


Abstract
Cancer-associated mutations in cancer genes constitute a diverse set of mutations associated with the disease. To gain insight into features of the set, substitution, deletion and insertion mutations were analysed at the nucleotide level, from the COSMIC database. The most frequent substitutions were c→t, g→a, g→t, and the most frequent codon changes were to termination codons. Deletions more than insertions, FS (frameshift) indels more than I-F (in-frame) ones, and single-nucleotide indels, were frequent. FS indels cause loss of significant fractions of proteins. The 5′-cut in FS deletions, and 5′-ligation in FS insertions, often occur between pairs of identical bases. Interestingly, the cut-site and 3′-ligation in insertions, and 3′-cut and join-pair in deletions, were each found to be the same significantly often (p < 0.001). It is suggested that these features aid the incorporation of indel mutations. Tumor suppressors undergo larger numbers of mutations, especially disruptive ones, over the entire protein length, to inactivate two alleles. Proto-oncogenes undergo fewer, less-disruptive mutations, in selected protein regions, to activate a single allele. Finally, catalogues, in ranked order, of genes mutated in each cancer, and cancers in which each gene is mutated, were created. The study highlights the nucleotide level preferences and disruptive nature of cancer mutations.
INTRODUCTION
Decades ago, it was shown that mutations in genes can cause cancer. Proto-oncogenes (PO) were shown to be activated by mutation to oncogenes, which triggered cancer (1,2). The existence of tumor-suppressor (TS) genes, their loss of function by a ‘two-hit’ scheme of mutation, and the cancer-promoting effect of loss of TS function have also been described (3–5). Years of subsequent research have led to a general acceptance of the paradigm that sequential accumulation of genetic errors or mutations in PO and TS eventually transforms a normal cell into a tumor cell (6). With the elucidation of the human genome, it is now possible to sequence every human gene in each cancer, and to examine the collective set of cancer-related mutations. Cancer genome sequencing projects have shown that about 47, 63, 90 and 90 mutations are observed in glioblastoma multiforme, pancreatic, breast and colorectal cancers, respectively (7,8). Thus, cancers are complex genetic diseases with numerous genes being affected, and drugs striking one or a few gene targets may not bring about a cure of the disease. Mutational data emerging from the cancer genome sequencing projects might reveal new paradigms for the disease as well as for treatment.
Cancer-associated genes and mutations, discovered and published over decades of research, have been organized into databases. Genes playing a causal role in cancer (cancer genes) have been compiled in the Cancer Gene Census (9). A gene has been considered to be causal if mutations in it were not attributable to chance and if it was likely that the mutations had been selected because they conferred a growth advantage on the tumor. Cancer-related mutations, reported in the literature, have been compiled in the COSMIC database [Catalogue of Somatic Mutations in Cancer (10–12)]. The aim of the present study is to analyse mutations observed in Cancer Gene Census genes and compiled in COSMIC.
Cancer mutations have been differentiated into drivers and passengers; while the former contribute to cancer development (as they have a functional impact), the latter do not [as they are functionally neutral (13,14)]. The sequencing of cancer genomes yields large collections of somatic mutations (7,13,15), which need to be differentiated into drivers and passengers. Computational methods have been developed for this purpose. Machine learning methods combine physicochemical, structural and conservation information of wild-type (WT) and mutant residues, and are trained to distinguish between known deleterious and neutral mutations. Thus, CanPredict and CHASM train random forest classifiers to discriminate between COSMIC mutations and either nsSNPs (16,17) or synthetically generated passenger mutations (18); likewise, a protein kinase-specific method trains a support vector machine to distinguish between known disease and common kinase nsSNPs (19). The trained classifiers are used to predict, in an unknown set of missense mutations, those that are likely to have a functional impact. ‘Direct methods’ use evolutionary conservation patterns to predict the functional impact of mutations on proteins (20–22). In Ref. (20), a functional impact score has been introduced and used to discriminate between COSMIC mutations and common polymorphisms. Protein structure and sequence analysis methods have been used to show that destabilization of protein 3D structure is the major molecular mechanism underlying driver mutations (23). The COSMIC database has been used to analyse patterns of mutation in cancers, to understand the crosstalk between cancer pathways and to examine the distribution of mutations in oncogenes and TS (24). A systems biology approach has also been taken and a network of cancer genes with co-occurring and mutually exclusive mutations has been constructed to study how the mutations contribute to tumorigenesis (25).
Many of the above-mentioned studies have focused on missense mutations and their effect at the protein level. Mutations in cancer are selected based on the growth advantage that the mutant protein confers on the cell. Nevertheless, mutations occur at the nucleotide (nt) level. In the present study, substitution, deletion and insertion mutations in cancer genes, sourced from COSMIC, have been analysed at the nt level. No effort has been made to classify mutations into drivers or passengers. Substitution mutations were analysed to determine the frequencies of base changes and of codon mutations. Frameshift (FS) and in-frame (I-F) deletions and insertions were analysed for their frequency of occurrence, length distributions, preferred starting and ending cut- or ligation-sites, locations in proteins and for the fraction of protein lost or gained due to them. The distribution of different types of mutations in, and their spread over the lengths of PO and TS were studied. Genes playing a role in each cancer, and cancers in which each gene was playing a role, were each ranked. Thus, the study examines, at the nt level, the variety and preferred kinds of mutations that occur over the time-scale of cancer (<100 years). It is hoped that the study will add perspective, in the effort to understand cancer mutations.
METHODS
Ranking genes in cancer of each tissue and ranking cancer tissues for each gene
Cancer genes and mutations were obtained from the COSMIC database. A description of the data set and data processing procedure is given in Supplementary Methods (i). The aim was to identify: (i) the various genes that were playing a role in cancer of each tissue and (ii) the various cancers in which each gene was playing a role. The number of samples analysed and mutated samples observed for each gene in each tissue were counted and used to calculate the fraction or proportion (p) of mutated samples, which was then used to calculate a rank score. As the numbers of samples analysed differed widely in the data set, the uncertainty in the estimated proportion (p) needed to be taken into account. This may be done by taking the 95% lower confidence limit on the estimate of the proportion as the ranking score. This approach has previously been used, and the confidence limit has been calculated using the (widely accepted) approximate formula, 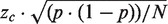 [cf., legend to Figure 1 in Ref. (10)]. In the present study, the more rigorous expression for the 95% lower confidence limit, using the same normal approximation to the binomial distribution, has been used (26), since it ensures that the ranking score never becomes negative. Thus, the 95% lower confidence limit or rank score has been calculated as follows:
[cf., legend to Figure 1 in Ref. (10)]. In the present study, the more rigorous expression for the 95% lower confidence limit, using the same normal approximation to the binomial distribution, has been used (26), since it ensures that the ranking score never becomes negative. Thus, the 95% lower confidence limit or rank score has been calculated as follows:
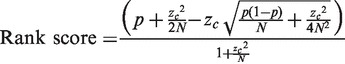 |
p = proportion of mutated samples (i.e. number of mutated samples/total number of samples analysed), zc = critical value = 1.6449, N = total number of samples analysed. As N becomes large, the score asymptotically approaches p. Results are given in Supplementary Table S1.
Figure 1.

Histograms showing the frequency of occurrence of each of the 12 possible base changes at pos1, pos2 and pos3 of codons in: (a) synonymous (b) missense and (c) nonsense substitutions. In each histogram, base changes are indicated along the x-axis, the number of times that each base change is observed (frequency) is indicated along the y-axis and the frequencies of base changes at pos1, pos2 and pos3 of codons are shown as separate series.
