


Abstract
In recent years, evidence has emerged for the existence of many diverse types of RNA, which play roles in a wide range of biological processes in all kingdoms of life. These molecules generally do not, however, act in isolation, and identifying which proteins partner with RNA is a major challenge. Many methods, in vivo and in vitro, have been used to address this question, including combinatorial or high-throughput approaches, such as systematic evolution of ligands, cross-linking and immunoprecipitation and RNA immunoprecipitation combined with deep sequencing. However, most of these methods are not trivial to pursue and often require substantial optimization before results can be achieved. Here, we demonstrate a simple technique that allows one to screen proteins for RNA-binding properties in a gel-shift experiment and can be easily implemented in any laboratory. This assay should be a useful first-pass tool for assessing whether a protein has RNA- or DNA-binding properties, prior to committing resources to more complex procedures.
INTRODUCTION
In recent years, interest in RNA has been consistently increasing as our understanding of the enormous diversity of RNA biology has developed. Besides the well-studied small nuclear and nucleolar RNA, ribosomal RNA and transfer RNA species, a plethora of other non-coding RNAs have emerged as important players in biological processes, including small interfering RNAs (1), micro RNAs (2,3) and long intergenic non-coding RNAs (4,5), underscoring the imperative for defining the biochemical functions of these versatile molecules.
Most RNA molecules act in concert with proteins and a variety of experimental strategies have been developed to define the RNA-binding properties of such proteins. Biophysical approaches, such as fluorescence anisotropy (6), surface plasmon resonance (7) and isothermal titration calorimetry (8,9) can provide detailed insight into interactions, kinetics and/or thermodynamics of protein–RNA binding, whereas NMR spectroscopy and X-ray crystallography allow the structural basis for RNA recognition to be defined. These approaches rely, however, on a prior knowledge of the target RNA sequence. Combinatorial methods, either in vivo or in vitro based, can allow the researcher to define the sequence preferences of a chosen RNA-binding protein. Systematic evolution of ligands by exponential enrichment (SELEX) is a well-established method for assessing the sequence preferences of a purified protein in vitro, and methods such as RNA-binding protein immunoprecipitation-microarray profiling (RIP-Chip) (10–12), RNA-binding protein immunoprecipitation coupled with high-throughput sequencing (RIP-seq) (13), in vivo cross-linking and immunoprecipitation (CLIP) (14,15) and its derivatives are high-throughput methods that provide a snapshot of the RNA-binding activity of a protein in a cellular context. These combinatorial methods, while powerful, can require substantial optimization before they yield reliable data and are often not trivial for an inexperienced laboratory to set up. Furthermore, while the cost of deep-sequencing analysis is declining, a considerable financial outlay is still required and can add to the activation barrier for a potential user.
Homopolymer pull-down experiments (in which homomeric ribopolymers such as polyA are attached to agarose or Sepharose beads and used to capture a protein of interest) and electrophoretic mobility shift assays (EMSAs) offer much simpler options for detecting RNA-binding activity in a target protein. A disadvantage of the former approach, however, is that only a very small range of RNA sequence space can be probed. Similarly, in a typical EMSA, one must choose the RNA sequence of interest, limiting the screening potential of this approach.
In previous work, we sought to create double-stranded DNA (dsDNA) sequences of maximum sequence diversity, reasoning that such sequences could be used as EMSA probes to screen new proteins for DNA-binding activity. We designed an algorithm that allowed the construction of a dsDNA sequence containing all possible n-base pair sequences in the shortest possible sequence (16). For example, we showed mathematically that 516 bp is the shortest theoretically possible length for an oligonucleotide that contains all 1024 5-bp combinations. We went on to show that such sequences of minimal length exist, and we constructed six overlapping 100-bp oligonucleotides (termed Pentaprobes) that contained all possible 5-bp sequences, based on the observation that many eukaryotic DNA-binding proteins recognize short sequence motifs. These oligonucleotides have proven to be a useful first-pass screening tool for the detection of DNA-binding activity.
In this work, we extend the Pentaprobe concept to single-stranded RNA (ssRNA). A substantial body of structural work has emerged in the last decade that provides insight into the molecular mechanisms by which proteins recognize ssRNA (17,18). A survey of the literature reveals that a similar pattern exists to that observed for dsDNA-binding proteins, in that many RNA-binding domains recognize sequences of ∼4–5 nt in length [Pumilio repeat domains are a notable exception (19,20)], with greater specificity being achieved through the use of tandem repeats of these recognition domains (18,21–23). Based on that observation, we have created vectors that allow the transcription of DNA Pentaprobes from a T7 promoter, yielding RNA Pentaprobes; the RNA-binding activity of a protein of interest can therefore be assessed in a straightforward way in an EMSA. These probes may prove to be a useful tool for laboratories seeking an initial indication of whether a target protein might act by recognizing ssRNA without the complication and expense associated with combinatorial screening or CLIP-type experiments.
MATERIALS AND METHODS
Pentaprobe design and construction
A sequence of minimum theoretical length (516 bp) containing every possible 5-nt long motif was designed computationally (16) and created as six double stranded, 100-bp long, overlapping sequences we termed Pentaprobes (PPs). These oligonucleotides were synthesized (Sigma-Aldrich®) as 12 ssDNAs (PP1–PP12; Supplementary Figure S1). PP1 is complementary to PP7, PP2–PP8 and so on.
To enable production of ssRNA Pentaprobes by in vitro transcription, PP 1–6 were each annealed with the complementary PP and each double-stranded probe was inserted into the pcDNA3.1 plasmid downstream of a T7 promoter site. Ligation was performed in a blunt end, bidirectional manner and clones selected to obtain 12 plasmids, each capable of transcribing one of the 12 PP sequences as ssRNA.
