


Abstract
The utilization of archived, formalin-fixed paraffin-embedded (FFPE) tumor samples for massive parallel sequencing has been challenging due to DNA damage and contamination with normal stroma. Here, we perform whole genome sequencing of DNA isolated from two triple-negative breast cancer tumors archived for >11 years as 5 µm FFPE sections and matched germline DNA. The tumor samples show differing amounts of FFPE damaged DNA sequencing reads revealed as relatively high alignment mismatch rates enriched for C·G > T·A substitutions compared to germline samples. This increase in mismatch rate is observable with as few as one million reads, allowing for an upfront evaluation of the sample integrity before whole genome sequencing. By applying innovative quality filters incorporating global nucleotide mismatch rates and local mismatch rates, we present a method to identify high-confidence somatic mutations even in the presence of FFPE induced DNA damage. This results in a breast cancer mutational profile consistent with previous studies and revealing potentially important functional mutations. Our study demonstrates the feasibility of performing genome-wide deep sequencing analysis of FFPE archived tumors of limited sample size such as residual cancer after treatment or metastatic biopsies.
INTRODUCTION
To date, massively parallel sequencing of cancer genomes has largely been performed using flash frozen tissue or immortalized cancer cell lines (1–6). These studies have provided tremendous insight into the types of mutations and genomic rearrangements that occur in cancer cells. However, limiting sequencing to flash frozen tissues restricts the types of important clinical questions that can be addressed (7). Since formalin fixation and paraffin embedding (FFPE) has been the standard sample preparation for pathologists for decades, the ability to perform massively parallel sequencing of FFPE samples would open up large archived tumor specimen collections. As these large archives frequently have historical records of patient progression and outcome, this would allow for powerful retrospective studies exploring DNA changes that influence disease progression.
RNA isolated from FFPE samples is commonly used for genome-wide expression studies (8–10), however performing whole-genome analyses of DNA isolated from FFPE samples has two major application-specific challenges. First is the fact that tumors of biological and clinical interest stored in blocks are often contaminated with normal stroma, and thus dissection, which is not easy to perform in blocks, is required to enrich for tumor material. A second challenge is the fact that formalin-fixed tissues exhibit a higher frequency of non-reproducible DNA sequence alterations than frozen tissues. This is likely due to formalin cross-linking of cytosine nucleotides on either strand, resulting in Taq polymerase during PCR not recognizing the cytosine and incorporating an adenine in place of a guanosine causing an artificial C > T or G > A mutation (11,12). Previous studies have successfully isolated DNA from FFPE tissue samples stored in paraffin blocks and performed targeted sequencing of single genes (13,14) or whole exome sequencing (15). In a few instances, sequencing was extended to the whole genome but was limited to copy number analysis or high-level mutational profile analysis (16,17). In the Kerick study, artificial mutations resulting from the formalin fixation process were observed in the sequence data by comparison to matched frozen tissues but methods for removing these false positive calls in the analysis steps of the sequence data were not presented.
In the work presented below, we sequenced DNA isolated from two FFPE triple-negative breast tumors archived as 5 µm sections as well as their matched germline DNA. As the tumor was mounted on slides, it was straightforward to identify and isolate DNA from areas containing >80–85% malignant cells. By characterizing the patterns of DNA mismatches in the FFPE tumor sequencing reads, we determined that one of the samples was more heavily damaged by fixation than the other and propose guidelines for a rapid FFPE integrity test. We then call somatic variants and implement original filters to remove false positive calls specifically resulting from the formalin fixation process, thus leading to a set of high-confidence somatic mutations in each of the tumors. Finally, we identify a set of mutations of potential functional importance in the progression of the disease (or lack thereof) in each of the two cases.
MATERIALS AND METHODS
Patient information
From the Women’s Healthy Eating and Living (WHEL) cohort (18), we identified two female non-Hispanic white patients (06408 and 02542) diagnosed with Stage III histologic Grade III infiltrating ductal triple negative breast cancer in 1999 and 1995 at the ages of 38 and 30 years, respectively. All patients provided written informed consent for enrollment in the WHEL Study and for related genomic studies. Triple negative breast cancer indicates that the estrogen and progresterone receptor staining on tumor tissue was negative and Her2/neu over-expression was not observed. Both patients received adjuvant chemotherapy and local regional radiation therapy. Patient 02542’s tumor metastasized 18 months after initial diagnosis and she died shortly afterwards. Patient 06408 is still alive without recurrence as of 2006. The patients underwent curative intent surgical resection and breast tumor material not needed for diagnosis was formalin fixed, embedded in paraffin, sectioned at 5 µm thickness and mounted on slides. Germline DNA was extracted from peripheral blood mononuclear cells (PBMC).
DNA isolation
Areas of tumor cells on a hematoxylin and eosin (H&E) stained 5 µm FFPE section were identified and marked by a pathologist allowing the collection of malignant cells with a >80–85% purity (Supplementary Figure S1). Additional tumor material from an adjacent unstained section was isolated by scraping the area corresponding to the marked section with a sterile scalpel. DNA was isolated from the FFPE specimens using BiOstic FFPE Tissue DNA Isolation kit (MO BIO, Carlsbad, CA, USA). The samples were heated at 55°C for an hour in an optimized wax melting buffer and Protease K to completely digest the tissue. Samples were heated at 90°C for 1 h to remove protein–DNA cross-links, purified on a silica spin filter and eluted with 10 mM Tris pH 8.0.
Tumor cell counting
The H&E stained slides were used to estimate the number of tumor cells from which DNA was isolated (Supplementary Figure S1). DNA was isolated from unstained 5 µm thick sections ∼1.0 and 2.0 cm2 areas for samples 06408 and 02542, respectively. We used a Nikon Eclipse E600 microscope to take images of the cells and processed the images with MetaMorph 7.7 (Molecular Devices, Sunnyvale, CA, USA). Six random fields within the marked areas were taken. We calculated the number of nuclei in each random field to get an approximate number of cells per slide. To count the cells, we first separated the constituent blue, red and green channels from each of the 24-bit RGB images. Only the blue channel was used to count the number of nuclei in the image. Nuclei were selected by setting the appropriate intensity threshold. The resulting nuclei were filtered by area to remove noise and counted using the morphometry tool in Metamorph. The number of nuclei was used to calculate the average cell density per image, which was used to extrapolate the number of cells used for sequencing. The area of each image was 1360 pixels by 1024 pixels, with 1 pixel = 0.334355 µm.
