

Abstract
Temporal lobe epilepsy (TLE) is the most common form of focal epilepsy. Previous research has demonstrated several trends in human tissue that, undoubtedly, contribute to the development and progression of TLE. In this study we examined resected human hippocampus tissue for a variety of changes including gliosis that may contribute to the development and presentation of TLE. The study subjects consisted of 6 TLE patients and 3 sudden-death controls. Clinicopathological characteristics were evaluated by H&E staining. Immunohistological staining and Western blotting methods were used to analyze the samples. Neuronal hypertrophy was observed in resected epileptic tissue. Immunohistological staining demonstrated that activation of astrocytes was significantly increased in epileptic tissue as compared corresponding regions of the control group. The western blot data also showed increased CX43 and AQP4 in the hippocampus and downregulation of Kir4.1, α-syntrophin, and dystrophinin, which are the key constituents of AQP4 multi-molecular complex. These tissues also demonstrated changes in inflammatory factors (COX-2, TGF-β, NFkB) suggesting that these molecules may play an important role in TLE pathogenesis. In addition we detected increases in metabotropic glutamate receptor (mGluR) 2/3, mGluR5 and kainic acid receptor subunits KA1 (Grik4) and KA2 (Grik5) in patients' hippocampi. We noted increased expression of the α1c subunit comprising Class C L-type Ca2+ channels and calpain expression in these tissues, suggesting that these subunits may have an integral role in TLE pathogenesis. These changes found in the resected tissue suggest that they may contribute to TLE and that the Kainic acid receptor (KAR) and deregulation of GluR2 receptor may play an important role in TLE development and disease course. This study identifies alterations in number of commonly studied molecular targets associated with astrogliosis, cellular hypertrophy, water homeostasis, inflammation, and modulation of excitatory neurotransmission in hippocampal tissue from TLE patients.
1.1 BACKGROUND
Epilepsy is a chronic brain disorder, defined by spontaneous recurrent seizures. Temporal lobe epilepsy (TLE), the most common form in adults, is generally characterized by a unilateral temporal lobe seizure foci (Cascino 2005; Sharma et al., 2007). Due to the lack of effective pharmacotherapies, a relatively large percentage of the TLE patients suffer from medically intractable seizures. As a result, invasive resection of the seizure focus is often recommended as a final alternative to improve quality of life. Analysis of hippocampal tissue resected from TLE patients and animal models of this disorder reveal a number of histopathological changes including neuronal loss in the CA1 and CA3 regions, sclerotic gliosis, and aberrant mossy fiber sprouting from dentate granule cells (Cascino 2005; Sharma et al., 2007; Bae et al. 2010; de Lanerolle et al. 2003; Epsztein et al. 2005; Melo 2010; Wieser 2004; Yang T, 2010). Despite intensive study, changes at the molecular level responsible for the histological aberrations observed in TLE patients are largely unknown. Further understanding of these molecular alterations may provide insight into disease pathology and novel therapeutic targets for the treatment of TLE.
Epileptogenesis is not strictly a result of neural pathway dysfunction, but may also occur due to aberrant glial cell function. However, the role of glial cells, particularly astrocytes, in TLE pathogenesis and progression remains understudied. Reactive astrogliosis generally serves as a non-specific marker of CNS scarring and has been noted in animal models of epilepsy where seizures cause their activation and subsequent pro-inflammatory cytokine production (Ridet et al., 1997; Ravizza et al., 2011; Choi et al., 2009; Lauri et al., 2011). Astrocytic activation results in varying outcomes following seizure because inflammatory mediators may protect or damage CNS cells (Raivich et al., 1999). Therefore, a delicate balance between pro-inflammatory and anti-inflammatory processes may determine the extent of residual cellular damage in affected brain regions of TLE patients.
In addition to regulating inflammatory activity in the epileptic brain, astroglia also act to buffer extracellular ions and neurotransmitter levels thus maintaining synaptic homeostasis (Shao and McCarthy 1994). Voltage-gated Ca2+ channels serve as one mechanism by which astrocytes respond to and regulate ion gradients across neuronal membranes (Dani et al., 1992; Papadopoulos et al., 2002). Manipulation of cell volume is another means by which astrocytes regulate both extracellular volume and ionic concentrations and has been shown to affect neuronal excitability (D'Ambrosio 2006). Aquaporin (AQP) 4, the predominant form of AQP expressed in astrocytes, is a membrane-associated water channel which plays an important role in maintaining the extracellular environment. Altered expression of AQP4 has been shown in response to a number of pathological conditions marked by astrocytic activation and/or blood-brain barrier (BBB) disruption (St. Hilarie et al., 2005; Taniguchi et al., 2000; Theis et al., 2004). These observations suggest that astroglial AQP4 may modulate neuronal excitability by regulating the extraneuronal and extrasynaptic environments. Connexin 43 (CX43), a major component protein of astrocyte gap junctions, also aids in astrocyte regulation of the extracellular environment in the CNS (Wolburg et al., 2009). As osmolarity within the astrocyte increases, solutes (such as neurotransmitters) diffuse through CX43-expressing astrocytic gap junctions allowing for greater neurotransmitter clearance from the extracellular milieu.
There is also physiological evidence that coupling of the inwardly rectifying K+ channel 4.1 (Kir4.1) to AQP4 may have a role in neuronal excitability. It is believed that Kir4.1 and AQP4 are key constituents of a multi-molecular complex in astrocytes that includes α-syntrophin, Dp71, dystrobrevin, α-dystroglycan and β-dystroglycan. This protein complex, typically localized to the perivascular astrocyte endfoot, permits efficient uptake of K+ and regulation of neuronal membrane potential (Aronica et al., 2000). Increased membrane expression of metabotropic glutamate receptors (mGluR)5 and mGluR2/3 has been demonstrated in reactive astrocytes of epileptic hippocampal tissue (Seifert et al., 2006). Activation of these glial mGluRs can regulate glial function and facilitate neuron-glia communication (Seifert et al., 2006). Furthermore, mGluR3 can be co-localized with AQP4, suggesting a role for astrocytic mGluRs in regulation of extracellular glutamate levels (Seifert et al., 2006; Jankowsky et al., 2001).
