. M. (2006). Proc. Natl Acad. Sci. USA, 103, 6712–6717. [DOI] [PMC free article] [PubMed] [Retracted]
- Ryan, R. P., Tolker-Nielsen, T. & Dow, J. M. (2012). Trends Microbiol. 20, 235–242. [DOI] [PubMed]
- Sauer, J.-D., Sotelo-Troha, K., von Moltke, J., Monroe, K. M., Rae, C. S., Brubaker, S. W., Hyodo, M., Hayakawa, Y., Woodward, J. J., Portnoy, D. A. & Vance, R. E. (2011). Infect. Immun. 79, 688–694. [DOI] [PMC free article] [PubMed]
- Schirmer, T. & Jenal, U. (2009). Nature Rev. Microbiol. 7, 724–735. [DOI] [PubMed]
- Simm, R., Morr, M., Kader, A., Nimtz, M. & Römling, U. (2004). Mol. Microbiol. 53, 1123–1134. [DOI] [PubMed]
- Slater, H., Alvarez-Morales, A., Barber, C. E., Daniels, M. J. & Dow, J. M. (2000). Mol. Microbiol. 38, 986–1003. [DOI] [PubMed]
- Smith, K. D., Shanahan, C. A., Moore, E. L., Simon, A. C. & Strobel, S. A. (2011). Proc. Natl Acad. Sci. USA, 108, 7757–7762. [DOI] [PMC free article] [PubMed]
- Smith, K. D., Lipchock, S. V., Ames, T. D., Wang, J., Breaker, R. R. & Strobel, S. A. (2009). Nature Struct. Mol. Biol. 16, 1218–1223. [DOI] [PMC free article] [PubMed]
- Sondermann, H., Shikuma, N. J. & Yildiz, F. H. (2011). Curr. Opin. Microbiol. 15, 140–146. [DOI] [PMC free article] [PubMed]
- Stols, L., Gu, M., Dieckman, L., Raffen, R., Collart, F. R. & Donnelly, M. I. (2001). Protein Expr. Purif. 25, 8–15. [DOI] [PubMed]
- Tal, R., Wong, H. C., Calhoon, R., Gelfand, D., Fear, A. L., Volman, G., Mayer, R., Ross, P., Amikam, D., Weinhouse, H., Cohen, A., Sapir, S., Ohana, P. & Benziman, M. (1998). J. Bacteriol. 180, 4416–4425. [DOI] [PMC free article] [PubMed]
- Tanaka, Y. & Chen, Z. J. (2012). Sci. Signal. 5, ra20. [DOI] [PMC free article] [PubMed]
- Tao, F., He, Y.-W., Wu, D.-H., Swarup, S. & Zhang, L.-H. (2010). J. Bacteriol. 192, 1020–1029. [DOI] [PMC free article] [PubMed]
- Tischler, A. D. & Camilli, A. (2004). Mol. Microbiol. 53, 857–869. [DOI] [PMC free article] [PubMed]
- Tuckerman, J. R., Gonzalez, G. & Gilles-Gonzalez, M.-A. (2011). J. Mol. Biol. 407, 622–639. [DOI] [PubMed]
- Witte, G., Hartung, S., Büttner, K. & Hopfner, K. P. (2008). Mol. Cell, 30, 167–178. [DOI] [PubMed]
- Woodward, J. J., Iavaron, A. T. & Portnoy, D. A. (2010). Science, 326, 1703–1705. [DOI] [PMC free article] [PubMed]
- Wu, Y.-Y., Chin, K.-H., Chou, C.-C., Lee, C.-C., Shr, H.-L., Gao, F. P., Lyu, P.-C., Wang, A. H.-J. & Chou, S.-H. (2005). Acta Cryst. F61, 902–905. [DOI] [PMC free article] [PubMed]
- Xiong, A.-S., Peng, R.-H., Zhuang, J., Gao, F., Li, Y., Cheng, Z.-M. & Yao, Q.-H. (2008). FEMS Microbiol. Rev. 32, 522–540. [DOI] [PubMed]