Sequencing
Purified tumor and germline DNA were directly used as starting material for SOLiD fragment library preparation (Life Technologies, Carlsbad, CA, USA) following manufacturer’s recommendation. DNA was sheared to ∼150 bp using the Covaris S2 system standard fragmentation conditions recommended in the SOLiD4 Library Prep User Guide. After DNA end-repair, P1 and P2 adaptors were ligated, the adaptor-ligated DNA underwent nick translation and then amplification with six and eight PCR cycles for germline and tumor DNA, respectively, using Library PCR primer 1 and 2, and Platinum PCR amplification mix. Purified library was quantified by TaqMan assay and used for preparing SOLiD templated beads. Each sequencing run resulted in ∼500 million raw 50-bp color-space reads per slide. The samples were sequenced over several runs each using both SOLiD3+ and SOLiD4 platforms generating between 1.3 and 3.1 billion total raw reads per sample (Supplementary Table S1).
Genotyping array data generation and analysis
Germline DNA was genotyped on the Illumina Omni 2.5 M array and processed using GenomeStudio (version 2010.3) using standard methods. Genotypes were exported into reference genome PLUS orientation (build hg19) based on HumanOmni2.5-4v1_D.bpm. As the content on this array contains new SNPs that are not present in dbSNP 132 and were not always named according to dbSNP identifiers, we verified that all positions were present and consistent with dbSNP 132. We converted 1000 Genomes Project (19) SNPs (kgp identifiers) to rsIDs by matching chromosome, position and alleles in dbSNP132. We excluded 17 959 1000 Genome Project SNPs that were duplicates of SNPs with rsID identifiers, 11 536 SNPs that had more than two alleles in dbSNP, and 405 516 SNPs that were not present in dbSNP 132. This resulted in a total of 2 016 729 SNPs. Since the sequencing analysis was performed in the hg18 reference, we converted the positions and orientation of the genotyped SNPs from hg19 to hg18 using the LiftOverVCF.pl script within GATK (20). The 2 015 517 SNPs with successful coordinate conversion were used in subsequent analysis.
Calculating concordance between genotyping array and sequencing data
To determine concordance, we used the genotypes of the 2 015 517 SNPs described above and the genotypes called in the sequencing data passing Filter 1.1 (see below) that had at least the indicated coverage (Supplementary Table S4). We calculated the total number of the genotypes (homozygous reference, heterozygous and homozygous alternate) called in the sequencing data that agreed with the genotypes called by the array and divided by the total number of genotypes called in both data sets.
Initial sequence data analysis
Alignment
All raw color-space reads were aligned to the human genome reference sequence (hg18), limited to chromosomes 1–22, X and Y, as well as mitochondrial genome. The alignment was carried out using BFAST v0.6.1c with default masks and parameters, except for −M = 384 and 10 in the match and local alignment steps, respectively, and −K = 100 in the match step (21). We identified reads originating from potential PCR duplicate fragments (referred to as duplicate reads) as mapping to the same location and showing an identical strand orientation and sequence in the first 40 nt. For all duplicate reads, we kept the read with the highest quality score. The reads were then subjected to local realignment using GATK IndelRealigner (20), to improve the detection of insertion–deletions (indel) and remove false positive single nucleotide variants (SNVs) within 200 bp of indels.
Merging of replicates
Two independent libraries were generated and sequenced for both tumor samples 06408 and 02542. The sequences generated from these technical replicates had similar alignment efficiencies and overall quality metrics (Supplementary Table S1) without any obvious bias, thus we merged the BAM files resulting from the alignments and used the consolidated data in the rest of our analysis.
Coverage
The coverage was calculated by using SAMtools v0.1.8-13 (22) ‘pileup’ command and custom perl scripts. The normalized coverage was calculated by dividing the coverage at each base by the average coverage across the genome for each sample (Supplementary Figure S2).
Mismatches
To look for potential DNA damage caused by the formalin fixation process, we analyzed the number of mismatches in the mapped reads (Figure 1A and Supplementary Table S2a). A mismatch is defined as any base substitution within an aligned read. The number of mismatches within realigned reads was calculated by using the MD field in the SAM file format (23) and custom programs. The MD field characterizes the location, number and type of mismatches, a read has with the reference sequence.
Figure 1.

(A) Frequency of mismatches within sequencing reads for germline and FFPE tumor samples. The distribution of reads with 0, 1, 2 or ≥3 mismatches to the reference genome is shown for all sequencing data (All) and a random subset of 50 M, 5 M and 1 M sequencing reads. (B) Read based global nucleotide mismatch rate for all base substitutions. (C) Read based global nucleotide mismatch rate for each substitution type.
Calculation of global nucleotide mismatch rates
We determined the global nucleotide mismatch rate profile for sequencing reads in each tumor sample across all 6 nt substitution types; A·T > C·G, A·T > G·C, A·T > T·A, C·G > A·T, C·G > G·C and C·G > T·A (Figure 2 and Supplementary Table S3). To do this, we investigated a set of high confidence homozygous reference sites, for each patient, derived from a random set of reference loci across the genome. These homozygous reference sites were chosen by first removing all variant positions passing Filter 1.2 in both matched germline and FFPE tumor samples (see below). We then removed all sites that are variant in dbSNP132 and/or the 1000 genomes project. From the remaining homozygous reference loci, we randomly selected four sets of 100 000 A, T, C and G sites that had at least 3× coverage in the sample, making a total of 400 000 random loci selected per sample. In each sample, the expected global nucleotide mismatch rate for each substitution type i → j, 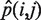 , was then calculated by summing the number of mismatches for a given substitution type and dividing it by the total coverage at the reference site. For example, for the substitution type A·T > C·G, we summed up the number of times we saw an A > C or T > G substitution, and then divided by the total coverage obtained by summing over all 200 000 reference A and T sites.
, was then calculated by summing the number of mismatches for a given substitution type and dividing it by the total coverage at the reference site. For example, for the substitution type A·T > C·G, we summed up the number of times we saw an A > C or T > G substitution, and then divided by the total coverage obtained by summing over all 200 000 reference A and T sites.
Figure 2.

Distribution of substitution types for variants passing Filter 2.1 in germline (G) and FFPE tumor (T) samples and called homozygous alternate (Alt) or heterozygous (Het). Variants identified in public SNP repository (Known) or novel for both patients in this study (Novel) or passing in both germline and FFPE tumor samples (Paired) or only in one sample (Unique) are distinguished. The fraction of novel heterozygous variants (C·G > T·A) called between the tumor and germline samples of patient 02542 is substantially different.
Somatic variant detection procedure
Step1: variant calling
In each sample, we called the variants from the consensus model generated by SAMtools v0.1.8-13 (22) with the following two modifications: (i) to correct for the under calling of homozygous alternate alleles, we set −r to 7.0 × 10−7 (1); and (ii) to scale the mapping quality to the BFAST standard, we set −M option to 255.
