

Abstract
Uncovering the origin and nature of phenotypic variation within species is the first step in understanding variation between species. Mouse models with altered activities of crucial signal pathways have highlighted many important genes and signal networks regulating the morphogenesis of complex structures, such as teeth. The detailed analyses of these models have indicated that the balanced actions of a few pathways regulating cell behavior modulate the shape and number of teeth. Currently, however, most mouse models studied have had gross alteration of morphology, whereas analyses of more subtle modification of morphology are required to link developmental studies to evolutionary change. Here, we have analyzed a signaling network involving ectodysplasin (Eda) and fibroblast growth factor 20 (Fgf20) that subtly affects tooth morphogenesis. We found that Fgf20 is a major downstream effector of Eda and affects Eda-regulated characteristics of tooth morphogenesis, including the number, size and shape of teeth. Fgf20 function is compensated for by other Fgfs, in particular Fgf9 and Fgf4, and is part of an Fgf signaling loop between epithelium and mesenchyme. We showed that removal of Fgf20 in an Eda gain-of-function mouse model results in an Eda loss-of-function phenotype in terms of reduced tooth complexity and third molar appearance. However, the extra anterior molar, a structure lost during rodent evolution 50 million years ago, was stabilized in these mice.
Keywords: Ectodysplasin, Edar, Fgf20, Tooth development, Evolution, Mouse
INTRODUCTION
The regulation of organ form and complexity is a key question in biology. The development of complex structures requires specific sets of genes to be activated and/or inhibited in a correct temporal and spatial pattern. Gene expression in all developing organs is regulated by conserved signals belonging to well characterized signaling molecule families such as Wnt, Fgf, Hedgehog (Hh), Bmp and Tgfβ. These signaling pathways are also of central importance for the morphogenesis of ectodermal appendages, such as hair, teeth and exocrine glands. In addition, the ectodysplasin (Eda) pathway, composed of the tumor necrosis factor-like ligand Eda-A1 (hereafter Eda) and its receptor Edar, is crucial for ectodermal organogenesis (Mikkola, 2009). Mutations in EDA and other components of the EDA pathway cause human ectodermal dysplasia syndromes characterized by impaired formation of ectodermal organs (Mikkola, 2009). The phenotypes include missing and abnormally shaped teeth, sparse hair, and impaired function of various exocrine glands. As shown in hair and teeth, Eda has a key function in the formation of ectodermal placodes, which function as signaling centers during the initiation of organogenesis (Mustonen et al., 2004). Recently, a similar role for Eda was shown in the formation of fins and scales in zebrafish (Harris et al., 2008), identifying a conserved function for Eda signaling throughout vertebrate evolution.
Mammalian teeth are ecologically important in food processing, and the type of diet that a species has adapted to eat is reflected in their tooth morphology. Mammalian molars show a substantial degree of phenotypic variation, which has been a long-standing interest of evolutionary biologists (Osborn, 1888; Bateson, 1894). Recently, genes and networks underlying dental variation have been intensively studied (Salazar-Ciudad and Jernvall, 2010). In particular, transgenic mouse models in which mutations in signaling pathways have caused phenotypic alterations in dental characteristics have provided insights into mechanisms possibly involved in evolutionary changes of dentition (Järvinen et al., 2006; Kangas et al., 2004; Kassai et al., 2005; Klein et al., 2006; Line, 2003).
Tooth shape characteristics are determined during embryonic development. The mouse incisors and molars are initiated in embryonic day (E) 11-12 embryos from ectodermal placodes that invaginate into the mesenchyme, forming epithelial buds. The bud grows and folds into a cap-shaped enamel organ surrounding the mesenchymal dental papilla. A central structure in tooth development, the primary enamel knot (pEK), is formed during the bud-to-cap transition in the epithelium. The pEK is a signaling center composed of non-proliferating cells (Jernvall et al., 1994) expressing a set of secreted signaling molecules and their antagonists including Shh, several Wnts, Bmps, Fgfs and follistatin (reviewed by Tummers and Thesleff, 2009). In multicuspid teeth, the pEK induces the formation of secondary enamel knots, which begin to form in mouse molars at E15. The balance between activators and inhibitors has been proposed to determine the spatial arrangement of the secondary enamel knots (Salazar-Ciudad and Jernvall, 2002; Salazar-Ciudad and Jernvall, 2010). The molecules secreted by the secondary enamel knots regulate epithelial proliferation and differentiation to specify the position and shape of the cusps and, thus, the shape of the tooth crown. Experimentally manipulating the level of gene expression in the enamel knots results in variation of molar morphologies. For example, modulation of Shh, follistatin and Wnt signaling have been shown to result in altered molar shapes and cusp patterns (Dassule et al., 2000; Harjunmaa et al., 2012; Järvinen et al., 2006; Liu et al., 2008; Wang et al., 2004).
Edar is expressed in the incisor and molar placodes as well as in the primary and secondary enamel knots and is activated by epithelially derived Eda (Laurikkala et al., 2001). Teeth are affected in spontaneous Eda pathway loss-of-function mouse mutants (Grüneberg, 1966; Pispa et al., 1999). Wild-type (WT) mice have, in each half of the jaw, one incisor, a toothless diastema region and three molars (m1-m3). Eda–/– mice have smaller teeth and reduced cusp numbers, particularly in the first molar. Tooth number can also vary and 17-55% of Eda–/– mice lack the third molar (m3) (Charles et al., 2009; Grüneberg, 1966; Pispa et al., 1999; Kangas et al., 2004) (our unpublished results). Inactivation of transcription factor NF-κB results in a phenotype that is very similar to the Eda pathway loss-of-function mutants and a multitude of evidence indicates that Eda signaling is mediated through NF-κB (Häärä et al., 2011; Mikkola, 2009; Schmidt-Ullrich et al., 2001). Overexpression of Eda, under control of the keratin 14 promoter (K14-Eda), leads to a subtle increase in molar complexity and the formation of an extra molar anterior to the molars, in the location of an ancestral premolar (Kangas et al., 2004; Mustonen et al., 2003). Tooth characteristics of both Eda–/– and K14-Eda mice are polymorphic in nature. By genetically manipulating Eda activity, several tooth characteristics, including size, shape and number, can be simultaneously affected.