Other plasmids
pGEX2T vectors containing ZRANB2 F132N (residues 1–41), ZRANB2 F2 (residues 65–95) and a pGEX6P vector encoding ZRANB2 F1232D (residues 1–95) were described previously (24).
hnRNP F qRRM12, qRRM1 F120A-Y180A, Fox-1 RRM and Fox-1 RRM F160A (25,26), each cloned into a pET28 vector, were generously donated by Frederic Allain’s laboratory (ETH, Zurich).
ZNF180ZF (residues 344–691) was generously donated by Seth Frietze, University of Southern California and cloned into a pMalp2X vector.
Lysozyme was purchased from Biomatik.
A pET11a vector containing GATA1 C-terminal zinc finger [residues 237–318 of human GATA1(27)] was generously donated by Jacqui Matthews, University of Sydney, Australia.
A pGEX2T vector containing the transcriptional co-regulator LMO2-LDB1LID (the complete coding sequence of mouse LMO2 fused to a 40 residue region of ldb1) (28) was generously donated by Jacqui Matthews, University of Sydney, Australia.
Probe preparation
Synthetic ssDNA probes were 5′-end labeled with [γ-32P] ATP (PerkinElmer), by adding 5 pmol oligonucleotide, 30 µCi [γ-32P] ATP, 1 µl 10× buffer (70 mM Tris–Cl, pH 7.6, 10 mM MgCl2 and 5 mM DTT) and 1 µl T4 polynucleotide kinase into 10 µl total volume and incubating the reaction for 30 min at 37°C. Double-stranded oligonucleotides were prepared by mixing labeled oligonucleotide (20 pmol) with the unlabeled complementary oligonucleotide in a 1:4 molar ratio in 50 μl of buffer (10 mM Tris–Cl, pH 8.0, 50 mM NaCl and 1 mM EDTA), heating for 1 min at 95°C and then annealing by cooling slowly to room temperature. Unincorporated [γ-32P] ATP was removed from all reactions by Sephadex® G-25 Quick Spin™ columns and labeled probes were stored at −20°C until needed.
Single-stranded RNA Pentaprobes were produced by linearizing the pcDNA3.1 plasmid, containing the Pentaprobe of interest, with ApaI and filling the resulting 3′ overhang with DNA polymerase I large (Klenow) fragment (New England Biolabs). ApaI was used due to its proximity to the 3′-end of the inserted Pentaprobe to terminate the transcription reaction after the insertion site and thus to prevent the addition of nucleotides to the constant region. Transcription was carried out using an in vitro transcription kit (RiboMAX™ Large Scale RNA production system-T7, Promega) in the presence of [α-32P] UTP (10 mCi ml−1) and subsequently the probes were gel purified under denaturing conditions by incubation at 95°C for 2 min followed by gel electrophoresis on a pre-equilibrated 6% Urea–TBE gel (Invitrogen™); this step eliminated truncations and unincorporated single nucleotides. Probes were stored at −20°C until needed.
Protein preparation
Plasmids containing hnRNP F qRRM12, hnRNP F qRRM1 F120A-Y180A, Fox-1 RRM, Fox-1 RRM F160A were transformed into Escherichia coli (BL21) and incubated in LB media containing Kanamycin at 37°C until OD600 of ∼0.6 was reached. Protein expression was induced using 0.4 mM IPTG and bacteria were cultivated at 20°C over night. Cells were harvested by centrifugation (5000 rpm, 30 min, 4°C) and resuspended in lysis buffer [50 mM Na2HPO4, pH 8, 10 mM imidazole, 0.5 mM PMSF and 200 mM NaCl (Fox-1) or 1 M NaCl (hnRNP F)]. In each case, over-expressed protein was released from the cells by gentle sonication and the soluble fraction was separated from the insoluble fraction through centrifugation (10 000 rpm, 30 min, 4°C). The soluble fraction was incubated with Ni-NTA Agarose beads (Invitrogen™) and unbound protein and contaminants were removed by washing with lysis buffer. Bound protein was eluted with an imidazole gradient (50 mM Na2HPO4, pH 8, 100 mM NaCl, 20–500 mM imidazole).
Plasmids containing ZRANB2 F12 (1–95) were transformed into E. coli (BL21-RIG) cells and incubated in LB media containing ampicillin and chloramphenicol at 37°C. Protein expression was induced when OD600 reached ∼0.8–1 by adding 0.2 mM IPTG and bacteria were incubated at 25°C for 12–16 h. Cells were harvested by centrifugation (5000 rpm, 30 min, 4°C), resuspended in lysis buffer (50 mM Tris–HCl, pH 8.0, 200 mM NaCl, 1% Triton®X-100, 2 mM PMSF, 1.4 mM β-mercaptoethanol, 0.1 µg ml−1 lysozyme, Complete™ protease inhibitor (used according to manufacturer instructions, Roche) and freeze–thawed once. Following centrifugation (13 000 rpm, 30 min, 4°C), the soluble fraction was bound to glutathione-Sepharose® 4B beads (GE Healthcare) and washed with wash buffer (50 mM Tris–HCl, pH 8.0, 300 mM NaCl, 10% glycerol, 2 mM PMSF, 0.1 μl ml−1 β-mercaptoethanol) to remove unbound protein and contaminants. Bound protein was eluted in a buffer containing 20 mM reduced glutathione, 50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1 mM DTT and 0.005% Triton®X-100.