Filter 1.1: SAMtools varFilters
Our first filter removes low confidence variants. Variants were filtered using samtools.pl varFilter command with the following parameters: (i) Minimum Root Mean Square of base quality (RMS) set to 43; (ii) Minimum consensus quality set to 20 and (iii) the SNP quality set to 50.
Filter 1.2: Coverage thresholds
We next filtered to remove false positives caused by too low or too high sequence coverage. To obtain the optimal minimum and maximum coverage thresholds for calling variants, we used the set of 2 015 517 loci assayed by the genotyping array to maximize the concordance between the array-based genotype calls and the sequence-based genotype calls, for each patient (both germline and tumor). Due to limited amount of FFPE DNA to carry out genotyping, we compared the tumor FFPE sequencing variant calls to the matched germline array genotypes. The results are presented in Supplementary Table S4a and S4b. We determined that removing positions with <5× and 10× for germline and FFPE tumor samples, respectively, and >100× depth of coverage optimized the concordance while still being able to call somatic variants in ∼80% of the FFPE tumor genomes (Supplementary Table S4a and S4b).
Step 2: identification of somatic variants
We used custom programs to compare the variants called in Step 1 from the germline and FFPE tumor samples for both patients. A variant was called somatic if it passed the following successive filters:
Filter 2.1: High quality in matched germline and tumor samples
This filter removes genomic positions of low quality in either germline or tumor samples. For each subject, we removed the genomic positions that did not pass Filters 1.1 and 1.2 in both germline and tumor samples. This step removes variants that cannot be confidently called somatic due to poor quality or coverage in either sample.
Filters 2.2 and 2.3 below remove potential germline variants.
Filter 2.2: Novel variants
This filter removes previously identified variants present in public databases. We filtered somatic variants in the tumor samples that correspond to known variants present in either dbSNP132 (updated on 18 March 2011) (24) or the 1000 genomes project (updated on July 2010) (19).
Filter 2.3: Somatic variants
This filter removes variants that either are in the germline sample or have supporting reads in the germline sample: (i) all loci called variant in both the FFPE tumor DNA and the matched germline DNA and (ii) all tumor variants for which 2 or more sequence reads carrying the alternate allele are present in the germline data.
Filter 2.4: High supporting read diversity
This filter removes variants with biased read diversity: Duplicate sequencing reads carrying an error can result in false positive calls. Although duplicate reads were initially removed after alignment, here we increase the stringency for reads supporting alternate alleles in candidate somatic variant positions. Filter 2.4 removes candidate somatic variants supported by reads with less than three different start positions.
Filter 2.5: Normal local mismatch rate
This filter removes variants in regions with significantly elevated local mismatch rate (LMR): The accuracy of Next Generation Sequencing data is very sensitive to sequence context (low-complexity, repeats, di/tri-nucleotide composition) as well as composition (percent GC). We empirically estimated the LMR at each somatic variant position (see ‘Testing for elevated LMR method’ section) and tested whether the alternate allele frequency (AAF) supporting the candidate somatic variant was significantly above the expected LMR (Q score). We removed any variant where a Q score was within the 90th percentile of the Q score distribution of a gold-standard set of heterozygous variants.
Filter 2.6: Unbiased global nucleotide mismatch profile
This filter uses the global nucleotide mismatch rates to remove variants supported by significantly biased calls. The formalin fixation introduces a bias in the type of nucleotide substitutions observed (11) (Figure 1C). We used the global nucleotide mismatch rate profiles to distinguish candidate somatic variants from random substitutions that result from the fixation procedure. For each genomic position passing Filter 2.5, we calculated a post hoc P-value of a i → j substitution using the binomial distribution Bin(x, n, 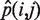 ), where n is the total number of reads covering the position, x is the number of reads with the alternate allele j, and
), where n is the total number of reads covering the position, x is the number of reads with the alternate allele j, and 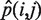 is the global nucleotide mismatch rate (see ‘Calculation of Global Nucleotide Mismatch Rates’ section above) for the given base substitution i → j. We removed all positions where the AAF is not significantly different from the expected global nucleotide mismatch rate using ranked P-values corrected for a false discovery rate (FDR) of 0.05 according to Benjamini and Hochberg (25).
is the global nucleotide mismatch rate (see ‘Calculation of Global Nucleotide Mismatch Rates’ section above) for the given base substitution i → j. We removed all positions where the AAF is not significantly different from the expected global nucleotide mismatch rate using ranked P-values corrected for a false discovery rate (FDR) of 0.05 according to Benjamini and Hochberg (25).
Testing for elevated LMR
For the set of candidate somatic variants passing filter 2.4, we calculated the AAF which is the ratio of alternate allele supporting reads to the total number of reads at that position. We then calculated the LMR from positions 10 bp upstream and 10-bp downstream of the candidate variant position LMR = m/(n + m), where (m) is the number of positions matching the reference and (n) the number of mismatched (excluding the candidate variant position itself). Notably, mismatches include nucleotide substitutions, insertions and deletions. For example, a deletion of 3 bp would result in three mismatch counts. Finally, we inferred a Q score = (AAF − LMR) at each position. We generated a gold standard set of heterozygous variant positions by selecting the 1 229 492 and 986 314 heterozygous SNPs from patient 06408 and 02542, respectively, that were called in the sequencing data and are present in dbSNP132 and/or the 1000 genomes project. We calculated the Q scores of these gold-standard variants in the tumor FFPE DNA and compared their distribution to the candidate somatic variants Q score (Filter 2.5, Supplementary Figure S3).
Estimation of alternate allele under-calling
To estimate the false negative rate in the sequencing data for each sample, we determined the fraction of genotyping array alternate allele sites not called in the sequencing data that passed Filter 2.1. The numerator (alternate allele sites not called) was calculated by summing the number of sites called as AB by the genotyping array and as AA in the sequencing data; plus the sites called as BB by the genotyping and AA or AB in the sequencing data. The denominator (number of possible sites with an alternate allele) was calculated by summing all AB and BB sites in the genotyping array excluding sites that were called missed variant (MV) or missed called (MC) in the sequencing data (Supplementary Table S5).
Annotation of somatic variants
We used the SeattleSeq Annotation server (http://gvs.gs.washington.edu/SeattleSeqAnnotation/) for functional annotation of somatic variants called in FFPE tumor samples 06408 and 02542. To identify genes carrying somatic mutations of potential importance for breast tumor initiation and progression, we downloaded the cancer gene census list, updated 22 March 2011, (26) consisting of 457 genes (27) and created a list of DNA damage repair genes from the Gene Ontology database (28). Briefly, by searching for ‘DNA damage repair’ in the GO terms of ‘Biological process’ we identified 5 GO terms and 1049 genes.
