

Abstract
The insect order Dermaptera, belonging to Polyneoptera, includes ∼2,000 extant species, but no dermapteran mitochondrial genome has been sequenced. We sequenced the complete mitochondrial genome of the free-living earwig, Challia fletcheri, compared its genomic features to other available mitochondrial sequences from polyneopterous insects. In addition, the Dermaptera, together with the other known polyneopteran mitochondrial genome sequences (protein coding, ribosomal RNA, and transfer RNA genes), were employed to understand the phylogeny of Polyneoptera, one of the least resolved insect phylogenies, with emphasis on the placement of Dermaptera. The complete mitochondrial genome of C. fletcheri presents the following several unusual features: the longest size in insects is 20,456 bp; it harbors the largest tandem repeat units (TRU) among insects; it displays T- and G-skewness on the major strand and A- and C-skewness on the minor strand, which is a reversal of the general pattern found in most insect mitochondrial genomes, and it possesses a unique gene arrangement characterized by a series of gene translocations and/or inversions. The reversal pattern of skewness is explained in terms of inversion of replication origin. All phylogenetic analyses consistently placed Dermaptera as the sister to Plecoptera, leaving them as the most basal lineage of Polyneoptera or sister to Ephemeroptera, and placed Odonata consistently as the most basal lineage of the Pterygota.
Introduction
Typical metazoan mitochondrial DNA (mtDNA) encodes for 13 proteins, 22 tRNAs, 2 rRNAs, and harbors a single non-coding control region that regulates the transcription and replication of the mtDNA [1], [2]. The size of the insect mitochondrial genome ranges from 14 to 19 kb [2], [3]; one exception is the 30–36 kb-long heteroplasmic genome of the bark weevil, which was determined by size estimation [4]. The source of the size variation is mainly the control region, and is due to the presence of variable lengths and numbers of tandem repeats [5], [6]. Another source is the presence of large intergenic spacers apart from the control region, an infrequent occurance in insects [4], [7]–[9]. These are composed mainly of a variable number of tandem repeats (e.g., a 1,448-bp long intergenic spacer composed of seven 202-bp tandem repeats and a partial 99-bp copy as found in Acyrthosiphon pisum; GenBank: FJ411411) or a non-repetitive sequence of substantial length (e.g., a 733-bp long non-coding sequence as found in Tetraleurodes acacia [9]). Either or both of these two types of large intergenic spacers substantially contribute to the whole genome size in some insects.
Because gene rearrangement is an important evolutionary event in the mitochondrial genome, it likely is regarded as a phylogenetic signal for the inference of evolutionary relationships, e.g. in bilaterian phylogeny [10]. Currently, 11 of 25 insect orders have shown mitochondrial gene rearrangement from the ancestral arrangement of insects, which is identical to that of Drosophila [11], [12]. However, among the 60 sequenced polyneopteran insects representing 9 orders, rearrangement is limited only to the Orthoptera where it involves 2 translocated tRNAs [13] and the Embioptera which shows substantial gene rearrangements [14]. Nevertheless, considering that full-length mitochondrial genome information for polyneopteran Dermaptera and Zoraptera is still unavailable, estimation of the magnitude and extension of gene rearrangement in this group remains far from completion.
Dermaptera are nocturnal insects with the characteristic feature of forceps-like, unsegmented cerci in adults that assists in predation and mating [15]. Dermaptera is divided into 3 extant suborders (Arixeniina, Forficulina, and Hemimerina) and one extinct suborder (Archidermaptera), with ∼ 2,000 extant species [16]. The history of Dermaptera dates back to the Late Triassic to Early Jurassic period of ∼208 million years ago (MYA), based on fossil records found in England and Australia [17]. It has been assumed that Dermaptera originated from Protelytroptera, which resembles modern Blattodea or cockroaches, but no fossil evidence of morphological change from Protelytroptera to Dermaptera has yet been unearthed [17]. The Grylloblattodea was once regarded as very closely related to Dermaptera [18], although a closer relationship of Phasmatodea, Embioptera, Plecoptera, or Dictyoptera to Dermaptera has also been posited [19].
Until now, diverse morphological characteristics [20]–[23] and molecular data from nuclear and mitochondrial genes have been employed to infer phylogenetic relationships among the polyneopteran orders [24]–[27]. From the morphological perspective, Hennig [20] and Wheeler et al. [28] placed Dermaptera as the sister to Dictyoptera on the basis of synapomorphic wing elevators and a basisternal fold, but Kukalová-Peck [22] obtained unresolved relationships among Dermaptera, Grylloblattodea, and Dictyoptera, placing this clade as the sister to the remainders of the Polyneoptera, based on comprehensive morphological data including wing and mouthpart structures. In terms of molecular data, 18S rDNA suggested that Dermaptera was the sister to Plecoptera as a basal lineage for Polyneoptera [26]. However, combined molecular and morphological data instead supports the placement of Dermaptera in an unresolved clade including Grylloblattodea, Zoraptera, and Dictyoptera [28] or a sister relationship between Dermaptera and Zoraptera [25].
In this study, we sequenced the complete mitochondrial genome of Challia fletcheri: the first complete mitochondrial genome for Dermaptera. Although its distribution includes Korea, Japan, and China, this species is listed as an endangered animal in Korea due to its annual decrease in numbers [29], [30]. The genomic sequence of the species is herein described and compared to other polyneopteran insects in terms of comparative genomics. In addition, the genomic sequence was employed for the inference of the phylogenetic position of Dermaptera among the polyenopteran orders.
Results and Discussion
General Features of the Genome
The complete mitochondrial genome of C. fletcheri (GenBank: JN651407) is 20,456-bp in size (Figure 1; Table 1). This is the largest size among the insect mitochondrial genomes sequenced so far, although a bark weevil mitochondrial genome has been estimated to be 30–36 kb [4]. Such a long mitochondrial genome is mainly attributed to the expansions of large non-coding regions and the A+T-rich region. The C. fletcheri contains the ancestral 37 genes, 9 PCGs and 17 tRNAs encoded in the major strand, and 4 PCGs, 5 tRNAs, and 2 rRNAs encoded in the minor strand (Figure 1).
Figure 1. Circular map of the mitochondrial genome of C. fletcheri.

tRNAs are denoted as one-letter symbols consistent with the IUPAC/IUB single letter codes for amino acids, with L = trnL(CUN); L* = trnL(UUR); S = trnS(AGN); S* = trnS(UCN). TRU indicates the tandem repeat unit. Gene names that are not underlined indicate a clockwise transcriptional direction, whereas underlines indicate a counter-clockwise transcriptional direction. Numbers show the sizes of intergenic spacers (positive values) and overlapping region between genes (negative values). The C. fletcheri mitochondrial genome was sequenced by 2 overlapping short (SF1 and SF2) and long (LF1 and LF2) fragments, as shown in a single line within the circle.
Table 1. Summary of mitochondrial genome of C. fletcheri.