Analyses of substitution, deletion and insertion mutations
A single mutation may be observed many times. The 1633G > A, E545K substitution in the PIK3CA gene, in breast tissue, for example, occurs 165 times. This is because a large number of breast PIK3CA samples have been studied and the mutation occurs frequently in them. Unless otherwise specified, a mutation occurring multiple times in a tissue has been considered only once (i.e. only unique mutations in a tissue have been considered), in order to avoid biases due to differing sample sizes. The same mutation occurring in multiple tissues, however, has been considered once in each tissue.
Substitution mutations
Single-base substitutions were sorted into synonymous, missense and nonsense ones, and each set was analysed separately [Supplementary Methods (i)a]. Multiple-base substitutions were also analysed. WT and mutant codons from all single-base substitutions, and from multiple-base ones in which 2 or 3 bases in a single codon were substituted, were used to generate a 64 × 64 WT codon—mutant codon pair frequency matrix (Supplementary Table S2).
Deletion and insertion mutations
Deletions and insertions were separated into I-F and FS ones, and each set was analysed separately [Supplementary Methods (i)b]. Results are given in Supplementary Tables S3 and S4, respectively.
RESULTS AND DISCUSSION
Results of the analysis of substitution mutations
In the set of cancer-related mutations, there were 6013 single-base-, 169 2-base-, 12 3-base-, 2 4-base- and 2 5-base substitutions (Table 1). Single-base substitutions consisted of 489 synonymous, 4555 missense, 964 nonsense and 5 no-stop ones. Substitution of a nt can occur at positions 1, 2 or 3 (pos1, pos2 or pos3) of a codon and, at each position, there are 12 substitution possibilities, because any of the 4 bases can be changed to any of the three remaining ones. Figure 1a shows that, out of 489 synonymous substitutions, 20, 0 and 469 (sums of values in each series; Table 1) arise from base changes at pos1, pos2 and pos3, respectively. Synonymous codons for the majority of amino acids differ by a base at pos3, the exceptions being some L and R codons, which differ at pos1 (codon table). Accordingly, synonymous substitutions are frequent at pos3. Arising due to the degeneracy of the genetic code, these substitutions provide some protection against the effect of mutations, but are thought not to confer any growth advantage on a tumor. However, replacing an efficient codon with a less efficient synonymous one can affect production of the protein (27). Out of 4555 missense substitutions, 1951, 2262 and 342 arise from base changes at pos1, pos2 and pos3, respectively (Figure 1b); base changes at pos2 and pos1 lead to missense substitutions more frequently than do those at pos3. No c→t or t→c changes are observed at pos3 because they lead only to synonymous mutations. Out of 964 nonsense substitutions, 672, 132 and 160 arise from base changes at pos1, pos2 and pos3, respectively (Figure 1c). The codon table shows that nine codons of five amino acids (Q, K, E, R, G), four codons of two amino acids (L, S) and five codons of three amino acids (Y, C, W) can change to stop codons by making base changes at pos1, pos2 and pos3, respectively. As more codons of more amino acids can change to a stop codon by base changes at pos1, nonsense substitutions at this position are more frequent. In general, substitutions occur more frequently at pos1 and pos2 than at pos3 (Table 1).
Table 1.
Summary of results for substitution mutations
| Numbers of 1-, 2-, 3-, 4-, 5-base substitutions: 6013, 169, 12, 2, 2 | ||||
| 1-base substitutions: | ||||
| Types | Observed | At pos1 | At pos2 | At pos3 |
| Synonymous | 489 | 20 | 0 | 469 |
| Missense | 4555 | 1951 | 2262 | 342 |
| Nonsense | 964 | 672 | 132 | 160 |
| No-stop | 5 | 0 | 3 | 2 |
| Total | 6013 | 2643 | 2397 | 973 |
| Most | c→t (1267) | c→t (713) | g→a (457) | g→a (189) |
| frequent | g→a (1239) | g→a (593) | c→t (375) | c→t (179) |
| base | g→t (697) | g→t (399) | a→g (320) | |
| changes | g→t (204) | |||
| Numbers of WT bases undergoing substitution: g(2288), c(1906), a(979), t(835) | ||||
| Numbers of mutant bases after substitution : g(1041), c(897), a(1820), t(2250) | ||||
| Amino acids undergoing the most substitutions: | ||||
| Synonymous G(65), L(60) | ||||
| Nonsense R(208), Q(206), E(191) | ||||
| Missense G(654) undergoes most mutations; C(294), K(262), N(211) generated in | ||||
| significant numbers; interesting mutations: P(239)→S(91),L(86); | ||||
| A(296)→T(108),V(94); Y(136)→C(69); E(286)→K(153) | ||||
| Most frequently occurring single-base substitutions: | ||||
| cga_R→tga_TER, 194; cag_Q→tag_TER, 158; gag_E→tag_TER, 102; gaa_E→taa_TER, 89; gag_E→aag_K, 87; ggt_G→gat_D, 75; gaa_E→aaa_K, 66; ggc_G→gac_D, 63; ggc_G→agc_S, 60; cgg_R→tgg_W, 59; tct_S→ttt_F, 58; tgg_W→tga_TER, 57; gtg_V→atg_M, 52 | ||||
| 2-base substitutions: | ||||
| WT bases substituted most frequently : gg (39), cc (31), tg (22), gc (16) | ||||
| Mutant bases observed most frequently: tt (51), aa (29), at (21), ct (13) | ||||
| Most frequently observed substitutions : cc→tt (28), gg→aa (13), gg→tt (10) | ||||
| Amino acid mutations resulting from 2- and 3-base substitutions: | ||||
| G → F6, V6, D5, L3, Y3, N3, E3, S2, P2(3-base), I2, K1; L → P6, R4, S2, K1, W1; P → L6, H2, F1; V → E5, D4, K2, R2, G1, C1, A1; Q → R5, L2; W → Ter5, K2(3-base), A1; R → P4(3-base), F1, L1, V1; D → F3, I2; S → Ter3, L2, N2, Q1, F1; A → F2, L2(3-base), V2, I1, N1, G1; K → L2, S1(3-base), P1(3-base); F → K1(3-base); I → K1, D1, C1; M → P1, T1, N1; T → I1, E1; Y → V1; E → M1, V1 | ||||
| To approximately how many mutant codons does a WT codon mutate, in cancer? | ||||
| 61 WT → six or more mutant | ||||
| 44 WT → 8–11 mutant | ||||
| 13 WT → 6–7 mutant | ||||
| 4 WT → 15–19 mutant (ctg_L → 15; ggc_G → 15; gtg_V → 16; ggt_G → 19) | ||||
| 1 WT TER codon → three non-TER mutant (no-stop mutations) | ||||
Figure 2 shows that, in the set of substitution mutations, the most frequently occurring base changes are c→t, g→a and g→t. Table 1 lists the most frequently occurring base changes at each position. Overall as well as at each position, c→t, g→a and g→t changes are preferred. The only exception is the occurrence, with some frequency, of a→g at pos2; this may be a result of coding requirements in mutant codons. The greater frequency of c→t and g→a mutations is related to an epigenetic modification of DNA that commonly occurs at cg (or CpG) dinucleotides: the methylation of cytosine at the 5-position. Methylated CpG di-nt are unusually mutable, undergo deamination to t and cause 1 c→t and 1 g→a transition [(28); Box 3 in (29)]. A significant number of mutations causing human genetic disease occur at methylated CpG di-nt and the majority of these are c→t and g→a transitions (30). Another effect of CpG hypermutation is the observed higher rate of c→t and g→a substitution in exons (at synonymous sites) as compared to non-coding DNA: owing to protein coding requirements, there is an over-abundance of synonymous exonic sites involved in CpG dinucleotides, which leads to the observed increased rate of substitution (31). The g→t transversion has also been shown to occur preferentially at methylated CpG sequences (by an unknown molecular mechanism) at sites of adduct formation by carcinogens and has been noted to be frequent in p53 in lung cancers from smokers (32). In the present study, g→t tranversions have been observed to be frequent in lung [103], large intestine [87] and haematopoietic-and-lymphoid [63] cancers. At methylated CpG sequences, the rates of transitions (c→t, g→a) and transversions (g→t) are elevated by ∼30-fold and a few-fold, respectively, relative to the average mutation rate (33). The number of times that each base undergoes substitution in the set of WT codons, and is the mutant base in the set of mutant codons was also counted (Table 1). The most substituted bases are g, c [2288, 1906] and the most frequently occurring mutant ones are t, a [2250, 1820]. Thus, substitutions tend to change g, c to t, a. Further, Figure 2 shows that base changes that result in an a or t are preferred over their reverse: c→t > t→c, g→a > a→g, g→t > t→g.