Mounting evidence also suggests that other glutamate receptors including kainate receptor subunits KA1 and KA2 are contributors to neuronal pre- and post-synaptic activity through direct modulation of intracellular Ca2+ level (Darstein et al., 2003). KA1 and KA2 are differentially expressed in the hippocampus with KA1 mRNA expression restricted to the CA3 region whereas KA2 mRNA is widespread (Lauri et al., 2011). Altered levels of KA1 or KA2 expression could have serious functional consequences for synaptic homeostasis.
In the current study, we examined surgically resected hippocampal tissue from TLE patients for a variety of changes that may contribute to the development and progression of TLE. GFAP immunoreactivity, generalized cellular swelling, and inflammatory mediators (COX-2, TGF-β, NFkB) were determined as measures of astrocyte activation. Levels of several known regulators of extracellular environment and neuronal excitability (AQP4, CX43, Kir4.1, mGluRs, KA1/2, etc.) were also measured to determine their potential involvement in TLE pathology. Findings presented in this study suggest that refractory TLE may be characterized by aberrant expression of osmoregulatory proteins, metabotropic glutamate receptors, and the Kir4.1 potassium channel. Furthermore, a number of associated pathological changes were noted, including: increased GFAP immunoreactivity, cellular swelling, and production of pro-inflammatory mediators that may be indicative of generalized astrocyte activation in the epileptic hippocampus. Together, the biochemical and cellular alterations described in this study may serve both as potential disease markers and targets for therapeutic intervention in TLE patients.
EXPERIMENTAL PROCEDURES
2.1 Acquisition of Human Epileptic Samples
Patient samples were collected following temporal lobe resection performed as treatment for refractory TLE. Patient consent was obtained for tissue use before surgery. All patients included in this study were diagnosed as epileptic according to the criteria defined by the International League Against Epilepsy (Commission of Classification and Terminology of the International League Against Epilepsy). Patients were evaluated based on clinical history, physical examination, ictal EEG recording, and MRI imaging. All patients were considered to have medication refractory TLE, and after careful review of the clinical information by the multi-disciplinary comprehensive epilepsy team at the Medical University of South Carolina, all patients were referred for anterior temporal lobectomy for treatment of epilepsy. All surgeries were performed by the same neurosurgeon (SG) and the surgical technique was equivalent for all cases. Tissue was collected and handled according to the Medical University of South Carolina procedures and with approval of the Institutional Review Board of the Medical University (IRB). Postmortem blocks of tissue to be used as controls in this study were obtained from the Harvard Brain Research Center and Medical Research Council Sudden Death Brain & Tissue Bank in Edinburgh, Scotland. To the best of our knowledge, control patients were not taking neuroactive medication at the time of death. Hippocampal samples were surgically removed from deceased patients, and within 15 min were processed and stored at −80°C.
2.2 H&E Staining
Sections of 8µm thickness were taken using a Rechert-Jung cryostat (Leica, Deerfield, IL, USA) and processed for hematoxylin and eosin (H&E) staining by standard methods. Three slides bearing a single section were made by taking slices from throughout each section. Six randomly acquired 400× images were taken from the stratum radiatum and dentate gyrus regions for each sample (three images per region × three sections per sample). Three control sudden death samples and six samples from TLE patients were compared for evidence of neuronal hypertrophy, a hallmark of chronic epilepsy.
2.3 Immunofluorescent Staining
Fresh frozen samples were cut at 8µm thickness and stained for glial fibrilary acidic protein (GFAP) to determine astrocyte reactivity. Sections were fixed in 95% EtOH for 12 min then washed in 1× PBS twice. Sections were blocked in 4% serum comprised of 2% goat and 2% horse sera (Sigma). Sections were then incubated overnight at 4°C with primary antibody in 4% serum (monoclonal mouse anti-GFAP @ 1:200 and polyclonal rabbit anti-AQP4; AbCam), washed in PBS, and exposed to Texas Red conjugated secondary horse anti-mouse IgG, (1:100; Vector Laboratories, Burlingame, CA) and FITC conjugated secondary goat anti-rabbit IgG (1:100; Vector Laboratories) for one hour at room temperature in 4% serum. Finally, slides were washed in PBS and mounted using Vectashield mounting media with DAPI (Vector Laboratories).
Three slides from throughout the tissue sample bearing a single tissue sections each were stained in the described manner and at least three 200× images were taken from the stratum radiatum and dentate gyrus regions (for a total of six images per section and 18 images per hippocampus specimen) using a Zeiss fluorescent microscope (Carl Zeiss, Inc, North America, USA). Exposure time was identical for each image captured and determined by the strongest staining epileptic tissue sample. Mean Texas red fluorescent intensity was measured by NIH ImageJ software and statistical analysis was performed using a T-test to compare epileptic tissue GFAP intensity against sudden death controls. Differences from the control were considered significant at P ≤ 0.05.
2.4 Extraction of Protein and Western Blot Analysis
Normal and epileptic hippocampus tissues (100mg each) were homogenized (Brinkmann Instruments, Westbury, NY) in ice-cold (4°C) 50 mM Tris-HCl buffer (pH 7.4) containing 0.32 M sucrose, 0.1 mM phenylmethylsulfonyl fluoride (PMSF) (Sigma Chemical Company, St. Louis, MO), and 1.0 mM EDTA. The concentration of total proteins was quantitated spectrophotometrically using Coomassie® Plus Protein Assay Reagent (Pierce, Rockford, IL). Procedures for Western blot analysis have been described previously (Das et al., 2010). Monoclonal antibody against β-Actin (Sigma) was used to standardize cytosolic protein loading on SDS–PAGE gels. Monoclonal mouse antibodies (CX43, GluR1, NFkB), rabbit polyclonal antibodies (AQP4, Calpain, COX-2) and goat polyclonal antibodies (KA1, KA2) were purchased from Santa Cruz Biotech and diluted 1:250 (Santa Cruz, CA, USA). All other primary IgG antibodies were purchased from Calbiochem and diluted 1:250 (Gibbstown, NG, USA). Secondary antibodies were horseradish peroxidase-conjugated goat anti-goat IgG (Sigma), horseradish peroxidase-conjugated goat anti-mouse IgG and horseradish peroxidase-conjugated goat anti-rabbit IgG (ICN Biomedicals, Solon, OH, USA). All secondary antibodies were dilute 1:1,000 prior to use.
