

Abstract
Although new research technologies are constantly used to look either for genes or biomarkers in the prediction of metabolic syndrome (MS), the pathogenesis and pathophysiology of this complex disease remains a major challenge. Interestingly, Cheng et al recently investigated possible pathways underlying MS by high-throughput metabolite profiling in two large and well characterized community-based cohorts. The authors explored by liquid chromatography and mass spectrometry the plasma concentrations of 45 distinct metabolites and examined their relation to cardiometabolic risk, and observed that metabolic risk factors such as obesity, insulin resistance (IR), high blood pressure, and dyslipidemia were associated with several metabolites, including branched-chain amino acids, other hydrophobic amino acids, tryptophan breakdown products, and nucleotide metabolites. In addition, the authors found a significant association of IR traits with glutamine, glutamate and the glutamine-to-glutamate ratio. These data provide new insight into the pathogenesis of MS-associated phenotypes and introduce a crucial role of glutamine-cycling pathway as prominently involved in the development of metabolic risk. We consider that the hypothesis about the role of abnormal glutamate metabolism in the pathogenesis of the MS is certainly challenging and suggests the critical role of the liver in the global metabolic modulation as glutamate metabolism is linked with aminotransferase reactions. We discuss here the critical role of the “liver metabolism” in the pathogenesis of the MS and IR, and postulate that before fatty liver develops, abnormal levels of liver enzymes, such as alanine and aspartate aminotransferases might reflect high levels of hepatic transamination of amino acids in the liver.
Keywords: Alanine, Aspartate, Glutamine, Glutamate, 2-oxoglutarate, Glycolysis, Pyruvate
INVITED COMMENTARY ON HOT ARTICLES
The metabolic syndrome (MS), a complex disorder associated with several metabolic disturbances and mostly characterized by insulin resistance (IR) in several tissues, results from a complex interplay between genetic and environmental factors[1]. Among the environmental factors, decreased physical activity, increased nutrient availability and over nutrition, play an important role and are also largely considered to be responsible for the modern epidemic of MS-related phenotypes, such as obesity, arterial hypertension and type 2 diabetes (T2D). Moreover, the pathogenesis of IR is strongly associated with the ability of the liver to suppress endogenous glucose production, suggesting that this organ is a key player in the pathophysiology of the MS. Some metabolic disturbances in the hepatic tissue, such as abnormal triglycerides accumulation observed in fatty liver, have been suggested as the trigger events and perhaps the causative factors of IR[2,3]. As such, nonalcoholic fatty liver disease (NAFLD) is now considered to be an additional component of the MS strongly associated with cardiovascular disease (CVD)[1,4-6].
Although significant efforts have been made in the last years and new research technologies are constantly used to look for either genes or biomarkers in the MS prediction, the pathogenesis and pathophysiology of this complex disease remains a major challenge.
We read with great interest the article by Cheng et al[7] recently published in Circulation. Interestingly, Cheng et al[7] investigated possible pathways underlying MS by high-throughput metabolite profiling in two large and well characterized community-based cohorts, including 1015 individuals from the Framingham Heart Study and 746 from the Malmö Diet and Cancer Study. By liquid chromatography and mass spectrometry, the authors explored the plasma concentrations of 45 distinct metabolites and examined their relation to cardiometabolic risk, and found that metabolic risk factors such as obesity, IR, high blood pressure, and dyslipidemia were associated with several metabolites, including branched-chain amino acids (BCAA), other hydrophobic amino acids, tryptophan breakdown products, and nucleotide metabolites. In addition, the authors observed a significant association of IR traits with glutamine, glutamate and the glutamine-to-glutamate ratio in individuals from both cohorts. They described for the first time that a high glutamine-to-glutamate ratio is associated with a lower risk of incident diabetes mellitus. The authors also followed up these findings by a dietary-intervention study in mice, and observed that administration of glutamine led to both increased glucose tolerance and decreased blood pressure[7]. Hence, the authors conclude that individuals with metabolic risk factors have higher circulating concentrations of glutamate and BCAA, and lower concentrations of glutamine, and suggest that glutamate may contribute to the development of the MS. Moreover, the authors observed that circulating levels of BCAA are not only associated with obesity and impaired glucose tolerance but also with dyslipidemia and blood pressure.
What can this metabolomic data tell us about the pathogenesis of MS?
These data open new perspectives about the pathogenesis of MS-associated phenotypes and introduce a crucial role of glutamine-cycling pathway as prominently involved in the development of metabolic risk.
Actually, the role of glutamine-cycle in the regulation of metabolic syndrome-related phenotypes was postulated many years ago, as Hermanussen et al[8] showed that chronic hyperglutamatemia may intoxicate arcuate nucleus neurons, thereby disrupting the hypothalamic signaling cascade of leptin action, causing hyperphagia, obesity and hyperleptinaemia. Surprisingly, glutamate has also been associated with metabolic programming and it was postulated that the thrifty phenotype, the epidemiological association between poor fetal and infant growth and the subsequent development of the MS, may be the consequence of fetal hyperglutamatemia[8].