Analysis of Illumina sequencing reads for FFPE DNA damage
We downloaded publicly available Illumina sequence data of 89 FFPE non-small cell lung tumors (29). The sequencing reads were aligned to the human reference genome (hg19) using BWA v5.9 (30) with default parameters, except for a seed length of 25. BWA is more stringent than BFAST in aligning reads that contain mismatches; therefore samples with high FFPE damage are expected to have fewer Illumina reads aligning to the genome. For this reason, to estimate the extent of DNA damage caused by FFPE we calculated the alignment rate and percent of aligned reads with greater than or equal to two mismatches. We used a k-means clustering algorithm on the alignment and mismatch rates to separate the 89 tumor samples into two groups; one group contained 11 samples and the other contained 78 samples.
RESULTS
We sequenced two triple-negative breast cancer tumors (WHEL Study samples 06408 and 02542) and matched patient germline DNA. The tumor samples had been formalin fixed and paraffin wax embedded (FFPE) and stored as 5 µm section for 11 and 16 years, respectively, before DNA was isolated for our study. DNA was isolated from approximately a 1-cm2 area of 85% tumor cellularity containing ∼5.4 × 105 cells from sample 06408 and from approximately a 2-cm2 area of 80% tumor cellularity containing about of 1.3 × 106 cells from sample 02542 (Supplementary Figure S1). We performed technical replicates (DNA isolation, library construction and sequencing) for both tumor samples 06408 and 02542. After read alignment, duplicate reads removal and local realignment, the data resulting from the technical replicates were checked for consistency before being merged into a single data set for further analysis (Supplementary Table S1). This resulted in a coverage depth, respectively, of 13× and 23× for patient 06408 germline and FFPE tumor DNA and 12× and 22× for patient 02542 germline and FFPE tumor DNA (Table 1). The coverage depth distribution across the genome was similar between FFPE tumor and germline samples (Supplementary Figure S2), indicating that the FFPE process did not create any large-scale bias affecting the ability to examine specific intervals of the genome for somatic variants.
Table 1.
Sequencing statistics
| Patient Sample | Sample 06408 |
Sample 02542 |
||
|---|---|---|---|---|
| Germline | FFPE tumor | Germline | FFPE tumor | |
| Raw color-space reads | 1 352 676 084 | 2 823 592 370 | 1 251 754 629 | 3 174 447 825 |
| Fraction of reads aligned to hg18 (%) | 67.2 | 59.3 | 65.8 | 54.5 |
| Fraction of uniquelya aligned readsb (%) | 70 | 63 | 70 | 60 |
| Average haploid coverage (×) | 12.6× | 23.4× | 11.7× | 22.2× |
| Fraction of genome covered (%) | 88 | 89 | 87 | 89 |
| Fraction of genome with ≥3× coverage (%) | 85 | 86 | 81 | 87 |
aReads with only one possible mapping location.
bReads after mapping, duplicate removal, local-realignment and merging technical replicates; excluding chrY.
Characterizing formalin fixation induced DNA damage
The DNA damage caused by the FFPE process is expected to lead to a high number of mismatches in the aligned sequencing reads (11,12) confounding the identification of DNA variants. However, the FFPE damage occurs at different nucleotide positions in different cells of the sample and thus has a random distribution across all DNA sequencing reads. By analyzing the combined signal of mismatches in sequence reads of the FFPE tumor sample, it is possible for the pattern of random FFPE-induced damage to be recognized, and then corrected for in the data analysis. Therefore, in order to comprehensively characterize FFPE induced errors, we analyzed mismatches in each read prior to consensus variant calling. The FFPE tumor DNA showed reduced alignment rates (54–61%) as compared to the germline (66–67%) (Supplementary Table S1). Moreover, the proportion of reads with ≥1 mismatch was greater in both of the FFPE tumor samples (∼32% in 06408 and ∼51% in 02542) when compared to their corresponding germline samples (∼21%) (Figure 1A and Supplementary Table S2). These data are all consistent with formalin fixation induced DNA damage resulting in the FFPE tumor aligned sequence reads having a higher number of mismatches.
Interestingly, FFPE tumor sample 02542 had 1.5 times more reads with ≥1 mismatches than FFPE tumor sample 06408. This greater number of mismatches was consistent across technical replicates (Supplementary Table S2), suggesting that the observation was not an artifact of the DNA isolation or library preparation process but that the extent of DNA damage due to formalin fixation is greater in the FFPE tumor sample 02542. Mismatch distribution differences between the two FFPE tumor samples were apparent by examining a random set of 50 million, 5 million and 1 million non-filtered sequence reads from the germline and FFPE tumor samples of both patients (Figure 1A and Supplementary Table S2). This implies that by sequencing as few as 1 million reads per sample, one can estimate the extent of DNA damage in a FFPE tumor from the mismatch distribution. To further investigate the ability to assess the extent of DNA damage caused by FFPE in low coverage data we downloaded publicly available Illumina sequence reads from 89 FFPE tumors (29); each sample has about 1 million reads. We aligned the sequence reads to the human reference genome and then calculated the fraction of reads that aligned and the mismatch rate of the aligned reads. Of the 89 samples, 11 had poor mismatch and alignment rates suggesting that they have a significant amount of DNA damage from FFPE processing (Supplementary Figure S4). The other 78 samples had moderate to good mismatch and alignment rates suggesting that the FFPE DNA damage was minimal. Overall these results suggest that low-coverage data sets can be used to assess the integrity of the FFPE tumor DNA and thus can serve as an important quality control step before performing costly whole genome sequencing.
We next determined the global nucleotide mismatch rate in the DNA sequencing reads (Figure 1B), as well as the profile of each of the six different types of substitutions (Figure 1C). To estimate the global nucleotide mismatch rate profiles, we focused on four sets of 100 000 sites each called as homozygous reference A, T, C and G in each patient’s germline genome (based on random high confidence reference sites across the genome) and had at least 3× coverage in the matched FFPE tumor. While the global nucleotide mismatch rates were similar in the germline DNA of the two patients (∼11 × 10−3), the global nucleotide mismatch rates in the FFPE samples were substantially higher (1.6 - and 2.9-fold higher than in the germline, for patients 06408 and 02542, respectively). The higher relative global nucleotide mismatch rate in the 02542 FFPE tumor sample compared to the 06408 FFPE tumor sample is consistent with a greater amount of DNA damage. Across the six substitution types, the FFPE tumor samples have a greater global nucleotide mismatch rate than the germline samples (Figure 1C). The increase in the global nucleotide mismatch rate was particularly prominent for C·G > T·A substitutions, which was 1.5 - and 1.8-fold higher than the other substitution types in tumor samples 06408 and 02542, respectively. This is consistent with the types of DNA sequence read mismatches expected to result from formalin induced cross-linking of cytosine nucleotides. The atypical global nucleotide mismatch rate profiles of the FFPE tumor sample suggests that the majority of the DNA sequence read mismatches are due to the formalin fixation process rather than the oncogenic process. Consequently, we used the atypical global nucleotide mismatch rate profiles in the FFPE tumor samples to better distinguish high-confidence somatic variants from formalin fixation induced mismatches (see Filter 2.6 in ‘Materials and Methods’ section and ‘Somatic variant calling and filtering’ section below).