Plasmids containing ZNF180ZF (344–691) were transformed into E. coli (BL21-RIG) cells and incubated in LB media containing ampicillin at 37°C. Protein expression was induced when OD600 reached ∼0.6–0.8 by adding 0.5 mM IPTG and bacteria were incubated at 37°C for 3 h. Cells were harvested by centrifugation (6000 rpm, 20 min, 4°C), resuspended in lysis buffer [10 mM Tris–HCl, pH 7.0, 90 mM KCl, 1 mM MgCl2, 90 μM ZnCl2, 1 mM DTT, 0.5 mM PMSF, 1 M NaCl and Complete™ protease inhibitor (used according to manufacturer instructions, Roche)] and freeze–thawed twice, followed by gentle sonication. After DNase I (10 μg ml−1) and RNase (5 μg ml−1) treatment and centrifugation (14 000 rpm, 20 min, 4°C) the soluble fraction was bound to amylose resin (New England Biolabs®) and washed with wash buffer [10 mM Tris–HCl, pH 7.0, 90 mM KCl, 1 mM MgCl2, 90 μM ZnCl2, 1 mM DTT, 0.5 mM PMSF, 1 M NaCl, 10% (w/v) glycerol] to remove unbound protein and contaminants. Bound protein was eluted in a buffer containing 10 mM Tris–HCl, pH 7.0, 90 mM KCl, 1 mM MgCl2, 90 μM ZnCl2, 1 mM DTT, 0.5 mM PMSF, 1 M NaCl, 40 mM maltose and 10 mM CaCl2.
Plasmids containing GATA1 C-finger were transformed into E. coli Rosetta 2 cells and incubated in LB media containing ampicillin and chloramphenicol at 37°C. Protein expression was induced when OD600 reached ∼0.6 by adding 0.4 mM IPTG and bacteria were incubated at 37°C for 3 h. Cells were harvested by centrifugation (5000 rpm, 30 min, 4°C), resuspended in lysis buffer (50 mM Tris–Cl, pH 8.0, 50 mM NaCl, 1% Triton X-100, 1.4 mM PMSF, 1.4 mM β-mercaproethanol) and over-expressed protein was released by gentle sonication. After centrifugation (14 000 g, 30 min), the soluble fraction was bound to Sepharose beads and washed with buffers containing increasing amounts of NaCl (50 mM HEPES, pH9, 50 mM NaCl, 1 mM DTT, with successive washes containing 200 mM of additional NaCl). The eluted fractions were equilibrated to a final concentration of 200 mM NaCl and applied to a cation exchange column (Uno S-1, Bio-Rad). Fractions were eluted with a NaCl gradient (50 mM HEPES, pH 9.0, 1 mM DTT, 50 mM–1 M NaCl). After determining the purity of the protein by SDS–PAGE (NuPage, 4–12% Bis–Tris, Invitrogen™), the eluted fractions were applied to a gel filtration column (Superdex 75 16/60, Pharmacia) and eluted in the elution buffer (50 mM HEPES, pH 9.0, 150 mM NaCl, 1 mM DTT) [for purification see also (29)].
LMO2-LDB1LID was purified as described in (28,30).
Lysosyme was resuspended in H2O.
Expression and purity of all proteins was monitored by SDS–PAGE (NuPage, 4–12% Bis–Tris, Invitrogen™). Protein concentrations were determined by absorbance at 280 nm.
RNA-binding assays
Nucleic acid probes (∼0.1 nM) were incubated with protein samples in gel shift buffer (10 mM MOPS, pH 7.0, 50 mM KCl, 5 mM MgCl2, 1 mM, DTT, 10% glycerol and 0.03 μg μl−1 heparin) at 4°C for 30 min. For antibody supershift experiments, 0.1 μl monoclonal α-GST antibody (mouse) was pre-incubated with the protein mixture before the addition of nucleic acid probe. The binding reactions were loaded onto 4–10% native acrylamide/bisacrylamide gel (19:1) and electrophoresed in 45 mM Tris, 45 mM boric acid at 200 V for 1–2 h either at 4°C or at room temperature. Gels were visualized on a Phosphor screen with a Typhoon PhosphoImager™ (FLA 9000).
RESULTS
Design and production of ssRNA Pentaprobes
We showed previously that a dsDNA sequence of 516 nt can encode all possible 5-nt motifs when the sequences on both strands are taken into account (16). In order to encode all possible motifs in ssRNA, a similar analysis shows that a single oligonucleotide with a minimum length of 1028 nt is required. We chose to approximate this requirement by using the 12 90–100 nt PP sequences (PP 1–12, Supplementary Figure S1) that we had synthesized for the dsDNA work. Twelve nucleotides of overlap were included between successive Pentaprobes to reduce possible issues with anomalous binding to end sequences. Each of PP 1–6 was annealed with its reverse complementary partner sequence (PP 7–12) and individually cloned in a bidirectional manner into a pcDNA3.1 vector downstream of a T7 promoter site, yielding a set of 12 vectors that each could be used as a template from which to transcribe one of the 12 PP sequences. Prior to transcription, the vector was linearized with ApaI directly after the insertion site to prevent the addition of nucleotides to the constant region and the consequent transcription of RNA artifacts. Figure 1 shows the important elements contained in each of these vectors.
Figure 1.

Schematic of ssRNA Pentaprobe production. pcDNA3.1 vector containing a dsDNA Pentaprobe sequence under the control of a T7 promoter site is linearized and the in vitro transcribed to produce a ssRNA sequence encoding the Pentaprobe sequence. Highlighted is the ApaI restriction site (purple), T7 promoter site (pink), the encoded DNA sequence (blue) and the resulting ssRNA probe sequence (blue).
To prepare the ssRNA probes, in vitro transcription was carried out using a standard RiboMAX™ Large Scale RNA production kit (Promega). Transcription was carried out in the presence of [α-32P] UTP (10 mCi ml−1) to allow incorporation of radioactive labeling into the resulting RNA probe. The reaction mixtures were purified on a denaturing polyacrylamide gel. Despite the purification step, however, subjecting probes alone to EMSAs revealed multiple bands in some cases, most likely reflecting the formation of alternative conformations (or multimers) that are in slow exchange on the timescale of the EMSA (hours). The secondary structures of each probe were predicted using Mfold (31) and are shown in Supplementary Figure S2. In an effort to reduce the multiple banding, we heated one of the probes that showed multiple bands to denature it and rapidly cooled it on ice. As shown in Figure 2, this step significantly reduced the occurrence of multiple bands in EMSAs, thereby improving the overall quality of the observed band.