In addition, previous evidences from a human study, including a metabolic profiling performed on 74 obese and 67 lean subjects, identified a cluster of obesity-associated changes in specific amino acid, acylcarnitine, and organic acid metabolites in obese compared to lean subjects that was associated with IR[9]. Newgard et al[9] tested the effect of supplementation of high fat diet with BCAA in an experimental study, and showed that this model was associated with decreased levels of circulating α-ketoglutarate and increased levels of glutamate, and speculated that the accumulation of glutamate increases the transamination of pyruvate to alanine, leading to the development of obesity-associated IR. Newgard et al[9] in fact extended this reasoning to that the increase in alanine, a highly gluconeogenic amino acid, contributes to the development of glucose intolerance in obesity, as circulating alanine levels are elevated in obese subjects.
Furthermore, a recent human study exploring metabolite predictors of deteriorating glucose tolerance in two Finnish population-based studies consisting of 1873 individuals and reexamination of 618 individuals after 6.5 years in one of the cohorts showed that alterations in BCAA metabolism precede hyperglycemia[10]. In addition, alanine, lactate, and pyruvate were predictive of post-challenge glucose[10]. A candidate gene association study in 9369 non-diabetic or newly diagnosed T2D Finnish men that explored the association of glycemia and 43 genetic risk variants showed that hyperglycemia and a variant of glucokinase (hexokinase 4) regulator (GCKR) are associated with the levels of eight amino acids, including alanine, leucine, isoleucine, tyrosine, and glutamine predicted incident T2D in a 4.7-year follow-up[11]. Among the 43 risk variants, only one single nucleotide polymorphism, the glucose-increasing major C allele of rs780094 of GCKR, was significantly associated with decreased levels of alanine and isoleucine and elevated levels of glutamine[11].
The role of the liver in glutamate metabolism: Aminotransferases and glutamate cycle
The hypothesis about the role of abnormal glutamate metabolism in the pathogenesis of the MS is certainly challenging and suggests the critical role of the liver in the global metabolic modulation as glutamate metabolism is linked with aminotransferase reactions. Actually, in the liver, the enzymes of glutamine metabolism critically determine the level of glutamine that is released to circulation[12]. Moreover, glutamate increases the transamination of pyruvate to alanine. In fact, the metabolism of almost all of the amino acids is initiated by aminotransferases, and the transfer of the amino group produces glutamate which may then be substrate of either glutamate dehydrogenase or aspartate aminotransferase[13].
The reactions of transamination mediate the synthesis of aspartate, asparagine, glutamate, and glutamine from ammonia and intermediate of the glycolysis pathway, and allow for the utilization of the carbon atoms from these four amino acids for glucose synthesis under fasting conditions. A short overview of alanine (ALT) and aspartate (AST) aminotransferases is shown in Table 1, and a comprehensive illustration of the AST and ALT pathway is shown in Figure 1.
Table 1.
Overview about liver aminotransferases alanine and aspartate
| ALT or GPT |
| Catalyzes the reversible transamination1 between alanine and 2-oxoglutarate to form pyruvate and glutamate: L-alanine + 2-oxoglutarate = pyruvate + L-glutamate |
| ALT has both degradative and biosynthetic roles in the glutamate cycling |
| ALT participates in cellular nitrogen metabolism and also in liver gluconeogenesis starting with precursors transported from skeletal muscles |
| ALT is present in tissues including liver, kidney, heart, and skeletal muscle. |
| AST or GOT |
| Catalyzes the reversible transamination between L-aspartate and 2-oxoglutarate to form oxaloacetate and glutamate: L-alanine + 2-oxoglutarate |
| L-aspartate + 2-oxoglutarate = oxaloacetate + L-glutamate |
| Cytosolic AST (GOT 1 catalyzes the reversible reaction of oxaloacetate and glutamate to form aspartate and 2-oxoglutarate (alpha-ketoglutarate) |
| AST has two isoforms: cytoplasmatic and mitochondrial |
Transaminase: A subclass of enzymes that catalyze the transfer of an amino group from a donor (generally an amino acid) to an acceptor (generally 2 keto acid) in a cyclic process using pyridoxal phosphate as a cofactor. ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; GPT: Glutamate pyruvate transaminase; GOT: Glutamate oxaloacetate transaminase.
Figure 1.

Schematic presentation of the role of aminotransferases alanine and aspartate in metabolic pathways. Alanine and aspartate pathways and their relationship with fatty acid and amino acid metabolism, glycolysis, citrate and urea cycle. Available at: KEGG Metabolic Pathways. http://www.biologie.uni-hamburg.de/b-online/kegg/kegg/Classes/dblinks_java/map/map00252.html.
