


Abstract
MicroRNAs are master gene regulators that can also be under the control of transcriptional regulation. We have previously shown that miR-145 is a tumor suppressor capable of silencing c-Myc and the tumor suppressor p53 induces miR-145 by directly binding to the miR-145 promoter, demonstrating the role of miR-145 in p53-mediated c-Myc repression. However, little is known as to why miR-145 is often downregulated in tumors. In this study, we identify CCAAT/enhancer binding protein beta (C/EBP-β) as a negative regulator for miR-145 expression by direct interaction with the putative C/EBP-β binding site in the miR-145 promoter. In the wild-type p53 background, C/EBP-β counteracts the ability of p53 to induce miR-145. Moreover, C/EBP-β is able to suppress miR-145 in the mutant p53 background, suggesting the p53 independent regulation of miR-145. Of interest, both the large isoform (LAP-2) and the small isoform (LIP) of C/EBP-β can exert suppressive function for miR-145. Finally, we further show that, like serum starvation and PI3K inhibitor LY29, the antioxidant resveratrol suppresses pAkt and phosphorylation of C/EBP-β and at the same time, it induces miR-145. Together, these results suggest a miR-145 regulatory system involving the Akt and C/EBP-β, which may contribute to the downregulation of miR-145 in cancer cells.
INTRODUCTION
The role of microRNAs in human malignancy has been intensively investigated (1). It becomes evident now that microRNAs can function as tumor suppressors or oncogenes and they are often dysregulated in tumors. In this regard, oncogenic microRNAs are frequently upregulated, whereas tumor suppressive microRNAs are downregulated in tumors. For instance, let-7 has been reported to be underexpressed in lung cancer and to target the oncogenic Ras (2); similarly, miR-15/miR-16 has been shown to be downregulated in chronic lymphocytic leukemia (3) and is able to target Bcl-2. In contrast, oncogenic microRNAs such as miR-21 are upregulated in variety of tumors (4–7). miR-145 is a tumor suppressive microRNA that is underexpressed in several types of tumors (8–10) and it suppresses cell growth and invasion by targeting a number of important genes such as c-Myc (11), IRS-1 (12) and mucin 1 (13) and others (14,15). Furthermore, miR-145 is able to target the pluripotency factors OCT4, SOX2 and KLF4 and functions as a key regulator of human embryonic stem cells (16) or promotes differentiation and repressing proliferation of smooth muscle cells (17), highlighting the significance of miR-145 as a key regulator of these biological events.
We have previously shown that miR-145 is a direct target for p53 that binds to the miR-145 promoter and transcriptionally induces its expression. Although several transcriptional factors such as Foxo (18) and RREB1 (19), in addition to p53, have been implicated in the regulation of miR-145, it is still unclear as to why miR-145 is frequently downregulated in many types of tumors, including those carrying a mutant p53. CCAAT/enhancer-binding protein-beta (C/EBP-β) is a transcription factor and plays a critical role in cell growth and differentiation. The importance of C/EBP-β also stems from the findings that it serves a mediator of cell survival and tumorigenesis. Three isoforms of C/EBP-β can be expressed in cells through alternative translation of the C/EBP-β mRNA (20). Evidence suggests that they can either activate transcription or represses transcription (21). However, their role in regulation of miR-145 expression has not been described yet.
In this study, we show that C/EBP-β functions as a negative regulator of miR-145. More importantly, C/EBP-β is not only able to counter the ability of p53 to induce miR-145 in the wild-type p53 background, but also suppress miR-145 expression in cancer cells carrying mutant p53 possibly through the Akt pathway.
MATERIALS AND METHODS
Cell culture
All cell lines were purchased from American Tissue Culture Collection (ATCC). Breast cancer cell lines BT-549, MDA-MB-231 and MCF-7 cells were grown in RPMI 1640 (Lonza, Walkersville, MD, USA) supplemented with 10% FBS (Sigma-Aldrich). Non-tumorigenic breast cell MCF-10A was grown in serum-free MEGM medium (Lonza). HEK-293T cells were cultured in DMEM (Lonza) supplemented with 10% FBS. All serum containing media were supplemented with 100 U of penicillin/ml and 100 µg of streptomycin/ml. Cells were incubated at 37°C and supplemented with 5% CO2 in the humidified chamber.
Reagents
Primary antibodies were purchased from the following vendors: C/EBP-β, p53 (C-terminal from Epitomics), Akt, p-Akt, p-C/EBP-β for western (Cell Signaling), p-C/EBP-β for immunocytochemistry (ICC) from Epitomics (Burlingame, CA, USA); Myc-tag from Applied Biological Materials (Vancouver, BC, Canada). Secondary antibodies conjugated with IRDye 800CW or IRDye 680 were purchased from LI-COR Biosciences (Lincoln, NE, USA). PCR primers were purchased from IDT (Coralville, IA, USA). C/EBP-β siRNAs and p53 siRNAs were from ThermoFisher Scientific and Cell Signaling, respectively. Resveratrol (RSV) was purchased from Sigma (St Louis, MO, USA). Biotin-labeled anti-miR-145-LNA probe was from Exiqon (Denmark).
Transfection
DNAfectin (Applied Biological Materials) was used for the transfection of plasmid DNA. Transfection with siRNAs was performed using RNAfectin reagent (Applied Biological Materials) following the manufacturer’s protocol.