Variant calling and initial quality assessment
As described in ‘Materials and Methods’ section we called variants using SAMtools v0.1.8-13 (22) and then applied two filters to remove low confidence variants (Filter 1.1) and to remove false positive variants caused by genomic regions with too low or too high sequence coverage (Filter 1.2). We used the genotype information obtained from the Illumina Omni 2.5 array analysis of each patient’s germline DNA to assess variant calling performance and optimize additional standard and novel filters. After applying Filters 1.1 and 1.2, we called 84–95% of the array’s SNP positions in all four samples using the sequencing data (Supplementary Table S5). Of note, this estimation of variant detection sensitivity is likely an overestimate as variants analyzed on genotyping arrays are easier to detect using next generation sequencing than variants not amendable to array analysis (31). The genotype concordance between the array and germline variants was 96.9% and 96.8%, respectively, in patients 06408 and 02542. For patient 06408, the corresponding FFPE tumor DNA sample had similar concordance with the genotyping array (96.6%); however for patient 02542 the FFPE tumor DNA sample had lower concordance (92.7%). This higher discordance is primarily the result of under-called alternate alleles, which is more prominent in the 02542 FFPE tumor sample (∼21%) than in the matching germline sample (∼8%) (Supplementary Table S5). For patient 06408, the rate of under-calling alternate alleles was similar between the FFPE tumor (∼9%) and the germline sample (∼8%). A variety of reasons likely underlie this increased under-calling of the alternate allele in the 02542 FFPE tumor sample including biological reasons, such as deletions resulting in loss of heterozygosity.
Because the amount of DNA isolated from the FFPE tumor samples was low, we examined whether or not contaminating DNA was introduced during the library preparation. For both patients, the FFPE tumor variants were more concordant with the genotyping array results of the matched germline sample than with the other patient’s germline sample (93–97% versus 69%, Supplementary Table S6). These data suggest that a contaminating DNA source was not introduced during library preparation as the cross-sample concordance between the germline array genotypes and the FFPE tumor sequence genotypes would have been lower than what we observed and likely have had an expected inflation of heterozygous calls (Supplementary Table S5). Thus, we are confident that we sequenced DNA isolated from the FFPE tumor 5 µm sections.
To characterize the bias in variant calling introduced by the formalin fixation process, we compared variants called in the germline and matched FFPE tumors. In each of the four samples, we identified ∼1.8–2.1 × 106 variants with high sequence quality (Figure 3, passing Filter 2.1). Consistent with the expected findings from the sequencing of a Caucasian individual (20), ∼95% of the germline variants have been previously observed and are in public databases (Figure 3, passing Filter 2.2). The 02542 FFPE tumor sample had a higher number of novel variants (3.8×) than the 06408 FFPE tumor sample or the matched germline samples. These variant data are in alignment with the observed higher global nucleotide mismatch rate suggesting that the 02542 FFPE tumor sample has extended damage from formalin fixation. Additionally, it is important to consider the fact that these higher number of novel variants may partially be due to an increased number of somatic mutations in the 02542 FFPE tumor sample. We also observed a marked difference in the distribution of the six nucleotide substitution types of the variants passing Filter 2.1 in the 02542 FFPE sample as compared to the matched germline and the 06408 FFPE tumor sample (Figure 2). While the variant substitution profiles in the 06408 FFPE tumor DNA is largely similar to that of the matched germline DNA for most categories of substitution types (Figure 2 and Supplementary Table S3), we noted a highly biased profile in the novel heterozygous variants present only in the FFPE tumor DNA of patient 02542; the proportion of C·G > T·A substitutions is 1.9 times higher than that observed in the matched germline and 2.7-fold higher than what is observed in 06408 FFPE tumor DNA. This biased C·G > T·A variant substitution rate is consistent with our previous observation of the increased global nucleotide mismatch rate profiles in the 02542 FFPE tumor (Figure 1C). We note that the transition to transversion ratio of the paired known variants (∼2.2) is close to the expected value (20) whereas heterozygous novel variants that are uniquely present in the tumor samples have a substantially lower value (∼0.8–1.7), indicative of a low-confidence for this latter class of variants (Figure 2).
Figure 3.

Flow diagram describing the number of variants passing each filtering step for both patients 06408 (blue) and 02542 (red).
Somatic variant calling and filtering
Following the above quality assessment, we devised several successive filters to derive a set of high-confidence somatic variant calls. After removing germline variants (Figure 3, Filter 2.3), there are 55 551 and 290 341 candidate somatic variants for tumor samples 06408 and 02542, respectively, which is substantially higher than previous reports in breast cancer (6,32). Despite removal of duplicate sequencing reads after alignment we noticed that a significant proportion of candidate somatic variants were supported by reads with fewer than three different start positions. We believe that the initial filter did not remove all duplicate reads due to the presence of variable insertions and deletions. A more stringent filtering of these duplicate reads (Figure 3, Filter 2.4) resulted in a further reduction in the number of somatic variants.
False positive mutations as well as real cancer somatic mutations are generally expected to be heterozygous in the tumor DNA. To further enhance our detection of high-confidence somatic mutations, we compared the alternate allele read frequency at all somatic variant positions to a standard set of germline heterozygous variants from the same patient. The alternate allele read frequency of germline heterozygous variants had a median of 42%, while the candidate somatic mutations alternate allele read frequency was 20–26% (Supplementary Figure S3A). Upon closer inspection, we noticed that the somatic mutations with relatively low alternate allele read frequencies were frequently located in regions with elevated LMRs, probably resulting from alignment or sequencing artifacts that were not corrected through local realignment (Supplementary Figure S3B). We confirmed that the LMR is higher for somatic variants than the standard set of variants (4.6 versus 3.1 × 10−2 on an average). We filtered the candidate somatic mutations for which the AAF was not significantly higher than the LMR calculated 10-bp upstream and downstream from the position considered (Figure 3, Filter 2.5). After applying this filter, the alternate allele read frequencies and LMRs for somatic mutations in both FFPE tumor samples is closer to the standard set of heterozygous variants (Supplementary Figure S3). This filtering step resulted in 19 750 and 35 733 candidate somatic variants in patient 06408 and 02542, respectively. Finally, Filter 2.6 takes advantage of the biased global nucleotide mismatch rate profiles that we observed in the FFPE tumor DNA sequence reads (Figure 1C) to identify a set of high-confidence somatic variants. Here, we filtered candidate somatic variants for which the alternate allele read frequency is not significantly different from the global nucleotide mismatch rate. This resulted in 19 176 and 22 524 high-confidence somatic variants in sample 06408 and 02542, respectively. Tumor samples are typically heterogeneous composed of a mixed population of different clones. Given that the minimum AAF needed to call a high-confidence somatic variant after applying Filter 2.6 is ∼18–21% with the mean around 32–34%, thus in the best case scenario we would be able to call a heterozygous mutation found in ∼50% of tumor cells in a sample with intra-tumor heterogeneity.