| Gene | Direction | Nucleotide number | Size | Anticodon | Start codon | Stop codon |
| trnM | F | 1–68 | 68 | 31–33 CAT | – | – |
| nad2 | F | 69–1085 | 1017 | – | ATT | TAA |
| trnI | F | 1217–1285 | 69 | 1247–1249 GAT | ||
| TRU | 1295–4150 | 2856 | – | |||
| trnW | F | 4174–4245 | 72 | 4206–4208 TCA | – | – |
| cox1 | F | 4284–5825 | 1542 | – | ATA | TAA |
| trnL(UUR) | F | 5850–5920 | 71 | 5884–5886 TAA | – | – |
| cox2 | F | 5918–6601 | 684 | – | ATG | TAA |
| trnK | F | 6615–6685 | 71 | 6645–6647 CTT | – | – |
| trnD | F | 6687–6760 | 74 | 6720–6722 GTC | – | – |
| atp8 | F | 6755–6925 | 171 | – | ATA | TAG |
| atp6 | F | 6919–7602 | 684 | – | ATG | TAA |
| cox3 | F | 7621–8406 | 786 | – | ATG | TAG |
| trnG | F | 8430–8500 | 69 | 8463–8465 TCC | – | – |
| nad3 | F | 8501–8854 | 354 | – | ATT | TAG |
| trnA | F | 8862–8935 | 74 | 8892–8894 TGC | – | – |
| trnN | F | 8947–9011 | 65 | 8978–8980 GTT | – | – |
| trnR | F | 9012–9078 | 67 | 9043–9045 TCG | – | – |
| trnS(AGN) | F | 9125–9191 | 67 | 9150–9152 GCT | – | – |
| trnY | F | 9199–9268 | 70 | 9228–9230 GTA | ||
| trnC | F | 9274–9340 | 67 | 9303–9305 GCA | – | – |
| trnQ | F | 9364–9432 | 69 | 9394–9396 TTG | – | – |
| trnE | R | 9441–9526 | 86 | 9494–9496 TTC | – | – |
| trnF | R | 9744–9812 | 69 | 9774–9776 GAA | – | – |
| nad5 | R | 9819–11570 | 1752 | – | ATC | TAA |
| trnH | R | 11568–11641 | 74 | 11597–11599 GTG | – | – |
| nad4 | R | 11641–12981 | 1341 | – | ATG | TAA |
| nad4L | R | 12981–13271 | 291 | – | ATA | TAA |
| trnT | F | 13275–13341 | 67 | 13308–13310 TGT | – | – |
| trnP | R | 13342–13422 | 81 | 13388–13390 TGG | – | – |
| nad6 | F | 13425–13949 | 525 | – | ATT | TAA |
| cytb | F | 14033–15169 | 1137 | – | ATG | TAG |
| trnS(UCN) | F | 15277–15347 | 71 | 15307–15309 TGA | – | – |
| nad1 | R | 15450–16388 | 939 | – | ATG | TAA |
| trnL(CUN) | R | 16389–16454 | 66 | 6422–16424 TAG | – | – |
| rrnL | R | 16455–17788 | 1334 | – | – | – |
| trnV | R | 17794–17862 | 69 | 17825–17827 TAC | – | – |
| rrnS | R | 17878–18640 | 763 | – | – | – |
| A+T-rich region | 18641–20456 | 1816 | – | – | – |
The nucleotide composition of the C. fletcheri mitochondrial genome is biased toward A/T nucleotides, which account for 72.5%, and this value is well within the range detected in mitochondrial genomes of Polyneoptera (Table S1). In terms of individual nucleotides, the compositions of A, T, and C in C. fletcheri are 28.2%, 40.3%, and 13.9%, respectively, and these values are well within the ranges found in Polyneoptera. However, the G content in C. fletcheri (17.6%) is slightly higher than in any other species (Table S2). The codon-based nucleotide composition of C. fletcheri indicates that the A/T content at the third codon position (79.3%) is higher than that of the second and first codon positions (63.9% and 62.5%, respectively) (Table S2).
All C. fletcheri PCGs start with typical invertebrate initiation codons; either isoleucine (cox1, atp8, nad2, nad3, nad4L, nad5, and nad6) or methionine (cox2, cox3, atp6, cytb, nad1, and nad4), and end with the complete termination codon, TAG (4 genes) or TAA (9 genes) (Table 1). Among 13 PCGs, 4 are neighbors to another PCG: atp8-atp6, atp6-cox3, nad4-nad4L, and nad6-cytb. These are either interspaced (atp6-cox3 and nad6-cytb) or overlapping (atp8-atp6 and nad4-nad4L) (Table 1). The secondary structure formed by transcribed polycistronic mRNA has been proposed to be responsible for cleavage of such neighboring proteins, which is analogous to the tRNA excision model [12], [31]. However, a more recent investigation has shown the importance of a base-pair mismatch, which causes a bulge located close to the start codon of a downstream protein in the stem-and-loop structure. The bulge was suggested to provide a physical attribute for the enzyme to recognize the cleavage site at the start codon of the downstream protein [32].
The complete set of 22 tRNAs (one specific for each amino acid, and 2 each for leucine and serine) found in metazoan mitochondrial genomes were identified in C. fletcheri, and they are interspersed throughout the genome (Table 1; Figure 2). Of the 22 tRNAs, 21 can be folded into a typical cloverleaf structure with a 7-bp amino-acyl stem, 5-bp anticodon arm, 7-bp anticodon loop, a variable loop, DHU arm, and a TψC arm. Unlike others, the trnS(AGN) lacks the DHU stem, and instead, has a 12-bp long DHU loop, as is commonly found in metazoan mitochondrial genomes [3]. A somewhat unusual feature found in trnE and trnP is the presence of much larger variable loops (20 and 19-bp long, respectively; Figure 2) than in other insects. The anticodons of C. fletcheri tRNAs (Table 1) are all identical to those of the respective tRNAs found in the other Polyneoptera (data not shown).
Figure 2. Predicted secondary structures for the 22 tRNA genes of C. fletcheri.

Dashes (–) indicate Watson-Crick base-pairing and centered dots (•) indicate G-U base-pairing. Arms of tRNAs (clockwise from top) are the amino acid acceptor (AA) arm, TψC (T) arm, the anticodon (AC) arm, and the dihydrouridine (DHU) arm.
The rrnL and rrnS genes are located between trnL (CUN) and trnV, and between trnV and the A+T-rich region, respectively. The lengths of rrnL and rrnS are 1,334 bp and 763 bp, and A/T contents are 70.9% and 70.2%, respectively (Table S1). These values are well within the ranges found in Polyneoptera.
The A+T-rich region of C. fletcheri is 1,816 bp long, is located between rrnS and trnM, and contains a high A/T content of 89.31% (Table S1). Such a long C. fletcheri A+T-rich region is attributable to 4 tandem repeats composed of 2 identical 297-bp copies, 2 nearly identical copies, plus a 60-bp partial copy of the beginning of the repeat (Figure 3A). Along with the tandem repeats, each of the 4 identical trnY-like, trnI-like, and trnD-like sequences were detected (Figures 3A and 3B). The sequence homology between the regular and tRNA-like sequence was 50% for trnY, 66% for trnI, and 59% for trnD, showing substantial sequence divergence between them. The long tandem repeats also have been found in several other polyneopteran A+T-rich regions, such as Mantodea, Isoptera, and Orthoptera [33]–[35]. For example, the Reticulitermes hageni (Isoptera) A+T-rich region harbored two 189-bp tandem repeats plus an 89-bp partial copy of the beginning of the repeat, and two 554-bp tandem repeats plus a 99-bp partial copy of the beginning of the repeat [34].
Figure 3. Tandem repeat units and tRNA-like sequences found in the A+T-rich region of C. fletcheri.

(A) The A+T-rich region sequence of C. fletcheri; (B) Predicted secondary structures for 3 tRNA-like sequences found in the A+T-rich region. The sequences covered by green and yellow are tandem repeat units; the single underline and double underlines indicate trnI-like and trnY-like sequences, respectively; the italic nucleotides indicate trnD-like sequences; the rectangular boxes indicate the respective anticodons, and the nucleotide position is indicated at the beginning and end sites of the sequence.
A total of 11 different stem-and-loop structures were detected on the major strand of the C. fletcheri A+T-rich region and 2 of them harbor the conserved flanking sequences TATA and G(A)nT at the 5′ and 3′ end, respectively (Figure S1). The stem-and-loop structure on the minor strand of the A+T-rich region was thought to be associated with the replication origin of the major strand, and the conserved flanking sequences at each end of the structure have regulatory roles for recognition of the replication origin [36]. However, more recently, Saito et al. [37] proposed that the poly-T stretch located on the minor strand near the trnI in holometabolous insects is involved in replication initiation of mtDNA. A 10-bp long analogous poly-T stretch (with one A insertion) was detected in the C. fletcheri A+T-rich region, but it is located near the 5′ end of the rrnS gene on the major strand. As for some other hemimetabolous insects (e.g., Orthoptera) which do not possess an obvious poly-T stretch in the A+T-rich region, the stem-and-loop structures were suggested to perform the role of the poly-T stretch [37].