To study the developmental functions of the Eda pathway in ectodermal organogenesis, we searched for Eda-responsive genes by microarray analysis (Fliniaux et al., 2008; Lefebvre et al., 2012). One of the putative target genes identified was Fgf20, a member of the fibroblast growth factor (Fgf) family. In this report, we have validated the regulation of Fgf20 by Eda and show that molars in Fgf20-null (Fgf20βGal/βGal) mice resemble Eda–/– molars. We provide evidence indicating that Fgf4 and Fgf9, which are also expressed in the pEK, function redundantly with Fgf20. Analysis of tooth morphogenesis in K14-Eda;Fgf20βGal/βGal mutants indicated that Fgf20 has an essential function in the Eda-mediated activator-inhibitor balance that regulates tooth size, number and cusp patterning. These studies define a gene interaction network that regulates dental variation and suggest that changes in the expression level of Eda target genes, such as Fgf20, might be a mechanism for the evolutionary adaptation of tooth number and shape.
MATERIALS AND METHODS
Animals
K14-Eda mice (Mustonen et al., 2003) were maintained in C57Bl/6 background, and Eda–/– (Tabby) mice in B6CBA background (Pispa et al., 1999). The generation of Fgf20βGal/βGal mice has been described previously (Huh et al., 2012) and they were maintained on a mixed C57Bl/6J-129Sv/J background. The appearance of the vaginal plug was taken as E0. The embryos were staged by limb morphology.
Organ cultures
Embryonic teeth were cultivated in a Trowell-type culture as described previously (Laurikkala et al., 2001; Närhi and Thesleff, 2010). The following proteins (all from R&D Systems) were used: Fgf20, Fgf4, Fgf9 and bovine serum albumin (BSA). Beads were incubated in a protein concentration of 100 ng/μl for 1 hour at 37°C prior to use. The samples were imaged under Olympus SZX9 or Olympus AX70 microscope. EdU proliferation assay (Invitrogen) was performed according to the manufacturer’s protocol using 1:1000 dilution and 1 hour EdU incorporation at 37°C.
Promoter analysis and luciferase assay
A 10 kb sequence upstream of the translation initiation site of Fgf20, as well as a 5 kb sequence spanning the first intron, were aligned with corresponding human and rat Fgf20 sequences and analyzed for the presence of potential NF-κB responsive elements. CONSITE program (http://asp.ii.uib.no:8090/cgi-bin/CONSITE/consite) with 80% conservation cut-off, window size 50 and score threshold 80% was used for analysis. To test whether the regions containing the NF-κB responsive elements were responsive to Edar, three different luciferase reporter vector constructs were generated (supplementary material Fig. S1). The luciferase reporter assay was carried out as previously described (Fliniaux et al., 2008), using pNF-κB luc as a positive control (Koppinen et al., 2001). Primer sequences are listed in supplementary material Table S1.
Histology, in situ hybridization and X-gal staining
The tissues were fixed in 4% paraformaldehyde (PFA) and taken through an ethanol series (50%, 70%, absolute) and xylene into paraffin and sectioned at 7 μm. For whole-mount in situ hybridization (ISH), embryonic tissues were fixed in 4% PFA for 24 hours at 4°C and dehydrated through a methanol series (25, 50, 75, 100%). Whole-mount ISH was performed with InSituPro robot (Intavis AG, Germany) and ISH on histological sections as previously described (Fliniaux et al., 2008), using specific probes for Runx2, Fgf3 and Fgf20 (D’Souza et al., 1999; Kettunen et al., 2000). Digoxigenin-labeled probes were detected with BM Purple AP Substrate Precipitating Solution (Boehringer Mannheim, Germany). Radioactive ISH on paraffin sections was carried out according to standard protocols using 35S-UTP labeling (Amersham) and probes specific to Fgf20, Spry2, Spry4, Wnt10b, ectodin (Sostdc1 – Mouse Genome Informatics), Shh and Edar (Åberg et al., 2004; Bitgood and McMahon, 1995; Laurikkala et al., 2001; Laurikkala et al., 2002; Sarkar and Sharpe, 1999; Zhang et al., 2001). X-gal staining was performed as described previously (Häärä et al., 2011).
Quantitative RT-PCR
Lower molars were dissected from E13 and E14 Eda–/– mice. Four to six molars from one side of the mandible were pooled and incubated for 4 hours in medium supplemented with 250 ng/ml recombinant Fc-Eda-A1 protein (Gaide and Schneider, 2003), whereas molars from the other side of the mandible were maintained in control medium. For RNA isolation, molars were harvested in 350 μl of RNeasy (Qiagen) lysis buffer as specified by the manufacturer. DNase-treated RNA (300 ng) was reverse transcribed with Superscript III (Invitrogen), and quantitative PCR was performed in a LightCycler480 (Roche). Expression of each gene was normalized against Ranbp1. Primer sequences are listed in supplementary material Table S1.
Morphometric analysis
For adult tooth shape analysis, the jaws were boiled in tap water, cleaned and imaged (Olympus SZX9 microscope). Morphometric parameters were measured with ImageJ. High-resolution three-dimensional laser-confocal imaging has been described previously (Evans et al., 2007).