Data from Western blots were analyzed to detect significant differences between subjects with TLE and controls by one-way analysis of variance followed by Fisher's post hoc tests using Statview software (Abacus Concepts, Berkeley, CA). Quantified blot intensities were averaged within groups and were expressed as the mean ± the standard error of the mean (SEM) of three or more independent experiments. Differences from the control were considered significant at P ≤ 0.05.
RESULTS
3.1 Demographic and clinical characteristics of subjects
The mean age of the epilepsy patients was 33.5 ± 8.73 years, with 3 men and 3 women in the experimental group (Table 1). The mean duration of seizure recurrence was 8.9 ± 3.06 years. Clinical information from all patients is summarized in table 2. Epilepsy patients experienced seizure recurrence for at least 5 years, with 3 in 6 having recurrences for more than 10 years. The sudden death control group consisted of 3 men, with an average age of 30.90 ± 8.65 years. Statistical analysis showed that there were no significant differences in age between the patients and controls (p <0.05).
Table 1.
List of relevant abbreviations and significance for epilepsy
| CX43: Connexin 43 | Gap junction component protein; found in astrocyte-to-astrocyte junctions |
| AQP4: Aquaporin 4 | Membrane protein that regulates water passage cell membrane; expressed in astrocytes; elevated in response to injury |
| AMPA 2-amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl)propanoic acid | AMPA receptor agonist; regulates glutamate receptor activation; generates fast excitatory postsynaptic potentials |
| mGluR2/3: Glutamate Receptor 2/3 | Excitatory neurotransmitter receptors found in astrocytes and neurons |
| mGluR5: Glutamate Receptor 5 | Excitatory neurotransmitter receptor found in astrocytes and neurons |
| TGF-β: Transforming Growth Factor Beta | Controls proliferation, cellular differentiation in mitotically active cell types; elevated in activated immune cells |
| COX-2: Cyclooxegenase-2 | Enzyme that is a common marker of cellular inflammatory response |
| NFkB: Nuclear Factor kappa Beta | Transcription factor that is elevated by cellular stressors (i.e. inflammation) |
| KA: Kainic Acid Receptor | General term for excitatory glutamate receptors which are responsive to KA excitation |
| KA1(GluK4): Kainic Acid Receptor Subunit 1 | Subunit of glutamate receptors responsive to KA; found in astrocytes and neurons pre- and post-synaptically |
| KA2(GluK5): Kainic Acid Receptor Subunit 2 | Subunit of glutamate receptors responsive to KA; found in astrocytes and neurons pre- and post-synaptically |
| Kir4.1: Inwardly rectifying K+ channel 4.1 | A K+ ion channel which allows inward flow of ions when open; allows for cellular regulation of membrane potential; highly expressed in neurons and also expressed in other cell types |
| cAMP: Cyclic Adenosine Monophosphate | Intracellular second messenger which is elevated in activated neurons and astrocytes; regulates Ca2+ passage through ion channels |
Table 2.
Background for collected patient hippocampi
| Patient ID | ID # 1 | ID #2 | ID #3 | ID #4 | ID #5 | ID #6 |
|---|---|---|---|---|---|---|
| Sex | F | F | M | M | F | M |
| Age | 38 | 36 | 31 | 23 | 47 | 26 |
| Semiology | Behavioral arrest, moves right automatisms, grunts picks at clothes | Staring spells, Bilateral shaking with awareness, Generalized convulsion | Rising sensation going through his abdomen and chest, chewing and lip smacking noise speaks non-words | Stomach ache, blank staring with right head version, sometimes fall down Jerking | Stop doing suddenly, Freeze up with a blank stare, Shaking right hand | Very typical for mesial temporal lobe onset of seizures |
| Current Medication(s) | Keppra; Vimpat | Keppra, Topiramate | Carbamaze pine, Zonisemide, Tranxene | Depakote, Keppra, Valium, Vimpat | Topamax, Trileptal | Topamax, Trileptal, Levetiracetam |
| Previous Medication(s) | Topamax, Lamictal, Tegretol, Dilantin | Dilantin, Tegretol, Trileptal | Levetiracetam (rash), phenytoin, Lamotrigine Topiramate, Valporate | Dilantin, Lamictal, Phenobarb ital, Topamax, | Dilantin, Depakote, Lyrica, Neuron tin, Lamictal, Keppra | Depakote |
| Prior surgical treatment | Non | Non | Non | None | None | None |
| Neurological Exam. | Near total blindness in both eyes | Unremarkable, except memory disturbance | Normal | Mild mental delay, otherwise normal | Normal | Ask repetitious questions and voices concerns. |
3.2 Neuronal Hypertrophic Morphology and Astrogliosis in the TLE Hippocampus
Characterization of neuronal hypertrophy and astroctye activation were performed histologically with H&E staining and GFAP-immunoreactivity, respectively (Figure 1). Morphologically hypertrophic neurons were easily visible at 400× in both the stratum radiatum and dentate gyrus of epileptic hippocampi (Figure 1A). Quantification of GFAP by Western blot and immunofluorescence using NIH Image J revealed significantly elevated GFAP expression in epileptic hippocampi when compared to sudden death controls (P<0.05; Figure 1, C & D).