Non-coding Regions
Except for the A+T-rich region, a total of 24 non-coding regions ranging in size from 1 to 2,888 bp (a total of 3,784 bp) were interspersed throughout the C. fletcheri mitochondrial genome (Figure 1), and the total length is the longest in Polyneoptera (data not shown). Among these non-coding regions, the longest one (2,888 bp) is located between trnI and trnW, spanning a 2,876 bp tandem repeat unit (TRU) without coding any protein. This TRU consists of twenty-one 135-bp tandem repeats plus a 21-bp partial copy of the beginning of the repeat (Figure 4A). Within the TRU, 20 identical trnL(UUR)-like and 21 identical trnA-like sequences, along with the repeats, were identified by tRNA structure search. The sequence similarity between the regular and tRNA-like sequence was 51% for trnL(UUR) and 60% for trnA. These tRNA-like sequences fold into cloverleaf structures harboring the corresponding anticodons (Figure 4B), but their functionality remains unknown. The 75.60% A/T content in the TRU is higher than that of the whole genome (13 PCGs, rrnL, and rrnS), but lower than that of the A+T-rich region (Table S1). To our knowledge, the size of this TRU is the longest reported in insects to date. In Hemiptera, a 1,513-bp long TRU located between trnE and trnF was found in the A. pisum mitochondrial genome (Unpublished, GenBank: FJ411411). This TRU contains seven 202-bp tandem repeats with the first repeat unit overlapping with the 3′ end of trnE, and a partial 99-bp copy of the beginning of the repeat. Similarly, a 1,724-bp long TRU has also been reported from the coleopteran, Pyrocoelia rufa [8]. In addition, a 409-bp long TRU including 4 identical trnL(UUR), which all were presumed to be functional was reported in the hymenopteran Abispa ephippium [38]. However, a similar TRU has never been found in the other mitochondrial genomes of Polyneoptera. Such TRUs in animal mitochondrial genomes could originate from slipped-strand mispairing, resulting in expanded repeat caused by an unequal crossing-over event due to the mispairing propensity of the simple tandem repeats [39].
Figure 4. Tandem repeat units (TRU) and secondary structures of trnL(UUR)-like and trnA-like sequences found in the TRU.

(A) TRU; (B) The secondary structures of trnL(UUR)-like and trnA-like sequences found in the TRU. The sequences covered by yellow and green indicate each repeat unit; the underlined sequences indicate trnL(UUR)-like sequences; the italic nucleotides indicate trnA-like sequences; the rectangular boxes indicate the respective anticodons, and the nucleotide position is indicated at the beginning and end sites of the sequence.
The second longest non-coding sequence (217 bp) is located between trnE and trnF (Figure 1), wherein 1 trnY-like sequence with a proper secondary structure and anticodon is found (Figure 5). This region is not homologous to any other gene or region of the genome. The sequence similarity between trnY-like and regular trnY gene is 58%. Another substantially large non-coding region (131 bp) is located between nad2 and trnI and can be labeled as an unidentified opening reading frame (UORF) because it comprises a start codon (TTG) infrequently described in many insects and an incomplete stop codon (TA), encoding 43 amino acids. The encoded peptide, however, is not similar to any known sequence, although an extensive GenBank database on NCBI has been searched through BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Triatoma dimidiate (Hemiptera) mitochondrial genome has been reported to have a UORF between trnS(UCN) and nad1, encoding 103 amino acids; the function of the UORF was presumed to be another origin of replication [7].
Figure 5. Predicted secondary structure for trnY-like sequence found in intergenic spacer between trnE and trnF.
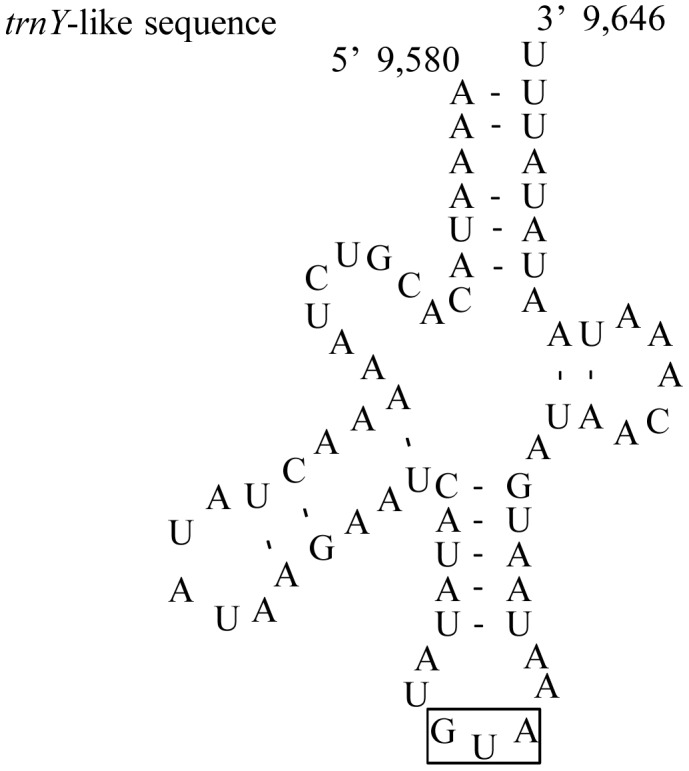
The rectangular boxes indicate the respective anticodons, and the nucleotide position is indicated at the beginning and end sites of the structures.
Reversal Strand Asymmetry
Nucleotide bias, measured in terms of base skewness of AT and GC in the whole C. fletcheri genome (measured from major strand) is −0.166 and 0.346, respectively (Table S1), indicating that more Ts and Gs are encoded in the whole genome of C. fletcheri. All the other Polyneoptera have more As and Cs measured from the major strand. C. fletcheri is T- and G-skewed (−0.177 and 0.117, respectively) for concatenated PCGs. All other species of Polyneoptera harbor more Ts, but the G-skew is not unanimous among species in concatenated PCGs (Table S3). As for the PCGs of C. fletcheri encoded in the major strand (9 PCGs), the AT- and GC-skewness was found to be −0.327 and 0.383, respectively, whereas those in the minor strand were 0.059 and −0.315, respectively. In other species of Polyneoptera, the minor strand is T- and G-skewed, the reverse of C. fletcheri, but the major strand is either A and C-skewed or T- and C-skewed (Table 2; Table S3). This pattern of strand asymmetry has also been found in many animals, including arachnids and lepidopterans [40], [41].
Table 2. The number of species presenting AT- or GC-skewness in whole genome, whole PCGs, major strand, and minor strand PCGs in Polyneoptera.
| Skewness | Categories | |||
| Whole genome | Whole PCGs | Major strand | Minor strand | |
| A- and C-skewed | 59 | 0 | 32 | 0 |
| T- and G-skewed | 0 | 12 | 0 | 59 |
| T- and C-skewed | 0 | 45 | 27 | 0 |
| T-skewed* | 0 | 2 | 0 | 0 |
GC-skewness is zero.
Francino and Ochman [42] and Hassanin et al. [43] proposed that asymmetrical mutation pressure on the mitochondrial genome causes biased occurrence of mutations between strands. During replication of mtDNA, the parental minor strand remains as a single strand for a longer time until ∼ 97% of the nascent minor strand is replicated using the major strand as a template. Therefore, the parental minor strand is easily damaged by hydrolysis, resulting in the deaminations of A and C [43]–[45]. This results in A- and C-skewness on the major strand and T- and G-skewness on the minor strand. As described above, however, a substantial number of Polyneoptera evidence deviation from this general rule in the major strand, particularly in C. fletcheri, which shows a unique reversal in both major and minor strands. One of the plausible explanations for this is the inversion of replication origin located in the A+T-rich region [43], [44], resulting in the major strand being single-stranded for longer than the minor strand and resulting in A- and C-skewness on the minor strand and T- and G-skewness on the major strand [43]. By examination of the regulatory elements in the A+T-rich region, Wei et al. [46] found inverted A+T-rich regions in several insect species and the resultant reversal of strand asymmetry in the major strand. Their reasoning for inversion was based on the finding of inverted regulatory elements (e.g., the stem-and-loop structure with conserved flanking sequences at each end and poly-T stretch). The stem-and-loop structures found in the C. fletcheri A+T-rich region, are located in a reversed direction on the complementary strand compared to those generally found in other insects. Nevertheless, reverse strand asymmetry resulting from the inversion of the A+T-rich region may not be obvious if the event is recent [43], suggesting that the inversion event in C. fletcheri may have occurred a long ago.