In summary, ALT not only plays a key role in the intermediary metabolism of glucose and amino acids, but can also be considered as a major contributor to the steady-state glutamate levels as the enzyme can simultaneously catabolize and synthesize glutamine[13].
Biological significance of high ALT and AST levels and cardiovascular risk: Is there any association with altered glutamate metabolism?
Serum activity levels of ALT are routinely used as a biomarker of liver injury caused by drug toxicity, viral infection, alcohol abuse and fatty liver. Nevertheless, several epidemiological studies showed that CVD and the MS are associated with abnormal liver enzymes, such as ALT, even in the absence of liver injury or steatosis. For instance, increased levels of ALT are associated with long-term development of multiple metabolic disorders among participants of the Framingham Offspring Heart Study[14]. Goessling and coworkers also demonstrated that higher ALT levels were significantly associated with an increased risk of T2D and CVD in age-sex adjusted analyses[14]. There was also a significant interaction between body mass index and ALT levels, and the follow-up study of these overweight and obese participants with highest ALT levels for 20 years showed a 30-fold increased risk for developing T2D[14].
The association of ALT with the risk of development MS was also evaluated in 1097 subjects from the population-based cohort of Caucasian men and women (Hoorn Study), and ALT was significantly associated with fasting plasma glucose at follow-up[15]. The 10-year risk of all-cause mortality, fatal and non-fatal CVD in relation to ALT was also assessed in 1439 subjects participating in the Hoorn Study, and the predictive value of ALT for coronary events, seems independent of traditional risk factors[16].
Moreover, findings from the Western Australian Health Department data linkage system, an Australian population-based cohort study, support a strong association between ALT levels and the MS independent of insulin resistance[17].
An overview about the epidemiological evidence of liver enzymes and cardiovascular outcomes was recently published[18].
In spite of the epidemiological evidences mentioned above, the research community is still inconclusive about the pathobiological meaning of the elevated ALT levels and CV risk. In fact, the question of whether abnormalities in ALT levels precede the development of MS, or whether the MS components themselves can lead to the increase of ALT levels is still unanswered[14]. Hence, the biological mechanisms responsible for the association between liver enzymes and the MS- related phenotypes are still poorly understood, and much of the speculations focus on the putative liver injury associated with fatty liver that frequently coexists with the MS.
The metabolomic data presented by Cheng et al[7] not only raised new questions about the role of glutamate-glutamine cycle in the pathogenesis of the MS, but also suggested a dramatic change in the paradigm of the meaning of elevated aminotransferase levels in the context of MS-related phenotypes. Thus, we speculate that abnormal levels of ALT and AST are associated with a deregulation of normal amino acid metabolism in the liver, including aromatic amino acid, and then special compounds such as glutamate are released into the general circulation. This hypothesis attempts to illustrate the critical role of the “liver metabolism” in the pathogenesis of the MS and IR, and postulates that before the liver becomes fatty, abnormal levels of liver enzymes might reflect high levels of hepatic transamination of amino acids in the organ.
Is there any experimental evidence for this? Stegink et al[19] have demonstrated that if a large proportion of glutamate is ingested, portal glutamate increases and this elevation results in increased hepatic glutamate metabolism, leading to release of glucose into systemic circulation, a physiopathogenic event that may perpetuate hyperglycemia.
Systems biology also provides a rational evidence for the association between liver transaminases and the metabolic abnormalities observed by Cheng et al[7].
We performed a functional association analysis that included protein and genetic interactions, pathways, co-expression, co-localization, and protein domain similarity using the bioinformatics resource GenMANIA[20]. Interestingly, several genes are regarded as direct “neighbors” of liver transaminases (GPT and GOT1/2, as described in Table 1), but including isocitrate dehydrogenases 1 (IDH1) and 2 (IDH2) and the transcription factor hepatic nuclear factor 4 alpha, because they are involved in the regulation of liver transaminases and glutamine synthetase[21] (Figure 2). Interestingly, the predicted genes (Table 2) belong to pathways that explain most of the findings of Cheng et al[7], such as glutamine family amino acid metabolic process, indolalkylamine catabolic process, indole-containing compound catabolic process, tryptophan catabolic process, tryptophan metabolic process, indolalkylamine metabolic process, indole-containing compound metabolic process, cellular biogenic amine catabolic process, among others (Table 3).
Figure 2.