Examining the six substitution types (Figure 4), reveals that this specific filter diminished the C·G > T·A substitution bias characteristic of formalin fixation induced DNA damage and resulted in a distribution of substitution types similar and more balanced for the 02542 FFPE tumor sample. While sample 06408 only had 3% of its candidate somatic variants filtered by Filter 2.6, 37% of candidate somatic variants in sample 02542 were removed (Figure 3). This supports our previous statement that sample 02542 had greater FFPE induced DNA damage causing an increase in the number of false positive somatic variants. On the other hand, both FFPE tumor samples had >50% of their candidate somatic variants filtered by Filter 2.5 which removes false positive variants caused by sequencing and alignment errors.
Figure 4.

Filters 2.5 and 2.6 remove false positive somatic variants due to formalin fixation and other systematic and random errors in the process. Shown is the fraction of substitution types for somatic variants after Filter 2.4, after Filter 2.5 and after Filter 2.6 for 06408 and 02542 FFPE tumors. After Filter 2.6 the novel somatic variants of substitution type C·G > T·A called in 02542 tumor have a similar profile to that observed for novel germline variants in the matched sample (Figure 2).
To further examine the effects of Filters 2.4, 2.5 and 2.6 on the total number of candidate somatic mutations and the distribution of substitution types, we applied these filters in different combinations and determined that all three filters are necessary (Supplementary Figure S6). These results show the importance of Filters 2.4, 2.5 and 2.6 as FFPE tumor samples have increased alignment errors compared to matched germline samples most likely due to both somatic mutations and formalin fixation induced DNA damage. The succession of filters (2.4–2.6) removed ∼65% and 92% of the candidate somatic variants in 06408 and 02542, respectively (Figure 3). In a recently published framework for somatic variant calling proposed by the Broad Institute, 62% of novel variants were filtered (20). The higher fraction of candidate somatic variants filtered in our study is expected, as our goal is to filter out false positive calls due to the formalin fixation induced DNA damage in both FFPE tumors samples.
Somatic coding variation
The final set of high-confidence somatic mutations contained 19 176 and 22 524 variants in tumor samples 06408 and 02542, respectively. Of those, 268 and 423 variants were coding or affect splice sites (Supplementary Figure S5; Supplementary Tables S7 and S8). These numbers are in agreement with previously sequenced whole genomes of breast cancer (6,32), which suggests our filtering process has adequate stringency.
We examined 457 genes from the Cancer Gene Census (27) and 1049 genes involved in DNA damage repair for somatic coding variants. Sample 06408 had 8 high-confidence somatic mutations in 8 genes (1 nonsense and 7 missense) whereas sample 02542 had 16 high-confidence somatic mutations in 16 genes (1 nonsense, 12 missense and 3 coding-synonymous) (Table 2). A number of these changes are of potential biological interest. Both patients carry variation in TP53: sample 06408 carries a heterozygous nonsense mutation in TP53, suggesting the inactivation of one copy of this tumor suppressor gene and sample 02542 carries a somatic missense mutation. Sample 06408 also carries a heterozygous missense mutation in NOTCH1 which has been shown to be a recurring mutation in chronic lymphocyte leukaemia, lung squamous cell carcinoma and breast cancer (5,33,34). The nonsense mutation in TP53 together with the missense mutation in NOTCH1 could be driver mutations for sample 06408’s tumorigenesis. Sample 02542 carries missense mutations in both MLL2 and MLL3 which together were recently found as significantly mutated in 16% of childhood medulloblastoma cases (35).
Table 2.
High-confidence FFPE tumor coding somatic variants within cancer associated genes and/or DNA damage repair genes
| Patient | Gene | NCBI ID | Chr | Position (hg18) | Germline | Tumor | Mutation type | Amino acid change |
|---|---|---|---|---|---|---|---|---|
| 06408 | ATRX | NM_000489 | chrX | 76735852 | A/A | A/C | Missense | L2027R |
| ELN | NM_000501 | chr7 | 73109920 | G/G | G/C | Missense | A458P | |
| KIAA1549 | NM_020910 | chr7 | 138253476 | T/T | T/C | Missense | Q429R | |
| MYH9 | NM_002473 | chr22 | 35040266 | T/T | T/A | Missense | K475M | |
| NOTCH1 | NM_017617 | chr9 | 138520141 | G/G | G/A | Missense | A1343V | |
| NUMA1 | NM_006185 | chr11 | 71417948 | C/C | C/G | Missense | V27L | |
| NUP214 | NM_005085 | chr9 | 132998395 | A/A | A/G | Missense | D270G | |
| TP53 | NM_000546 | chr17 | 7517747 | G/G | G/A | Nonsense | R306STOP | |
| 02542 | AKT1 | NM_001014431 | chr14 | 104312544 | A/A | A/G | Missense | F161L |
| BLM | NM_000057 | chr15 | 89105082 | T/T | T/A | Missense | F492Y | |
| CREBBP | NM_001079846 | chr16 | 3772787 | G/G | G/A | Missense | P453L | |
| EXT1 | NM_000127 | chr8 | 118886256 | G/G | G/T | Missense | D647E | |
| GNA11 | NM_002067 | chr19 | 3070205 | A/A | A/G | Missense | N246S | |
| JARID1A | NM_001042603 | chr12 | 297581 | G/G | G/C | Missense | T950R | |
| LPP | NM_005578 | chr3 | 190066803 | G/G | G/T | Missense | G511V | |
| MLL2 | NM_003482 | chr12 | 47722022 | T/T | T/C | Missense | K2043R | |
| MLL3 | NM_170606 | chr7 | 151504320 | G/G | G/C | Missense | Q3051E | |
| PDGFRA | NM_006206 | chr4 | 54824777 | G/G | G/A | Missense | G185E | |
| RET | NM_020630 | chr10 | 42921884 | G/G | G/T | Missense | G308W | |
| RPN1 | NM_002950 | chr3 | 129823703 | G/G | G/T | Nonsense | C545STOP | |
| RUNX1 | NM_001001890 | chr21 | 35181094 | C/C | C/T | Coding-synonymous | NA | |
| STK11 | NM_000455 | chr19 | 1171708 | G/G | G/A | Coding-synonymous | NA | |
| TP53 | NM_000546 | chr17 | 7519259 | C/C | C/A | Missense | K132N | |
| ZNF521 | NM_015461 | chr18 | 21060818 | C/C | C/G | Coding-synonymous | NA |
DISCUSSION