Expression vectors
Sequences of all primers for cloning were listed in Supplementary Table S1. For ectopic expression, we cloned C/EBP-β, c-Jun and c-Fos, respectively, into a modified pCDH-CMV-copGFP (System Biosciences) which carried an N-terminal Myc tag using the Cold Fusion cloning kit (System Biosciences). For easy monitoring under a fluorescence microscope, we also cloned different isoforms of C/EBP-β into an expression vector carrying red fluorescent protein (RFP). PCR primers for LAP-2 were LAP2-Myc-R1-5.1 (sense) and LAP2-IRES-RED-3.1 (antisense); and primers for LIP were LIP-Myc-R1-5.1 (sense) and LAP2-IRES-RED-3.1 (anti-sense). Primers for p53 were p53-Myc-R1-5.1 (sense) and p53-IRES-RED-3.1 (antisense). The luciferase reporter containing the putative miR-145 promoter in pGL3 basic vector (Promega, Madison, WI, USA) was previously described (11). At the same time, the same fragment was also cloned into pGZ m CMV reporter (System Biosciences) by replacing the CMV promoter at Cla1 and BamH1 sites by standard PCR amplification and ligation methods to generate a miR-145 promoter GFP reporter. Primers used were miR-145p-Cla1-5.1 (sense) and miR-145p-BamH1-3.2 (antisense). A mutant luciferase reporter in the potential C/EBP-β binding site was generated by two-step PCR, followed by the procedure described previously (4), using primers miR-145p-Kpn1-5.1 (sense) (11) and miR-145p-CEPBm-3.1 (antisense); miR-145p-CEPBm-5.1(sense) and miR-145p-Xho1-3.2 (11). All PCR products were verified by DNA sequencing.
Luciferase assays
Luciferase assays were carried out in 293T cells. First, cells were transfected with appropriate plasmids or siRNAs in 12-well plates. Then they were harvested and lysed for luciferase assays 24 h after transfection. Luciferase assays were performed using a luciferase assay kit (Promega) according to the manufacturer’s protocol. Renilla luciferase was used for normalization.
PCR/RT–PCR and real-time RT–PCR
PCRs were performed to amplify the different isoforms of C/EBP-β, miR-145 promoter and deletion and mutation constructs according to the standard three-step procedure. Expression of the mature miR-145 was determined by TaqMan real-time RT–PCR (22).
Western blot
Cells were harvested and protein was extracted from transfected or infected cells by standard methods, as previously described (4,11).
Immunocytochemistry
Immunocytochemistry (ICC) was used to detect C/EBP-β induction by RSV in cells grown on the coated coverslips as described previously (13). After RSV treatment, the cells were first fixed and then permeabilized by 1% Triton-100 according to the standard methods. Signals were revealed using Histostain Plus kit (Invitrogen) according to the manufacturer’s instruction.
In situ hybridization
Detection of miR-145 in MDA-MB-231 cells after treatment of RSV was essentially same as described previously (23) except that biotin-labeled anti-miR-145-LNA probe was used in these assays.
Chromatin immunoprecipitation assays
Procedure for chromatin immunoprecipitation (ChIP) assays was previously described (11). Primers miR145-ChIP-5.3 and miR145-ChIP-3.3 (P1 and P2) covered the potential C/EBP-β binding site of miR-145 promoter as indicated in Figure 6E. Primers miR145-ChIP-5.3 and miR145-ChIP-3.3 (P3 and P4) served as a negative control.
Figure 6.

Suppression of miR-145 by Lap-2 involves DNA binding domain. (A) Schematic description of domain structure of C/EBP-β. DB: DNA binding domain; DD: dimerization domain. (B) Effect of Lap-2 deletion on the miR-145 promoter luciferase activity. The 293T cells were co-transfected with the miR-145 promoter luciferase reporter and Lap-2-WT or deleted Lap-2 expression vectors (Lap-2-del or Lap-2-del1). Luciferase assays were performed 24 h after transfection. (C) Effect of Lap-2 deletion on expression of the endogenous miR-145. MDA-MB-231 cells were infected with Lap-2-WT, Lap-2-del or Lap-2-del1. RNA was extracted from the infected cells for detection of miR-145 by real-time RT–PCR. (D) C/EBP-β directly interacts with the miR-145 promoter in MDA-MB-231 cells as detected by ChIP assays, following the previously described procedure (11). Values in (B) and (C) are mean ± SE, n = 3. **P < 0.01 compared with vector control.
RESULTS
Expression of miR-145 in response to RSV
We (11) and others (24) have previously shown that the DNA-damaging agent doxorubicin (doxo) activates p53 which in turn induces miR-145 expression. However, the mutant p53 at the DNA binding domain has no such effect on miR-145 (11). We found here that miR-145 was not always negatively correlated with p53 status. For example, although both non-tumorigenic MCF-10A and cancer cell line MCF-7 carry wild-type p53, with a different level of expression (Supplementary Figure S1A), the miR-145 level was very different between them (Supplementary Figure S1B), suggesting that factor(s) other than p53 is also involved in miR-145 regulation. Therefore, we tested three breast cancer cell lines for their response to resveratrol (RSV), a well-known chemoprevention and therapeutic agent (25,26). MCF-7 is non-invasive and carries wild-type p53, whereas MDA-MB-231 and BT-549 are invasive, and carry mutant p53 at the DNA binding domain (27). Since p53 has been implicated in the RSV-induced cellular effect (20,28), we asked whether RSV is able to induce miR-145 expression depending on p53 status. As shown in Figure 1A, although there was little induction of p53 in MDA-MB-231 or BT-549 cells at 30 µM of RSV, we detected a significant upregulation of miR-145 in both MDA-MB-231 and BT-549 cells (Figure 1B). For MCF-7 cells, neither p53 activation nor miR-145 induction was seen until the concentration of RSV was increased to 100 µM (Figure 1A and B). We further confirmed the RSV-mediated upregulation of miR-145 in MDA-MB-231 cells by in situ hybridization (ISH; Figure 1C). These results suggest that RSV can induce miR-145 expression in a p53-dependent as well as p53-independent manner.