Figure 1.

Sclerosis in the epileptic hippocampus. Characteristic swelling of neurons (white arrows; A) in the chronic epileptic hippocampus (black arrow indicates normal neuron). GFAP staining of representative captures from control and epileptic tissue (GFAP-red; DAPI-blue; B). Quantification of Western blot and immunofluorescent GFAP optical density (C & D, respectively). * P < 0.05.
3.3 Astrogliosis is Associated with Changes in Expression of Aquaporin 4 and Associated Proteins
AQP4 plays a major role in astrocytic regulation of extracellular contents and volume. Western blot analysis demonstrated an approximate two-fold increase in AQP4 expression among chronic TLE patients (P<0.05; Figure 2). Immunfluorescence was also utilized to determine changes in localization within the cell membrane. Co-labeling of astrocytes with GFAP and AQP4 demonstrated expression of AQP4 at the astrocyte endfoot as well more generalized distribution along the astrocyte membrane (Figure 2).
Figure 2.

Aquaporin and associated markers of astrocytic swelling. Representative images taken from 8µm frozen sections stained for AQP4 (green), GFAP (red) and DAPI (blue) from sudden death controls and resected sclerotic hippocampi (A). Elevated levels of AQP4 are apparent in epileptic tissue compared to control. Representative Western blots for AQP4, CX43, and Kir4.1 complex markers are shown (B). Quantification of blot signals relative to β-actin confirmed significantly elevated AQP4 and CX43 concurrent with a significant downregulation of Kir4.1 and associated dystophin and α-syntrophin (C). Data taken from 3 controls, and 6 epileptic hippocampus samples; *P<0.05.
Since AQP4 directly associates with the Kir4.1 channel in astrocytes to regulate extracellular K+ homeostasis and thus neuronal membrane potential, we also examined protein levels of Kir4.1. Interestingly, our analysis indicated ~50% reduction in Kir4.1 expression in TLE patients, suggesting possible dysregulation of K+ homeostasis in the epileptic hippocampus. We also examined changes in members of the dystrophin associated protein complex (DAPC), including dystrophin and α-syntrophin, which is responsible for anchoring of both AQP4 and Kir4.1 to the astrocyte membrane. Western blot analysis demonstrated significantly reduced expression of both dystrophin and α-syntrophin in TLE patients. These findings may indicate dysfunction of both anchoring and localization of the AQP4/Kir4.1 complex to the astrocyte membrane, especially in light of results from our GFAP/AQP co-labeling experiments.
The astrocytic gap junction protein CX43 works in concert with AQP4 to regulate the extracellular ionic environment. Given their associated roles in ion homeostasis, we also determined levels of CX43 in the epileptic hippocampus. Western blot demonstrated an approximate 1.5-fold increase in the expression of CX43 in epileptic patients when compared to control. This finding is likely to demonstrate aberrant maintenance of ionic gradients in TLE hippocampi.
3.4 Association of Astrocyte Activation and Pro-inflammatory Changes in TLE
In order to better understand whether astrocyte activation in the TLE hippocampus results in activation of inflammatory cascades, we examined expression of COX-2, NFkB, and TGF-β in patient sample. Western blot results demonstrated significant increases in COX-2 (~2.7 fold), NFkB (~2.4 fold), and TGF-β (~1.6 fold) in the TLE hippocampus (Figure 3). These findings suggest that inflammatory events and cytokine production may play a role in TLE pathology, but do not indicate whether this is a protective or damaging alteration.
Figure 3.
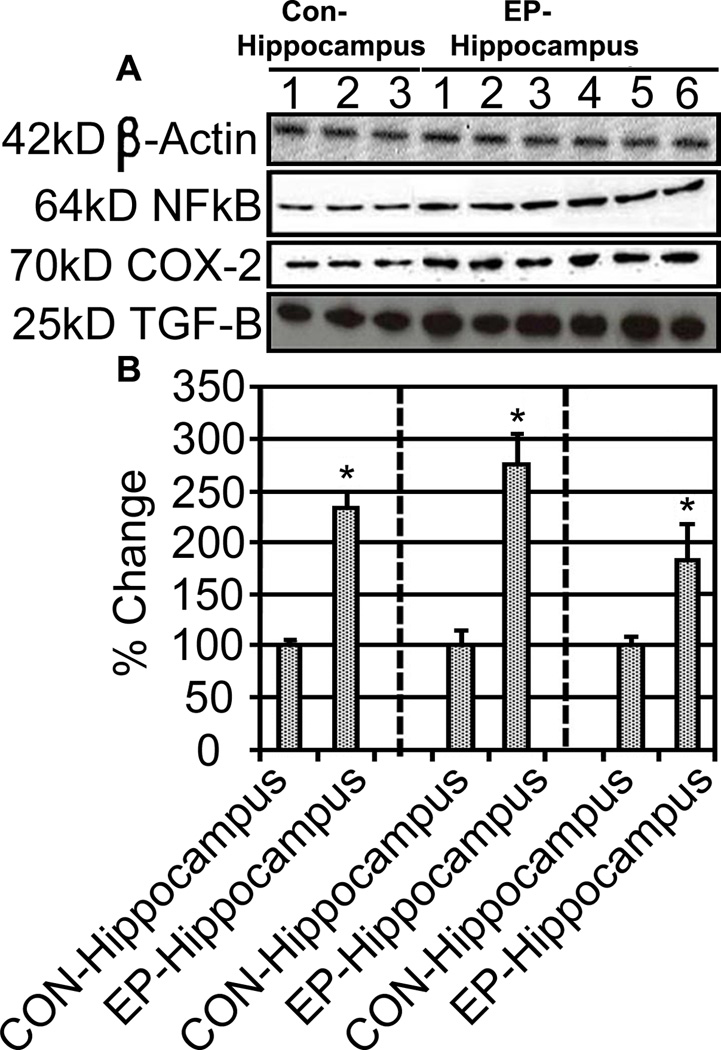
Inflammatory markers upregulation is concurrent with TGF-β increases. Representative Western blots showing inflammatory markers NFκB and COX-2 as well as TGF-β (A). Quantification of 3 control and 6 epileptic hippocampi samples is shown (B). NFκB, COX-2 and TGF-β were significantly elevated in epileptic hippocampi compared to sudden death controls; *P<0.05.