RESULTS
Fgf20 expression is confined to localized domains in dental epithelium throughout tooth morphogenesis and correlates with Edar expression
In situ hybridization in E10-16 mouse embryos was used to map Fgf20 expression during molar development (Fig. 1). At the onset of tooth morphogenesis (E10-11) Fgf20 was expressed in the thickened oral epithelium at the site of tooth initiation (Fig. 1A,B). At the onset of epithelial budding, expression was restricted to the signaling center in the tooth placode epithelium (E12) and subsequently to the tip of the bud in the forming enamel knot (E13) (Fig. 1C,D). Fgf20 expression was strongly localized in the fully formed pEK at E14 (Fig. 1E), and subsequently in the secondary enamel knots at E16 and E18 (Fig. 1F; data not shown). Interestingly, this pattern of expression correlated with the expression of Edar during molar development (Tucker et al., 2000; Laurikkala et al., 2001).
Fig. 1.
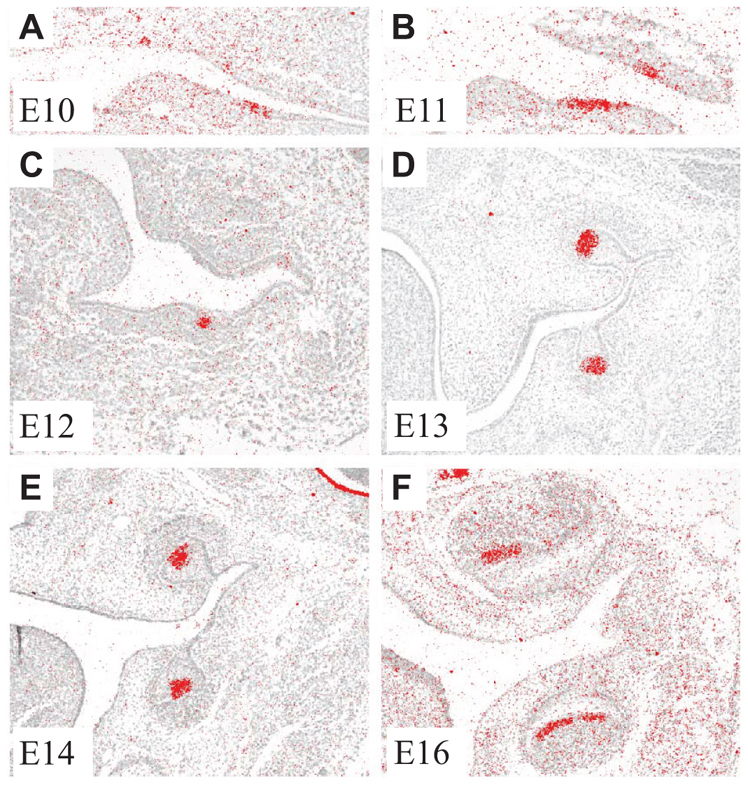
Fgf20 expression in mouse molar development. (A-D) Fgf20 is expressed in the dental epithelium: at initiation (A,B), placode (C) and bud (D). (E,F) At the cap stage, expression is concentrated in the primary enamel knot (E) and at the bell stage in the secondary enamel knots (F).
Fgf20 expression is regulated by the Eda pathway
Microarray analysis revealed that Fgf20 is one of the most rapidly induced genes by Eda in hair placodes and, thus, is a putative transcriptional target of Eda (Fliniaux et al., 2008; Lefebvre et al., 2012). To test whether Eda regulates Fgf20 in developing teeth, we treated E13 Eda–/– molar buds with recombinant Eda protein (250 ng/ml) for 4 hours. This led to a ∼4.5-fold increase in the Fgf20 mRNA levels compared with controls (Fig. 2A; n=14; Wilcoxon signed rank test, P<0.05). To validate this observation in vivo, we compared the intensity of Fgf20 expression by whole-mount in situ hybridization in the jaws of Eda–/–, WT and K14-Eda mice at E12, E13 and E14. Fgf20 expression was significantly decreased in the Eda–/– molars whereas the K14-Eda mice showed increased Fgf20 expression compared with WT mice (Fig. 2B-J). We also confirmed the correlation of Fgf20 expression level with Eda activity by analyzing Fgf20 expression using an Fgf20-β-galactosidase (βGal, encoded by lacZ) knock-in allele and quantifying βGal staining in compound mutants of Eda–/–;Fgf20+/βGal, Fgf20+/βGal and K14-Eda;Fgf20+/βGal mice at E14.5, E15.5 and E16.5 (supplementary material Fig. S1). Significant differences in the size of βGal staining domains were observed between the mutants at all stages. In addition, in K14-Eda mice, the forming extra molar expressed Fgf20-βGal (supplementary material Fig. S1). From these results, we conclude that Fgf20 lies downstream of Eda signaling in tooth development.
Fig. 2.

Fgf20 expression is regulated by Eda. (A) Treatment of E13 Eda–/– mouse molars with Eda protein upregulates Fgf20 transcription after 4 hours of culture (mean ± s.d.). *P<0.05. (B-J) Expression of Fgf20 at E12 (B-D), E13 (E-G) and E14 (H-J) in developing teeth of Eda–/–, WT and K14-Eda mice. i, incisor; m, molar. Asterisk indicates extra molar.
To examine further the function of Eda/Edar/NF-κB signaling in activating Fgf20 transcription, we searched for putative NF-κB binding sites in Fgf20 promoter and intronic regions. Although several potential NF-κB binding sites were identified, in promoter-reporter assays neither a 5.2 kb promoter region nor intron 1 sequences encompassing putative NF-κB sites were responsive to Edar (supplementary material Fig. S2), suggesting either that an additional factor is needed to activate Fgf20 transcription, or that Fgf20 is regulated through a distant enhancer sequence yet to be identified.