Figure 2.

Histogram showing the frequency of occurrence of each of the 12 possible base changes when all substitution mutations (synonymous, missense, nonsense), occurring at pos1, pos2 and pos3 of codons, are considered. Base changes are indicated along the x-axis and their frequencies are indicated along the y-axis.
Substitution mutations were also examined at the amino acid level (Supplementary Figure S1). G, L undergo the most synonymous mutations, followed by S, P, T, A, all amino acids with four to six codons (S1a); R, with six codons, however, shows fewer mutations. R, Q, E undergo the most nonsense mutations (S1b), using codons which make c→t, g→t changes at pos1 (R: cga→tga, 194; Q: cag→tag, 158; E: gag→tag, 102; gaa→taa, 89). G undergoes the most missense mutations; C, K, N appear as mutant amino acids in significant numbers (S1c). Missense mutations undergone by each amino acid were also examined (S1d–h); some of the more striking ones, where particular mutant amino acids are frequently observed, are listed in Table 1. G, the smallest amino acid, often undergoes mutation to R, the largest one (S1h). In all mutations, amino acid codons are mutated to neighbours differing by a single base.
In the data set of cancer-related mutations, 169 2-base substitutions were also present. They were of the types, ‘12-’, ‘-23’, ‘--3 1--’, where the first two or the last two bases in a codon, or the last base in one codon and the first base in the adjacent codon were substituted, respectively. There were 66, 63 and 40 substitutions of the three types, and none, 8 and 19 of them, respectively, formed termination (TER) codons. The preferred way of forming a TER codon was by a ‘--3 1--’ type substitution in which the mutant base at pos1 was t; the TER codon was also formed by substitutions at codon positions 2 and 3, but never by those at positions 1 and 2. Supplementary Figure S2 shows that WT base pairs undergoing substitution most frequently are: gg, cc, tg, gc, and the most frequently occurring mutant pairs are: tt, aa, at, ct; cg never occurred as a mutant base pair. There were 65 types of WT → mutant pairs, the most frequently occurring ones being cc→tt, gg→aa, gg→tt (Table 1). Thus, 2-base substitutions, like single-base ones, tended to change g, c to t, a. The 12 3-base, 2 4-base and 2 5-base substitutions observed in the data set were also examined. All three bases in a codon have been substituted in three of the 3-base ones, and in all 4- and 5-base ones. Mutations in which two adjacent nt have been substituted, referred to as tandem base mutations, have been observed in skin cancer in BRAF [gt→ag, aa; tg→aa; (34)]. The cc→tt tandem substitution is a signature of mutagenesis due to UV exposure and is observed in TP53 and PTCH genes in sun-exposed skin cancer (35). In the present study, the largest numbers of 2-base mutations were observed in skin [54], haematopoietic-and-lymphoid [24] and lung [19] cancers.
A WT codon—mutant codon pair frequency matrix (Supplementary Table S2) was generated using WT and mutant codons from 6013 single-base, 129 2-base and 7 3-base substitutions. The arrangement of single-base substitutions (turquoise, tan, green boxes) in diagonals across the matrix reflects the fact that mutations occur between codon neighbours differing by a single base. The large number of empty boxes corresponds to mutations that are not observed because they require multiple-base substitutions, which occur rarely. A comparison of this matrix with an empirical evolutionary codon substitution matrix (36) shows that the entire latter matrix is well-populated. However, by calculating mutation scores (36), it has been shown that, in the latter matrix, synonymous substitutions are more likely to occur than missense ones, and that >1 nt substitutions are less likely to occur than single-nt ones. Single-base substitutions leading to the formation of TER codons are the most frequent, followed by E→K and G→D mutations (Table 1). In contrast to the codon substitution matrix, where mutations to TER codons are not observed (despite the long time-scales of evolution), in the present matrix, they are the most frequently occurring mutations (time-scale, <100 years). Thus, mutation selection occurs differently in evolution and cancer. The matrix may also be used to obtain an idea of the approximate number of mutations that a codon may undergo in cancer. Counting the number of mutant codons observed for each WT codon (the number of coloured boxes in each row) showed that 61 WT, non-TER codons each mutated to six or more codons (Table 1), with the majority [44] each mutating to 8–11 codons; ctg_L, ggc_G, gtg_V and ggt_G mutated to the largest numbers of codons [15, 15, 16, 19], and one TER codon (tga) underwent three no-stop mutations.
Amino acid mutations resulting from 2-, 3-base substitutions are summarized in Table 1. Codons of G undergo the largest number and variety of 2-base substitutions. L and P undergo substitutions to one another, L and Q to R, and R to P. V undergoes substitution to acidic and basic residues, and W and S to TER. Some 2-base substitutions are conspicuously rare. In the Ser/Thr kinase, BRAF, T598 and S601 occur in the kinase activation segment. Mutation of either residue to a negatively charged one would mimic phosphorylation, cause unregulated kinase activation and promote tumor growth. However, such mutations, despite their advantage to the tumor, are rare, because 2-base changes that mutate T,S→D,E are rare (37). In the matrix, while no S→D,E substitution is observed, a single T→E one does occur, in the Tyr kinase, KIT. The occurrence of the rare 2-base mutation becomes less surprising upon consideration that the mutation occurs at a mutational hotspot (38).
Results of the analyses of deletion and insertion mutations
Below, indels has sometimes been used while referring to insertions and deletions. Supplementary Figure S3 shows the different possible ways in which nt can be deleted and inserted in I-F and FS deletions and insertions. While no change in the gene reading frame occurs in I-F indels (nt lost or gained in multiples of three), in FS ones, the reading frame changes (odd or even numbers of nt, that are not multiples of three, lost or gained). There are three types of I-F (1-3, 2-1, 3-2) and six types of FS (1-1, 2-2, 3-3, 1-2, 2-3, 3-1) indels. The first number indicates the position in the codon at which the indel begins, and the second, the position in the same or a downstream codon at which the indel ends; for example, 2-1 indicates that the indel constitutes the segment running from pos2 of one codon to pos1 of the adjacent or a downstream codon (figure legend).