3.5 Changes in Expression of Ionotropic and Metabotropic Glutamate Receptors in TLE Hippocampal Tissue
Since glutamate, the major excitatory neurotransmitter, and its receptors have been identified as potential targets in the enhanced neuronal activity characteristic of TLE, we examined expression of both metabotropic and ionotropic glutamate receptors. GluR5 is found post-synaptically in neurons and in astrocytes and is thought to enhance excitatory neurotransmission by IP3-mediated release of intracellular Ca2+ stores. In contrast, GluR2/3 is expressed in pre-synaptic terminals and astrocytes and negatively regulates neuronal excitation. Western blot analysis indicated increased expression of mGluR2/3 and mGluR5 receptors in epileptic hippocampi compared to sudden death controls (Figure 4). This represents potential dysregulation of metabotropic glutamate receptors as a mechanism of epileptogenesis and disease progression in TLE.
Figure 4.

Ionotropic and Metabotropic Glutamate Receptors are elevated in TLE. Representative Western blots showing mGlur2/3 and mGlur5 as well as KA1, and KA2 (A). Quantification of 3 control and 6 epileptic hippocampi samples is shown (B). All receptors were significantly elevated in epileptic hippocampi compared to sudden death controls; *P<0.05.
Similar changes were also noted in expression of ionotropic glutamate receptor subunits in TLE patients. Higher levels of both KA1 and KA2 kainate receptor subunits were noted in the hippocampus of TLE patients (Figure 4). Interestingly, we also noted altered expression of the GluR2 subunit of AMPA receptors in the TLE hippocampus. GluR2-containing AMPA receptors are generally considered to be impermeable to calcium. Deregulation of GluR2 may thus indicate a shift towards AMPA receptors capable of increasing intracellular Ca2+ levels. Together, these changes in iGluRs may favor increased Ca2+ levels and enhanced neuronal excitability.
3.6 Increased Expression of Calcium-associated Proteins in TLE Patients
Because we noted changes in both mGluRs and iGluRs favoring increased intracellular Ca2+ in the TLE hippocampus, we also aimed to determine expression of other calcium-associated proteins in epileptic samples. The L-type voltage-gated Ca2+ channel (VGCC) opens during membrane depolarization to facilitate entry of Ca2+ from the extracellular space and may be involved in the hyperexcitable state of neurons in TLE. As expected, our results demonstrated increased expression of the principal α1c subunit of the L-type Ca2+ channel (Figure 5). This finding further supports the idea that changes in intracellular Ca2+ dynamics may have a role in epileptic activity. Finally, we also noted increased expression of m-calpain, a calcium-dependent cysteine protease, in TLE samples. Along with indicating increases in intracellular Ca2+, high levels of m-calpain may also be suggestive of ongoing pro-inflammatory and degenerative events in the TLE hippocampus.
Figure 5.

L-type Ca2+ channel α1C expression increases along with m-calpain expression. Representative Western blots from 3 control and 6 epileptic hippocampi samples are shown (A). L-type Ca2+ channel and m-calpain were significantly elevated in epileptic hippocampi compared to sudden death controls (B); *P<0.05.
4.1 DISCUSSION
Histological and molecular changes associated with TLE are complex and still poorly defined. Neuronal hypertrophy and increased expression of pro-inflammatory factors are hallmarks of TLE and generally suggest astrocytic activation. In this regard, our work has confirmed previous findings. Changes in extracellular osmotic and ionic gradients have been implicated as a mechanism by which dysfunctional astrocytes may promote neuronal hyperexcitability in TLE (Yang T, 2010). Hypoosmotic conditions predispose TLE patients to seizure activity by increasing cell size and initiating a series of downstream events. In particular, astrocyte swelling causes release of astroglial glutamate into the extracellular milieu (D'Ambrosio 2006). This released glutamate has the capacity to induce extra-synaptic currents and may facilitate synchronization in local neurons (Olsson et al., 2006; Benarroch et al., 2009). Furthermore, reduced extracellular volume secondary to more generalized cell swelling in TLE patients likely contributes to epileptiform spike generation by increasing neuronal sensitivity to field effects (Olsson et al., 2006; Benarroch et al., 2009). Thus, astrogliosis observed in this study and others is likely a major contributor to both disease development and severity in TLE.
Astrogliosis observed in this study was demonstrated by increased GFAP staining. Astrocyte activation has been associated with increased expression of pro-inflammatory factors which might contribute to epileptogenesis and disease progression in TLE patients (Ravizza et al., 2011; Choi et al.,2009; Lauri et al., 2011). Therefore, one aim of the present study was to determine the expression of well-known inflammatory factors (NFkB, COX-2, and TGF-β) in chronic TLE. Many experimental studies suggest that NFkB, a transcription factor involved in acute inflammation, and cytokines participate in neuronal excitability and/or glial scar formation in epilepsy. In this report, we examined the expression of NFκB in hippocampi surgically removed from patients with TLE. Our results showed that NFkB-p65 was significantly over-expressed in TLE hippocampi when compared to control specimens. COX-2 is another important inflammatory mediator which may have a role in epilepsy (Desjardins et al., 2003). COX-2 expression in the brain is dramatically increased in a wide range of neurological disorders including cerebral ischemia, Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS) (Westenbroek et al., 1998). Our results demonstrated induction of COX-2 (Figure 4) in TLE patients. We also assessed expression of TGF-β, a cytokine with pleiotropic actions in the CNS (Zhu et al., 2000). Interestingly, TGF-β has been implicated as a mediator of pro-inflammatory events following BBB disruption. Since BBB disruption may contribute to epileptogenesis, TGF-β is of particular interest in TLE. As expected, significantly increased TGF-β expression was noted in the epileptic hippocampus. These results suggest that in sclerotic hippocampi typical of long-standing TLE, inflammatory processes may be chronically activated and may contribute to disease pathology and progression.