Fgf20βGal/βGal mice have smaller molars with mildly altered anteroconid cusp morphology
To elucidate the role of Fgf20 in tooth development, we examined the tooth phenotype in the lower jaw of Fgf20-null (Fgf20βGal/βGal) mice. No differences were found between the teeth of heterozygote Fgf20+/βGal and those of WT mice. The number of teeth was normal in Fgf20βGal/βGal mice but the molars were smaller than in WT littermates (Fig. 3A-B′, Eda–/– for comparison in 3C). Both mesio-distal length (Fig. 3D) and bucco-lingual width (Fig. 3E) were significantly reduced, and thus the crown area was significantly reduced in all Fgf20βGal/βGal molars (n=14 tooth rows) compared with WT littermate controls (n=15) (Fig. 3F; Student’s t-test, P<0.001). The jaws of the Fgf20βGal/βGal and WT littermates were the same size (Fig. 3G) with no apparent morphological differences. The incisors of Fgf20βGal/βGal mice appeared similar in size and shape as in littermate controls with no apparent enamel defects (supplementary material Fig. S3).
Fig. 3.

Adult Fgf20βGal/βGal molars are small and show defects in anteroconid cusp patterning. (A-C) Molars in Fgf20βGal/βGal mice are smaller compared with WT, resembling Eda–/– mice. The cusp pattern is affected in the anterior part of the first molar (dashed line). (D-G) All molars (m1, m2, m3) are shorter (D) and narrower (E), and thus smaller in two-dimensional size (F) compared with WT teeth. The width of the whole jaw is unaffected (G). (H) The distance (normalized to molar diameter) between the cusps in the first cusp pair (1) is shorter, whereas the distances in other cusp pairs (2-7) show no differences compared with the WT molars (cusp pairs indicated in A). Mean ± s.d. *P<0.05, **P<0.01, ***P<0.001.
We next analyzed the cusp patterns and morphology of molars (Fig. 3H, see 3A for corresponding cusp pairs). Although the overall cusp pattern of the Fgf20 mutants appeared normal, the anterior cusps were mildly affected: the mean distance (normalized to bucco-lingual width of corresponding molar) between the first pair of cusps was reduced (cusp pair 1 in Fig. 3H). The relative size of the molars (m2/m1 and m3/m1) was similar in Fgf20βGal/βGal and WT mice (data not shown). These results suggest that Fgf20 is involved in the regulation of tooth size and can fine-tune the patterning of the anterior cusps.
Anteroconids are absent or smaller in developing Fgf20βGal/βGal molars resembling Eda–/– molar development
To study the developmental basis of tooth abnormalities in Fgf20βGal/βGal mice, we analyzed embryonic molar development in ex vivo cultures. Lower molars were dissected at E13 and cultured for seven days. The molars of Fgf20 heterozygotes (n=18; not shown) and WT littermates (n=12) appeared to be similar before and during culture (Fig. 4). Fgf20βGal/βGal molars were small compared with WT (P<0.01), with the most anterior cusp, the anteroconid, reduced in relative size (5/13) or lacking completely (6/13). In addition, the posterior part of the molar, the talonid, was represented by a single cusp instead of the normal two or three (5/13). This reduction in size and cusp number is similar to what is seen in cultured Eda-null molars (Harjunmaa et al., 2012), though dissimilar in that the effect was not quite as severe, and that the anteroconid was reduced more often than the talonid. In vivo histological analysis supported the findings from organ culture experiments. At E14, Fgf20βGal/βGal molars formed a cap, but this was dome-shaped, thus resembling Eda–/– molars (Pispa et al., 1999). At E18, Fgf20βGal/βGal molars appeared histologically normal, but the shape of the anteroconid cusp varied more in Fgf20βGal/βGal compared with control molars (supplementary material Fig. S4). Additionally, the overall size of the molars was smaller in Fgf20βGal/βGal mutants. At E14, the expression of well-characterized genes in tooth development, Edar and Wnt10b in the pEK, and ectodin in mesenchyme and epithelium excluding the pEK, were found to be patterned similarly in Fgf20βGal/βGal and WT embryos (supplementary material Fig. S5). The embryonic analysis is consistent with the adult Fgf20βGal/βGal tooth phenotype (Fig. 3) and suggests a role for Fgf20 in the regulation of tooth size and anteroconid cusp patterning.
Fig. 4.

Fgf20βGal/βGal molars are reduced in size and cusp number during development. (A,B) In tissue culture conditions, WT mouse molars (A) typically form five cusps, lacking the most posterior cusp, the hypoconulid. Eda–/– molars (B) typically form only three cusps, lacking the most anterior cusp, the anteroconid (white arrow). The two most posterior cusps, the talonid cusps (black arrows), are represented by only one cusp. (C-E) Fgf20βGal/βGal molars show a variety of phenotypes ranging from WT-like five cusps (C) to Eda-null-like four cusps (D). Most commonly, the anteroconid is abnormally small (E) or missing completely. (F) Measurement of tooth area at E13+7 days. Mean ± s.d. **P<0.01. NS, non-significant.
Fgf4 and Fgf9 might have redundant functions with Fgf20 during molar development
As the changes in the Fgf20-null tooth phenotype were relatively subtle, we examined the potential redundancy between Fgf20 and other Fgf ligands. Fgf4 and Fgf9 are co-expressed with Fgf20 in the pEK (Kettunen and Thesleff, 1998). We first studied the effects of recombinant Fgf20, Fgf4 and Fgf9 proteins on dental mesenchyme isolated from E13 WT molars. All three Fgfs induced Fgf3 expression, whereas the control BSA beads did not (Fig. 5A-D). Fgf4 and Fgf9 have previously been shown to induce Runx2 expression, which is required for the induction of Fgf3 in dental mesenchyme (Åberg et al., 2004). Also Fgf20 induced the expression of Runx2 (Fig. 5E,F). We next analyzed the capacity of Fgf20 to promote proliferation of dental mesenchymal cells, as previously reported for Fgf4 and Fgf9 (Kettunen et al., 1998). When isolated E13 mesenchyme was cultured with Fgf20 beads, proliferation was induced in the cells underlying and surrounding the bead (Fig. 5I,J). BSA beads had no effect (Fig. 5G,H).