Figure 1.
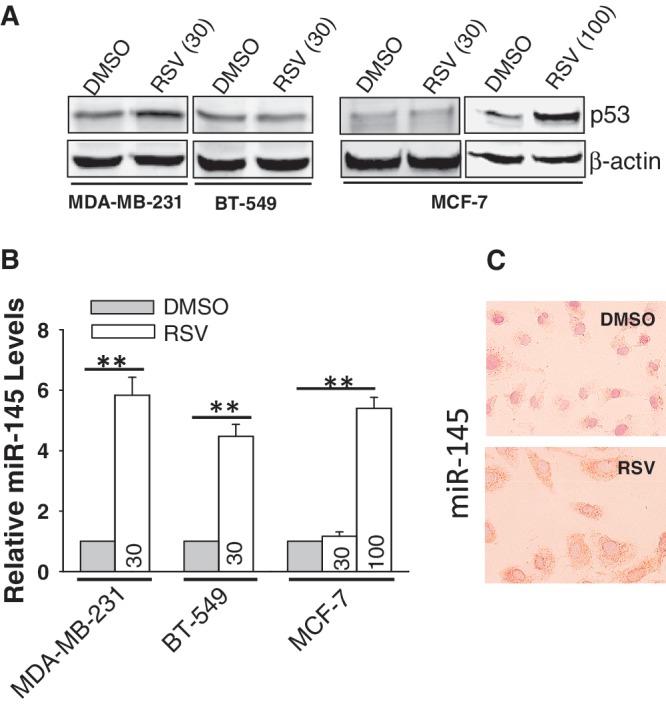
Induction of miR-145 by resveratrol (RSV). (A) Expression of p53 in MCF-10A, 231 and BT-549, MCF-7 and T47D by western. (B) RSV at 30 µM induced miR-145 in MDA-MB-231 and BT-549 cells, respectively. However, in MCF-7 cells the same concentration of RSV did not induce miR-145 until the concentration was increased to 100 µM. Cells were treated with RSV for 24 h before RNA extraction, followed by real time RT–PCR. (C) In situ hybridization also detected an increase of miR-145 after RSV treatment (30 µM) in MDA-MB-231 cells. Values in (B) are mean ± SE, n = 3. **P < 0.01 compared with vector control.
Since MDA-MB-231 cells carry a mutant p53 at the DNA binding domain (27), we asked whether this mutant p53 is still functional to induce miR-145 expression in response to RSV, compared with doxo treatment. Although both RSV and doxo caused upregulation of p53 (Figure 2A), we only detected miR-145 upregulation in RSV-treated cells (over 5-fold) but not in the doxo-treated cells (Figure 2B). These results suggest that the mutant p53 plays no role in miR-145 expression in MDA-MB-231 cells. To further define the role of p53 in the regulation of miR-145 expression in response to RSV, we suppressed p53 by RNAi. Both siRNA-1 and -2 suppressed p53 in MCF-7 and MDA-MB-231 cells (Supplementary Figure S2A) as well as some other cell lines (Supplementary Figure S2B). Of interest, knockdown of p53 was also detected in cells treated with RSV (Supplementary Figure S3). Although RSV-induced p53 in both MDA-MB-231 (30 µM) and MCF-7 (100 µM), p53-siRNA caused a substantial reduction of RSV-mediated miR-145 induction in only MDA-MB-231 cells (Figure 2C), implying that wild-type p53 is critical to this induction of miR-145. In contrast, the same p53 siRNAs did not affect the RSV-induced miR-145 in MDA-MB-231 cells (Figure 2C), further supporting the notion that factor(s) other than p53 may also be involved in the regulation of miR-145 expression.
Figure 2.

Induction of miR-145 in a p53 independent manner by RSV. (A) Expression of p53 in response to RSV and doxo. MDA-MB-231 cells were treated with either RSV (30 µM) or doxo (1 µg/ml) for 24 h and then harvested for protein extraction and western. (B) Detection of miR-145 by real-time RT–PCR after treatment of RSV or doxo. The same treatment was done as in (A) before extraction of total RNA. (C) Effect of suppression of p53 on the RSV-mediated induction of miR-145 as detected by real-time RT–PCR. Cells were first transfected with control siRNA (Ctrl) or p53-siRNA and were subsequently treated with RSV (30 µM for MDA-MB-231; 100 µM for MCF-7). RNA was extracted 24 h after RSV treatment. Values in (B) and (C) are mean ± SE, n = 3. **P < 0.01 compared with vector control.