Astrocytes also possess the necessary machinery to maintain proper homeostasis of both neurotransmitters and ions between the intra- and extracellular compartments within the CNS. Changes in AQP4 function have been implicated in epileptogenesis (Araújo et al., 2008). Astrocytes selectively express AQP4, a channel important for transport of both water and solutes across the plasma membrane (St. Hilarie et al., 2005; Taniguchi et al., 2000; Theis et al., 2004). Normal AQP4 activity is critical for maintenance of both cell volume and ionic gradients in the CNS. Interestingly, studies in AQP4 knockout mice have also suggested a role for this channel in astrocyte migration and neural transmission. Our findings indicate an overall increase in AQP4 protein levels in epileptic hippocampi when compared to control. It is likely that this change in AQP4 expression may be at least partially responsible for the astrocyte hypertrophy noted in this study.
AQP4 expressed in astroctyes also plays an important role in K+ homeostasis through coupling to the Kir4.1 channel. The Kir4.1-AQP4 protein complex acts to remove K+ from the extracellular space and thus plays a major role in establishing the hyperpolarized neuronal membrane potential. Our experiments demonstrate reduced expression of Kir4.1 channels in TLE hippocampi compared to control, suggesting dysregulation of normal maintenance of K+ homeostasis by astrocytes. Increased extracellular K+ resulting from absence of Kir channels is likely to contribute to neuronal hyperexcitability in TLE patients. The present study also examined expression of other proteins associated with AQP4 including α-syntrophin, dystrophin, and CX43. Dystrophin and α-syntrophin are members of the dystrophin associated protein complex (DAPC) responsible for anchoring AQP4 at the perivascular astrocyte endfoot. Our results indicated that expression of both dystrophin and α-syntrophin were significantly down-regulated when compared to normal controls (Figure 2). Interestingly, co-localization with GFAP suggested re-distribution of AQP4 from astrocytic endfeet to a more generalized pattern of membrane expression (Figure 2A). Taken together, these findings point to dysfunction in localization and anchoring of AQP4 to the astrocyte endfoot. Although unknown, it is likely that reduced expression of α-syntrophin and dystrophin in TLE patients disrupts AQP4 activity and may contribute to cellular swelling. In addition, we have shown increased expression of CX43 in TLE hippocampi. CX43 is an essential component of astrocytic gap junctions and has been shown to regulate a variety of factors including glia-glia and neuron-glia communication, nutrient transport, uptake of extracellular glutamate, and maintenance of ionic gradients (Fatemi et al., 2008). Although mechanisms by which Cx 43 may contribute to epileptogenesis remain unknown, our findings indicate a potential role for this protein in TLE pathogenesis. It must be noted, however, that changes in Cx 43 expression both at the mRNA and protein levels remain a topic of considerable debate because some studies have failed to show altered levels in hippocampal samples from chronic TLE patients (Elisevich et al., 1997).
Both ionotropic and metabotropic glutamate receptor subtypes (iGluRs and mGluRs) including kainate (KA) receptors, AMPA receptors, and members of the mGluR2/3 and mGluR1/5 families are expressed widely by neurons and astrocytes in the hippocampus. These receptors contribute, by varying mechanisms, to modulation in excitatory signaling and changes in intracellular ion concentrations, particularly levels of Ca2+. Metabotropic GluR5 receptors are heavily expressed in astrocytes and within the post-synaptic region. GluR5 is coupled to Gq and thus facilitates release of intracellular Ca2+ stores by activation of the phospholipase-C cascade. Thus, GluR5 is an important mediator of calcium-dependent glutamate release in astrocytes and may promote neuronal excitability. Similarly, stimulation of neuronal GluR5 may enhance postsynaptic activity by increasing Ca2+. The present study demonstrated increased protein levels of GluR5 in hippocampal tissue from TLE patients, suggesting a potential role for its overexpression in aberrant neuronal firing. In contrast, mGluR2/3 located in astrocytes and pre-synaptic terminals is thought to negatively regulate excitatory neurotransmission via alterations in intracellular cAMP levels. Interestingly, our findings indicate that mGluR2/3 expression was significantly increased in TLE patients when compared to controls. Although uncertain, this may represent a compensatory response aimed at reducing pathologic neuronal activity.
Activation of ionotropic glutamate receptors in neurons also promotes membrane depolarization and firing by facilitating influx of cations, particularly Ca2+ and Na+. In order to determine whether iGluRs may be involved in the pathogenesis of TLE, we measured expression of kainate (KA1 and KA2) and AMPA (GluR2) receptor subunits. Our findings suggest that expression of both KA1 and KA2 kainate receptor subunits is elevated in the TLE hippocampus. Kainate receptors are characterized by selective permeability for Na+ and K+ and play a minor role in post-synaptic excitatory signaling. In addition, Western blot analysis of the GluR2 AMPA receptor subunit demonstrated reduced expression in TLE patients. This is of particular interest since presence or absence of the GluR2 subunit is a major determinant of Ca2+ permeability for AMPA receptors. GluR2-lacking AMPARs are generally permeable to Ca2+. Thus, altered expression of GluR2 in the TLE hippocampus may represent a shift toward specific AMPARs able to facilitate increases in intracellular calcium. Interestingly, expression of two other calcium-associated proteins was also increased in TLE patients when compared to controls. First, we noted increased expression of the α1c subunit comprising Class C L-type Ca2+ channels. L-type Ca2+ channels are expressed on both neurons and astrocytes and respond to membrane depolarization (i.e. voltage-dependency) by allowing for increased influx of Ca2+. Studies suggest that L-type channels are commonly upregulated following a variety of CNS insults and may represent a necessary mechanism for calcium-mediated cytotoxic events (Shao et al., Science 1984; Leski et al., 1999). Finally, we have also shown increased expression of milli-calpain (m-calpain) in the TLE hippocampus. Up-regulation of m-calpain is likely to represent the effects of chronically elevated intracellular (neuronal and astrocytic) calcium. Calpain may also serve as another marker of pro-inflammatory events and activation of pathways for cell death. Thus, calpain may be a target of interest in seizure-induced neurodegeneration (Stucki et al., 2004). However, further work is necessary to characterize the role of calpain in epilepsy pathogenesis and progression. Taken together, our findings indicate dysregulation of both glutamate receptor expression and normal ionic homeostasis occurs in chronic TLE.