Fig. 5.

Fgf20 has similar effects on embryonic dental mesenchyme as Fgf4 and Fgf9 and acts redundantly with Fgf9. (A-D) Beads supplemented with Fgf4, Fgf9 and Fgf20, but not BSA, induce the expression of Fgf3 in WT molar mesenchyme after 24 hours. (E,F) Fgf20 induces Runx2 expression around the bead. Asterisk indicates endogenous expression of Runx2 in an island of osteogenic mesenchyme. (G-J) Fgf20 protein stimulates cell proliferation in isolated dental mesenchyme (EdU-positive cells; red) after 24 hours of culture, whereas BSA has no effect. The position of the protein-releasing bead is indicated (dashed line). (K-N) Fgf9–/–;Fgf20βGal/βGal molars have developed into cap stage at E14.5, similar to WT control and Fgf9 and Fgf20 single mutants, and express Shh in the enamel knot (red). (O) However, the enamel knot is significantly shorter in Fgf9–/–;Fgf20βGal/βGal compared with single mutants and controls. (P) Expression of Fgf4 and Fgf9 is not induced after 4 hours Eda treatment in E13 (Fgf9) or E14 (Fgf4) Eda–/– molars. Mean ± s.d. *P<0.05, **P<0.01. NS, non-significant.
To analyze further the possible compensatory actions of Fgf9 and Fgf20 in vivo, we crossed Fgf9 and Fgf20 mutant mice to create Fgf9–/–;Fgf20βGal/βGal compound mutants. Because Fgf9–/– mice die at birth, we studied the embryonic development. We analyzed the phenotype of E14.5 cap stage molars from serial histological sections and the expression of Shh as a marker of the pEK. The molars appeared morphologically normal but the enamel knot length, measured on serial sections, indicated that the Fgf9–/–;Fgf20βGal/βGal enamel knot was shorter compared with controls (Fig. 5K-N), including Fgf9–/–;Fgf20+/βGal (P<0.01) and Fgf20βGal/βGal genotypes (Fig. 5O; P<0.05; Mann-Whitney U-test with Bonferroni correction). Hence, removal of both Fgf9 and Fgf20 had a significant additive effect on enamel knot length. Conditional deletion of Fgf4 function in dental epithelium with the Nestin-Cre line did not affect either the shapes or sizes of adult molars or the histological appearance of cap stage embryonic molars (T.Å., X. Sun, G. Martin and I.T., unpublished). The deletion of the floxed exon in cap stage molars was confirmed by in situ hybridization.
As Fgf4 and Fgf9 are co-expressed in the enamel knot with Fgf20 and Edar, we next analyzed by qRT-PCR whether they are induced by Eda. However, in contrast to Fgf20 (Fig. 2A), Fgf4 (data not shown) and Fgf9 (Fig. 5P) expression were unaltered in E13 molars after 4 hours Eda treatment. As Fgf4 expression level was very low at E13, we analyzed Fgf4 expression also at E14 and found no effect of Eda (Fig. 5P).
Inactivation of Fgf20 in K14-Eda mice leads to reduced tooth size, altered tooth number and aberrant cusp morphology
K14-Eda mice have a more complex cusp pattern in all molars compared with WT mice, and, in addition, ∼50% of individuals (in the FVB background) have an extra molar anterior to the first molar in the position of an ancestral premolar (Kangas et al., 2004; Mustonen et al., 2003). To assess the role of Fgf20 in the phenotype of K14-Eda mice, we generated compound K14-Eda;Fgf20βGal/βGal mutants. Measurements of molar dimensions indicated that m1, m2 and m3 were smaller in K14-Eda;Fgf20βGal/βGal mice (n=16) compared with K14-Eda littermates (n=16; one-way ANOVA with Bonferroni post hoc, P<0.001; Fig. 6). In K14-Eda;Fgf20+/βGal mice (n=34) only m2 was statistically significantly smaller compared with K14-Eda (P<0.01), but all three molars were significantly larger compared with K14-Eda;Fgf20βGal/βGal mice (P<0.001).
Fig. 6.

The number, size and shape of K14-Eda molars are affected by deletion of Fgf20. (A,B) K14-Eda mice have an extra molar (EM) anterior to m1 in ∼50% of tooth rows (A with an EM; B without an EM, black arrow). (C,G) Inactivation of one Fgf20 allele (K14-Eda;Fgf20+/βGal) results in a slightly smaller m2, without otherwise affecting the K14-Eda phenotype (mean ± s.d.). (D-F) In complete knockout of Fgf20 (K14-Eda;Fgf20βGal/βGal), m1-m3 are smaller and the cusp pattern is abnormal, with reduced cusp number and complexity. (H) Tooth size in K14-Eda;Fgf20βGal/βGal mice shows a trend towards increased reduction of more posterior molars compared with K14-Eda. (I) Also, the relative size of molars within a tooth row is changed: m2/m1 and m3/m1 are altered in K14-Eda;Fgf20βGal/βGal mice compared with K14-Eda (linear regression of means). Upper table: mean molar proportions (m2/m1 and m3/m1) ± s.d. Lower table: Frequency of the EM is increased in K14-Eda;Fgf20+/βGal and, more clearly, in K14-Eda;Fgf20βGal/βGal tooth rows compared with K14-Eda mice. Also, m3 is lost in 31% of the K14-Eda;Fgf20βGal/βGal tooth rows. *P<0.05, **P<0.01, ***P<0.001.