Suppression of miR-145 by C/EBP-β
In light of these findings, we searched for factors that might be responsible for the regulation of miR-145 expression. Based on bioinformatics analysis using the Genomatix MatInspector (http://www.genomatix.de), there are several putative transcription factors that may bind to the miR-145 promoter (Figure 3A). For example, in addition to previously demonstrated p53, AP-1 and C/EBP-β could potentially regulate miR-145. Therefore, we generated two miR-145 promoter reporters carrying either luciferase (pMir-145p-Luc) or GFP (pMir-145p-GFP). Experiments with ectopic expression of c-Jun + c-Fos (binding to the AP-1 site), or C/EBP-β along with these reporters showed that only C/EBP-β was able to suppress the miR-145 promoter activity for both reporters (Figure 3B and C). Consistent with these results, real-time RT–PCR detected a significant reduction of miR-145 in the cells transfected with C/EBP-β (Figure 3D), suggesting that C/EBP-β is a negative regulator of miR-145.
Figure 3.

Identification of C/EBP-β as a potential negative regulator of miR-145. Experiments were performed in transfected 293T cells. (A) Schematic description of the putative miR-145 promoter with potential transcription factor binding sites and pre-miR-145 start site (an arrow). (B) Luciferase assays using pMir-145p-Luc reporter. (C) GFP expression using pMir-145p-GFP. (D) Effect of C/EBP-β on miR-145, as detected by real-time RT–PCR. Values in (B) and (D) are mean ± SE, n = 3. **P < 0.01 compared with vector control.
Although C/EBP-β is transcribed as a single mRNA, it can be translated into three isoforms from alterative translation initiation sites, two large isoforms (LAP-1, a full-length protein, and LAP-2) and a small isoform LIP (21,29) (Figure 4A). Different names have been used to describe these isoforms (30); they may have distinctive functions as a transcription activator or repressor. To delineate which isoform(s) is responsible for the suppression of miR-145, we first examined the endogenous level of C/EBP-β isoforms. As shown in Figure 4B, we detected a predominant LIP isoform in MCF-10A, MDA-MB-231 and BT-549 cells, but this form was barely seen in MCF-7 cells. On the other hand, although the level of LAP-2 is relatively low compared with LIP, this band was readily detectable in these cells except for MCF-10A cells. No LAP-1 was detected in any of them, consistent with the previous finding (30). Of note, the phosphorylation at Thr 235 has been shown to activate the transcription of C/EBP-β (27); this band was very weak in MCF-10A cells, compared with that in other three cancer cell lines (Figure 4B, top), which may highlight the importance of phosphorylation of C/EBP-β in cancer cells. Therefore, LAP-2 and LIP were further investigated for their role in miR-145 expression in this study. We first ectopically expressed LAP-2 and LIP, respectively, and then tested the effect of each of them on suppression of the miR-145 promoter activity as well as the endogenous miR-145. Although it was reported that the LAP isoforms have active transcription activity whereas the LIP form has repressive activity (21), our study indicated that both LAP-2 and LIP suppressed the promoter activity by ∼60% (Figure 4C). To further determine the role of C/EBP-β isoforms in the suppression of miR-145, we knocked down both isoforms in MDA-MB-231 and BT-549 cells (Figure 4D), and demonstrated that this knockdown increased miR-145 expression as well as miR-145 promoter activity (Figure 4E and F). These results provide further evidence that C/EBP-β is a negative regulator of miR-145.
Figure 4.

Both Lap-2 and Lip isoforms of C/EBP-β suppress the endogenous miR-145 and the miR-145 promoter activity. (A) Schematic description of Lap-1, Lap-2 and Lip with their predicted molecular weights. DB: DNA binding domain; DD: dimerization domain. (B) Expression of C/EBP-β isoforms in various cell lines. Note that only Lap-2 and Lip were detectable in these cells by western. (C) Effect of Lap-2 and Lip on miR-145. MDA-MB-231 cells were infected with vector alone or Lap or Lip expression vector. RNA from these cells was extracted and real-time RT–PCR was performed as described in ‘Materials and Methods’ section. (D) Suppression of Lap-2 and Lip by C/EBP-β-siRNAs in MDA-MB-231 and BT-549 cells. Cells were transfected with negative control (Ctrl) or C/EBP-β siRNAs as described in ‘Materials and Methods’ section; 24 h after transfection, the cells were harvested for western. (E) Effect of C/EBP-β siRNAs on miR-145 as detected by real-time RT–PCR. The same cells as in (D) were used for RNA extraction. (F) Effect of C/EBP-β siRNAs on promoter activity. 293T cells were first transfected with the miR-145 promoter luciferase reporter and subsequently with C/EBP-β siRNAs. The cells were extracted for luciferase assays 24 h after transfection. Values in (C), (E) and (F) are mean ± SE, n = 3. **P < 0.01; *P < 0.05 compared with vector control.