Overall, this study identifies alterations in number of commonly studied molecular targets associated with astrogliosis, cellular hypertrophy, water homeostasis, inflammation, and modulation of excitatory neurotransmission in hippocampal tissue from TLE patients. More sensitive studies identifying cell type, localization, and functionality associated with molecular markers including glutamate receptors, ion channels, and AQPs may be vital for true understanding of the contribution of these factors to disease. In conclusion, the findings presented here suggest a complex series of alterations both at the cellular and molecular level in chronic TLE. Our study indicates that inflammatory processes triggered by reactive astrocytes may promote upregulation of many detrimental factors or pathways contributing to disease pathogenesis and severity. Thus, inflammation merits serious attention as a therapeutic target for the treatment of epilepsy. Future studies will place particular emphasis on the role on inflammation in TLE pathology.
We evaluated the astrocytic and inflammatory involvement in human epilepsy samples
High GFAP is associated with increased AQP4, CX43, mcalpain, & L-type Ca2+ channels
Kir4.1, Dystrophin, α-syntrophin, are decreased in human epilepsy
Inflammatory markers and glutamate receptors/subunits are also increased
Astrocyte-related inflammation may be a significant target for new epilepsy therapy
ACKNOWLEDGEMENTS
This work was supported in part from the National Institute of Neurological Disorders and Stroke (NS-31622, NS-38146, and NS-41088)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent.
REFERENCES
- Araújo IM, Gil JM, Carreira BP, Mohapel P, Petersen A, Pinheiro PS, Soulet D, Bahr BA, Brundin P, Carvalho CM. Calpain activation is involved in early caspase-independent neurodegeneration in the hippocampus following status epilepticus. J Neurochem. 2008;105:666–676. doi: 10.1111/j.1471-4159.2007.05181.x. [DOI] [PubMed] [Google Scholar]
- Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH, Gorter JA. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci. 2000;12:2333–2344. doi: 10.1046/j.1460-9568.2000.00131.x. [DOI] [PubMed] [Google Scholar]
- Bae EK, Jung KH, Chu K, Lee ST, Kim JH, Park KI, Kim M, Chung CK, Lee SK, Roh JK. Neuropathologic and clinical features of human medial temporal lobe epilepsy. J Clin Neurol. 2010;6:73–80. doi: 10.3988/jcn.2010.6.2.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE. Astrocyte-neuron interactions: implications for epilepsy. Neurology. 2009;73:1323–1327. doi: 10.1212/WNL.0b013e3181bd432d. [DOI] [PubMed] [Google Scholar]
- Broberg M, Pope KJ, Lewis T, Olsson T, Nilsson M, Willoughby JO, et al. Cell swelling precedes seizures induced by inhibition of astrocytic metabolism. Epilepsy Res. 2008;80:132–141. doi: 10.1016/j.eplepsyres.2008.03.012. [DOI] [PubMed] [Google Scholar]
- Cascino GD. Temporal lobe epilepsy: more than hippocampal pathology. Epilepsy Curr. 2005;5:187–189. doi: 10.1111/j.1535-7511.2005.00059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Nordli DR, Jr, Alden TD, DiPatri A, Jr, Laux L, Kelley K, Rosenow J, Schuele SU, Rajaram V, Koh S. Cellular injury and neuroinflammation in children with chronic intractable epilepsy. J Neuroinflammation. 2009;6:38. doi: 10.1186/1742-2094-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio R. Does glutamate released by astrocytes cause focal epilepsy? Epilepsy Curr. 2006;6:173–176. doi: 10.1111/j.1535-7511.2006.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8:429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- Darstein M, Petralia RS, Swanson GT, Wenthold RJ, Heinemann SF. Distribution of kainate receptor subunits at hippocampal mossy fiber synapses. J Neurosci. 2003;23:8013–8019. doi: 10.1523/JNEUROSCI.23-22-08013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, McDowell M, O'Dell CM, Busch ME, Smith JA, Ray SK, Banik NL. Post-treatment with voltage-gated Na(+) channel blocker attenuates kainic acid-induced apoptosis in rat primary hippocampal neurons. Neurochem Res. 2010;35:2175–2183. doi: 10.1007/s11064-010-0321-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Williamson A, Spencer SS, Zaveri HP, Eid T, Spencer DD. A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: evidence for distinctive patient subcategories. Epilepsia. 2003;44:677–687. doi: 10.1046/j.1528-1157.2003.32701.x. [DOI] [PubMed] [Google Scholar]
- Desjardins P, Sauvageau A, Bouthillier A, Navarro D, Hazell AS, Rose C, Butterworth RF. Induction of astrocytic cyclooxygenase-2 in epileptic patients with hippocampal sclerosis. Neurochem Int. 2003;42:299–303. doi: 10.1016/s0197-0186(02)00101-8. [DOI] [PubMed] [Google Scholar]
- Elisevich K, Rempel SA, Smith BJ, Edvardsen K. Hippocampal connexin 43 expression in human complex partial seizure disorder. Exp Neurol. 1997;145:154–164. doi: 10.1006/exnr.1997.6467. [DOI] [PubMed] [Google Scholar]