Mitochondrial Gene Rearrangement
In contrast to the ancestral arrangement commonly found in insect mitochondrial genomes, C. fletcheri has a substantial number of tRNA rearrangements (Figure 6). These include 3 translocations resulting from 2 duplication/random deletion events (trnI, trnN, and trnR), 3 shuffling with remote inversions (trnQ, trnY, and trnC), and 1 local inversion (trnE). Several mechanisms responsible for mitochondrial gene rearrangement have been proposed: (1) translocation either by intramitochondrial recombination or by gene duplication/random loss, (2) inversion by intramitochondrial recombination, and (3) shuffling with remote inversion (translocation and inversion) either by combined intramitochondrial genome recombination and duplication/random loss of gene block, or by 2 separate intramitochondrial genome recombinations [47]–[51]. A schematic illustration of each possible event for C. fletcheri mitochondrial genome is presented in Figure 6. The translocation of trnI is probably caused by duplication of gene block trnI-trnQ-trnM-nad2, resulting in the arrangement trnI-trnQ-trnM-nad2-trnI-trnQ-trnM-nad2. A subsequent random loss of trnI in the first copy and trnQ-trnM-nad2 in the second copy may have resulted in trnQ-trnM-nad2-trnI. Likewise, the duplication/random loss of the gene blocks trnC-trnY and trnR-trnN may also have resulted in an intermediate arrangement of trnY-trnC and the current arrangement of trnN-trnR, respectively (Figure 6). The local inversion of trnE may have been caused by the breakage of the mitochondrial genome at trnE and recombination of the trnE on the other strand at the same position [48], [52]. In the case of the shuffling with remote inversion of 3 tRNAs (trnQ, trnY, and trnC), this may be caused by 2 separate intramitochondrial recombinations. The first inversion event may include recombination of trnQ and the gene block trnY-trnC in opposite directions, and the second translocation event may be caused by recombination of trnQ and trnY-trnC in the same directions, resulting in shuffling with remote inversion. Nevertheless, as each rearrangement event was independent, there is no way of determining the actual order of events.
Figure 6. Schematic illustration of each event for mitochondrial gene rearrangement in C. fletcheri.

Gene sizes are not drawn to scale. Gene names that are not underlined indicate a forward transcriptional direction, whereas underlines indicate a reverse transcriptional direction. tRNA genes are abbreviated using the one-letter amino acid code, with L* = trnL(UUR); S = trnS(AGN). White boxes represent genes with the same relative position as in the ancestral insect arrangement pattern. Yellow boxes represent gene translocations; red boxes represent gene inversions; green boxes represent gene shuffling with remote inversions compared to the ancestral insect arrangement. The grey boxes represent gene deletions. The remaining genes and gene orders identical to the ancestral insect are omitted. Each rearrangement event was independent, so there is no way of determining the order of event.
Boore [2] reported that the regions flanking the control region and between nad3 and nad5 are the hot spots for gene rearrangements in Arthropoda. In the C. fletcheri mitochondrial genome, most rearrangements occurred within these regions. For example, the rearranged trnI and trnQ are located in the region flanking the A+T-rich region in the ancestral arrangement, while the translocation of trnR and trnN and the local inversion of trnE occurred within the region between nad3 and nad5 (Figure 6).
All Polyneoptera except Orthoptera and Embioptera have shown mitochondrial gene arrangement identical to the ancestral type. In Orthoptera, 27 of 28 available species belonging to the suborder Caelifera have shown only 1 identical tRNA translocation, resulting in the order trnD-trnK instead of the ancestral order of trnK-trnD, and this rearrangement has been suggested to be synapomorphic for Acridomorpha [13]. In the case of another orthopteran suborder, Ensifera, 1 of 10 species has shown common translocation of 1 tRNA between trnN and trnE compared to the ancestral type (Figure S2). Owing to a paucity of rearrangements in Polyneoptera, it is difficult to speculate the possible mechanisms that might be responsible for the extensive mitochondrial genome rearrangement of C. fletcheri.
Phylogeny of Polyneoptera
To study the phylogeny of Polyneoptera, we performed Bayesian Inference (BI) and Maximum Likelihood (ML) analyses based on several datasets such as all codon positions of 13 PCGs plus all RNAs (rRNAs and tRNAs) (PCG123RNA) and 1st +2nd codon positions of 13 PCGs plus all RNAs (PCG12RNA), along with 2 partitioning strategies for BI analyses. Here, we only present 2 phylogenetic topologies obtained with the datasets PCG123RNA and PCG12RNA (Figure 7), because the analyses based on partitioning strategies also supported the either of 2 topologies presented (data not shown).
Figure 7. Phylogeny of polyneopteran orders. (.

A) Bayesian inference phylogeny obtained with the dataset PCG123RNA; (B) Bayesian inference phylogeny obtained with the dataset PCG12RNA. The numbers associated with the nodes are posterior probabilities obtained by BI analysis with the dataset PCG12RNA (first) or bootstrap values obtained by ML analysis with the dataset PCG12RNA (second) and ML analysis with the dataset PCG123RNA (third). The species of Collembola and Diplura were utilized as outgroups to root the trees. The scale bar indicates the number of substitutions per site.
Phylogenetic analyses yielded unequivocal tree topology for the position of Dermaptera in Polyneoptera (Figure 7), placing as the sister to Plecoptera. This group was located either as the sister to Ephemeroptera (Figure 7A) or as the most basal lineage of the remaining polyneopteran orders (Figure 7B). The nodal support for sister group Dermaptera and Plecoptera was 0.93 ∼ 1 by BI and 85% ∼ 87% by ML analyses (Figure 7). This result is consistent with the previous findings from other molecular data. For example, Kjer [24] reported a sister relationship between Dermaptera and Plecoptera, with this group identified as the basal lineage of Neoptera (Polyneoptera, Paraneoptera, and Holometabola) on the basis of 18S rDNA. The same result also was obtained in another study using data obtained from 2 nuclear and 2 mitochondrial rRNAs, and amino acid sequences from 4 PCGs by the weighted parsimony method [27]. Another study that included newly sequenced 18S rDNA of Zoraptera also showed a sister relationship between Dermaptera and Plecoptera, placing them as the basal lineage of Neoptera [26]. However, on the basis of comprehensive morphological characteristics, Dermaptera was found to be closely related to Dictyoptera and Grylloblattodea, instead of Plecoptera [20]–[23]. Nevertheless, when the wing base structure data were analyzed independently, the sister relationship between Dermaptera and Plecoptera was supported by 2 non-homoplasious apomorphies, such as ventral basisubcostale and articulation between the antemedian total wing process and the first axillary sclerite [23]. In addition to the wing base structure, the possible synapomorphies supporting Dermaptera and Plecoptera as sister groups include 3 segmented tarsi, lack of male gonostyli and a functional ovipositor, and paired male gonopores [53].