Figure 2.
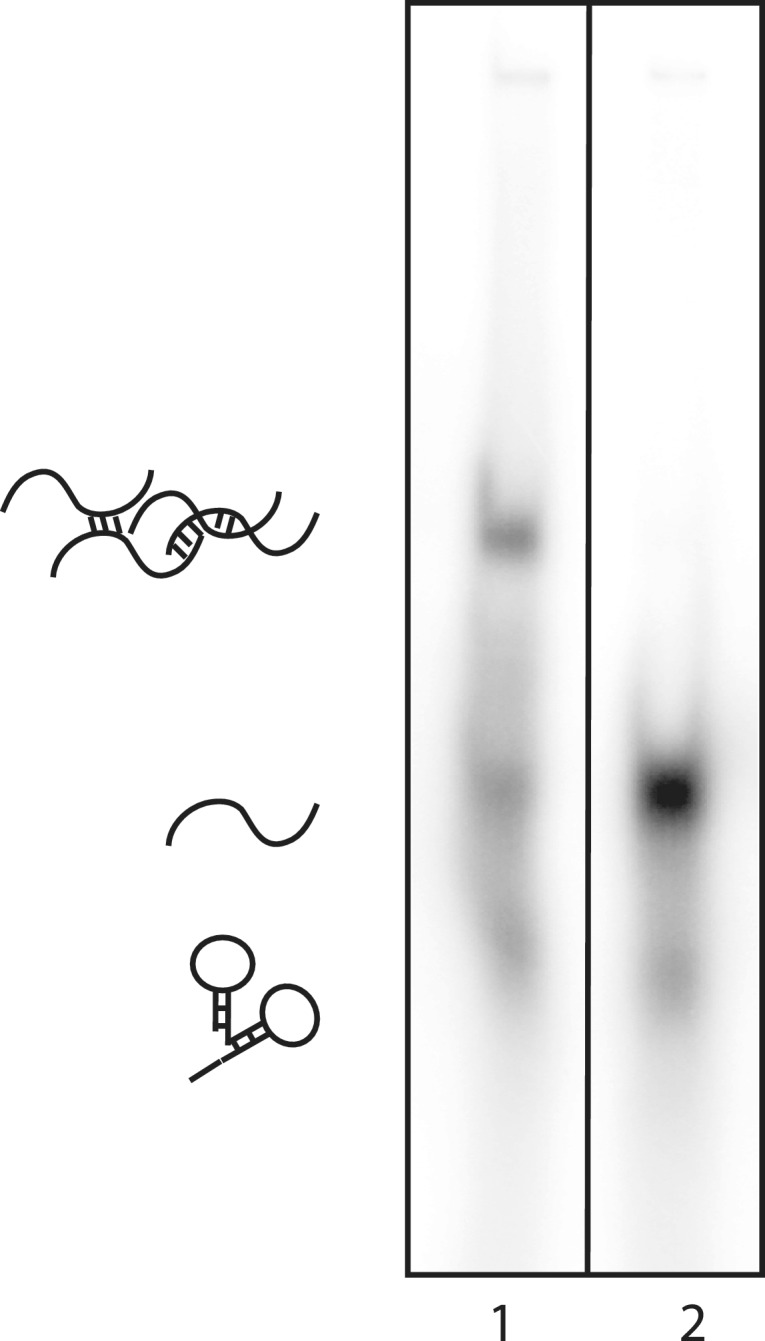
Rapid heating and cooling reduces the formation of multiple conformations in a ssRNA Pentaprobe. 32P-labeled PP3 was subjected to denaturation and rapid cooling and then analysed on a 4% polyacrylamide gel; for comparison, a non-heated probe was also analysed. Lane 1: probe PP3 pre-denaturation, showing multiple bands that most likely reflect different secondary structure or oligomerization states. Lane 2: PP3 post denaturation. Structures shown are for illustrative purposes only.
Testing the RNA Pentaprobes with known RNA-binding proteins
To assess the functionality of the RNA Pentaprobes, we exposed them to well-characterized RNA-binding proteins containing RNA-binding domains from several structural classes. The first protein tested was ZRANB2, a Ser-Arg rich (SR)-like protein that contains two RanBP2-type zinc-finger domains at its N-terminus. The zinc fingers of ZRANB2 have been shown to interact with ssRNA in a sequence-specific manner, each zinc finger binding to an 5′-AGGUAA-3′ motif in its target (24). The function of ZRANB2 is not well understood, but its interaction with important spliceosomal proteins and ability to alter splicing of reporter genes suggest a role in pre-mRNA splicing (32,33).
Figure 3 shows GST-ZRANB2 F12 (1–95) exposed to dsDNA and ssRNA forms of one of the Pentaprobes. No retarded bands can be observed when the protein is incubated with dsDNA Pentaprobe (Figure 3A, lane 4). Lane 2 shows a positive control, in which the C-terminal zinc finger of GATA-1 (a known dsDNA-binding protein) (34) was mixed with the dsDNA probe and lane 3 shows a negative control in which GST alone was used to show that the interaction observed in lane 4 is not mediated by the GST tag. Figure 3B shows GST-ZRANB2 F12 exposed to one of the 12 ssRNA Pentaprobes, and a clear shifted band is observed. To verify the specificity of the interaction and exclude the possibility of showing interactions to contaminants, an anti-GST antibody was used to supershift the complex (lane 7).
Figure 3.