- Epsztein J, Represa A, Jorquera I, Ben-Ari Y, Crépel V. Recurrent mossy fibers establish aberrant kainate receptor-operated synapses on granule cells from epileptic rats. J Neurosci. 2005;5:8229–8239. doi: 10.1523/JNEUROSCI.1469-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Reutiman TJ, Lee S. Expression of astrocytic markers aquaporin 4 and connexin 43 is altered in brains of subjects with autism. Synapse. 2008;62:501–507. doi: 10.1002/syn.20519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Patterson PH. The role of cytokines and growth factors in seizures and their sequelae. Prog Neurobiol. 2001;63:125–149. doi: 10.1016/s0301-0082(00)00022-8. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Taira T. Role of kainate receptors in network activity during development. Adv Exp Med Biol. 2011;717:81–91. doi: 10.1007/978-1-4419-9557-5_8. [DOI] [PubMed] [Google Scholar]
- Leski ML, Valentine SL, Coyle JT. L-type voltage-gated calcium channels modulate kainic acid neurotoxicity in cerebellar granule cells. Brain Res. 1999;828:27–40. doi: 10.1016/s0006-8993(99)01270-6. [DOI] [PubMed] [Google Scholar]
- MacVicar BA. Voltage-dependent calcium channels in glial cells. Science. 1984;226:1345–1347. doi: 10.1126/science.6095454. [DOI] [PubMed] [Google Scholar]
- Melo T, Bigini P, Sonnewald U, Balosso S, Cagnotto A, Barbera S, Uboldi S, Vezzani A, Mennini T. Neuronal hyperexcitability and seizures are associated with changes in glial-neuronal interactions in the hippocampus of a mouse model of epilepsy with mental retardation. J Neurochem. 2010;115:1445–1454. doi: 10.1111/j.1471-4159.2010.07048.x. [DOI] [PubMed] [Google Scholar]
- Olsson T, Broberg M, Pope KJ, Wallace A, Mackenzie L, Blomstrand F, Nilsson M, Willoughby JO. Cell swelling, seizures and spreading depression: an Impedance study. Neuroscience. 2006;140:505–515. doi: 10.1016/j.neuroscience.2006.02.034. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Krishna S, Verkman AS. Aquaporin water channels and brain edema. Mt Sinai J Med. 2002;69:242–248. [PubMed] [Google Scholar]
- Raivich G, Jones LL, Werner A, Blüthmann H, Doetschmann T, Kreutzberg GW. Molecular signals for glial activation: pro- and anti-inflammatory cytokines in the injured brain. Acta Neurochir Suppl. 1999;73:21–30. doi: 10.1007/978-3-7091-6391-7_4. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Balosso S, Vezzani A. Inflammation and prevention of epileptogenesis. Neurosci Lett. 2011;497:223–230. doi: 10.1016/j.neulet.2011.02.040. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Seifert G, Schilling K, Steinhäuser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- Shao Y, McCarthy KD. Plasticity of astrocytes. Glia. 1994;11:147–155. doi: 10.1002/glia.440110209. [DOI] [PubMed] [Google Scholar]
- Sharma AK, Reams RY, Jordan WH, Miller MA, Thacker HL, Snyder PW. Mesial temporal lobe epilepsy: pathogenesis, induced rodent models and lesions. Toxicol Pathol. 2007;35:984–999. doi: 10.1080/01926230701748305. [DOI] [PubMed] [Google Scholar]
- St. Hilarie C, Vargas D, Prado CA, Gincel D, Mann J, Rothstein JD, McArthur JC, Conant K. Aquaporin 4 is increased in association with human immunodeficiency virus dementia: Implications for disease pathogenesis. J Neurovirol. 2005;11:535–543. doi: 10.1080/13550280500385203. [DOI] [PubMed] [Google Scholar]
- Stucki BL, Bolognani F, Perrone-Bizzozero NI, Banik NL, Griesemer DA, Ray SK. Upregulation of calpain in neuronal apoptosis in kainic acid induced seizures in rats. J Neurochem. 2004;90:8. [Google Scholar]
- Taniguchi M, Yamashita T, Kumura E, Tamatani M, Kobayashi A, Yokawa T, Maruno M, Kato A, Ohnishi T, Kohmura E, Tohyama M, Yoshimine T. Induction of aquaporin-4 water channel mRNA after focal cerebral ischemia in rat. Brain Res Mol Brain Res. 2000;78:131–137. doi: 10.1016/s0169-328x(00)00084-x. [DOI] [PubMed] [Google Scholar]
- Theis M, Speidel D, Willecke K. Astrocyte cultures from conditional connexin43-deficient mice. Glia. 2004;46:130–141. doi: 10.1002/glia.10350. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Bausch SB, Lin RC, Franck JE, Noebels JL, Catterall WA. Upregulation of L-type Ca2+ channels in reactive astrocytes after brain injury, hypomyelination, and ischemia. J Neurosci. 1998;18:2321–2334. doi: 10.1523/JNEUROSCI.18-07-02321.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieser HG. ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia. 2004;45:695–714. doi: 10.1111/j.0013-9580.2004.09004.x. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009;335:75–96. doi: 10.1007/s00441-008-0658-9. [DOI] [PubMed] [Google Scholar]
- Yang T, Zhou D, Stefan H. Why mesial temporal lobe epilepsy with hippocampal sclerosis is progressive: uncontrolled inflammation drives disease progression? J Neurol Sci. 2010;296:1–6. doi: 10.1016/j.jns.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Roth-Eichhorn S, Braun N, Culmsee C, Rami A, Krieglstein J. The expression of transforming growth factor-beta1 (TGF-beta1) in hippocampal neurons: a temporary upregulated protein level after transient forebrain ischemia in the rat. Brain Res. 2000;866:286–298. doi: 10.1016/s0006-8993(00)02240-x. [DOI] [PubMed] [Google Scholar]