Regarding the phylogenetic relationships among polyneopteran orders and palaeopteran orders, Odonata was always placed as the basal lineage of the Pterygota (Figure 7). This position of Odonata was also supported in recent mitochondrial genome-based phylogenetic studies on 13 basal hexapod orders and 9 basal pterygotan orders [54], [55], as well as several molecular and/or morphological studies [24], [26], [27], [56]. On the contrary, the position of Ephemeroptera was variable (Figure 7). Both BI and ML analyses based on PCG12RNA and ML analysis based on PCG123RNA supported the sister relationships between Ephmeroptera and the Polyneoptera (Figure 7B), whereas the BI analysis of PCG123RNA instead supported the sister relationships between Ephmeroptera and the group composed of (Dermaptera + Plecoptera), placed as the basal lineage of Polyneoptera (Figure 7A). Several previous studies using molecular markers and/or morphological characters have shown that Ephemeroptera is the sister to Polyneoptera, placing Odonata the sister to this group [24], [26], [27], [56]. This relationship was also supported by synapomorphic character, direct sperm transfer, shared by Ephemeroptera and Polyneoptera, while Odonata and primitive wingless hexapods possessed symplesiomorphic character, indirect sperm transfer mechanism [21]. However, recent studies using mitochondrial genomes by 1st +2nd codon position [54] and amino acid sequences of 13 PCGs [54], [55] without Dermaptera supported (Ephemeroptera + Plecoptera), similar to the results obtained by some of our data. With regard to the phylogenetic relationships among polyneopteran orders, the relationships within Dictyoptera (Isoptera, Mantodea, and Blattodea) were variable depending on datasets (Figure 7). Both BI and ML analyses based on dataset PCG12RNA and the ML analysis based on dataset PCG123RNA supported (Mantodea + (Isoptera + Blattodea)) (Figure 7B), whereas the BI analysis of PCG123RNA instead supported (Isoptera + (Mantodea + Blattodea)) (Figure 7A). The nodal support for both of the 2 relationships was similar (Figure 7). Both relationships have been proposed in previous studies using morphological and/or molecular data [20], [21], [26]–[28].
To clarify the 2 topologies presenting the conflicting relationships among polyneopteran and palaeopteran orders and among dictyopteran orders, topology tests [57] were performed with the 2 representative datasets, applying the GTR [58] + I + G model. The 6 statistical tests performed (ELW, BP, KH, SH, WSH, and AU) for topological characterization demonstrated that both topologies had confidence values well within the range of the 95%, suggesting that both topologies have an equal possibility, although the PCG12RNA dataset, which supports ((Ephemeroptera + Polyneoptera) + Odonata) and ((Isoptera + Blattodea) + Mantodea) (Figure 7B) obtained somewhat higher statistical confidence levels in ELW, BP, and AU tests (Table S4). Thus, a decisive conclusion on their phylogenetic relationships should be postponed until additional information from polyneopteran orders and species become available.
Except for those conflicts, the interordinal relationships within Polyneoptera were identical in all analyses (Figure 7). Dictyoptera (Isoptera + Blattodea + Mantodea), the most widely accepted superordinal group within Polyneoptera (e.g., [20], [21], [23], [28], [53], [59]), was strongly supported (1 by BI and 100% by ML analyses) as being monophyletic across all analyses. Also, the group consisting of ((Mantophasmatodea + Phasmatodea) + Grylloblattodea) was strongly supported as being monophyletic by all analyses (0.98 ∼ 1 by BI and 88% ∼ 89% by ML analyses), as first proposed by Cameron et al. [33], Kjer et al. [27], Ma et al. [35], and Gullan and Cranston [15]. This group was placed as sister to Dictyoptera in all analyses (Figure 7), which is consistent with Kjer et al. [27] and Ma et al. [35]. Nevertheless, several other studies placed either Zoraptera, Dermaptera, or Grylloblattodea together with Mantophasmatodea as the sister group to Dictyoptera using morphological characteristics and/or molecular markers [20], [21], [25], [26].
Orthoptera was supported as the sister to the group composed of (((Mantophasmatodea + Phasmatodea) + Grylloblattodea) + Dictyoptera) in all analyses with high nodal supports (1 by BI and 97% ∼ 98% by ML analyses) (Figure 7). Previous phylogenetic studies proposed a somewhat widely accepted sister relationship between Orthoptera and Phasmatodea [21]–[23], [27], [28], [59]. However, Kjer et al. [27] have shown that Orthoptera clustered together with the remaining Neoptera, excluding the very weakly supported (Dermaptera + Plecoptera) by the weighted parsimony method. This result is similar to ours in that Ortheroptera was placed as the sister to the remaining Polyenoptera, excluding Dermaptera plus Plecoptera.
Materials and Methods
Ethics Statement
No experiments involving vertebrate samples were performed in this study. An ethics statement is not required for the experiment which only involves an insect. The collection of the free-living earwig was permitted by the Han River Basin Environmental Office, Republic of Korea (permission number 2008–04).
Specimens and DNA Extraction
An adult C. fletcheri was collected from Bukhan Mountain, Seoul, Republic of Korea in June, 2008. All necessary permits were obtained for the described field studies. DNA was extracted from a hind leg with a Wizard™ Genomic DNA Purification Kit in accordance with the manufacturer’s instructions (Promega, Madison, WI, USA).
Generation of Sequence Data
The complete mitochondrial genome of C. fletcheri was sequenced with each 2 short and 2 long overlapping fragments. Each 400–600 bp of C. fletcheri cox3 and rrnS (SF1 and SF2 in Figure 1, respectively) was sequenced using the primers designed from the alignment of several full-length mtDNA sequences of polyneopteran insects. The primer sequences are listed in Table 3. A polymerase chain reaction (PCR) was conducted under the following conditions: denaturation at 94°C for 7 min; 35 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min; and a final extension at 72°C for of 7 min. Based on the sequence information of SF1 and SF2, two pairs of primers were designed for the amplification of 2 overlapping long fragments, LF1 and LF2 (Figure 1; Table 3). Long PCR was performed using LA Taq™ from Takara Biomedical (Otsu, Shiga, Japan) under the following conditions: 94°C for 1 min; 30 cycles of 94°C for 30 sec and 58°C for 15 min; and a final extension step at 72°C for 12 min. The 2 long PCR fragments were then employed in the construction of a shotgun library after purification using the QIAquick PCR Purification Kit reagents (Qiagen, Germantown, MD, USA). DNA sequencing was conducted using the ABI PRISM® BigDye® Terminator ver. 3.1 Cycle Sequencing Kit and the ABI PRISM™ 3100 Genetic Analyzer from PE Applied Biosystems (Carlsbad, CA,USA). All fragments were sequenced from both strands.
Table 3. List of primers used to amplify long and short fragments of C. fletcheri mitochondrial genome.
| Fragment | Primer name | Direction | Primer sequence (5′ to 3′) | Position* |
| SF1 | Der-CO3F1 | F | CWATAATYCAATGATGACG | 7778 ∼ 7796 |
| Der-CO3R1 | R | GTCCATGRAATCCTGTTGCTA | 8216 ∼ 8236 | |
| SF2 | Der-srRNAF1 | F | CTACTWTGTACGACTTATCTC | 17892 ∼ 17913 |
| Der-srRNAR1 | R | TAAACTAGGATTAGATACCCC | 18297 ∼ 18317 | |
| LF1 | CF-CO3F2 | F | GCATCAGGGGTTACAGTTACTTG | 8035 ∼ 8057 |
| CF-srRNAR1 | R | GAAAATGAATTCTCGACGAATAC | 18009 ∼ 18031 | |
| LF2 | CF-srRNAF3 | F | AATCTTAAGACGACGGTATACAAGC | 18134 ∼ 18158 |
| CF-CO3R2 | R | CACAAACAGGAGAAAGACTTC | 7931 ∼ 7951 |
Position of primer sequences corresponds to C. fletcheri mitogenome; F, forward; and R, reverse.
Genome Annotation
Sequenced fragments were assembled into a single contig with overlapping ends using the Phred+Phrap+Consed package [60]. Raw sequence data obtained were manually proof checked for indels or ambiguous base calls. The boundary of each of 13 PCGs, 2 rRNA genes, and the A+T-rich region of C. fletcheri was delimitated by the alignment with homologous regions of polyneopteran mitochondrial genome sequences using CLUSTAL X [61]. The nucleotide sequences of 13 PCGs were translated into amino acid sequences based on the invertebrate mtDNA genetic code using the Transseq program available at the EBI web site (http://www.ebi.ac.uk/Tools/emboss/transeq/). Delimitation and prediction of secondary structure of tRNAs, except for trnS(AGN), were performed by tRNAscan-SE 1.21 using invertebrate mitochondrial codon predictors and a cove score cut off of 1 [62]. The trnS(AGN) was determined via sequence comparison with published polyneopteran mitochondrial genomes, and its secondary structure was predicted manually. The sequence data is deposited in the GenBank database (GenBank: JN651407).