In the data set of cancer-related mutations, there were 2021 FS and 588 I-F deletions, and 903 FS and 347 I-F insertions. Figure 3 shows the frequencies with which the six and three types of FS and I-F deletions (first series) and insertions (second series) occur in the data set. FS indels (both series) of the types, 1-1, 2-2, 3-3, are observed the most frequently. Complete codon I-F deletions (type 1-3) are more frequent than mid-codon I-F ones (types 2-1, 3-2); in I-F insertions, a similar, but less marked, trend is observed. All three types of I-F insertions are more frequent than 1-2, 2-3, 3-1 type FS ones; only complete codon I-F deletions (type 1-3) are more frequent than 1-2, 2-3, 3-1 type FS ones. Figure 4 shows the length distributions of the different types of deletions (4A) and insertions (4B). Clearly, single nt insertions and deletions are the most frequent. FS indels of the types 1-1, 2-2, 3-3 are most frequently 1 nt in length (i.e. a single nt is lost or gained at positions, 1, 2 or 3 of a codon), and those of the types 1-2, 2-3, 3-1 are frequently 2 nt in length. The lengths of deletions vary more than those of insertions. Among I-F indels, complete codon (type 1-3) ones are often preferred over mid-codon (types 2-1, 3-2) ones, especially among deletions (high frequencies observed for 3, >15 nt deletions of type 1-3). In general, short indels (1, 2, 3, 4 nt) are more common than longer ones; however, I-F indels of longer lengths (>15, >9 nt) are more frequent than FS ones of corresponding lengths.
Figure 3.

Histogram showing the frequency of occurrence of each type of FS and I-F deletion and insertion (for nomenclature, see Supplementary Figure S3). The first series shows the frequency distribution for deletions, the second for insertions.
Figure 4.

Length distributions of the different types of FS and I-F: (a) deletions and (b) insertions. The lengths of indels (in nt) and their frequency of occurrence are given along the x- and y-axes, respectively. The three series, for each length, give the frequencies of the three types of deletions or insertions specified along the x-axis; for example, deletions of length 1 nt result due to FS deletions of types 1-1, 2-2, 3-3, whose frequencies, respectively, are 305, 332, 337. The first three bars give the frequencies of 1-1, 2-2, 3-3 type FS indels (1 nt), the next three give the frequencies of 1-2, 2-3, 3-1 type FS indels (2 nt) and the next three give the frequencies of 1-3, 2-1, 3-2 type I-F indels (3 nt). The cycle then repeats, with the next three bars again giving the frequencies of 1-1, 2-2, 3-3 type FS indels (4 nt), and so on.
The occurrence of 2609 deletions versus 1250 insertions suggests that deletions are the preferred mode of mutation; the occurrence of more FS than I-F indels [2924, 935] suggests that the former are preferred, perhaps because they are easier to generate. A study of small indels in the genomes of 79 humans has shown that healthy humans harbour a number of indels in coding exons (coding indels), and 53.5 and 46.5% of these are FS and I-F ones, respectively (39). The indels are believed to create the genetic variation necessary for biological function in some gene families, to create biological and phenotypic diversity, to have negative effects on gene function and to cause diseases. In the present study, 75.7 and 24.2% of indels are FS and I-F ones, respectively; thus, in comparison with the distribution of coding indels in healthy humans, in cancer genes, FS indels are preferred over I-F ones.
Deletion of one or more nt in a gene involves two cuts and a ligation (Table 2); for example, in the sequence, cct|cgatctct|attt, deletion of the central segment involves a 5′- or start-cut (between t|c) and a 3′- or end-cut (between t|a) and, after the deletion, a ligation or join-pair (between t-a). Insertion of one or more nt into a gene involves a cut and two ligations. Thus, in the sequence, gcttctta|aa|cgccgtc, if the central segment is the insertion, the cut-site in the WT sequence occurs between a-c and, after insertion, in the mutant sequence, the 5′-ligation occurs between a|a and the 3′-ligation between a|c. Deletion start- and end-cuts and insertion 5′- and 3′-ligations can occur between any of 16 possible pairs of adjacent nt. The frequency with which each nt pair is cut at the start and end of FS and I-F deletions, and forms 5′- and 3′-ligations in FS and I-F insertions was analysed [Figure 5; χ2 tests in Supplementary Methods (iv)]. The starting cut in FS deletions prefers to occur between a-a, c-c, g-g, t-t, and the 5′-ligation in FS insertions, between a-a, t-t, c-c (Table 2); i.e. they prefer to occur between pairs of identical nt. The ending cut in FS deletions prefers to occur between a-g, and the 3′-ligation in FS insertions, between t-g, a-c, a-g; i.e. they prefer to occur between pairs of dissimilar nt. No start- or end-cut and no 5′- or 3′-ligation preferences were observed for I-F indels. Thus, while FS indels show preferences in their selection of start- and end-cut sites, or 5′- and 3′-ligation sites, I-F indels are less discriminating.
Table 2.
Cut- and join-sites in indels
| Deletions | Insertions |
|---|---|
| FS I-F | FS I-F |
| 2021 588 | 903 347 |
| Nomenclature: | Nomenclature: |
| gtggac|g|acagg | gagccctt|a|actgccc |
| cct|cgatctct|attt | gcttctta|aa|cgccgtc |
| Start-cut: c|g, t|c | Cut-site : t-a, a-c |
| End-cut : g|a, t|a | 5′-Ligation: t|a, a|a |
| Join-pair: c-a, t-a | 3′-Ligation: a|a, a|c |
| Start-, end-cut preferences: | 5′-, 3′-Ligation preferences: |
| FS: | FS: |
| Start-cut : a-a, c-c, g-g, t-t | 5′-Ligation: a-a, t-t, c-c |
| End-cut : a-g | 3′-Ligation: t-g, a-c, a-g |
| I-F: | I-F: |
| No start-, end-cut preferences | No 5′-, 3′-ligation preferences |
| Start-, end-cut, join-pair | Cut-site, 5′-, 3′-ligation |
| combination preferences: | combination preferences: |
| FS, I-F: | FS, I-F: |
| End-cut, join-pair often same | Cut-site, 3′-ligation often same |
Figure 5.

Histograms showing the frequency with which each of 16 pairs of adjacent nt are cut at the start and end of FS deletions (a and b), and occur as 5′- and 3′-ligations in FS insertions (c and d). In (a), the two series show the frequencies with which each nt pair (e.g. a-a) is cut at the start and end of FS deletions [212, 93]; the difference between the two frequencies for each nt pair [119] is given in (b). In (c), the two series show the frequencies with which each nt pair (e.g. a-a) forms 5′- and 3′-ligations in FS insertions [137, 69]; the difference between the two frequencies for each nt pair [68] is given in (d).
An examination of insertions such as, aaag|g|aaaag, ttttt|t|aaag, aga|ga|tatcaa, ttacc|c|tgtg, tataa|taa|cact, suggested that the cut-site and 3′-ligation are often the same (g-a, g-a; t-a, t-a; a-t, a-t; c-t, c-t; a-c, a-c). Thus, for FS and I-F insertions, an attempt was made to count combinations of cut-site, 5′- and 3′-ligations, in which none, two or all three sites are the same; similarly, for FS and I-F deletions, combinations of start-cut, end-cut and join-pair, in which none, two or all three pairs are the same, were counted (Figure 6, Supplementary Table S6). Out of 903 and 347 FS and I-F insertions, in more than half, only one combination was observed: ‘only cut, 3′- same’ [498, 205]; the other combinations were observed in smaller numbers. Two combinations (‘only cut, 3′- same’ and ‘cut, 5′-, 3′- same’) occur significantly more frequently [p < 0.001; χ2 tests in Supplementary Methods (v)]. Thus, it is reported here that in FS and I-F insertions, the 3′-ligation is often the same as the cut-site. There are 2011 FS and 550 I-F deletions in which start-cut, end-cut and join-pair are present (deletions of N- and/or C-termini have been left out because they lack one or more of the sites). One combination was observed significantly more frequently: ‘only end, join same’ [887, 194; p < 0.001]. Thus, in deletions, the join-pair is often the same as the end-cut. The 5′-ligation in insertions or the start-cut in deletions prefer to occur between pairs of identical bases (Figure 5). Inserting or deleting a base that is identical to the adjoining one, may fail to trigger a corrective response from the cell’s copyediting machinery. Further, the cut-site in an insertion and the end-cut in a deletion are often replaced by an identical 3′-ligation and join-pair, respectively. Replacing a cut by an identical ligation may also be a mechanism to confuse the cell’s copyediting machinery into accepting the mutation.