Genomic translational research faces a scarcity of properly stored and annotated clinical samples. Archived formalin-fixed tissues in paraffin blocks offer a unique opportunity to study thousands of samples with extensive clinical records and follow-up information. In our study, we show that it is possible to obtain enough DNA from a single 5 µm FFPE slide (∼1–2 cm2) to perform whole genome sequencing of sufficient coverage depth to identify potentially important mutations. The FFPE process combined with long storage times is known to result in DNA fragmentation. We show that for the two breast tumor samples analyzed DNA fragmentation did not produce large biases in coverage depth distribution (Supplementary Figure S2). However, we observed a higher global nucleotide mismatch rate within aligned reads from FFPE tumor samples when compared to matched germline (Figure 1A) and a higher base substitution rate across all 6 different substitution types (Figure 1C). Consistent with damage due to formalin fixation, we observed this increase was biased towards C·G > T·A mismatches. Interestingly the two samples studied were differentially affected by the formalin fixation, tumor 02542 showing a 1.8-fold increase in the global nucleotide mismatch rate and greater C·G > T·A bias compared to tumor 06408. This discrepancy can be explained by the absence of strict standards in the formalin fixation step, where tissue samples are routinely fixed between 24 and 48 h (11) but sometimes can be fixed for considerably longer times. The time of the formalin fixation step is not known for the studied samples and not generally included in pathology reports. Another possible explanation could be the size of the tumor tissue, or its density, which also affects the fixation procedure. As formalin fixation-induced DNA damage could potentially be so great as to inhibit the ability to analyze an FFPE sample by next generation sequencing we have established a relatively simple test to assess the integrity of FFPE samples. By simply sequencing from 500 000 to 1 million raw reads from a single FFPE tumor, one can determine the extent of DNA damage and identify the best preserved samples to conduct larger, more expensive whole genome sequencing (Figure 1A and Supplementary Figure S4).
Using a set of innovative filters (Filter 2.4–2.6), we establish a successful method for filtering false positive somatic variants caused by the FFPE damage to the tumor DNA, thus increasing our confidence in the final set of called somatic mutations. It is important to compare our novel filters to existing post-alignment filtering methods such as GATK (20). Existing methods filter for poor base quality with a stringent threshold; this is due to the fact that incorrectly called variants are typically caused by low quality sequence data. The fact that FFPE causes random damage, the ‘errors’ do not have poor base quality. Our method filters on the AAF without using a threshold for all substitution types; but rather it uses a mismatch error rate across the genome of the given sample. This is important as the amount of FFPE DNA damage varies from sample to sample. To achieve the same goal as our novel post-alignment filters, one could propose applying more stringent criteria to align the reads. Aligners that trim the reads when their mismatch rate becomes too high have been implemented (36,37). As a result, the global nucleotide mismatch rate would improve, but at the cost of a reduced effective sequencing coverage depth. Such strategies could also remove bona fide somatic mutations surrounded by extensive DNA damage therefore limiting the sensitivity to call variants. A second potential alternate approach for achieving a set of high-confidence somatic mutations in FFPE samples would be to sequence to greater coverage depth. Since formalin fixation is performed on the resected tumor sample and will generally randomly affect different DNA locations in different cells, elevated global nucleotide mismatch rates in DNA sequencing reads should still lead to accurate variant calls at sufficiently high sequencing coverage depth. In our study, the global nucleotide mismatch rate was indeed higher than the variant calling rate, especially in FFPE tumors (18–32 × 10−3 versus 10–11 × 10−4). In a recent study of whole-exome sequencing of FFPE tumors, 40-fold coverage was insufficient to filter false positives due to formalin fixation DNA damage identified by the substitution profile and discordance with matched frozen tissue (15). Indeed, the authors estimate that 80× coverage is required to obtain accurate variant calling in the presence formalin fixation DNA damage. However, for samples such as 02542 in our study with substantial amounts of formalin fixation induced DNA damage, the coverage depths required to overcome the global nucleotide mismatch rates in the sequencing reads to achieve accurate variant calls could be even greater. Thus, applying our series of standard and novel filters will likely have utility for identifying high-confidence somatic mutations in FFPE tumor samples even when there is relatively low sequence coverage depth.
In our study, we have not analyzed the tumors for somatic events such as chromosomal translocations or large copy number alterations (CNA). Methods developed for this purpose (38–40), rely more on the correct mapping of read pairs than accurate sequence. We have only sequenced single reads, and were thus not able to perform this analysis. We believe that the vast majority of the reads mapped in our FFPE tumor samples are mapped at the correct location. However, it is possible that the sensitivity of translocation or CNA detection would be affected as a greater number of reads might have ambiguous mappings due to the mismatches introduced by the FFPE damage. Various distributions of insert size in read pairs, especially large ones (1–10 kb) obtained through mate-pair libraries, can also improve the sensitivity of the detection of large deletions. However, the FFPE process fragments the DNA and therefore would not be adequate for such studies.
Overall, our study demonstrates that a methodical characterization and analysis of the sequencing data can reduce the noise resulting from formalin fixation induced DNA damage and lead to calling a high-confidence set of somatic mutations. This opens up the possibility of sequencing huge archives of stored clinical FFPE samples of a variety of cancers. Furthermore, we demonstrate that a limited amount of DNA can be used for a genome-wide deep sequencing analysis, which enables studies on small clusters of tumor cells such residual cancer after treatment or dormant metastases.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–8, Supplementary Figures 1–6 and Supplementary Programs.
FUNDING
National Center for Research Resources, Center for Translational Science Award [1UL1RR031980-01]; National Cancer Institute [grants 1R21CA152613-01, 1R21CA155615-01A1, CA69375]; Safeway Foundation, Breast Cancer Research Foundation and University of California Office of the President [grant 6762 Subaward 6067sc to Athena Breast Health Network]. Funding for open access charge: National Institute of Health.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Kersi Pestonjamasp, Ph.D., Manager of the UCSD Cancer Center Microscopy Core, who helped isolate cell nuclei from stained FFPE slides for cell counting. We would like to thank Brian Coullahan from Life Technologies for support and assistance with library preparation and SOLiD sequencing.