Comparative Sequence Analysis
The A/T-content in 13 PCGs, 2 rRNAs, the A+T-rich region, and whole genome, were calculated using the EditSeq program in the Lasergene software package (www.dnastar.com). The compositional skew for whole PCGs, and major-strand and minor-strand encoded PCGs was calculated with the following formula: GC-skewness = (G - C)/(G + C) and AT-skewness = (A - T)/(A + T), where C, G, T and A are the frequencies of each nucleotide [63]. The nucleotide composition at each codon position of the PCGs was calculated using PAUP ver. 4.0b10 [64]. The overlapping regions and intergenic spacers between genes were counted manually. Tandem repeat units in the genome were searched using Tandem Repeats Finder program [65]. The stem-and-loop structures in the A+T-rich region were predicted by the web server Mfold [66].
Phylogenetic Analysis
A total of 13 polyneopteran mitochondrial genomes representing 9 orders were employed to reconstruct the phylogenetic relationships of Polyneoptera using BI and ML algorithms. The mitochondrial genome of embiopteran species, Aposthonia japonica [14] was not included in the analyses because it just became available during final phase of our manuscript. With the consideration that the Ephemeroptera has been proposed to be the sister to Plecoptera [54], [55] under the controversial relationships among Neoptera, Ephemeroptera, and Odonata [21], [27], [28] and that the monophyly of Ectognatha (Pterygota, Archaeognatha, and Zygentoma) is widely accepted [67], the two palaeopteran orders Odonata and Ephemeroptera, and each 2 species of Archaeognatha [68], [69] and Zygentoma [70], [71] were also included as ingroup taxa in the analyses. 2 species of Diplura [72], [73] and 3 species of Collembola [74]–[76], which have been used to root pterygotan phylogeny [54] were utilized as outgroups,. Considering the previous suggestions that either all mitochondrial genome data (all PCGs, rRNAs, and tRNAs) or 1st +2nd codon positions of 13 PCGs are better utilized to infer ordinal relationships of holometabolous orders or basal pterygotan orders [54], [77], the datasets PCG123RNA (all codon positions of 13 PCGs plus all rRNAs and tRNAs) and PCG12RNA (1st +2nd codon positions plus all rRNAs and tRNAs) were generated for phylogenetic analyses for Polyneoptera and Palaeoptera. Both datasets were divided into either 2 partitions dividing 13 PCGs into one and RNAs into another partition or 14 partitions dividing each PCGs and one for RNAs for BI analyses.
To obtain codon-based alignment for each PCG, the nucleotide sequence of each PCG was subjected to RevTrans ver. 1.4 [78]. The well-aligned conserved blocks of each PCG were selected using GBlocks 0.91b [79]. Substitution model selection was conducted via comparison of Akalike Information Criterion (AIC) scores [80] using Modeltest ver. 3.7 [81]. For datasets PCG123RNA and PCG12RNA, the GTR [58] + I + G was selected as the best-fit model for both ML and BI analyses. The ML and BI analyses were conducted using RAxML [82] and MrBayes ver. 3.1 [83], respectively, as previously described [54], [84]. When the partition option was employed, each partition was unlinked with the model selected by ModelTest [81] applied (data not shown).
Because discordant topologies were obtained depending on datasets, those topologies were subjected to topology tests using the ML method incorporated in Treefinder [57], applying the GTR [58] + I + G model. The statistical confidence values of each topology were determined via 6 statistical tests, each with 1,000 replications: ELW [85], BP [86], KH [87], SH [88], WSH [88], and AU [89].
Supporting Information
Stem-and-loop structures found in the A+T-rich region of C. fletcheri mitochondrial genome. The underline and dashed lines indicate the identical and similar flanking sequences in the stem-and-loop structures, respectively, that have conservatively been found in Orthoptera and Diptera. The numbers in parenthesis indicate the number of redundant structures. The nucleotide position is indicated at the beginning and end sites of the structures.
(PDF)
Mitochondrial gene arrangement in Polyneoptera. Gene sizes are not drawn to scale. tRNA genes are abbreviated using the one-letter amino acid code, with L = trnL(CUN); L* = trnL(UUR); S = trnS(AGN); S* = trnS(UCN). Gene names that are not underlined indicate a forward direction, whereas underlines indicate a reverse transcriptional direction. CR indicates the A+T-rich region.
(PDF)
Nucleotide composition and skewness of mitochondrial genomes of Polyneoptera.
(PDF)
Nucleotide composition at each codon position of the concatenated 13 PCGs in Polyneoptera.
(PDF)
Composition and skewness of mitochondrial PCGs in Polyneoptera.
(PDF)
Results of topological tests for 2 datasets, showing values from 6 statistical tests performed.
(PDF)
Acknowledgments
We thank three anonymous reviewers for comments on the manuscript.
Funding Statement
This work was supported by research grants from the National Institute of Biological Resources – “The Genetic Evaluation of Important Biological Resources” awarded to Iksoo Kim. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Brown WM (1985) The mitochondrial genome of animals. In: Macintyre RJ, editor. Molecular Evolution Genetics. New York: Plenum. [Google Scholar]
- 2. Boore JL (1999) Animal mitochondrial genomes. Nucleic Acids Res 27: 1767–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wolstenholme DR (1992) Animal mitochondrial DNA: structure and evolution. Int Rev Cytol 141: 173–216. [DOI] [PubMed] [Google Scholar]
- 4. Boyce TM, Zwick ME, Aquadro CF (1989) Mitochondrial DNA in the bark weevils: size, structure and heteroplasmy. Genetics 123: 825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fauron CMR, Wolestenholme DR (1980) Extensive diversity among Drosophilla species with respect to nucleotide sequences within the adenine + thymine-rich region of mitochondrial DNA molecules. Nucleic Acids Res 8: 2439–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Inohira K, Hara T, Matsuura ET (1997) Nucleotide sequence divergence in the A+T-rich region of mitochondrial DNA in Drosophila simulans and Drosophila mauritiana . Mol Biol Evol 14: 814–822. [DOI] [PubMed] [Google Scholar]
- 7. Doston EM, Beard CB (2001) Sequence and organization of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiate . Insect Mol Biol 10: 205–215. [DOI] [PubMed] [Google Scholar]
- 8. Bae JS, Kim I, Sohn HD, Jin BR (2004) The mitochondrial genome of the firefly, Pyrocoelia rufa: complete DNA sequence, genome organization, and phylogenetic analysis with other insects. Mol Phylogenet Evol 32: 978–985. [DOI] [PubMed] [Google Scholar]
- 9. Thao ML, Baumann P (2004) Evidence of multiple acquisition of Arsenophonus by whitefly species (Sternorryncha: Aleyrodidae). Curr Microbiol 48: 140–144. [DOI] [PubMed] [Google Scholar]
- 10. Lavrov DV, Lang BF (2005) Poriferan mtDNA and animal phylogeny based on mitochondrial gene arrangements. Syst Biol 54: 651–659. [DOI] [PubMed] [Google Scholar]
- 11. De Bruijn MHL (1983) Drosophila melanogaster mitochondrial DNA, a novel organization and genetic code. Nature 304: 234–241. [DOI] [PubMed] [Google Scholar]
- 12. Clary DO, Wolstenholme DR (1985) The mitochondrial DNA molecule of Drosophila yakuba: Nucleotide sequence, gene organization and genetic code. J Mol Evol 22: 252–271. [DOI] [PubMed] [Google Scholar]
- 13. Sheffield NC, Hiatt KD, Valentine MC, Song H, Whiting MF (2010) Mitochondrial genomics in Orthoptera using MOSAS. Mitochondr DNA 21: 87–104. [DOI] [PubMed] [Google Scholar]
- 14. Kômoto N, Yukuhiro K, Tomita S (2012) Novel gene rearrangements in the mitochondrial genome of a webspinner, Aposthonia japonica (Insecta: Embioptera). Genome 55: 222–233. [DOI] [PubMed] [Google Scholar]
- 15. Hass F, Gorb SN, Wootton RJ (2000) Elastic joints in dermapteran hind wings: materials and wing folding. Arthropod Struct Dev 29: 137–146. [DOI] [PubMed] [Google Scholar]
- 16.Gullan PJ, Cranston PS (2010) The insect: An Outline of Entomology. 4th edition. Oxford: John Wiley & Sons. [Google Scholar]
- 17. Carpenter FM (1992) Superclass Hexapoda. In: Kaesler RL, editor. Treatise on Invertebrate Paleontology Part R, Arthropoda 4. Kansas: Geological Society of America, Boulder, Colorado, & University of Kansas. 279–655. [Google Scholar]
- 18. Giles ET (1963) The comparative external morphology and affinities of the Dermaptera. Trans Royal Entomol Soc Lond 115: 95–164. [Google Scholar]
- 19.Cedric G (2005) Entomology. 3rd edition. Dordrecht: Springer. [Google Scholar]
- 20.Hennig W (1969) Die Stammesgeschichte der Insekten. Frankfurt am Main: Kramer.