Figure 6.

Joint frequencies of cut- and join-sites in deletions and insertions. There are four groups of bars; the first two are for FS and I-F deletions, the last two for FS and I-F insertions. The first bar in each group gives the total number of mutations (FS or I-F deletions or insertions) that have cut- and join-sites. In the first two groups of bars (FS and I-F deletions), the second, third, fourth and fifth bars, respectively, give the number of times that: (i) start-cut, end-cut, join-pair are same, (ii) start-cut, end-cut, join-pair are different, (iii) only start-cut, join-pair are same and (iv) only end-cut, join-pair are same. In the last two groups of bars (FS and I-F insertions), the second, third, fourth and fifth bars, respectively, give the number of times that: (i) cut-site, 5′-ligation, 3′-ligation are same, (ii) cut-site, 5′-ligation, 3′-ligation are different, (iii) only cut-site, 5′-ligation are same and (iv) only cut-site, 3′-ligation are same.
The locations in proteins at which indels occur were analysed (Supplementary Figure S4). The start and end codon numbers of each deletion in the WT protein, and of each insertion in the mutant protein were used to identify the locations of indels in proteins. Each protein was divided into three parts—first (or N-terminal), second (or middle), third (or C-terminal)—and indels occurring in each part were identified. The largest, second- and third-largest numbers of I-F and FS deletions occur in the middle, N- and C-terminal regions of proteins (S4A), and of FS and I-F insertions occur in the middle-, C- and N-terminal regions of proteins (S4B). Premature termination codons (PTCs) occurring in the last, or 3′ 50-nt of the penultimate exons of genes, are likely to produce mRNAs that encode proteins with altered functions; PTCs occurring at other exon positions may cause the mRNA to be targeted for nonsense-mediated decay and may abolish gene function (39). Thus, in S4A, S4B, while indels occurring in the N-terminal region may abolish protein function, those occurring in the C-terminal region are likely to modify it. The greater frequency of occurrence of: (i) deletions in the N- rather than C-terminal regions suggests that deletions often abolish protein function (S4A) and (ii) of insertions in the C- rather than N-terminal regions suggests that insertions often modify protein function (S4B). Indels occurring in the middle of the protein are the preferred way to alter or disrupt protein function. In I-F deletions, but never in FS ones, the entire protein may be lost (S4A). In S4B, the last two sets of bars show that the majority of FS and I-F insertions cause the WT protein to become shorter and longer, respectively.
The fraction of protein lost as a result of each deletion, and gained or lost as a result of each insertion was calculated for all FS and I-F indels (Figure 7). A deletion may result in the loss of a few amino acids or in the introduction of a PTC, resulting in the loss of a part of the protein. Figure 7a shows that the fraction of protein lost was <0.1 for 87% of I-F deletions, and was ≥0.1 for 91%, ≥0.2 for 84% and ≥0.4 for 60% of FS deletions; thus, larger portions of the protein are lost as a result of FS than I-F deletions. Figure 7b shows that 96% of I-F insertions increase protein length by a fraction between 0 and 0.1; the number of amino acids added ranged from 1 to 64. The longest I-F insertion (in PTEN) increased protein length by 103 amino acids (a fraction of 0.26). On the other hand, only 9% of FS insertions increase protein length by a fraction between 0 and 0.1; 91% decrease protein length. The fraction of protein lost was between −0.3 and −1.0 for 65% of FS insertions. FS insertions may lead to the introduction of a PTC, which causes loss of a part of the protein. The longest segments lost (in BRCA2) and gained (in CEBPA) consisted of 3387 and 21 codons, respectively. Supplementary Figure S5 shows distributions of the lengths of protein lost due to FS and I-F deletions (S5A), and gained or lost due to I-F and FS insertions (S5B). As a result of FS indels, while protein segments 100 or fewer residues in length are often lost, the loss of longer segments (more than 100 residues) is more frequent. On the other hand, the majority of I-F deletions cause modest decreases, and the majority of I-F insertions cause modest increases in protein length (105 or less residues). After the point of FS deletion or insertion, a change in the gene reading frame occurs. The length distributions of corrupted protein sequences resulting from FS indels [Supplementary Methods (i)b] are shown in Supplementary Figure S6; while sequences of shorter lengths (1–10 amino acid residues) are the most frequent, those of longer lengths are also common. The figure provides insight into the stretches of protein corrupted by FS indels.
Figure 7.

(a) Histogram showing the fractions of protein lost as a result of FS [2021] and I-F [588] deletions (first and second series). Fractions are given as intervals along the x-axis, and the number of deletions occurring in each interval is given along the y-axis. The fraction of protein lost due to each deletion was calculated as: (number of codons lost)/(number of codons in WT protein). The fraction was <0.1 for 87% (510/588) of I-F deletions, and ≥0.1 for 91% [(2021−178 = 1843)/2021), ≥0.2 for 84% (1705/2021) and ≥0.4 for 60% of FS deletions. (b) Histogram showing the fractions of protein gained or lost as a result of FS [903] and I-F [347] insertions (first and second series). Fractions are given as intervals along the x-axis (range, 0.3 through −1.0). Fractions >0 indicate increase, and <0 indicate decrease in protein length. The number of observations in each interval is given along the y-axis. The fraction of protein gained or lost due to each insertion was calculated as: (number of codons in mutant protein–number of codons in WT protein)/(number of codons in WT protein). Nearly 96% (333/347) of I-F insertions caused increase, and 91% [(903−81 = 822)/903] of FS insertions caused decrease in protein length.
Mutations in PO and TS
The three types of substitution, two types of deletion and two types of insertion mutations were sorted gene-wise and tissue-wise (Supplementary Table S5). Genes in which at least one type of mutation had a value more than nine in at least one tissue were short-listed (Table 3); these were genes in which multiple unique mutations (more than nine) of at least one type were observed in at least one tissue. Greater the number of unique mutations detected in a gene, greater its significance for cancer (20), and genes with more than nine unique mutations have a definite significance for cancer. Genes were classified into PO and TS (Table 3, legend). The table differs from Table 4 in ref. (24) because only unique mutations in each tissue have been considered, and because FS and I-F deletions and insertions have been considered separately.
Table 3.