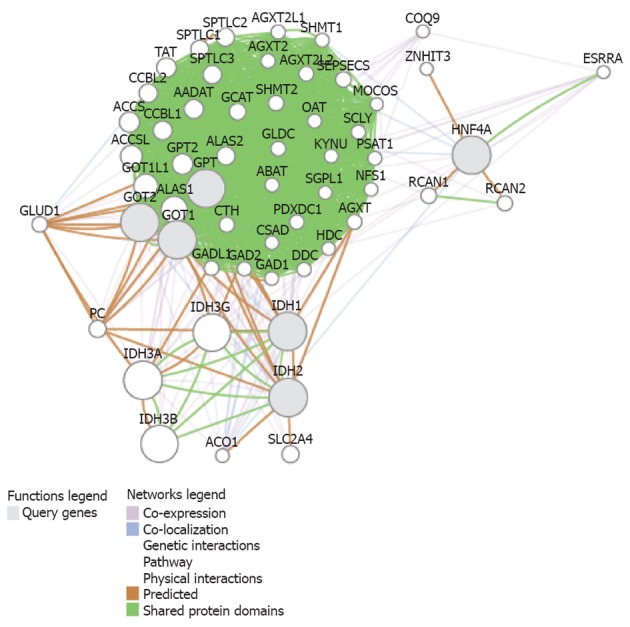
Integrated functional association analysis of protein and genetic interactions on alanine and aspartate. Pathways, co-expression, co-localization, and protein domain similarity were analyzed by the bioinformatics resource GenMANIA (genemania.org) for the 5 candidate genes [alanine also known as glutamate pyruvate transaminase (GPT) and GPT2], aspartate [also known as glutamate oxaloacetate transaminase (GOT)1 and GOT2, (Table 1)], isocitrate dehydrogenases 1 (IDH1), IDH2 and HNF4 (gray circles) and the predicted related genes by systems biology (open circles). List of gene symbol and gene function is shown in Table 2. Predicted functional pathways and Q values are shown in Table 3.
Table 2.
Candidate gene list (in bold) and 50 predicted genes by systems biology
| Symbol | Description | Score |
| HNF4A | Hepatocyte nuclear factor 4, alpha [source: HGNC symbol; Acc: 5024] | 66.77 |
| IDH2 | Isocitrate dehydrogenase 2 (NADP+), mitochondrial [source: HGNC symbol; Acc: 5383] | 62.04 |
| IDH1 | Isocitrate dehydrogenase 1 (NADP+), soluble [source: HGNC symbol; Acc: 5382] | 61.96 |
| GPT | Glutamic-pyruvate transaminase (alanine aminotransferase) [source: HGNC symbol; Acc: 4552] | 58.97 |
| GOT1 | Glutamic-oxaloacetic transaminase 1, soluble (aspartate aminotransferase 1) [source: HGNC symbol; Acc: 4432] | 54.78 |
| GOT2 | Glutamic-oxaloacetic transaminase 2, mitochondrial (aspartate aminotransferase 2) [source: HGNC symbol; Acc: 4433] | 54.48 |
| IDH3A | Isocitrate dehydrogenase 3 (NAD+) alpha [source: HGNC symbol; Acc: 5384] | 2.68 |
| IDH3G | Isocitrate dehydrogenase 3 (NAD+) gamma [source: HGNC symbol; Acc: 5386] | 2.6 |
| IDH3B | Isocitrate dehydrogenase 3 (NAD+) beta [source: HGNC symbol; Acc: 5385] | 2.56 |
| ALAS1 | Aminolevulinate, delta-, synthase 1 [source: HGNC symbol; Acc: 396] | 1.57 |
| GOT1L1 | Glutamic-oxaloacetic transaminase 1-like 1 [source: HGNC symbol; Acc: 28487] | 1.35 |
| ACCSL | 1-aminocyclopropane-1-carboxylate synthase homolog (Arabidopsis)(non-functional)-like [source: HGNC symbol; Acc: 34391] | 1.14 |
| GPT2 | Glutamic pyruvate transaminase (alanine aminotransferase) 2 [source: HGNC symbol; Acc: 18062] | 1.03 |
| ACCS | 1-aminocyclopropane-1-carboxylate synthase homolog (Arabidopsis)(non-functional) [source: HGNC symbol; Acc: 23989] | 0.98 |
| TAT | Tyrosine aminotransferase [source: HGNC symbol; Acc: 11573] | 0.93 |
| AADAT | Aminoadipate aminotransferase [source: HGNC symbol; Acc: 17929] | 0.88 |
| CCBL1 | Cysteine conjugate-beta lyase, cytoplasmic [source: HGNC symbol; Acc: 1564] | 0.87 |
| CCBL2 | Cysteine conjugate-beta lyase 2 [source: HGNC symbol; Acc: 33238] | 0.83 |
| SPTLC3 | Serine palmitoyltransferase, long chain base subunit 3 [source: HGNC symbol; Acc: 16253] | 0.82 |
| ALAS2 | Aminolevulinate, delta-, synthase 2 [source: HGNC symbol; Acc: 397] | 0.8 |
| SPTLC2 | Serine palmitoyltransferase, long chain base subunit 2 [source: HGNC symbol; Acc: 11278] | 0.79 |
| SPTLC1 | Serine palmitoyltransferase, long chain base subunit 1 [source: HGNC symbol; Acc: 11277] | 0.75 |