- 21. Boudreaux HB (1979) Arthropod phylogeny. New York: John Wiley & Sons. [Google Scholar]
- 22.Kukalová-Peck J (1991) Fossil history and the evolution of hexapod structures. In: Naumann ID, Carne PB, Lawrence JF, Nielsen ES, Spradberry JP, Taylor RW, Whitten MJ, Littlejohn MJ, editors. The Insects of Australia: A Textbook for Students and Research Workers. Paris: Melbourne Univ Press. 141–179. [Google Scholar]
- 23. Yoshizawa K (2011) Monophyletic Polyneoptera recovered by wing base structure. Syst Entomol 6: 377–394. [Google Scholar]
- 24. Kjer KM (2004) Aligned 18S and insect phylogeny. Syst Biol 53: 506–514. [DOI] [PubMed] [Google Scholar]
- 25. Terry MD, Whiting MF (2005) Mantophasmatodea and phylogeny of the lower neopterous insects. Cladistics 21: 240–257. [Google Scholar]
- 26. Yoshizawa K, Johnson KP (2005) Aligned 18S for Zoraptera (Insecta): phylogenetic position and molecular evolution. Mol Phylogenet Evol 37: 572–580. [DOI] [PubMed] [Google Scholar]
- 27. Kjer KM, Carle FL, Litman J, Ware J (2006) A molecular phylogeny of Hexapoda. Arthropod Syst Phylogeny 64: 35–44. [Google Scholar]
- 28. Wheeler WC, Whiting M, Wheeler QD, Carpenter JM (2001) The phylogeny of the extant hexapod orders. Cladistics 17: 113–169. [DOI] [PubMed] [Google Scholar]
- 29. Yoon IB, Moon TY (1996) Habitats and distribution of rare endangered earwigs Challia fletcheri Burr in Korea. Korean J Entomol 26: 403–409. [Google Scholar]
- 30. Adachi K (2003) Insects collected on Danjo-gunto Islands (part I). Shin Tsukushi no Konchu 7: 41–74. [Google Scholar]
- 31. Ojala D, Montoya J, Attardi G (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature 290: 470–474. [DOI] [PubMed] [Google Scholar]
- 32. Fenn JD, Cameron SL, Whiting MF (2007) The complete mitochondrial genome sequence of the Mormon cricket (Anabrus simplex: Tettigoniidae: Orthoptera) and an analysis of control region variability. Insect Mol Biol 16: 239–252. [DOI] [PubMed] [Google Scholar]
- 33. Cameron SL, Barker SC, Whiting MF (2006) Mitochondrial genomics and the new insect order Mantophasmatodea. Mol Phylogenet Evol 38: 274–279. [DOI] [PubMed] [Google Scholar]
- 34. Cameron SL, Whiting MF (2007) Mitochondrial genomic comparisons of the subterranean termites from the genus Reticulitermes (Insecta: Isoptera: Rhinotermitidae). Genome 50: 188–202. [DOI] [PubMed] [Google Scholar]
- 35. Ma C, Liu C, Yang P, Kang L (2009) The complete mitochondrial genomes of two band-winged grasshoppers, Gastrimargus marmoratus and Oedaleus asiaticus . BMC Genomics 10: 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang DX, Szymura JM, Hewitt GM (1995) Evolution and structural conservation of the control region of insect mitochondrial DNA. J Mol Evol 40: 382–391. [DOI] [PubMed] [Google Scholar]
- 37. Saito S, Tamura K, Aotsuka T (2005) Replication origin of mitochondrial DNA in insects. Genetics 171: 1695–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cameron SL, Dowton M, Castro LR, Ruberu K, Whiting MF, et al. (2008) Mitochondrial genome organization and phylogeny of two vespid wasps. Genome 51: 800–808. [DOI] [PubMed] [Google Scholar]
- 39. Levinson G, Gutman GA (1987) Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol 4: 203–221. [DOI] [PubMed] [Google Scholar]
- 40. Masta SE, Longhorn SJ, Boore JL (2009) Arachnid relationships based on mitochondrial genomes: asymmetric nucleotide and amino acid bias affects phylogenetic analyses. Mol Phylogenet Evol 50: 117–128. [DOI] [PubMed] [Google Scholar]
- 41. Kim MJ, Wan X, Kim KG, Hwang JS, Kim I (2010) Complete nucleotide sequence and organization of the mitochondrial genome of endangered Eumenis autonoe (Lepidoptera: Nymphalidae). Afr J Biotechnol 9: 735–754. [Google Scholar]
- 42. Francino MP, Ochman H (1997) Strand asymmetries in DNA evolution. Trends Genet 13: 240–245. [DOI] [PubMed] [Google Scholar]
- 43. Hassanin A, Leger N, Deutch J (2005) Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Syst Biol 54: 277–298. [DOI] [PubMed] [Google Scholar]
- 44. Reyes A, Gissi C, Pesole G, Saccone C (1998) Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol 15: 957–966. [DOI] [PubMed] [Google Scholar]
- 45. Saccone C, De Giorgi C, Gissi C, Graziano P, Reyes R (1999) Evolutionary genomics in Metazoa: the mitochondrial DNA as a model system. Gene 238: 195–209. [DOI] [PubMed] [Google Scholar]
- 46. Wei SJ, Shi M, Chen XX, Sharkey MJ, van Achterberg C, et al. (2010) New views on strand asymmetry in insect mitochondrial genomes. PLoS ONE 5: e12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Macey JR, Larson A, Ananjeva NB, Fang Z, Papenfuss TJ (1997) Two novel gene orders and the role of light-strand replication in rearrangement of the vertebrate mitochondrial genome. Mol Biol Evol 14: 91–104. [DOI] [PubMed] [Google Scholar]
- 48. Dowton M, Campbell NJH (2001) Intramitochondrial recombination: is it why some mitochondrial genes sleep around? Trends Ecol Evol 16: 269–271. [DOI] [PubMed] [Google Scholar]
- 49. Silvestre D, Arias MC (2006) Mitochondrial tRNA gene translocations in highly eusocial bees. Genet Mol Biol 29: 572–575. [Google Scholar]
- 50. Dowton M, Cameron SL, Dowavic JI, Austin AD, Whiting MF (2009) Characterization of 67 mitochondrial tRNA gene rearrangements in the Hymenoptera suggests that mitochondrial tRNA gene position is selectively neutral. Mol Biol Evol 26: 1607–1617. [DOI] [PubMed] [Google Scholar]
- 51. Wei SJ, Shi M, He JH, Sharkey M, Chen XX (2009) The complete mitochondrial genome of Diadegma semiclausum (Hymenoptera: Ichneumonidae) indicates extensive independent evolutionary events. Genome 52: 308–319. [DOI] [PubMed] [Google Scholar]
- 52. Dowton M, Austin A (1999) Evolutionary dynamics of a mitochondrial rearrangement “hot spot” in the Hymenoptera. Mol Biol Evol 16: 298–309. [DOI] [PubMed] [Google Scholar]
- 53.Kristensen NP (1991) Phylogeny of extant hexapods. In: Naumann ID, Carne PB, Lawrence JF, Nielsen ES, Spradberry JP, Taylor RW, Whitten MJ, Littlejohn MJ, editors. The Insects of Australia: A Textbook for Students and Research Workers. Carlton: Melbourne Univ Press. 125–140. [Google Scholar]
- 54. Lin CP, Chen MY, Huang JP (2010) The complete mitochondrial genome and phylogenomics of a damselfly, Euphaea formosa support a basal Odonata within the Pterygota. Gene 468: 20–29. [DOI] [PubMed] [Google Scholar]
- 55. Zhang YY, Xuan WJ, Zhao JL, Zhu CD, Jiang GF (2010) The complete mitochondrial genome of the cockroach Eupolyphaga sinensis (Blattaria: Polyphagidae) and the phylogenetic relationships within the Dictyoptera. Mol Biol Rep 37: 3509–3516. [DOI] [PubMed] [Google Scholar]