REFERENCES
- 1.Clark MJ, Homer N, O’Connor BD, Chen Z, Eskin A, Lee H, Merriman B, Nelson SF. U87MG decoded: the genomic sequence of a cytogenetically aberrant human cancer cell line. PLoS Genet. 2010;6:e1000832. doi: 10.1371/journal.pgen.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, Stinson J, Yue P, Zhang Y, Pant KP, Bhatt D, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010;465:473–477. doi: 10.1038/nature09004. [DOI] [PubMed] [Google Scholar]
- 3.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman C, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, Escaramis G, Jares P, Beà S, González-Díaz M, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 7.Pikor LA, Enfield KS, Cameron H, Lam WL. DNA extraction from paraffin embedded material for genetic and epigenetic analyses. J. Vis. Exp. 2011;4:e2763. doi: 10.3791/2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.April C, Klotzle B, Royce T, Wickham-Garcia E, Boyaniwsky T, Izzo J, Cox D, Jones W, Rubio R, Holton K, et al. Whole-genome gene expression profiling of formalin-fixed, paraffin-embedded tissue samples. PLoS One. 2009;4:e8162. doi: 10.1371/journal.pone.0008162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farragher SM, Tanney A, Kennedy RD, Paul Harkin D. RNA expression analysis from formalin fixed paraffin embedded tissues. Histochem. Cell Biol. 2008;130:435–445. doi: 10.1007/s00418-008-0479-7. [DOI] [PubMed] [Google Scholar]
- 10.Kibriya MG, Jasmine F, Roy S, Paul-Brutus RM, Argos M, Ahsan H. Analyses and interpretation of whole-genome gene expression from formalin-fixed paraffin-embedded tissue: an illustration with breast cancer tissues. BMC Genomics. 2010;11:622. doi: 10.1186/1471-2164-11-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srinivasan M, Sedmak D, Jewell S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am. J. Pathol. 2002;161:1961–1971. doi: 10.1016/S0002-9440(10)64472-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams C, Pontén F, Moberg C, Söderkvist P, Uhlén M, Pontén J, Sitbon G, Lundeberg J. A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am. J. Pathol. 1999;155:1467–1471. doi: 10.1016/S0002-9440(10)65461-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ausch C, Buxhofer-Ausch V, Oberkanins C, Holzer B, Minai-Pour M, Jahn S, Dandachi N, Zeillinger R, Kriegshauser G. Sensitive detection of KRAS mutations in archived formalin-fixed paraffin-embedded tissue using mutant-enriched PCR and reverse-hybridization. J. Mol. Diagn. 2009;11:508–513. doi: 10.2353/jmoldx.2009.090022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Solassol J, Ramos J, Crapez E, Saifi M, Mange A, Vianes E, Lamy PJ, Costes V, Maudelonde T. KRAS mutation detection in paired frozen and formalin-fixed paraffin-embedded (FFPE) colorectal cancer tissues. Int. J. Mol. Sci. 2011;12:3191–3204. doi: 10.3390/ijms12053191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerick M, Isau M, Timmermann B, Sültmann H, Herwig R, Krobitsch S, Schaefer G, Verdorfer I, Bartsch G, Klocker H, et al. Targeted High Throughput Sequencing in Clinical Cancer Settings: Formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med. Genomics. 2011;4:68. doi: 10.1186/1755-8794-4-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schweiger MR, Kerick M, Timmermann B, Albrecht MW, Borodina T, Parkhomchuk D, Zatloukal K, Lehrach H. Genome-wide massively parallel sequencing of formaldehyde fixed-paraffin embedded (FFPE) tumor tissues for copy-number- and mutation-analysis. PLoS One. 2009;4:e5548. doi: 10.1371/journal.pone.0005548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wood HM, Belvedere O, Conway C, Daly C, Chalkley R, Bickerdike M, McKinley C, Egan P, Ross L, Hayward B, et al. Using next-generation sequencing for high resolution multiplex analysis of copy number variation from nanogram quantities of DNA from formalin-fixed paraffin-embedded specimens. Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkq510. e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierce JP, Faerber S, Wright FA, Rock CL, Newman V, Flatt SW, Kealey S, Jones VE, Caan BJ, Gold EB, et al. A randomized trial of the effect of a plant-based dietary pattern on additional breast cancer events and survival: the Women’s Healthy Eating and Living (WHEL) Study. Control Clin. Trials. 2002;23:728–756. doi: 10.1016/s0197-2456(02)00241-6. [DOI] [PubMed] [Google Scholar]
- 19.Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Homer N, Merriman B, Nelson SF. BFAST: an alignment tool for large scale genome resequencing. PLoS One. 2009;4:e7767. doi: 10.1371/journal.pone.0007767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19:1124–1132. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R. Stat. Soc. B. Met. 1995;57:289–300. [Google Scholar]
- 26.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordóñez GR, Bignell GR, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belvedere O, Berri S, Chalkley R, Conway C, Barbone F, Pisa F, MacLennan K, Daly C, Alsop M, Morgan J, et al. A computational index derived from whole-genome copy number analysis is a novel tool for prognosis in early stage lung squamous cell carcinoma. Genomics. 2012;99:18–24. doi: 10.1016/j.ygeno.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harismendy O, Ng PC, Strausberg RL, Wang X, Stockwell TB, Beeson KY, Schork NJ, Murray SS, Topol EJ, Levy S, et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiao X, Wood LD, Lindman M, Jones S, Buckhaults P, Polyak K, Sukumar S, Carter H, Kim D, Karchin R, et al. Somatic mutations in the notch, NF-KB, PIK3CA, and hedgehog pathways in human breast cancers. Genes Chromosomes Cancer. 2012;51:480–489. doi: 10.1002/gcc.21935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, Leigh IM, Collisson EA, Gordon PB, Jakkula L, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl Acad. Sci. USA. 2011;108:17761–17766. doi: 10.1073/pnas.1114669108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.David M, Dzamba M, Lister D, Ilie L, Brudno M. SHRiMP2: sensitive yet practical SHort Read Mapping. Bioinformatics. 2011;27:1011–1012. doi: 10.1093/bioinformatics/btr046. [DOI] [PubMed] [Google Scholar]
- 37.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medvedev P, Fiume M, Dzamba M, Smith T, Brudno M. Detecting copy number variation with mated short reads. Genome Res. 2010;20:1613–1622. doi: 10.1101/gr.106344.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koehler R, Issac H, Cloonan N, Grimmond SM. The uniqueome: a mappability resource for short-tag sequencing. Bioinformatics. 2011;27:272–274. doi: 10.1093/bioinformatics/btq640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.