| PC | Pyruvate carboxylase [source: HGNC symbol; Acc: 8636] | 0.69 |
| SLC2A4 | Solute carrier family 2 (facilitated glucose transporter), member 4 [source: HGNC symbol; Acc: 11009] | 0.69 |
| GCAT | Glycine C-acetyltransferase [source: HGNC symbol; Acc: 4188] | 0.66 |
| RCAN1 | Regulator of calcineurin 1 [source: HGNC symbol; Acc: 3040] | 0.56 |
| SEPSECS | Sep (O-phosphoserine) tRNA: Sec (selenocysteine) tRNA synthase [source: HGNC symbol; Acc: 30605] | 0.56 |
| GLUD1 | Glutamate dehydrogenase 1 [source: HGNC symbol; Acc: 4335] | 0.55 |
| SHMT2 | Serine hydroxymethyltransferase 2 (mitochondrial) [source: HGNC symbol; Acc: 10852] | 0.54 |
| CTH | Cystathionase (cystathionine gamma-lyase) [source: HGNC symbol; Acc: 2501] | 0.52 |
| AGXT2L2 | Alanine-glyoxylate aminotransferase 2-like 2 [source: HGNC symbol; Acc: 28249] | 0.49 |
| RCAN2 | Regulator of calcineurin 2 [source: HGNC symbol; Acc: 3041] | 0.49 |
| AGXT | Alanine-glyoxylate aminotransferase [source: HGNC symbol; Acc: 341] | 0.48 |
| GLDC | Glycine dehydrogenase (decarboxylating) [source: HGNC symbol; Acc: 4313] | 0.48 |
| GADL1 | Glutamate decarboxylase-like 1 [source: HGNC symbol; Acc: 27949] | 0.47 |
| PDXDC1 | Pyridoxal-dependent decarboxylase domain containing 1 [source: HGNC symbol; Acc: 28995] | 0.45 |
| AGXT2 | Alanine--glyoxylate aminotransferase 2 [source: HGNC symbol; Acc: 14412] | 0.45 |
| GAD2 | Glutamate decarboxylase 2 (pancreatic islets and brain, 65 kDa) [source: HGNC symbol; Acc: 4093] | 0.43 |
| SCLY | Selenocysteine lyase [source: HGNC symbol; Acc: 18161] | 0.43 |
| AGXT2L1 | Alanine-glyoxylate aminotransferase 2-like 1 [source: HGNC symbol; Acc: 14404] | 0.42 |
| ABAT | 4-aminobutyrate aminotransferase [source: HGNC symbol; Acc: 23] | 0.42 |
| DDC | Dopa decarboxylase (aromatic L-amino acid decarboxylase) [source: HGNC symbol; Acc: 2719] | 0.42 |
| KYNU | Kynureninase [source: HGNC symbol; Acc: 6469] | 0.41 |
| OAT | Ornithine aminotransferase [source: HGNC symbol; Acc: 8091] | 0.4 |
| SHMT1 | Serine hydroxymethyltransferase 1 (soluble) [source: HGNC symbol; Acc: 10850] | 0.4 |
| PSAT1 | Phosphoserine aminotransferase 1 [source: HGNC symbol; Acc: 19129] | 0.39 |
| GAD1 | Glutamate decarboxylase 1 (brain, 67 kDa) [source: HGNC symbol; Acc: 4092] | 0.38 |
| CSAD | Cysteine sulfinic acid decarboxylase [source: HGNC symbol; Acc: 18966] | 0.38 |
| NFS1 | NFS1 nitrogen fixation 1 homolog (S. cerevisiae) [source: HGNC symbol; Acc: 15910] | 0.38 |
| ACO1 | Aconitase 1, soluble [source: HGNC symbol; Acc: 117] | 0.37 |
| SGPL1 | Sphingosine-1-phosphate lyase 1 [source: HGNC symbol; Acc: 10817] | 0.36 |
| HDC | Histidine decarboxylase [source: HGNC symbol; Acc: 4855] | 0.36 |
| MOCOS | Molybdenum cofactor sulfurase [source: HGNC symbol; Acc: 18234] | 0.31 |
| ZNHIT3 | Zinc finger, HIT-type containing 3 [source: HGNC symbol; Acc: 12309] | 0.3 |
| COQ9 | Coenzyme Q9 homolog (S. cerevisiae) [source: HGNC symbol; Acc: 25302] | 0.3 |
| ESRRA | Estrogen-related receptor alpha [source: HGNC symbol; Acc: 3471] | 0.3 |
IDH: Isocitrate dehydrogenases; GPT: Glutamate pyruvate transaminase; GOT: Glutamate oxaloacetate transaminase.