K14-Eda;Fgf20βGal/βGal mice showed a trend of decreasing molar size towards the posterior molars compared with K14-Eda, and thus had reduced m2/m1 and m3/m1 ratios (Fig. 6H,I; P<0.05). K14-Eda;Fgf20+/βGal molars showed a similar, although milder, trend. In addition to reduced absolute and relative size, a characteristic feature of K14-Eda;Fgf20βGal/βGal molars was reduced molar complexity with the absence of the extra cusps typical of K14-Eda and with the buccal cusps becoming fused in m1 and m2 (Fig. 6; supplementary material Fig. S6). The aberrant development of cusps was visible prior to tooth eruption (E18), excluding enamel defects as the cause of the reduced crown complexity observed in adult K14-Eda;Fgf20βGal/βGal mice (supplementary material Fig. S6). The cusp pattern of K14-Eda;Fgf20+/βGal was more similar to K14-Eda than to K14-Eda;Fgf20βGal/βGal. However, some of the extra cusps present in K14-Eda were absent in K14-Eda;Fgf20+/βGal and the overall shape of m1 and m2 resembled more WT than K14-Eda (supplementary material Fig. S6). This suggests that partial loss of Fgf20 signaling in K14-Eda might rescue the cusp pattern close to WT phenotype, but the complete loss of Fgf20 signaling leads to highly reduced cusp number and flattened shape of the existing cusps, resembling Eda–/– molars.
The frequency of the anterior extra molar (EM) was increased after removal or reduction of Fgf20 in the K14-Eda background: 50% of K14-Eda, but 76% of K14-Eda;Fgf20+/βGal and 88% of K14-Eda;Fgf20βGal/βGal tooth rows had the EM (Fisher’s exact test, P<0.05; Fig. 6I). Moreover, 31% of K14-Eda;Fgf20βGal/βGal mice lacked m3 (P<0.05).
Altogether, K14-Eda;Fgf20βGal/βGal molars showed alterations in number, size, shape and molar proportions compared with the molars of K14-Eda mice. Thus, the loss of Fgf20 in the K14-Eda background had a more severe and variable effect than its loss in otherwise WT mice. The phenotype of K14-Eda;Fgf20+/βGal was intermediate between K14-Eda and K14-Eda;Fgf20βGal/βGal for all tooth characteristics measured.
Fgf20 downregulation stabilizes the Shh-expressing foci in the anterior end (AE) of m1 but Eda upregulation is required for their development into EMs In the mouse mutants developing EM, such as K14-Eda, Sostdc1–/–, Sprouty2–/– and Sprouty4–/–, a Shh expression focus (also called rudimentary bud or diastemal bud in the literature) is detected in the AE of m1 at E13.5 or E14.5 (Kangas et al., 2004; Kassai et al., 2005; Klein et al., 2006). To study the effect of Fgf20 signaling on this Shh expression domain, we analyzed Shh expression by whole-mount ISH in lower jaws of K14-Eda;Fgf20 mutant mice. Shh expression was detected at E13.5 in ∼30% of WT jaws, in ∼50% of Fgf20+/βGal and in all Fgf20βGal/βGal jaws studied (Fisher’s exact test, P<0.05; Fig. 7). Similar to Fgf20-null embryos, in K14-Eda, K14-Eda;Fgf20+/βGal and K14-Eda;Fgf20βGal/βGal embryos, the Shh expression focus in the AE was stabilized in >90% of jaws (P<0.01). At E14.5, this Shh expression was lost in WT mice but sustained in >80% of Fgf20+/βGal and in all Fgf20βGal/βGal, K14-Eda, K14-Eda;Fgf20+/βGal and K14-Eda;Fgf20βGal/βGal jaws (Fig. 7G-L; data not shown; P<0.05). The Shh domain was also enlarged in K14-Eda;Fgf20βGal/βGal compared with K14-Eda embryos (Fig. 7N). From these observations we conclude that Fgf20 deletion stabilizes the Shh foci in the AE, but is not sufficient to induce EM formation. This is consistent with the previous observation by Kangas et al. (Kangas et al., 2004) who detected Shh positive foci anterior to m1 Shh expression in all K14-Eda jaws, even though only half of them developed extra molars. The requirement of Eda upregulation for the AE to develop EM was clearly seen in Fgf20 X-gal staining in E15.5 molars (supplementary material Fig. S7). In Fgf20βGal/βGal m1, an anterior extension of epithelial X-gal activity was observed but this did not grow down to the mesenchyme to form EM. However, in K14-Eda;Fgf20+/βGal and K14-Eda;Fgf20βGal/βGal mice, the AE grew down to the mesenchyme and was separated from m1 and gave rise to EM (supplementary material Fig. S7).
Fig. 7.

Eda upregulation and Fgf20 inhibition independently stabilize the foci of Shh signaling in the anterior end of m1. (A-L) Shh localization in E13.5 and E14.5 mandibles by whole-mount in situ hybridization. In WT mice, a small dot of Shh expression (blue, black arrow) is detected in the anterior end (AE) of m1 in one third of the E13.5 jaws (A). This Shh expression focus has disappeared by E14.5 (G). However, partial (Fgf20+/βGal, B,H) or complete (Fgf20βGal/βGal, C,I) loss of Fgf20 stabilize the Shh focus at E13-14.5. Eda upregulation alone (K14-Eda, D,J) or combined to partial (K14-Eda;Fgf20+/βGal, E,K) or complete (K14-Eda;Fgf20βGal/βGal, F,L) loss of Fgf20 also stabilize the Shh expressing foci. (M) Percentage of Shh-expressing foci present or absent in E13.5 half mandibles and the number of samples. (N) In K14-Eda;Fgf20βGal/βGal, the Shh foci are larger compared with K14-Eda;Fgf20+/βGal at E14.5 *P<0.05, **P<0.01. Error bars represent mean ± s.d.