Distribution of substitution, deletion and insertion mutations in 29 TS and 24 PO. The second column gives the basis of the classification of each gene; ts and po refer to the classification of the gene by Swiss-prot (40), F refers to the classification given in Table 4 in ref. (24), V refers to that given in Table 1 in ref. (6) and N refers to that obtained via internet searches. The total numbers of mutations [6517, 2900] and missense mutations [2156, 2138], observed in the sets of TS and PO, are given
| Gene names | Classifications | Synonymous | Missense | Nonsense | I-F deletions | FS deletions | I-F insertions | FS insertions |
|---|---|---|---|---|---|---|---|---|
| TS: | ||||||||
| GATA1 | tsF | 2 | 9 | 8 | 4 | 31 | 1 | 37 |
| ATM | ts | 2 | 106 | 16 | 6 | 21 | 1 | 9 |
| BRCA2 | tsN | 2 | 13 | 3 | 12 | 5 | ||
| MLH1 | ts | 2 | 16 | 3 | 1 | 9 | 1 | |
| MSH2 | ts | 5 | 10 | 10 | 1 | 9 | 1 | |
| MSH6 | tsF | 8 | 20 | 7 | 9 | 6 | ||
| TP53 | ts | 4 | 324 | 58 | 11 | 39 | 4 | 12 |
| SMARCA4 | tsN | 14 | 2 | 8 | ||||
| SMARCB1 | ts | 2 | 11 | 28 | 2 | 29 | 1 | 19 |
| NOTCH1 | ts/poN | 7 | 62 | 23 | 12 | 14 | 49 | 40 |
| RUNX1 | ts/poN | 5 | 35 | 5 | 22 | 7 | 23 | |
| CDH1 | tsF | 6 | 52 | 12 | 11 | 35 | 14 | |
| HNF1A | tsN | 33 | 7 | 3 | 16 | 1 | 6 | |
| NF1 | ts | 2 | 22 | 31 | 4 | 48 | 5 | |
| NF2 | ts | 5 | 25 | 73 | 26 | 241 | 26 | |
| VHL | ts | 37 | 232 | 37 | 49 | 306 | 5 | 78 |
| FBXW7 | tsF/tsN | 68 | 18 | 2 | 6 | 1 | 10 | |
| SMAD4 | tsF | 4 | 88 | 25 | 2 | 16 | 11 | |
| SOCS1 | tsN | 1 | 3 | 6 | 15 | 1 | ||
| APC | ts | 11 | 109 | 166 | 2 | 400 | 2 | 151 |
| CDC73 | ts | 2 | 7 | 1 | 15 | 3 | ||
| CDKN2A | ts | 68 | 310 | 75 | 43 | 103 | 4 | 35 |
| CEBPA | tsF/poN | 9 | 19 | 13 | 28 | 104 | 76 | 92 |
| MEN1 | tsF | 31 | 17 | 10 | 57 | 1 | 14 | |
| PTCH1 | ts | 10 | 74 | 45 | 5 | 37 | 2 | 18 |
| PTEN | ts | 20 | 366 | 107 | 42 | 257 | 5 | 110 |
| RB1 | po/tsF/tsV | 4 | 23 | 68 | 5 | 46 | 1 | 14 |
| STK11 | tsF | 9 | 46 | 19 | 10 | 27 | 10 | |
| WT1 | ts/poF/tsV | 33 | 16 | 2 | 28 | 1 | 76 | |
| 2156 | 6517 | |||||||
| PO: | ||||||||
| PTPN11 | poF | 2 | 45 | |||||
| JAK2 | po | 8 | 35 | 6 | 5 | |||
| NPM1 | po/tsF | 2 | 52 | |||||
| BRAF | po | 16 | 152 | 2 | 6 | 1 | 3 | |
| MPL | po | 10 | 1 | |||||
| ABL1 | po | 1 | 24 | |||||
| ALK | po | 3 | 22 | 1 | ||||
| CSF1R | po | 6 | 14 | 3 | ||||
| CTNNB1 | poF | 21 | 402 | 5 | 91 | 4 | 2 | |
| EGFR | ts/poF/poN | 19 | 194 | 4 | 37 | 2 | 35 | 2 |
| ERBB2 | po | 3 | 32 | 10 | ||||
| FGFR3 | poF | 14 | 45 | 5 | 3 | 1 | ||
| FLT3 | poF | 4 | 30 | 6 | 1 | 82 | 1 | |
| GNAS | po | 21 | ||||||
| HRAS | po | 3 | 101 | 1 | 1 | 1 | ||
| KIT | po | 30 | 151 | 3 | 106 | 4 | 30 | |
| KRAS | po | 13 | 272 | 1 | 1 | 2 | 8 | |
| MET | po | 3 | 25 | 1 | 5 | |||
| NRAS | po | 3 | 121 | 1 | ||||
| PDGFRA | poF | 6 | 25 | 14 | 1 | 2 | 1 | |
| PIK3CA | ts/poF/poN | 14 | 332 | 3 | 11 | 3 | ||
| RET | po | 6 | 33 | 11 | ||||
| SMO | poF | 2 | 12 | 1 | ||||
| TSHR | poN | 38 | 2 | |||||
| 2138 | 2900 |
FS indels are more frequent in TS than PO. The NPM1 gene is an exception because, inspite of being a PO, it undergoes a large number of FS insertions [52]. An explanation for this might be that NPM1 can function as both PO and TS (41,42). While in PO, I-F indels are observed more frequently than FS ones, in TS, FS indels are more frequent. I-F deletions and I-F insertions are also observed in TS, but in lesser numbers, with the former being more frequent than the latter. In the TS, NOTCH1 and CEBPA, a significant number of I-F insertions are observed; this may be because these genes function as both TS and PO (43,44). Nonsense substitutions are observed far more frequently in TS than PO. Disruptive mutations—FS indels and nonsense substitutions—occur more frequently in TS, than PO. This is consistent with the requirement that TS, which inhibit cell proliferation, have to be inactivated for unrestrained cell division and cancer to occur. In PO, on the other hand, I-F indels are preferred. As these mutations modify, rather than disrupt, protein function, they are well-suited to activate PO (cellular genes that promote cell proliferation) to oncogenes (which promote excessive cell division and cancer). Mutations in TS cause loss of suppression activity by destabilizing protein structure; mutations in PO also destabilize protein structure, but gain of function results because either the less active form of the protein or the transition to it is destabilized, which increases the population of the active, disease causing state (23). The most frequently occurring mutations in PO are missense mutations, which are also well-suited to modifying function. The 24 PO and 29 TS, in the table, undergo 2138 and 2156 missense mutations, respectively; the average number of missense mutations observed per gene is higher for PO [89] than TS [74].
The total number of mutations undergone by TS genes [6517] is much larger than that undergone by PO [2900]. TS genes undergo large numbers of FS indels, missense and nonsense substitutions, as well as smaller numbers of I-F indels; PO, on the other hand, mainly undergo missense substitutions, and also smaller, but significant, numbers of I-F indels. Thus, TS genes undergo larger numbers and a greater variety of mutations than PO. One reason for this might be the requirement that both alleles that code for a TS gene be inactivated for tumor formation to occur; to inactivate two alleles, more mutations are recruited. Further, inactivating a protein by mutation is probably easier and less constraining than modifying its activity; therefore, a variety of mutations are employed for the purpose. On the other hand, for a PO, activation of a single allele is sufficient to turn it into an oncogene. Moreover, activation of a protein requires precise and specific mutations. Hence, the number of ways in which a PO can be mutated into an oncogene is limited. As the types of mutations that target PO (e.g. KIT) differ from those that target TS genes (e.g. APC), mutation probably operates in different ways on PO and TS.