C/EBP-β represses the p53-mediated induction of miR-145
To determine the role of C/EBP-β in the suppression of miR-145 in relation to p53, we first examined their effect on the miR-145 promoter activity. As in the case for the endogenous miR-145, both LAP-2 and LIP suppressed the miR-145 promoter activity (Figure 5A). In contrast, p53 induced the miR-145 promoter activity by about a 5-fold. This finding was further supported by the miR-145 promoter GFP reporter (Figure 5B). Moreover, suppression of C/EBP-β by RNAi caused the upregulation of miR-145 promoter activity, especially with co-transfection of p53 (Figure 5C), suggesting that C/EBP-β can counteract with the ability of p53 to induce miR-145. This result is consistent with the previous finding that C/EBP-β can functionally interact with and suppress p53 activity (31). We then generated a miR-145 promoter reporter containing mutations in the C/EBP-β binding site (miR-145p-m). Mutations of the C/EBP-β binding site caused further upregulation of miR-145 promoter activity, i.e. over four relative units higher than that of miR-145p-WT in the presence of p53 (Figure 5D). To further determine the role of C/EBP-β in the suppression of p53-mediated miR-145 promoter activity, we transfected cells with p53 along various amounts of LAP-2 expression vector and demonstrated that an equal amount of LAP-2 expression vector was sufficient to suppress its ability to induce miR-145 promoter activity (Figure 5E), consistent with the previous report that there is a negative cross-talk between p53 and C/EBP-β (32). Thus, this negative cross-talk between p53 and C/EBP-β may ultimately affect the expression of their targeted genes such as miR-145.
Figure 5.

Effect of C/EBP-β on the p53-mediated induction of miR-145 promoter activity. (A) While miR-145 promoter luciferase activity is suppressed by Lap-2 and Lip, p53 induces the luciferase activity. The 293T cells were co-transfected with both the luciferase reporter (pMir-145p-Luc) along with Lap-2 or Lip, or p53 expression vector. Luciferase assays were carried out 24 after transfection. (B) Detection of the effect of C/EBP-β and p53 on the miR-145 promoter activity by the GFP reporter (pMir-145p-GFP). The 293T cells were transfected with pMir-145p-GFP along with Lap-2, Lip or p53 and were examined under a fluorescence microscope 24 h after transfection. The RFP carrying vector was used to ectopically express Lap-2, Lip and p53, respectively. GFP pictures were taken at the same exposure time for all of them. Decreases or increases in GFP intensity represent reductions or inductions of miR-145 promoter activity. (C) Effect of C/EBP-β siRNAs on luciferase activity combined with ectopic expression of p53. 293T cells were co-transfected with the luciferase reporter and p53 expression vector, and subsequently transfected with negative control or C/EBP-β siRNAs. Luciferase assays were performed 24 h after transfection of siRNAs. (D) The C/EBP-β binding site of the miR-145 promoter is important for C/EBP-β to suppress miR-145. The 293T cells were co-transfected with a luciferase reporter (miR-145p-WT or miR-145p-m) along with expression vectors as indicated. Luciferase assays were performed 24 h after transfection. (E) Lap-2 represses p53-mediated induction of miR-145 promoter activity. The 293T cells were co-transfected with the miR-145 promoter luciferase reporter and p53 expression vector along with various amounts of Lap-2 expression vector. Values in (A), (C), (D) and (E) are mean ± SE, n = 3. **P < 0.01 compared with vector control.
CEBP-β directly binds to the CEBP-β binding site in the miR-145 promoter
These results above indicate that LIP is sufficient to suppress p53-mediated miR-145 induction. Since LIP consists of two domains, DNA binding domain and dimerization domain, we asked which domain is required for this suppression by deletion of either two domains (LAP-2-del) or dimerization domain alone (LAP-2-del1; Figure 6A). Deletion of both domains completely abolished the suppression activity as detected by the miR-145 promoter luciferase reporter (Figure 6B) as well as by the miR-145 prompter GFP reporter (Supplementary Figure S4). On the other hand, deletion of dimerization domain partially impaired this suppression activity (Figure 6B). Similarly, real-time RT–PCR also indicated that the DNA binding domain is critical (Figure 6C). Finally, ChIP assays confirmed that C/EBP-β directly interacted with the miR-145 promoter (Figure 6D).
RSV suppresses phosphorylation of C/EBP-β through Akt pathway
We have previously shown that miR-145 is a downstream target of Akt; suppression of Akt by serum starvation or PI3-K inhibitor LY29 induces miR-145 expression in a p53-dependent manner (11). However, this study showed that RSV was able to induce miR-145 in breast cancer cells carrying mutant p53 (Figure 1B). Moreover, RSV has been previously shown to suppress Akt activity (33). Therefore, we determined whether Akt is a molecular link underlying the RSV-induced miR-145 expression in tumor cells carrying mutant p53. We first examined the constitutive levels of Akt and pAkt in MDA-MB-231, BT-549 and MCF-7 cells; BT-549 cells expressed a relatively higher level of pAkt than the other two cell lines (Figure 7A). As expected, RSV caused downregulation of pAkt in BT-549 and MDA-MB-231 cells (Figure 7B); however, no obvious reduction was seen in MCF-7 cells in 50 or 100 µM. Importantly, we detected downregulation of p-LAP-2 in both MDA-MB-231 and BT-549 cells; this reduction in MCF-7 cells (Figure 7B, right) was not as obvious as in the other two cell lines (Figure 7B, left). Immunocytochemistry also showed downregulation of p-C/EBP-β by RSV in MDA-MB-231 cells (Figure 7C). Since c-Myc is a major target for miR-145 (11), which may in part account for miR-145 as a tumor suppressor, we also examined the effect of RSV on Myc expression. As expected, RSV reduced c-Myc level, associated with downregulation of p-C/EBP-β (Figure 7D), further suggesting the role of p-C/EBP-β in suppression of miR-145. Finally, ChIP assays indicated that both LY29 and RSV suppressed C/EBP-β binding to the miR-145 promoter (Figure 7E). Together, these results suggest that RSV induces miR-145 through suppression of pAkt and phosphorylation of C/EBP-β in the mutant p53 background.