Table 3.
Gene ontology annotation of predicted biological process
| Gene ontology annotation | Q value | Genes in network | Genes in genome |
| Transaminase activity | 4.86E-31 | 14 | 16 |
| Transferase activity, transferring nitrogenous groups | 6.17E-30 | 14 | 18 |
| Mitochondrial matrix | 3.10E-15 | 16 | 220 |
| Cellular amino acid catabolic process | 7.40E-15 | 12 | 77 |
| Amine catabolic process | 1.13E-14 | 12 | 81 |
| Dicarboxylic acid metabolic process | 1.75E-14 | 9 | 24 |
| Cellular amino acid biosynthetic process | 5.90E-13 | 10 | 54 |
| Carboxylic acid catabolic process | 2.55E-12 | 12 | 131 |
| 2-oxoglutarate metabolic process | 2.55E-12 | 7 | 13 |
| Organic acid catabolic process | 2.55E-12 | 12 | 131 |
| Amine biosynthetic process | 8.11E-12 | 10 | 72 |
| Glutamate metabolic process | 1.08E-10 | 6 | 10 |
| Carboxylic acid biosynthetic process | 4.53E-10 | 11 | 154 |
| Organic acid biosynthetic process | 4.53E-10 | 11 | 154 |
| Small molecule catabolic process | 4.87E-10 | 12 | 211 |
| Small molecule biosynthetic process | 2.21E-9 | 12 | 241 |
| Cofactor metabolic process | 2.24E-8 | 10 | 163 |
| Vitamin B6 binding | 9.53E-8 | 5 | 12 |
| Glutamine family amino acid metabolic process | 9.53E-8 | 6 | 27 |
| Cellular aromatic compound metabolic process | 9.53E-8 | 9 | 135 |
| Pyridoxal phosphate binding | 9.53E-8 | 5 | 12 |
| Cofactor binding | 6.48E-7 | 7 | 69 |
| Aromatic amino acid family catabolic process | 1.24E-6 | 5 | 19 |
| Coenzyme metabolic process | 1.46E-6 | 8 | 126 |
| Aromatic compound catabolic process | 3.14E-6 | 5 | 23 |
| Aromatic amino acid family metabolic process | 3.14E-6 | 5 | 23 |
| Vitamin binding | 8.74E-6 | 5 | 28 |
| Indolalkylamine catabolic process | 1.08E-5 | 4 | 11 |
| Indole-containing compound catabolic process | 1.08E-5 | 4 | 11 |
| Tryptophan catabolic process | 1.08E-5 | 4 | 11 |
| Tryptophan metabolic process | 1.48E-5 | 4 | 12 |
| Indolalkylamine metabolic process | 1.48E-5 | 4 | 12 |
| Indole-containing compound metabolic process | 1.48E-5 | 4 | 12 |
| Cellular biogenic amine catabolic process | 3.93E-5 | 4 | 15 |
| Serine family amino acid metabolic process | 5.07E-5 | 4 | 16 |
| Acetyl-CoA catabolic process | 1.23E-4 | 4 | 20 |
| Tricarboxylic acid cycle | 1.23E-4 | 4 | 20 |
| Coenzyme catabolic process | 1.23E-4 | 4 | 20 |
| Cofactor catabolic process | 3.67E-4 | 4 | 26 |
| Aerobic respiration | 4.88E-4 | 4 | 28 |
| Cellular biogenic amine metabolic process | 6.98E-4 | 5 | 71 |
| Lyase activity | 8.37E-4 | 5 | 74 |
| Acetyl-CoA metabolic process | 1.01E-3 | 4 | 34 |
| Transferase activity, transferring acyl groups other than amino-acyl groups | 1.41E-3 | 5 | 83 |
| Gluconeogenesis | 3.05E-3 | 4 | 45 |
| Aspartate family amino acid catabolic process | 3.67E-3 | 3 | 15 |
| Generation of a signal involved in cell-cell signaling | 3.67E-3 | 6 | 178 |
| Signal release | 3.67E-3 | 6 | 178 |
| Transferase activity, transferring acyl groups | 3.67E-3 | 5 | 103 |
| Sulfur amino acid metabolic process | 4.26E-3 | 3 | 16 |
| Neurotransmitter secretion | 5.20E-3 | 4 | 53 |
| Hexose biosynthetic process | 5.50E-3 | 4 | 54 |
| Water-soluble vitamin metabolic process | 6.24E-3 | 4 | 56 |
| Monosaccharide biosynthetic process | 1.04E-2 | 4 | 64 |
| Pteridine-containing compound metabolic process | 1.05E-2 | 3 | 22 |
| Neurotransmitter transport | 1.14E-2 | 4 | 66 |
| Carbon-carbon lyase activity | 1.48E-2 | 3 | 25 |
| Aspartate family amino acid metabolic process | 1.48E-2 | 3 | 25 |
| Regulation of neurotransmitter levels | 1.60E-2 | 4 | 73 |
| Sphingolipid metabolic process | 1.82E-2 | 4 | 76 |
| Alcohol biosynthetic process | 1.82E-2 | 4 | 76 |
| Pigment biosynthetic process | 1.96E-2 | 3 | 28 |
| Membrane lipid metabolic process | 2.35E-2 | 4 | 82 |
| Sphingolipid biosynthetic process | 2.35E-2 | 3 | 30 |
| Cofactor biosynthetic process | 2.53E-2 | 4 | 84 |
| Membrane lipid biosynthetic process | 2.77E-2 | 3 | 32 |
| Cellular modified amino acid metabolic process | 2.81E-2 | 4 | 87 |
| Cellular carbohydrate biosynthetic process | 4.06E-2 | 4 | 96 |
| Pigment metabolic process | 4.80E-2 | 3 | 39 |
| Vitamin metabolic process | 4.80E-2 | 4 | 101 |
Q value stands for the P value corrected for multiple testing. In addition, number of genes in the network and in the whole genome belonging to the biological process is depicted. The candidate genes are listed in Table 2, which are involved in functional enrichment analysis using the GeneMANIA tool (genemania.org).