Distribution of mutation positions over the lengths of TS and PO
An attempt was made to examine if mutation positions in each protein (sites of one or more mutations) preferred to occur in certain regions of it or if they were randomly distributed over its entire length [Supplementary Methods (ii)]. Figure 8 shows the distribution of mutation positions in each of 40 proteins (PO and TS). Proteins in which the second bar is >70% and the first bar <40% (>70% of mutation positions occur in <40% of the protein) were those in which the majority of mutation positions occurred in a portion (less than two-fifth) of the protein. The first 11 proteins meet this criterion (GATA1 to FGFR3); of these, seven are PO and four TS. In five more proteins, all PO (KIT, PIK3CA, NRAS, KRAS, HRAS), the majority of mutation positions occur in less than the entire protein (>80% of mutation positions occur in <55% of the protein). Thus, mutations in PO tend to occur in selected regions, rather than throughout the length of the protein. On the other hand, in the last 12 proteins (SOCS1 to CDKN2A), all TS, >90% of mutation positions occur in >90% of the protein (mutations are distributed over the entire protein length). The four TS that occur to the left, among the PO, are exceptions (discussed below). In four more proteins (RUNX1 to CDH1), which function either as TS, or both PO and TS, >80% of mutation positions are distributed over >60% of the protein.
Figure 8.

Distribution of mutation positions over the lengths of proteins. Genes [40] are listed along the x-axis and each gene name is prefixed by po, ts or b, which indicate, respectively, whether the gene functions as a PO, a TS or as both. For each gene, there is a pair of bars which are related to each other. The %fraction of the protein given in the first bar contains the %fraction of mutation positions given in the second bar [Supplementary Methods (ii)]. For example, in the PO, CTNNB1, 89% of all mutation positions (second bar) occur in 13% of the protein length (first bar). A tall second bar and a short first bar indicate that the majority of mutations occur in a small segment of the protein; first and second bars of nearly equal length indicate that the mutations occur over the entire length of the protein.
Supplementary Figure S7 (a through g) shows the distribution of mutation positions for each mutation type. GATA1, NOTCH1, APC and CDC73 are TS, but occur to the left in Figure 8, among PO, and undergo mutations in selected regions of, rather than throughout the lengths of the proteins. The different types of mutations undergone by these genes are: GATA1, FS indels (S7e,g); NOTCH1, missense, nonsense, I-F, FS insertions (S7a,b,f,g), APC, missense, nonsense, FS indels (S7a,b,e,g); CDC73, FS deletions (S7e). Each type of mutation occurs in specific regions of, rather than throughout the gene. It is possible that the genes have an intrinsic tendency to undergo mutations in these regions; i.e. positions in the regions may be mutational hotspots (45). In the PO, CTNNB1, I-F deletions, missense and even synonymous substitutions (S7d,a,c) are restricted to certain regions. On the other hand, in the TS, CDKN2A, VHL and PTEN, each of the different types of mutations occurs throughout the protein. Thus, different genes undergo different patterns of mutations, with TS preferring to mutate over the entire length, and PO preferring to mutate in specific regions. Mutations in selected regions of the gene are well-suited to activate PO, and those occurring over the entire length are suitable for inactivating the two alleles of a TS gene.
A catalogue of genes which play a role in each cancer
An attempt was made to rank genes playing a role in each cancer and to rank the cancers in which each gene was playing a role (see ‘Methods’ section). Cancer of each tissue was considered and genes showing mutations in the cancer were arranged by rank score; likewise, each gene was considered and cancers in which mutations in the gene were observed were ranked (Supplementary Table S1a and b). @@ or ** marked gene-tissue pairs constitute a more proven list of genes playing a role in the different cancers because more than one study has indicated their significance [Supplementary Methods (iii)].
Supplementary Table S1a and b are useful because they list out, in one place, the majority of genes that play a role in cancer of each tissue, and the different cancers in which a gene plays a role. Supplementary Table 1a shows that cancer is a multiple gene disease: multiple genes undergo mutations, resulting in mal-functioning proteins, which cause cancer. In most cancers, PO and TS play a role. Considering only marked genes, the largest number play a role in cancers of haematopoietic-and-lymphoid tissue, the reason being the variety of cancers associated with the different cell types of this tissue (leukaemias, lymphomas); different genes play a role in the different cancers. Further, more PO than TS play a role in cancers of this tissue.
Genes that have been recognized as playing a role in specific cancers are present in Supplementary Table S1b. For example, the TS, APC, MEN1, NF1, NF2, RB1, VHL and WT1 have been shown to play roles in colorectal carcinomas, multiple endocrine neoplasia type I, neurofibromatosis types I and II, retinoblastoma, renal cell carcinoma and paediatric kidney cancer (6), respectively; in the table, appropriately, they appear associated with cancers of the large intestine, pancreas, soft tissue, soft tissue and meninges, eye, kidney and kidney, respectively. Similarly, the PO, ABL1, EGFR, KIT and RET, are known to play roles in chronic myelogenous leukaemia, squamous cell carcinoma, sarcoma and thyroid cancer, respectively; in the table, appropriately, they appear associated with cancers of haematopoietic-and-lymphoid tissue, lung, soft tissue and thyroid.
In Supplementary Table S1b, genes with a large number of mutated samples (third number), a high proportion of mutated samples (second number) and with high ranks (first number) may, with confidence, be considered to be playing an important role in the corresponding cancer. Many of the marked genes meet these criteria: APC: large intestine; BRAF: skin; CDKN2A: pancreas; CTNNB1: soft tissue, pancreas; FGFR3: urinary tract; GATA1, JAK2: haematopoietic and lymphoid tissue; KRAS: pancreas, large intestine, biliary tract; NF2: soft tissue; PTCH1: skin; PTEN: endometrium; RB1: eye; SMARCB1: soft tissue; VHL: kidney. The table also corroborates the well-recognized fact that TP53 plays an important role in a wide range of cancers (6). Some other genes which play an important role in several cancers include: BRAF, CDKN2A, CTNNB1, KRAS and PIK3CA. Some genes appear to be predominantly associated with cancer of a single tissue. ABL1, ATM, CEBPA, GATA1, JAK2, MPL, NOTCH1, NPM1, PTPN11, RUNX1 are all associated only with cancers of haematopoietic-and-lymphoid tissue, TSHR is associated only with thyroid cancer. Genes which, in a tissue, undergo few (less than ten) unique mutations but which undergo a particular mutation repeatedly, are marked @@ in the tables. Examples of such gene-tissue pairs include: BRAF: thyroid, FGFR3: skin, FOXL2: ovary, GNAS: pituitary, IDH1: central nervous system, PDGFRA: small intestine. Mutations in the metabolic enzyme, IDH1, have been linked to glioma and other cancers (46,47). The 676 mutated IDH1 samples contain only five unique missense mutations; a single mutation, R132H, frequent in human glioma, is observed 606 times. Most cancer genes play a role in more than one cancer and, in most cancers, more than one gene plays a role. Thus, the scenario is far from the one in which different genes play roles in different cancers. The puzzle also remains as to why genes which function in all tissues, cause cancer only in certain tissues.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Methods, Supplementary Tables 1–6, Supplementary Figures 1–7 and Supplementary Reference [48].
FUNDING
Department of Science and Technology, Government of India, Women Scientists Scheme [SR/WOS-A/LS-82/2008 to P.I.]. Funding for open access charge: Waived by the Oxford University Press.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The author is grateful to Prof. N.V. Joshi for help with statistical analysis and to Prof. P. Balaram for helpful discussions during the course of this work.