Figure 7.

Suppression of phosphorylated Akt (p-Akt) and phosphorylated Lap-2 (p-Lap-2) by RSV. (A) Expression of Akt and pAkt in MDA-MB-231, BT-549 and MCF-7 cells. (B) RSV suppresses both p-Akt and p-C/EBP-β in MDA-MB-231, BT-549 and MCF-7 cells. (C) Induction of p-C/EBP-β by RSV in MDA-MB-231 cells as detected by ICC. Cells were treated with RSV at 30 µM for 24 h and then were subject to ICC analysis. (D) RSV suppresses the miR-145 target c-Myc in MDA-MB-231 and BT-549 cells, which is associated with suppression of p-Lap-2. (E) Both RSV and LY29 reduce the binding of C/EBP-β to the miR-145 promoter in MDA-MB-231 cells as detected by ChIP assays. Note that the PCR band in C/EBP-β lane was decreased in both RVS and LY29 treatment, compared with DMSO.
DISCUSSION
It is well known that microRNAs can play a critical role in human malignancy. As a tumor suppressor, miR-145 is able to suppress tumor growth and metastasis by targeting a number of oncogenic genes (11–13). Moreover, miR-145 has been implicated in repression of pluripotency in human embryonic stem cells by negative regulation of key transcription factors such as OCT4, SOX2 and KLF4 (34). In embryonic stem cells, miR-145 is expressed at a low level and then is elevated in differentiated epithelial cells and becomes downregulated again in neoplastic cells. However, the underlying mechanism of miR-145 regulation, especially in cancer, is elusive. We present evidence that C/EBP-β is a negative regulator of miR-145 which may be in part responsible for downregulation of miR-145 in tumor cells.
As a transcriptional repressor, C/EBP-β has been shown to transcriptionally repress a variety of genes including let-7i (35). Thus, identification of C/EBP-β as a negative regulator of miR-145 expands the growing list of C/EBP-β-regulated genes. More importantly, we show that phosphorylation of C/EBP-β is critical to this negative effect on miR-145. In this regard, we identify Akt as a potential upstream regulator of CEBP-β, as supported by several lines of evidence. First, activation of Akt correlates with phosphorylation of C/EBP-β; second, this positive correlation has been previously reported in clinical specimens and third, RSV induces miR-145 and at the same time, it decreases pAkt as well as phosphorylation of C/EBP-β. Therefore, these results suggest a possible pathway leading to miR-145 induction through C/EBP-β.
The ability of C/EBP-β to suppress miR-145 appears to function through counteraction of the ability of p53 to induce miR-145 in the wild-type p53 background. We show that p53 increases the endogenous miR-145 level as well as the miR-145 promoter activity; on the other hand, ectopic expression of C/EBP-β suppressed the p53-mediated induction of miR-145 and the miR-145 promoter activity. It is known that p53 interacts with C/EBP-β, and such interaction is functional because C/EBP-β has been shown to represses p53 to promote cell survival (36) or subsequently suppress p53 downstream genes (37). Thus, our study provides further evidence that miR-145 is under the control of a complex regulatory system involving p53 and C/EBP-β. The dysregulation of p53-C/EBP-β cross-talk is expected to play an important role in tumorigenesis. For example, enhanced expression of C/EBP-β may antagonize the tumor-suppressive role of p53. On the other hand, an increased level of p53 would keep the tumor-promoting role of C/EBP-β in check.
It remains to be determined regarding the mechanism by which C/EBP-β can suppress miR-145 in the mutant p53 background. One possibility is that C/EBP-β may act alone or counteract with other unknown activator(s) to suppress miR-145 expression. In support of this possibility, it has been shown that C/EBP-β itself can play a repressive role (38,39), and thus, we expect that increased levels of C/EBP-β would suppress its targeted genes such as miR-145, because C/EBP-β is frequently upregulated in cancer cells (40–42). Although three isoforms of C/EBP-β might be formed, the cell lines we tested only express LAP-2 and/or LIP. LIP is considered to be a dominant-negative regulator of the large isoforms because of the lack of a transactivation domain. Our study further suggests that both LAP-2 and LIP, in fact, play the same suppressive role for miR-145 expression. This finding is also supported by two reports that the large isoforms not only function as a transcriptional activator but can also act as a transcriptional repressor to inhibit gene transcription, such as PPARβ and Cox-2 (38,43). The C-terminal region of C/EBP-β carries the DNA binding domain and the dimerization domain. Deletion analysis indicates that the DNA domain of C/EBP-β is critical to its suppression of miR-145. ChIP assays further suggest that C/EBP-β directly interacts with the DNA region through the C/EBP-β binding site, implying that binding to the C/EBP-β site in the miR-145 may be essential for this suppression ability.