Fgf20 regulates the expression of Spry2 and Spry4
The inhibitors of Fgf signaling sprouty 2 and sprouty 4 (encoded by Spry2 and Spry4) are expressed at E14.5 in the dental epithelium and mesenchyme, respectively (Zhang et al., 2001). As loss of function of either Spry2 or Spry4 can result in the formation of an extra anterior molar in the diastema (Klein et al., 2006), we analyzed whether Spry expression is affected by Fgf20. Fgf20 beads, but not BSA beads, induced Spry2 and Spry4 in the epithelium and mesenchyme, respectively (Fig. 8A-D). As loss of function of either Spry2 or Spry4 induces the formation of the EM, and because the same feature was observed in K14-Eda;Fgf20βGal/βGal jaws, we hypothesized that Spry2 or Spry4 might be aberrantly expressed in the AE of K14-Eda;Fgf20βGal/βGal molars. To address this, we compared the expression of Spry2 and Spry4 in tissue sections of K14-Eda and K14-Eda;Fgf20βGal/βGal molars by in situ hybridization. The expression levels of both Spry2 and Spry4 were reduced in the AE of K14-Eda;Fgf20βGal/βGal m1, and also in the middle of m1 (cap), compared with K14-Eda (Fig. 8E-L). Etv5 (also known as Erm), a direct target of Fgf signaling (Roehl and Nüsslein-Volhard, 2001), was highly expressed in the AE epithelium in the molars of the E14.5 K14-Eda;Fgf20βGal/βGal mice but was downregulated in the mesenchyme compared with K14-Eda (Fig. 8M,N). Reduced Spry2 expression in the epithelium is thus not caused by reduced epithelial Fgf signaling, whereas reduced Etv5 expression in the mesenchyme might be due to the loss of Fgf20 function. From these results we conclude that Fgf20 can regulate the expression of Spry2 and Spry4 and that the reduction in the expression levels of Spry2, and possibly Spry4, in the K14-Eda;Fgf20βGal/βGal molars might facilitate the formation of the EM.
Fig. 8.
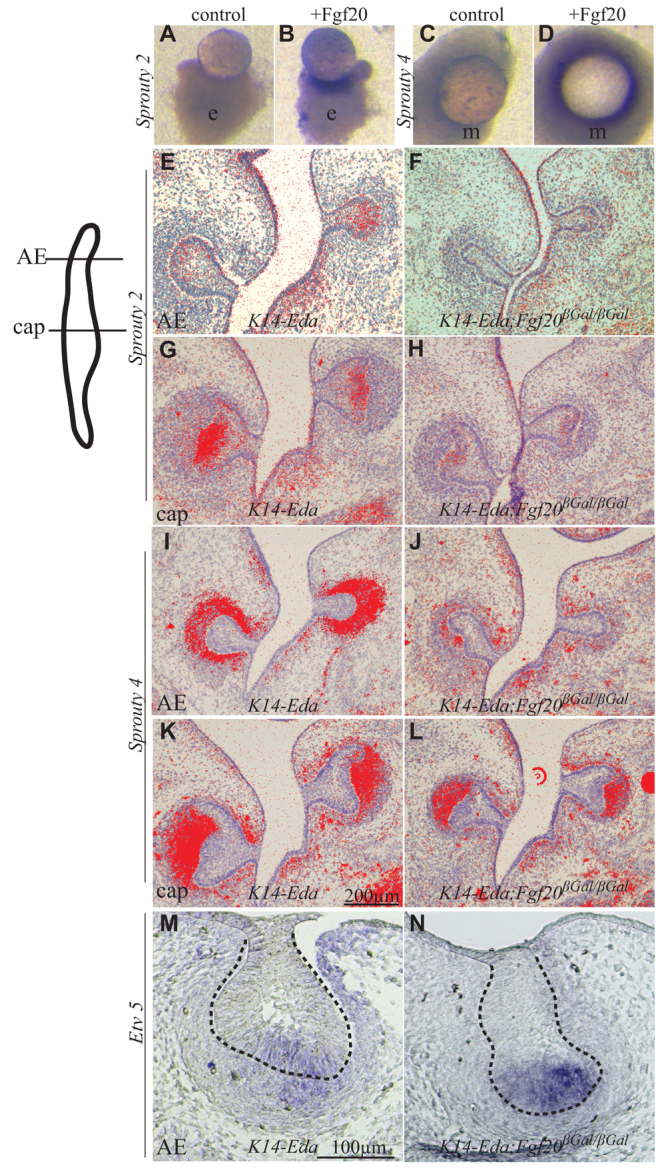
Fgf20 regulates the expression of Spry2 and Spry4. (A-D) E14.5 molar (m1) epithelium (A,B) or mesenchyme (C,D) cultured with heparin beads soaked in Fgf20 recombinant protein or BSA. Fgf20 induces Spry2 (blue; B) in the epithelium and Spry4 in the mesenchyme (D), whereas BSA does not (A,C). (E-L) Sections of E14.5 molar (m1) from the anterior end (AE) and the largest area of the cap (as illustrated in the schematic). Spry2 is expressed in the epithelium in the AE and cap of K14-Eda molar (E,G) and is reduced in K14-Eda;Fgf20βGal/βGal especially in AE (F,H). Spry4 expression is confined to mesenchyme (I,K), and is reduced in AE in K14-Eda;Fgf20βGal/βGal (J,L) compared with K14-Eda. (M,N) Etv5 expression is high in the AE of K14-Eda;Fgf20βGal/βGal in the epithelium but reduced in the mesenchyme (N) compared with the AE of K14-Eda (M). Dashed line indicates the margin of the epithelium.