Pentaprobe nucleic acid binding by the zinc fingers of ZRANB2. GST, GST-F1, GST-F2 and GST-F12 (∼15 μM) were incubated with 32P-labeled Pentaprobe in the form of (A) dsDNA and (B) ssRNA. All complexes were analysed on 6% polyacrylamide native gels. (A) ssDNA mixed with: Lane 1: buffer; Lane 2: GATA 1 C-terminal finger; Lane 3: GST; Lane 4: ZRANB2 GST-F12. (B) ssRNA mixed with: Lane 1: buffer; Lane 2: GST; Lane 3: ZRANB2 GST-F1; Lane 4: ZRANB2 GST-F2; Lane 5 and 6: decreasing concentrations of ZRANB2 GST-F12; Lane 7: ZRANB2 GST-F12 in complex with a monoclonal anti-GST antibody; Lane 8: buffer.
We next tested Fox-1, a tissue-specific regulator of alternative splicing that contains a single RNA-binding domain (RNA recognition motif, RRM). This protein recognizes GCAUG motifs in ssRNA in a sequence-specific manner (25). To test binding selective activity in our assay, the RRM of Fox-1 protein and a point mutant thereof, F160A, were expressed and purified using Ni-NTA affinity chromatography. The point mutant significantly reduces binding affinity to target RNA sequences (25). Figure 4 shows that incubating purified Fox-1 RRM with different Pentaprobe RNA sequences results in the appearance of retarded bands in an EMSA, compared to the probe alone, demonstrating that Fox-1 binds to the ssRNA probe. As expected, Fox-1 RRM F160A exhibits weaker binding activity, with a higher concentration of protein necessary to give rise to a shifted band and a reduced retardation of bands in comparison to the wild-type protein. The difference between wild-type and mutant was more pronounced with PP4 (Figure 4B). Examination of the motifs contained in each probe shows that PP10 contains a GCAUA and other closely related motifs, which are very similar to the previously described binding motif of Fox-1. PP4 (Figure 4B), on the other hand, contains only one such motif, resulting in different mobilities for the two complexes (Figure 4A and B). The decreased mobility of the shifted bands observed at higher protein concentrations suggests that multiple proteins are binding to a single Pentaprobe.
Figure 4.

Binding of Fox-1 RRM to RNA Pentaprobe sequences. EMSAs show different 32P-labeled RNA Pentaprobes incubated with increasing concentrations of Fox-1 RRM and the Fox-1 RRM F160A mutant. (A) PP10 mixed with: Lane 1: buffer; Lane 2–5: increasing amounts of Fox-1 RRM (1, 5, 10 and 50 µM); Lane 6–9: increasing amounts of Fox-1 RRM F160A (1, 5, 10 and 50 µM). (B) PP4 mixed with: Lane 1: buffer; Lane 2–5: increasing amounts of Fox-1 RRM (1, 5, 10 and 50 µM); Lane 6–9: increasing amounts of Fox-1 RRM F160A (1, 5, 10 and 50 µM). All samples were separated on 4% polyacrylamide gels.
In order to assess the activity of the whole family of RNA-Pentaprobes, we carried out REMSAs of Fox-1 with every probe (Supplementary Figure S3). Shifts were observed for every probe, illustrating the robustness and sequence diversity contained within the Pentaprobe system. Some differences were observed in the bands produced by different Pentaprobes, which most likely reflect differences in the abundance and accessibility of acceptable target sites. For example, only PP12 contains the full 5-nt recognition sequence GCAUG, and all other probes other than PP1, 4 and 8 contain sites that differ from GCAUG by only one base.
We also tested the quasi-RNA recognition motif (qRRM) of heterogeneous nuclear ribonucleoprotein F (hnRNP F), a member of the hnRNP H family that is involved in the regulation of alternative splicing and polyadenylation. hnRNP F contains three qRRMs that bind poly-G sequences (G-tracts) in ssRNA in a sequence-specific manner (26). To examine RNA binding, in our assay, we used a construct containing hnRNP F qRRM12. Figure 5 shows ssRNA Pentaprobes PP3 and PP9 exposed to increasing concentrations of hnRNP qRRM12; retarded bands were detected using both probes. Note that multiple bands appear in the PP3 probe-only lane (Figure 5A, Lane 1), which are probably due to either intermolecular interactions between probes or the formation of secondary structures. Binding is observed to both populations of RNA, which may indicate that the two pools are in equilibrium with each other.
Figure 5.

hnRNP F RRM binding to RNA Pentaprobe sequences. REMSAs showing different Pentaprobes incubated with increasing amounts of hnRNP F qRRM12. (A) PP3 mixed with: Lane 1: buffer; Lane 2–7: increasing amounts of hnRNP F qRRM12 (0.1, 0.5, 1, 5, 10 and 50 µM). (B) PP9 mixed with: Lane 1: buffer; Lane 2–4: increasing amounts of hnRNP F qRRM12 (0.5, 5 and 50 µM). All samples were separated on 4% polyacrylamide gels. Structures shown are for illustrative purposes only.
Testing the RNA Pentaprobes with a putative RNA-binding protein
Finally, we used ssRNA Pentaprobe to assess the RNA-binding properties of a protein with no described RNA-binding activity. ZNF180 is a poorly characterized multi-zinc-finger protein that is predicted to contain 12 classical zinc-finger domains and a KRAB domain. We expressed and purified a polypeptide corresponding to the 12 classical zinc fingers of this protein and tested its ability to recognize ssRNA by EMSA. Figure 6A shows that this protein is able to recognize ssRNA, providing a starting point from which the hypothesis that this protein plays a role in post-transcriptional gene regulation could be tested using more sophisticated approaches such as CLIP-seq. Note that the EMSA shown in Figure 6A was carried out using the probe that had been heat treated in Figure 2; an EMSA carried out under identical conditions except that the probe was not pre-treated is shown in Figure 6B, demonstrating the improvement given by the denaturation step.