Clinical perspective
To conclude, liver transaminases should not be considered as mere biomarkers of liver damage but central players in the pathophysiology of the NAFLD in particular or the MS components in general. Further research has to be done to define whether the elevation of these enzymes is an adaptive or a causative process of the disease.
In particular, because many confounding issues are implied in a pathogenetic relationship with liver, heart and kidney, it is time to look at multi-organ pathogenetic interactions, as recently revised by Bonora et al[22].
Finally, mutations in IDH1 and IDH2 seem to be critically involved in the generation of certain types of cancers because they create “neoenzymes” that produce 2-hydroxyglutarate, which are required for tumor cell growth from α-ketoglutarate (α-KG)[23]. α-KG is derived from glutamine through its conversion to glutamate by glutaminase. This process may explain the glutamine dependency of the cancer cell growth[24]. Then, it is tempting to speculate that glutamate excess as observed in the MS and NAFLD is an appropriate milieu for cancer development, which may explain the high prevalence of hepatocellular carcinoma in these patients[25], which offers, at the same time, new avenues for its treatment.
Footnotes
Supported by Grants PICT 2008-1521 and 2010-0441, from Agencia Nacional de Promoción Científica y Tecnológica; and UBACYT CM04, from Universidad de Buenos Aires
Peer reviewers: Ferruccio Bonino, MD, PhD, Professor of Gastroenterology, Director of Liver and Digestive Disease Division, Department of Internal Medicine, University of Pisa, Director of General Medicine 2 Unit University Hospital of Pisa, Via Roma 67, 56124 Pisa, Italy; Andrzej S Tarnawski, MD, PhD, DSc (Med), Professor of Medicine, Chief Gastroenterology, VA Long Beach Health Care System, University of California, Irvine, CA, 5901 E. Seventh Str., Long Beach, CA 90822, United States
S- Editor Cheng JX L- Editor Ma JY E- Editor Li JY
References
- 1.Sookoian S, Pirola CJ. Metabolic syndrome: from the genetics to the pathophysiology. Curr Hypertens Rep. 2011;13:149–157. doi: 10.1007/s11906-010-0164-9. [DOI] [PubMed] [Google Scholar]
- 2.Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, Okunade A, Klein S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci USA. 2009;106:15430–15435. doi: 10.1073/pnas.0904944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kotronen A, Yki-Järvinen H. Fatty liver: a novel component of the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:27–38. doi: 10.1161/ATVBAHA.107.147538. [DOI] [PubMed] [Google Scholar]
- 4.Sookoian S, Pirola CJ. Non-alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: a systematic review. J Hepatol. 2008;49:600–607. doi: 10.1016/j.jhep.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 5.Sookoian S, Gianotti TF, Rosselli MS, Burgueño AL, Castaño GO, Pirola CJ. Liver transcriptional profile of atherosclerosis-related genes in human nonalcoholic fatty liver disease. Atherosclerosis. 2011;218:378–385. doi: 10.1016/j.atherosclerosis.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 6.Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med. 2010;363:1341–1350. doi: 10.1056/NEJMra0912063. [DOI] [PubMed] [Google Scholar]
- 7.Cheng S, Rhee EP, Larson MG, Lewis GD, McCabe EL, Shen D, Palma MJ, Roberts LD, Dejam A, Souza AL, et al. Metabolite profiling identifies pathways associated with metabolic risk in humans. Circulation. 2012;125:2222–2231. doi: 10.1161/CIRCULATIONAHA.111.067827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hermanussen M, Tresguerres JA. Does the thrifty phenotype result from chronic glutamate intoxication? A hypothesis. J Perinat Med. 2003;31:489–495. doi: 10.1515/JPM.2003.075. [DOI] [PubMed] [Google Scholar]