As a well-known antioxidant, RSV has been reported to extend lifespan in cell culture (44). Moreover, RSV is able to inhibit the development of cancer (25). The cancer preventative activity of RSV is believed to be related to its ability to mediate anti-inflammatory effect or inhibit enzymes involved in carcinogenesis (26). Despite of these efforts, our understanding of how RSV target cellular pathways leading to suppression of tumor cells is still limited. Our study provides new insight into the function of RSV against cancer. We demonstrate that RSV induces miR-145 in both p53 wild-type and mutant p53 breast cancer cell lines. In particular, in the mutant p53 background, this is likely through suppression of the Akt pathway and reduction of phosphorylation of C/EBP-β. Of considerable interest, invasive cancer cell lines MDA-MB-231 and BT-549 appeared to be more sensitive to RSV for its induction of miR-145. Since miR-145, as a tumor suppressor, is often downregulated in a variety of tumors, identification of miR-145 as a target for RSV highlights the significance of RSV as an agent for cancer prevention and therapy.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–4.
FUNDING
Funding for open access charge: National Institutes of Health [R01 CA154989]; Susan G. Komen for the Cure [KG100027].
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat. Rev. Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 2.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 3.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl Acad. Sci. USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1) J. Biol. Chem. 2007;282:14328–14336. doi: 10.1074/jbc.M611393200. [DOI] [PubMed] [Google Scholar]
- 5.Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR-21-mediated tumor growth. Oncogene. 2007;26:2799–2803. doi: 10.1038/sj.onc.1210083. [DOI] [PubMed] [Google Scholar]
- 6.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 7.Michael MZ, O' Connor SM, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol. Cancer Res. 2003;1:882–891. [PubMed] [Google Scholar]
- 8.Sempere LF, Christensen M, Silahtaroglu A, Bak M, Heath CV, Schwartz G, Wells W, Kauppinen S, Cole CN. Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res. 2007;67:11612–11620. doi: 10.1158/0008-5472.CAN-07-5019. [DOI] [PubMed] [Google Scholar]
- 9.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 10.Schepeler T, Reinert JT, Ostenfeld MS, Christensen LL, Silahtaroglu AN, Dyrskjot L, Wiuf C, Sorensen FJ, Kruhoffer M, Laurberg S, et al. Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res. 2008;68:6416–6424. doi: 10.1158/0008-5472.CAN-07-6110. [DOI] [PubMed] [Google Scholar]
- 11.Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K, Mo YY. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl Acad. Sci. USA. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi B, Sepp-Lorenzino L, Prisco M, Linsley P, deAngelis T, Baserga R. Micro RNA 145 targets the insulin receptor substrate-1 and inhibits the growth of colon cancer cells. J. Biol. Chem. 2007;282:32582–32590. doi: 10.1074/jbc.M702806200. [DOI] [PubMed] [Google Scholar]
- 13.Sachdeva M, Mo YY. MicroRNA-145 suppresses cell invasion and metastasis by directly targeting mucin 1. Cancer Res. 2010;70:378–387. doi: 10.1158/0008-5472.CAN-09-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang B, Guo H, Zhang Y, Chen L, Ying D, Dong S. MicroRNA-145 regulates chondrogenic differentiation of mesenchymal stem cells by targeting Sox9. PLoS One. 2011;6:e21679. doi: 10.1371/journal.pone.0021679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Q, Liu LZ, Qian X, Chen Q, Jiang Y, Li D, Lai L, Jiang BH. MiR-145 directly targets p70S6K1 in cancer cells to inhibit tumor growth and angiogenesis. Nucleic Acids Res. 2011;40:761–774. doi: 10.1093/nar/gkr730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu T, Wang XY, Gong RG, Li A, Yang S, Cao YT, Wen YM, Wang CM, Yi XZ. The expression profile of microRNAs in a model of 7,12-dimethyl-benz[a]anthrance-induced oral carcinogenesis in Syrian hamster. J. Exp. Clin. Cancer Res. 2009;28:64. doi: 10.1186/1756-9966-28-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gan B, Lim C, Chu G, Hua S, Ding Z, Collins M, Hu J, Jiang S, Fletcher-Sananikone E, Zhuang L, et al. FoxOs enforce a progression checkpoint to constrain mTORC1-activated renal tumorigenesis. Cancer Cell. 2010;18:472–484. doi: 10.1016/j.ccr.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kent OA, Chivukula RR, Mullendore M, Wentzel EA, Feldmann G, Lee KH, Liu S, Leach SD, Maitra A, Mendell JT. Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev. 2010;24:2754–2759. doi: 10.1101/gad.1950610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.She QB, Bode AM, Ma WY, Chen NY, Dong Z. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinases and p38 kinase. Cancer Res. 2001;61:1604–1610. [PubMed] [Google Scholar]
- 21.Descombes P, Schibler U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell. 1991;67:569–579. doi: 10.1016/0092-8674(91)90531-3. [DOI] [PubMed] [Google Scholar]