Figure 6.

ZNF180 binding to RNA Pentaprobe sequences. REMSAs showing PP3 incubated with increasing amounts of ZNF180ZF. (A) Heat-treated PP3 mixed with: Lane 1: buffer; Lane 2–8: increasing amounts of ZNF180 (0.1, 1, 5, 10, 20, 30 and 40 µM). (B) Untreated PP3 mixed with: Lane 1: buffer; Lane 2–8: increasing amounts of ZNF180 (0.1, 0.2, 0.5, 1, 5, 10 and 20 µM). All samples were separated on 4% polyacrylamide gels.
Heparin reduces non-specific binding to Pentaprobes and improves clarity of EMSAs
In the case of hnRNP F, Fox-1 and ZNF180ZF, the addition of increasing amounts of protein results in the appearance of multiple retarded bands, which correspond most likely to multiple protein molecules binding to one molecule of RNA. Similar observations were also made for ZRANB2 with several of the probes. Such multiple species can give rise to smearing of the bands that correspond to protein:RNA complexes (Figure 4B, lane 3), reducing the clarity of the assay. We therefore trialed the addition of heparin to our EMSAs in an effort to reduce this effect. Heparin, a highly sulfated glycosaminoglycan that mimics the high negative charge of nucleic acids, has been shown to act as a DNA or RNA competitor and has been used widely as such (35). As shown in Figure 7, the addition of increasing amounts of heparin to an EMSA using ssDNA or ssRNA Pentaprobe gradually clarifies the gel shift, dampening the binding of protein to lower affinity partial sites and resulting in a single, more highly focused band with improved intensity.
Figure 7.

The effect of heparin on Pentaprobe EMSAs. EMSAs showing that the binding of GST-ZRANB2 F12 to ssDNA and ssRNA can be modulated by the addition of heparin. (A) ssDNA Pentaprobe mixed with: Lane 1: buffer; Lane 2–6: increasing amounts of GST-ZRANB2 F1232D (1, 2, 4, 8 and 16 µM); Lane 7: buffer; Lane 8–11: 15 µM GST-ZRANB2 F1232D in the presence of 0, 2.3, 23 and 230 µg ml−1 heparin. (B) Internally 32P labeled ssRNA Pentaprobe mixed with: Lane 1–2: buffer; Lane 3–6: increasing amounts of GST-ZRANB2 F1232D (1.2, 3.5, 11 and 22 µM); Lane 7–9: 11 µM GST-ZRANB2 F1232D in the presence of 2.3, 23 and 230 µg ml−1 heparin. All samples were separated on a 6% polyacrylamide gel.
Finally, we addressed the question of whether the Pentaprobes can effectively distinguish between genuine and irrelevant interactions by carrying out REMSAs with three proteins that are not known to bind RNA in vivo. We chose lysozyme, the C-terminal zinc finger of GATA1 (a dsDNA-binding domain) (27) and the LIM-domain protein LMO2 [a transcriptional co-regulator (36), expressed as a fusion with a short peptide from its binding partner ldb1]. The pIs of these proteins are 11.4, 9.0 and 5.7, respectively. As shown in Figure 8, no robust bands are observed for any of the three proteins, although there is substantial precipitation in the wells containing high concentrations of lysozyme. For comparison, the same range of protein concentrations is shown for Fox-1 (Figure 8D), highlighting that it is straightforward to distinguish between genuine interactions and non-specific aggregation/precipitation. It is worth noting that at lower heparin concentrations (25 µg ml−1), more precipitation is observed for all proteins (including Fox-1) other than LMO2-ldb1, due most likely to adventitious electrostatic interactions. The addition of heparin must therefore represent a balance between the abrogation of such interactions and the possibility that genuine interactions might be suppressed to below the detection limit of the assay. The robust bands observed for Fox-1 in the presence of 250 µg ml−1 heparin argues that genuine interactions are readily detectable under such conditions.
Figure 8.

Lysozyme, GATA 1 C-finger, Fox 1 and LMO2-LDB1LID binding to RNA Pentaprobe sequences. REMSAs showing different Pentaprobes incubated with increasing amounts of lysozyme (A), GATA 1 C-finger (B), Fox 1 (C) and LMO2-LDB1LID (D), respectively, in the presence of 250 µg ml−1 heparin. (A) PP7 mixed with: Lane 1: buffer; Lane 2–6: increasing amounts of lysozyme (0.1, 0.25, 0.5, 0.75 and 1 µM). (B) PP7 mixed with: Lane 1: buffer; Lane 2–6: increasing amounts of GATA1 C-finger (1, 2.5, 5, 7.5 and 10 µM). (C) PP7 mixed with: Lane 1: buffer; Lane 2–6: increasing amounts of Fox-1 (0.1, 0.25, 0.5, 0.75 and 1 µM). (D) PP2 mixed with: Lane 1: buffer; Lane 2–6: increasing amounts of LMO2-LDB1LID (0.1, 0.5, 1, 5 and 10 µM); Lane 7: Fox-1 (0.5 µM). All samples were separated on 4% polyacrylamide gels.
DISCUSSION
In this study, we have demonstrated that the RNA version of Pentaprobe provides a straightforward first-pass tool to examine RNA-binding activity in proteins. The relatively high concentrations of protein that can be used (up to tens of micromolar) allow for relatively weak interactions to be detected, as demonstrated by the observation of a shifted band for the Fox-1 point mutant, which has previously been shown to have severely impaired RNA-binding ability. In general, we would suggest an upper limit for protein concentrations of 1–10 μM to reduce the chance of observing non-physiological interactions or precipitation—although of course this upper limit will differ substantially for different proteins. Additionally, each Pentaprobe contains numerous motifs allowing proteins to bind to high- and low-affinity binding sites simultaneously. While this property can provide a measure of the promiscuity of the protein in question, the presence of multiple bands reduces the clarity of the gel. The use of heparin as a competitor can improve the overall appearance of the gel, to a point that, at least in some cases, most likely represents a 1:1 interaction between protein and RNA. Similarly, heat treatment of the probe prior to the experiment (Figure 6A) can improve the overall quality of the assay by reducing the number of RNA conformers present during the binding reaction.