- 56. Simon S, Strauss S, von Haeseler A, Hadrys H (2009) A phylogenomic approach to resolve the basal pterygote divergence. Mol Biol Evol 26: 2719–2730. [DOI] [PubMed] [Google Scholar]
- 57. Jobb G, von Haeseler A, Strimmer K (2004) TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol Biol 4: 18. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59. Hennig W (1981) Insect Phylogeny. New York: John Wiley & Sons. [Google Scholar]
- 60. Gordon D, Abajian C, Green P (1998) Consed: A graphical tool for sequence finishing. Genome Res 8: 195–202. [DOI] [PubMed] [Google Scholar]
- 61. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24: 173–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Perna NT, Kocher TD (1995) Patterns of nucleotide composition at four fold degenerate sites of animal mitochondrial genomes. J Mol Evol 41: 353–358. [DOI] [PubMed] [Google Scholar]
- 64. Swofford DL (2002) PAUP* Phylogenetic analysis using parsimony (*and other methods) ver 410b. Sunderland: Sinauer Associates. [Google Scholar]
- 65. Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Willmann R (2004) Phylogenetic relationships and evolution of insects. In: Cracraft J, Donoghue MJ, editors. Assembling the Tree of life. NewYork: Oxford University Press. 330–344. [Google Scholar]
- 68. Podsiadlowski L, Bartolomaeus T (2006) The mitochondrial genome of the bristletail Petrobius brevistylis (Archaeognatha: Machilidae). Insect Mol Biol 15: 253–258. [DOI] [PubMed] [Google Scholar]
- 69. Zhang JY, Song DX, Zhou KY (2008) The complete mitochondrial genome of the bristletail Pedetontus silvestrii (Archaeognatha: Machilidae) and an examination of mitochondrial gene variability within four bristletails. Ann Entomol Soc Am 101: 1131–1136. [Google Scholar]
- 70. Cook CE, Yue Q, Akam M (2005) Mitochondrial genomes suggest that hexapods and crustacean are mutually paraphyletic. Proc R Soc B 272: 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Comandi S, Carapelli A, Podsiadlowski L, Nardi F, Frati F (2009) The complete mitochondrial genome of Atelura formicaria (Hexapoda: Zygentoma) and the phylogenetic relationships of basal insects. Gene 439: 25–34. [DOI] [PubMed] [Google Scholar]
- 72. Carapelli A, Nardi F, Dallai R, Boore JL, Liò P, et al. (2005) Relationships between hexapods and crustaceans based on four mitochondrial genes. Crustacean Issues 16: 295–306. [Google Scholar]
- 73. Podsiadlowski L, Carapelli A, Nardi F, Dallai R, Koch M (2006) The mitochondrial genomes of Campodea fragilis and Campodea lubbocki (Hexapoda: Diplura): high genetic divergence in a morphologically uniform taxon. Gene 381: 49–61. [DOI] [PubMed] [Google Scholar]
- 74. Nardi F, Carapelli A, Fanciulli PP, Dallai R, Frati F (2001) The complete mitochondrial DNA sequence of the basal hexapod Tetrodontophora bielanensis: evidence for heteroplasmy and tRNA translocations. Mol Biol Evol 18: 1293–1304. [DOI] [PubMed] [Google Scholar]
- 75. Nardi F, Spinsanti G, Boore JL, Carapelli A, Dallai R, et al. (2003) Hexapod origins: monophyletic or paraphyletic? Science 299: 1887–1889. [DOI] [PubMed] [Google Scholar]
- 76. Carapelli A, Liò P, Nardi F, van der Wath E, Frati F (2007) Phylogenetic analysis of mitochondrial protein coding genes confirms the reciprocal paraphyly of Hexapoda and Crustacea. BMC Evol Biol 7 (Suppl 2)S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cameron SL, Sullivan J, Song H, Miller KB, Whiting FW (2009) A mitochondrial genome phylogeny of the Neuropterida (lace-wings, alderflies and snakeflies) and their relationship to the other holometabolous insect orders. Zool Scr 38: 575–590. [Google Scholar]
- 78. Wernersson R, Pedersen AG (2003) Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res 31: 3537–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17: 540–552. [DOI] [PubMed] [Google Scholar]
- 80. Akaike H (1974) A new look at the statistical model identification. IEEE Trans Autom Contr 19: 716–723. [Google Scholar]
- 81. Posada D, Crandal KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14: 817–818. [DOI] [PubMed] [Google Scholar]
- 82. Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690. [DOI] [PubMed] [Google Scholar]
- 83. Huelsenbeck JP, Ronquist F (2001) MrBayes: Bayesian inference of phylogeny. Bioinformatics 17: 754–755. [DOI] [PubMed] [Google Scholar]
- 84. Kim MJ, Kang AR, Jeong HC, Kim KG, Kim I (2011) Reconstructing intraordinal relationships in Lepidoptera using mitochondrial genome data with the description of two newly sequenced lycaenids, Spindasis takanonis and Protantigius superans (Lepidoptera: Lycaenidae). Mol Phylogenet Evol 61: 436–445. [DOI] [PubMed] [Google Scholar]
- 85. Strimmer K, Rambaut A (2002) Inferring confidence sets of possibly misspecified gene trees. Proc Biol Sci 269: 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17: 368–376. [DOI] [PubMed] [Google Scholar]
- 87. Kishino H, Hasegawa M (1989) Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data and the branching order in Hominoidea. J Mol Evol 29: 170–179. [DOI] [PubMed] [Google Scholar]
- 88. Shimodaira H, Hasegawa M (1999) Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol 16: 1114–1116. [Google Scholar]
- 89. Shimodaira H (2002) An approximately unbiased test of phylogenetic tree selection. Syst Biol 51: 492–508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Stem-and-loop structures found in the A+T-rich region of C. fletcheri mitochondrial genome. The underline and dashed lines indicate the identical and similar flanking sequences in the stem-and-loop structures, respectively, that have conservatively been found in Orthoptera and Diptera. The numbers in parenthesis indicate the number of redundant structures. The nucleotide position is indicated at the beginning and end sites of the structures.
(PDF)
Mitochondrial gene arrangement in Polyneoptera. Gene sizes are not drawn to scale. tRNA genes are abbreviated using the one-letter amino acid code, with L = trnL(CUN); L* = trnL(UUR); S = trnS(AGN); S* = trnS(UCN). Gene names that are not underlined indicate a forward direction, whereas underlines indicate a reverse transcriptional direction. CR indicates the A+T-rich region.
(PDF)
Nucleotide composition and skewness of mitochondrial genomes of Polyneoptera.
(PDF)
Nucleotide composition at each codon position of the concatenated 13 PCGs in Polyneoptera.
(PDF)
Composition and skewness of mitochondrial PCGs in Polyneoptera.
(PDF)
Results of topological tests for 2 datasets, showing values from 6 statistical tests performed.
(PDF)