REFERENCES
- 1.Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170–173. doi: 10.1038/260170a0. [DOI] [PubMed] [Google Scholar]
- 2.Tabin CJ, Bradley SM, Bargmann CI, Weinberg RA, Papageorge AG, Scolnick EM, Dhar R, Lowy DR, Chang EH. Mechanism of activation of a human oncogene. Nature. 1982;300:143–9. doi: 10.1038/300143a0. [DOI] [PubMed] [Google Scholar]
- 3.Harris H, Miller OJ, Klein G, Worst P, Tachibana T. Suppression of malignancy by cell fusion. Nature. 1969;223:363–368. doi: 10.1038/223363a0. [DOI] [PubMed] [Google Scholar]
- 4.Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl Acad. Sci. USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinberg RA. Tumor suppressor genes. Science. 1991;254:1138–46. doi: 10.1126/science.1659741. [DOI] [PubMed] [Google Scholar]
- 6.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat. Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 7.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 8.Hayden EC. Cancer complexity slows quest for cure. Nature. 2008;455:148. doi: 10.1038/455148a. [DOI] [PubMed] [Google Scholar]
- 9.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forbes S, Clements J, Dawson E, Bamford S, Webb T, Dogan A, Flanagan A, Teague J, Wooster R, Futreal PA, et al. COSMIC 2005. Br. J. Cancer. 2006;94:318–322. doi: 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. The Catalogue of Somatic Mutations in Cancer (COSMIC) Curr. Protoc. Hum. Genet. 2008 doi: 10.1002/0471142905.hg1011s57. Chapter 10, Unit 10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, Kok CY, Jia M, Ewing R, Menzies A, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010;38:D652–D657. doi: 10.1093/nar/gkp995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin M-L, Ordóñez GR, Bignell GR, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaminker JS, Zhang Y, Waugh A, Haverty PM, Peters B, Sebisanovic D, Stinson J, Forrest WF, Bazan JF, Seshagiri S, et al. Distinguishing Cancer-Associated Missense Mutations from Common Polymorphisms. Cancer Res. 2007;67:465–473. doi: 10.1158/0008-5472.CAN-06-1736. [DOI] [PubMed] [Google Scholar]
- 17.Kaminker JS, Zhang Y, Watanabe C, Zhang Z. CanPredict: a computational tool for predicting cancer-associated missense mutations. Nucleic Acids Res. 2007;35:W595–W598. doi: 10.1093/nar/gkm405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carter H, Chen S, Isik L, Tyekucheva S, Velculescu VE, Kinzler KW, Vogelstein B, Karchin R. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 2009;69:6660–6667. doi: 10.1158/0008-5472.CAN-09-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torkamani A, Schork NJ. Prediction of cancer driver mutations in protein kinases. Cancer Res. 2008;68:1675–1682. doi: 10.1158/0008-5472.CAN-07-5283. [DOI] [PubMed] [Google Scholar]
- 20.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas PD, Kejariwal A. Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects. Proc. Natl Acad. Sci. USA. 2004;101:15398–15403. doi: 10.1073/pnas.0404380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi Z, Moult J. Structural and functional impact of cancer-related missense somatic mutations. J. Mol. Biol. 2011;413:495–512. doi: 10.1016/j.jmb.2011.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeang CH, McCormick F, Levine A. Combinatorial patterns of somatic gene mutations in cancer. FASEB J. 2008;22:2605–2622. doi: 10.1096/fj.08-108985. [DOI] [PubMed] [Google Scholar]
- 25.Cui Q. A network of cancer genes with co-occurring and anti-co-occurring mutations. PLoS ONE. 2010;5:e13180. doi: 10.1371/journal.pone.0013180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spiegel MR, Schiller J, Srinivasan RA. Schaum’s Outlines Probability and Statistics, chapter 6. 2nd edn. New York: McGraw-Hill; 2000. p. 218. problem 6.27. [Google Scholar]
- 27.Carlini DB, Stephan W. In vivo introduction of unpreferred synonymous codons into the Drosophila Adh gene results in reduced levels of ADH protein. Genetics. 2003;163:239–243. doi: 10.1093/genetics/163.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bird AP. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980;8:1499–1504. doi: 10.1093/nar/8.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnheim N, Calabrese P. Understanding what determines the frequency and pattern of human germline mutations. Nat. Rev. Genet. 2009;10:478–488. doi: 10.1038/nrg2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease. Human Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- 31.Subramanian S, Kumar S. Neutral substitutions occur at a faster rate in exons than in noncoding DNA in primate genomes. Genome Res. 2003;13:838–844. doi: 10.1101/gr.1152803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon J-H, Smith LE, Feng Z, Tang M, Lee C-S, Pfeifer GP. Methylated CpG dinucleotides are the preferential targets for G-to-T transversion mutations induced by benzo[a]pyrene diol epoxide in mammalian cells: similarities with the p53 mutation spectrum in smoking-associated lung cancers. Cancer Res. 2001;61:7110–7117. [PubMed] [Google Scholar]
- 33.Hodgkinson A, Eyre-Walker A. Variation in the mutation rate across mammalian genomes. Nat. Rev. Genet. 2011;12:756–766. doi: 10.1038/nrg3098. [DOI] [PubMed] [Google Scholar]
- 34.Thomas NE, Alexander A, Edmiston SN, Parrish E, Millikan RC, Berwick M, Groben P, Ollila DW, Mattingly D, Conway K. Tandem BRAF mutations in primary invasive melanomas. J. Invest. Dermatol. 2004;122:1245–1250. doi: 10.1111/j.0022-202X.2004.22523.x. [DOI] [PubMed] [Google Scholar]
- 35.Skinner AM, Dan C, Turker MS. The frequency of CC to TT tandem mutations in mismatch repair-deficient cells is increased in a cytosine run. Mutagenesis. 2008;23:87–91. doi: 10.1093/mutage/gem047. [DOI] [PubMed] [Google Scholar]
- 36.Schneider A, Cannarozzi GM, Gonnet GH. Empirical codon substitution matrix. BMC Bioinformatics. 2005;6:134. doi: 10.1186/1471-2105-6-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 38.Dixit A, Yi L, Gowthaman R, Torkamani A, Schork NJ, Verkhiver GM. Sequence and structure signatures of cancer mutation hotspots in protein kinases. PLoS ONE. 2009;4:e7485. doi: 10.1371/journal.pone.0007485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mills RE, Pittard WS, Mullaney JM, Farooq U, Creasy TH, Mahurkar AA, Kemeza DM, Strassler DS, Ponting CP, Webber C, et al. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. 2011;21:830–839. doi: 10.1101/gr.115907.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O'Donovan C, Phan I, et al. The Swiss-Prot protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat. Rev. Cancer. 2006;6:493–505. doi: 10.1038/nrc1885. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Sejas DP, Burma S, Chen DJ, Pang Q. Nucleophosmin suppresses oncogene-induced apoptosis and senescence and enhances oncogenic cooperation in cells with genomic instability. Carcinogenesis. 2007;28:1163–1170. doi: 10.1093/carcin/bgm025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanlon L, Avila JL, Demarest RM, Troutman S, Allen M, Ratti F, Rustgi AK, Stanger BZ, Radtke F, Adsay V, et al. Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res. 2010;70:4280–4286. doi: 10.1158/0008-5472.CAN-09-4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chapiro E, Russell L, Radford-Weiss I, Bastard C, Lessard M, Struski S, Cave H, Fert-Ferrer S, Barin C, Maarek O, et al. Overexpression of CEBPA resulting from the translocation t(14;19)(q32;q13) of human precursor B acute lymphoblastic leukemia. Blood. 2006;108:3560–3563. doi: 10.1182/blood-2006-03-010835. [DOI] [PubMed] [Google Scholar]
- 45.Rogozin IB, Pavlov YI. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Mutat. Res. 2003;544:65–85. doi: 10.1016/s1383-5742(03)00032-2. [DOI] [PubMed] [Google Scholar]
- 46.Parsons DW, Jones S, Zhang X, Lin JCH, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. New Engl. J. Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum. Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.