- 9.Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–326. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Würtz P, Tiainen M, Mäkinen VP, Kangas AJ, Soininen P, Saltevo J, Keinänen-Kiukaanniemi S, Mäntyselkä P, Lehtimäki T, Laakso M, et al. Circulating Metabolite Predictors of Glycemia in Middle-Aged Men and Women. Diabetes Care. 2012;35:1749–1756. doi: 10.2337/dc11-1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stancáková A, Civelek M, Saleem NK, Soininen P, Kangas AJ, Cederberg H, Paananen J, Pihlajamäki J, Bonnycastle LL, Morken MA, et al. Hyperglycemia and a Common Variant of GCKR Are Associated With the Levels of Eight Amino Acids in 9,369 Finnish Men. Diabetes. 2012;61:1895–1902. doi: 10.2337/db11-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelly A, Stanley CA. Disorders of glutamate metabolism. Ment Retard Dev Disabil Res Rev. 2001;7:287–295. doi: 10.1002/mrdd.1040. [DOI] [PubMed] [Google Scholar]
- 13.Brosnan ME, Brosnan JT. Hepatic glutamate metabolism: a tale of 2 hepatocytes. Am J Clin Nutr. 2009;90:857S–861S. doi: 10.3945/ajcn.2009.27462Z. [DOI] [PubMed] [Google Scholar]
- 14.Goessling W, Massaro JM, Vasan RS, D’Agostino RB, Ellison RC, Fox CS. Aminotransferase levels and 20-year risk of metabolic syndrome, diabetes, and cardiovascular disease. Gastroenterology. 2008;135:1935–1944, 1944.e1. doi: 10.1053/j.gastro.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schindhelm RK, Dekker JM, Nijpels G, Stehouwer CD, Bouter LM, Heine RJ, Diamant M. Alanine aminotransferase and the 6-year risk of the metabolic syndrome in Caucasian men and women: the Hoorn Study. Diabet Med. 2007;24:430–435. doi: 10.1111/j.1464-5491.2007.02100.x. [DOI] [PubMed] [Google Scholar]
- 16.Schindhelm RK, Diamant M, Dekker JM, Tushuizen ME, Teerlink T, Heine RJ. Alanine aminotransferase as a marker of non-alcoholic fatty liver disease in relation to type 2 diabetes mellitus and cardiovascular disease. Diabetes Metab Res Rev. 2006;22:437–443. doi: 10.1002/dmrr.666. [DOI] [PubMed] [Google Scholar]
- 17.Olynyk JK, Knuiman MW, Divitini ML, Davis TM, Beilby J, Hung J. Serum alanine aminotransferase, metabolic syndrome, and cardiovascular disease in an Australian population. Am J Gastroenterol. 2009;104:1715–1722. doi: 10.1038/ajg.2009.229. [DOI] [PubMed] [Google Scholar]
- 18.Ghouri N, Preiss D, Sattar N. Liver enzymes, nonalcoholic fatty liver disease, and incident cardiovascular disease: a narrative review and clinical perspective of prospective data. Hepatology. 2010;52:1156–1161. doi: 10.1002/hep.23789. [DOI] [PubMed] [Google Scholar]
- 19.Stegink LD, Filer LJ, Baker GL. Effect of carbohydrate on plasma and erythrocyte glutamate levels in humans ingesting large doses of monosodium L-glutamate in water. Am J Clin Nutr. 1983;37:961–968. doi: 10.1093/ajcn/37.6.961. [DOI] [PubMed] [Google Scholar]
- 20.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–W220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanulović VS, Kyrmizi I, Kruithof-de Julio M, Hoogenkamp M, Vermeulen JL, Ruijter JM, Talianidis I, Hakvoort TB, Lamers WH. Hepatic HNF4alpha deficiency induces periportal expression of glutamine synthetase and other pericentral enzymes. Hepatology. 2007;45:433–444. doi: 10.1002/hep.21456. [DOI] [PubMed] [Google Scholar]
- 22.Bonora E, Targher G. Increased risk of cardiovascular disease and chronic kidney disease in NAFLD. Nat Rev Gastroenterol Hepatol. 2012;9:372–381. doi: 10.1038/nrgastro.2012.79. [DOI] [PubMed] [Google Scholar]
- 23.Borodovsky A, Seltzer MJ, Riggins GJ. Altered cancer cell metabolism in gliomas with mutant IDH1 or IDH2. Curr Opin Oncol. 2012;24:83–89. doi: 10.1097/CCO.0b013e32834d816a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70:8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–1832. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]