Existing data indicate that most ssRNA-binding proteins are constructed from domains that each display binding to relatively short RNA sequences, at least in vitro. We believe that, under our conditions (the presence of high concentrations of protein and RNA), the full coverage of 5-nt sequences represented in Pentaprobe should be sufficient to mediate binding to most, if not all, of these domains. Pentaprobes can readily be used to assess not only the binding of full-length proteins, but also of individual domains, in order to obtain a rapid assessment of which domains in a multi-domain protein are more or less important for RNA recognition. It remains possible, however, that some proteins that recognize longer target sites, such as the Pumilio repeat proteins, might exhibit reduced binding to our probes, given that their recognition sequences are not fully represented in Pentaprobes.
All of our Pentaprobes exhibit some secondary structure formation, which in some cases could obstruct the binding of a protein to RNA or mask a potential binding site, but so far we have no evidence that the presence of such structures interferes greatly with our assay. Indeed, the formation of secondary structure could even be beneficial, given that some proteins target RNA secondary structures. An obvious additional application in this regard for our method would be the determination of dsRNA-binding activity. Pentaprobe sequences can be easily dimerized (PP 1–6 are complementary to PP 7–12) and run side-by-side with ssRNA probes to gain an initial impression of whether a target protein prefers ssRNA or dsRNA. Furthermore, RNAse protection assays could be attempted, to allow more specific determination of a binding site comprised in the present sequence.
We also foresee other possible applications for the ssRNA Pentaprobe reagent. So far, just highly purified protein domains have been tested in our assay to determine interactions. However, one or even a mixture of Pentaprobes could be utilized as a broad-spectrum yet chemically defined bait with which to pull down RNA-binding proteins from cellular extracts for further analysis. Finally, these Pentaprobes also constitute a useful probe from which the RNA-cleaving activity of RNAse enzymes can be determined; this application has been successfully demonstrated in the case of a bacterial toxin (Vickery Arcus, personal communication).
In conclusion, we have described a reliable method for the detection of RNA-binding properties in target proteins that is inexpensive, fast and easy to implement in any standard life sciences laboratory.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–3.
FUNDING
Funding for open access charge: National Health and Medical Research Council [L3038 N7381].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Frederic Allain (Institute of Molecular Biology and Biophysics, Zürich, Switzerland) for providing all clones of hnRNP and Fox-1 protein constructs, Craig Plambeck (Morris laboratory, Physiology, University of Sydney, Australia) for providing us with plasmids encoding ZRANB2, Seth Frietze (University of Southern California, United States) for providing us with a plasmid encoding ZNF180ZF and the support and input from everyone in the Mackay and Matthews laboratories.
REFERENCES
- 1.Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 2.Aigner A. MicroRNAs (miRNAs) in cancer invasion and metastasis: therapeutic approaches based on metastasis-related miRNAs. J. Mol. Med. 2011;89:445–457. doi: 10.1007/s00109-010-0716-0. [DOI] [PubMed] [Google Scholar]
- 3.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat. Rev. Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 4.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl Acad. Sci. USA. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsai MC, Spitale RC, Chang HY. Long intergenic noncoding RNAs: new links in cancer progression. Cancer Res. 2011;71:3–7. doi: 10.1158/0008-5472.CAN-10-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zucconi BE, Ballin JD, Brewer BY, Ross CR, Huang J, Toth EA, Wilson GM. Alternatively expressed domains of AU-rich element RNA-binding protein 1 (AUF1) regulate RNA-binding affinity, RNA-induced protein oligomerization, and the local conformation of bound RNA ligands. J. Biol. Chem. 2010;285:39127–39139. doi: 10.1074/jbc.M110.180182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim MY, Jeong S. In vitro selection of RNA aptamer and specific targeting of ErbB2 in breast cancer cells. Nucleic Acid Ther. 2011;21:173–178. doi: 10.1089/nat.2011.0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Recht MI, Ryder SP, Williamson JR. Monitoring assembly of ribonucleoprotein complexes by isothermal titration calorimetry. Methods Mol. Biol. 2008;488:117–127. doi: 10.1007/978-1-60327-475-3_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feig AL. Studying RNA-RNA and RNA-protein interactions by isothermal titration calorimetry. Methods Enzymol. 2009;468:409–422. doi: 10.1016/S0076-6879(09)68019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galgano A, Gerber AP. RNA-binding protein immunopurification-microarray (RIP-Chip) analysis to profile localized RNAs. Methods Mol. Biol. 2011;714:369–385. doi: 10.1007/978-1-61779-005-8_23. [DOI] [PubMed] [Google Scholar]
- 11.Jain R, Devine T, George AD, Chittur SV, Baroni TE, Penalva LO, Tenenbaum SA. RIP-Chip analysis: RNA-binding protein immunoprecipitation-microarray (Chip) profiling. Methods Mol. Biol. 2011;703:247–263. doi: 10.1007/978-1-59745-248-9_17. [DOI] [PubMed] [Google Scholar]