- 22.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta A, Mo YY. Detection of microRNAs in cultured cells and paraffin-embedded tissue specimens by in situ hybridization. Methods Mol. Biol. 2011;676:73–83. doi: 10.1007/978-1-60761-863-8_6. [DOI] [PubMed] [Google Scholar]
- 24.Kandel ES, Skeen J, Majewski N, Di Cristofano A, Pandolfi PP, Feliciano CS, Gartel A, Hay N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol. Cell Biol. 2002;22:7831–7841. doi: 10.1128/MCB.22.22.7831-7841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res. 2004;24:2783–2840. [PubMed] [Google Scholar]
- 26.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 27.Bartek J, Iggo R, Gannon J, Lane DP. Genetic and immunochemical analysis of mutant p53 in human breast cancer cell lines. Oncogene. 1990;5:893–899. [PubMed] [Google Scholar]
- 28.Tang HY, Shih A, Cao HJ, Davis FB, Davis PJ, Lin HY. Resveratrol-induced cyclooxygenase-2 facilitates p53-dependent apoptosis in human breast cancer cells. Mol. Cancer Ther. 2006;5:2034–2042. doi: 10.1158/1535-7163.MCT-06-0216. [DOI] [PubMed] [Google Scholar]
- 29.Xiong W, Hsieh CC, Kurtz AJ, Rabek JP, Papaconstantinou J. Regulation of CCAAT/enhancer-binding protein-beta isoform synthesis by alternative translational initiation at multiple AUG start sites. Nucleic Acids Res. 2001;29:3087–3098. doi: 10.1093/nar/29.14.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eaton EM, Hanlon M, Bundy L, Sealy L. Characterization of C/EBPbeta isoforms in normal versus neoplastic mammary epithelial cells. J. Cell Physiol. 2001;189:91–105. doi: 10.1002/jcp.1139. [DOI] [PubMed] [Google Scholar]
- 31.Ewing SJ, Zhu S, Zhu F, House JS, Smart RC. C/EBPbeta represses p53 to promote cell survival downstream of DNA damage independent of oncogenic Ras and p19(Arf) Cell Death Differ. 2008;15:1734–1744. doi: 10.1038/cdd.2008.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider-Merck T, Pohnke Y, Kempf R, Christian M, Brosens JJ, Gellersen B. Physical interaction and mutual transrepression between CCAAT/enhancer-binding protein beta and the p53 tumor suppressor. J. Biol. Chem. 2006;281:269–278. doi: 10.1074/jbc.M503459200. [DOI] [PubMed] [Google Scholar]
- 33.Aziz MH, Nihal M, Fu VX, Jarrard DF, Ahmad N. Resveratrol-caused apoptosis of human prostate carcinoma LNCaP cells is mediated via modulation of phosphatidylinositol 3′-kinase/Akt pathway and Bcl-2 family proteins. Mol. Cancer Ther. 2006;5:1335–1341. doi: 10.1158/1535-7163.MCT-05-0526. [DOI] [PubMed] [Google Scholar]
- 34.Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009;137:647–658. doi: 10.1016/j.cell.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 35.O'Hara SP, Splinter PL, Gajdos GB, Trussoni CE, Fernandez-Zapico ME, Chen XM, LaRusso NF. NFkappaB p50-CCAAT/enhancer-binding protein beta (C/EBPbeta)-mediated transcriptional repression of microRNA let-7i following microbial infection. J. Biol. Chem. 2010;285:216–225. doi: 10.1074/jbc.M109.041640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ewing SJ, Zhu S, Zhu F, House JS, Smart RC. C/EBPbeta represses p53 to promote cell survival downstream of DNA damage independent of oncogenic Ras and p19(Arf) Cell Death Differ. 2008;15:1734–1744. doi: 10.1038/cdd.2008.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang W, Li Q, Bagchi IC, Bagchi MK. The CCAAT/enhancer binding protein beta is a critical regulator of steroid-induced mitotic expansion of uterine stromal cells during decidualization. Endocrinology. 2010;151:3929–3940. doi: 10.1210/en.2009-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di-Poi N, Desvergne B, Michalik L, Wahli W. Transcriptional repression of peroxisome proliferator-activated receptor beta/delta in murine keratinocytes by CCAAT/enhancer-binding proteins. J. Biol. Chem. 2005;280:38700–38710. doi: 10.1074/jbc.M507782200. [DOI] [PubMed] [Google Scholar]
- 39.Burkart AD, Mukherjee A, Sterneck E, Johnson PF, Mayo KE. Repression of the inhibin alpha-subunit gene by the transcription factor CCAAT/enhancer-binding protein-beta. Endocrinology. 2005;146:1909–1921. doi: 10.1210/en.2004-0842. [DOI] [PubMed] [Google Scholar]
- 40.Zahnow CA, Younes P, Laucirica R, Rosen JM. Overexpression of C/EBPbeta-LIP, a naturally occurring, dominant-negative transcription factor, in human breast cancer. J. Natl Cancer Inst. 1997;89:1887–1891. doi: 10.1093/jnci/89.24.1887. [DOI] [PubMed] [Google Scholar]
- 41.Bundy LM, Sealy L. CCAAT/enhancer binding protein beta (C/EBPbeta)-2 transforms normal mammary epithelial cells and induces epithelial to mesenchymal transition in culture. Oncogene. 2003;22:869–883. doi: 10.1038/sj.onc.1206216. [DOI] [PubMed] [Google Scholar]
- 42.Kim MH, Fields J. Translationally regulated C/EBP beta isoform expression upregulates metastatic genes in hormone-independent prostate cancer cells. Prostate. 2008;68:1362–1371. doi: 10.1002/pros.20801. [DOI] [PubMed] [Google Scholar]
- 43.Wang WL, Lee YC, Yang WM, Chang WC, Wang JM. Sumoylation of LAP1 is involved in the HDAC4-mediated repression of COX-2 transcription. Nucleic Acids Res. 2008;36:6066–6079. doi: 10.1093/nar/gkn607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.