- 12.Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M, Jr, Tuschl T, Ohler U, Keene JD. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol. Cell. 2011;43:327–339. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell. 2010;40:939–953. doi: 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat. Methods. 2011;8:559–564. doi: 10.1038/nmeth.1608. [DOI] [PubMed] [Google Scholar]
- 15.Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 16.Kwan AH, Czolij R, Mackay JP, Crossley M. Pentaprobe: a comprehensive sequence for the one-step detection of DNA-binding activities. Nucleic Acids Res. 2003;31:e124. doi: 10.1093/nar/gng124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Font J, Mackay JP. Beyond DNA: zinc finger domains as RNA-binding modules. Methods Mol. Biol. 2010;649:479–491. doi: 10.1007/978-1-60761-753-2_29. [DOI] [PubMed] [Google Scholar]
- 18.Cléry A, Blatter M, Allain FH. RNA recognition motifs: boring? Not quite. Curr. Opin. Struct. Biol. 2008;18:290–298. doi: 10.1016/j.sbi.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, McLachlan J, Zamore PD, Hall TM. Modular recognition of RNA by a human pumilio-homology domain. Cell. 2002;110:501–512. doi: 10.1016/s0092-8674(02)00873-5. [DOI] [PubMed] [Google Scholar]
- 20.Filipovska A, Razif MF, Nygård KK, Rackham O. A universal code for RNA recognition by PUF proteins. Nat. Chem. Biol. 2011;7:425–427. doi: 10.1038/nchembio.577. [DOI] [PubMed] [Google Scholar]
- 21.Hall TM. Multiple modes of RNA recognition by zinc finger proteins. Curr. Opin. Struct. Biol. 2005;15:367–373. doi: 10.1016/j.sbi.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 22.Lewis HA, Musunuru K, Jensen KB, Edo C, Chen H, Darnell RB, Burley SK. Sequence-specific RNA binding by a Nova KH domain: implications for paraneoplastic disease and the fragile X syndrome. Cell. 2000;100:323–332. doi: 10.1016/s0092-8674(00)80668-6. [DOI] [PubMed] [Google Scholar]
- 23.Schmitzova J, Rasche N, Dybkov O, Kramer K, Fabrizio P, Urlaub H, Luhrmann R, Pena V. Crystal structure of Cwc2 reveals a novel architecture of a multipartite RNA-binding protein. EMBO J. 2012;31:2222–2234. doi: 10.1038/emboj.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loughlin FE, Mansfield RE, Vaz PM, McGrath AP, Setiyaputra S, Gamsjaeger R, Chen ES, Morris BJ, Guss JM, Mackay JP. The zinc fingers of the SR-like protein ZRANB2 are single-stranded RNA-binding domains that recognize 5' splice site-like sequences. Proc. Natl Acad. Sci. USA. 2009;106:5581–5586. doi: 10.1073/pnas.0802466106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Auweter SD, Fasan R, Reymond L, Underwood JG, Black DL, Pitsch S, Allain FH. Molecular basis of RNA recognition by the human alternative splicing factor Fox-1. EMBO J. 2006;25:163–173. doi: 10.1038/sj.emboj.7600918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dominguez C, Allain FH. NMR structure of the three quasi RNA recognition motifs (qRRMs) of human hnRNP F and interaction studies with Bcl-x G-tract RNA: a novel mode of RNA recognition. Nucleic Acids Res. 2006;34:3634–3645. doi: 10.1093/nar/gkl488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clore GM, Bax A, Omichinski JG, Gronenborn AM. Localization of bound water in the solution structure of a complex of the erythroid transcription factor GATA-1 with DNA. Structure. 1994;2:89–94. doi: 10.1016/s0969-2126(00)00011-3. [DOI] [PubMed] [Google Scholar]
- 28.Deane JE, Sum E, Mackay JP, Lindeman GJ, Visvader JE, Matthews JM. Design, production and characterization of FLIN2 and FLIN4: the engineering of intramolecular ldb1:LMO complexes. Protein Eng. 2001;14:493–499. doi: 10.1093/protein/14.7.493. [DOI] [PubMed] [Google Scholar]
- 29.Wilkinson-White L, Gamsjaeger R, Dastmalchi S, Wienert B, Stokes PH, Crossley M, Mackay JP, Matthews JM. Structural basis of simultaneous recruitment of the transcriptional regulators LMO2 and FOG1/ZFPM1 by the transcription factor GATA1. Proc. Natl Acad. Sci. USA. 2011;108:14443–14448. doi: 10.1073/pnas.1105898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryan DP, Sunde M, Kwan AH, Marianayagam NJ, Nancarrow AL, Vanden Hoven RN, Thompson LS, Baca M, Mackay JP, Visvader JE, et al. Identification of the key LMO2-binding determinants on Ldb1. J. Mol. Biol. 2006;359:66–75. doi: 10.1016/j.jmb.2006.02.074. [DOI] [PubMed] [Google Scholar]
- 31.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams DJ, van der Weyden L, Mayeda A, Stamm S, Morris BJ, Rasko JE. ZNF265–a novel spliceosomal protein able to induce alternative splicing. J. Cell Biol. 2001;154:25–32. doi: 10.1083/jcb.200010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Chen XH, Xiao PJ, Li L, Lin WM, Huang J, Xu P. Expression pattern and splicing function of mouse ZNF265. Neurochem Res. 2008;33:483–489. doi: 10.1007/s11064-007-9461-3. [DOI] [PubMed] [Google Scholar]
- 34.Merika M, Orkin SH. DNA-binding specificity of GATA family transcription factors. Mol. Cell Biol. 1993;13:3999–4010. doi: 10.1128/mcb.13.7.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loziński T, Bolewska K, Wierzchowski KL. Equivalence of Mg2+ and Na+ ions in salt dependence of the equilibrium binding and dissociation rate constants of Escherichia coli RNA polymerase open complex. Biophys. Chem. 2009;142:65–75. doi: 10.1016/j.bpc.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 36.El Omari K, Hoosdally SJ, Tuladhar K, Karia D, Vyas P, Patient R, Porcher C, Mancini EJ. Structure of the leukemia oncogene LMO2: implications for the assembly of a hematopoietic transcription factor complex. Blood. 2011;117:2146–2156. doi: 10.1182/blood-2010-07-293357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.