DISCUSSION
Fgf20 is an important mediator of Eda signaling in teeth
The mechanisms by which Eda regulates tooth size, shape and number can be understood by identifying signaling pathways that function downstream of Eda. Eda signaling is a key regulator of morphogenesis of ectodermal organs in all vertebrates and regulates the transcription of downstream genes via the transcription factor NF-κB. Here, we report that one of the Fgf family ligands, Fgf20, is regulated by the Eda pathway in the developing tooth. We demonstrated that Eda rapidly induced Fgf20 expression and that the expression levels of Fgf20 correlated with Eda activity in vivo. The intimate link between Fgf20 and Eda was further supported by the similarities of Fgf20βGal/βGal and Eda–/– molar phenotypes. The polymorphism in cusp development is milder in Fgf20βGal/βGal than in Eda–/– mice, but the tendency towards a smaller or lacking anteroconid cusp is a shared feature in both mutants. Taken together, our results indicate that Fgf20 is a key mediator and is likely to be a direct target of Eda signaling. Fgf20 is also regulated by Wnt/β-catenin signaling (Chamorro et al., 2005). The residual Fgf20 expression observed in Eda-null teeth might, thus, be induced by the canonical Wnt pathway, which is known to be active in tooth placodes and in the enamel knot (Järvinen et al., 2006; Liu et al., 2008).
Although Eda/Edar/NF-κB signaling is crucial for normal tooth development, its target genes in teeth have remained elusive. Candidate gene and microarray profiling studies on embryonic mouse skin have shown that Eda signaling targets several components of major signal pathways including Bmp, Wnt and Hh (Mou et al., 2006; Pummila et al., 2007; Fliniaux et al., 2008; Häärä et al., 2011; Lefebvre et al., 2012). The addition of the Fgf pathway to the list of evolutionarily conserved signaling pathways regulated by Eda underlines the function of Eda as a signal pathway modulator in ectodermal organ formation.
Fgf20 acts redundantly with Fgf4 and Fgf9 and is part of the Fgf signaling loop between the epithelium and the mesenchyme
Fgfs regulate the morphogenesis of all organs and, typically, epithelial and mesenchymal Fgfs function in signaling loops across tissues (Kettunen et al., 2000; Sun et al., 2000; Klein et al., 2008; Ornitz and Yin, 2012). Fgf20 belongs to a class of Fgfs that are usually expressed in epithelium and that usually signal to mesenchyme. Fgf20 is co-expressed in tooth placodes and/or enamel knots with Fgf3, Fgf4, Fgf9 and Fgf15 (Åberg et al., 2004; Kettunen and Thesleff, 1998; Porntaveetus et al., 2011), and we showed that Fgf20 has similar effects on dental mesenchyme as Fgf4 and Fgf9. We also showed that Fgf20 directly affects the dental epithelium, similar to Fgf4, which was the first Fgf to be discovered in the enamel knot (Niswander and Martin, 1992) and has been suggested to regulate cusp formation (Jernvall et al., 1994). The overlapping in vitro functions of Fgf20, Fgf4 and Fgf9 suggest redundancy between these ligands, as tooth phenotypes in the Fgf20, Fgf4 and Fgf9 single null mutants are mild. The more severe embryonic molar phenotype of the Fgf20;Fgf9 compound mutant compared with single mutants directly demonstrates redundant functions of the two Fgfs.
The coordinated growth of tooth compartments is at least partially controlled by an Fgf signaling loop. Epithelial Fgfs regulate the expression of mesenchymal Fgfs, such as Fgf3 and Fgf10, which act redundantly to support growth and patterning of molars mainly via epithelial Fgf receptor 2b (Fgfr2b) (Wang et al., 2007; De Moerlooze et al., 2000). The molars are small in Fgf3–/–;Fgf10+/– mice (Wang et al., 2007), similar to Fgf20βGal/βGal reported here, suggesting that reduction of either epithelial or mesenchymal Fgf signaling cause similar effects on tooth growth. Fgf10 protein partially rescued the Eda–/– molar phenotype in vitro (Pispa et al., 1999), which, together with our results, indicates that one of the functions of Eda in tooth development is to sustain and/or balance the Fgf signaling loop (Fig. 9).
Fig. 9.
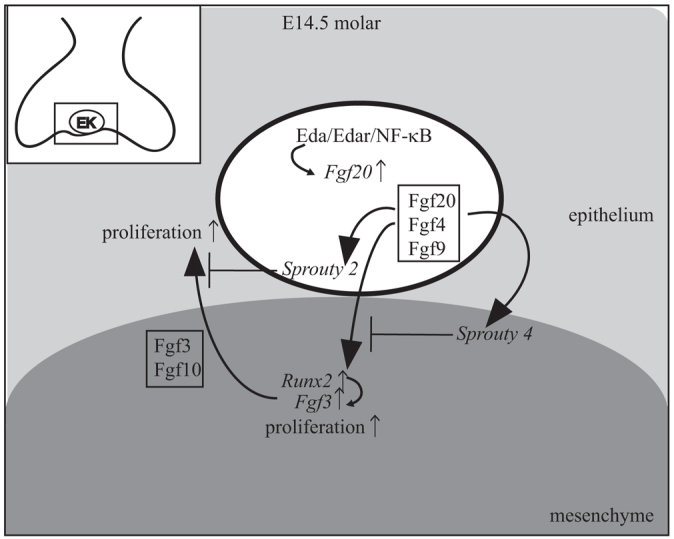
Integration of Eda/Edar/NF-κB signaling in the enamel knot with an Fgf signal loop regulates tooth crown development. Schematic based on our findings and reported data. Activation of Edar receptor by Eda in the enamel knot (EK) leads to activation of NF-κB and Fgf20 transcription. Fgf20, together with Fgf4 and Fgf9, moves to mesenchyme, induces the transcription of Runx2 and Fgf3 and stimulates cell proliferation. Fgf3 and Fgf10 signal back to the epithelium and stimulate proliferation. Fgf20 induces Spry4 expression in the mesenchyme and Spry2 expression in the epithelium. Spry4 and Spry2 inhibit Fgf signaling in the mesenchyme and epithelium, respectively.
The inhibitory functions of Spry2 and Spry4 on Fgf signaling are thought to keep the diastema toothless (Klein et al., 2006), a characteristic feature of mouse dentition for over 50 million years. We showed that Fgf20 can induce the expression of Spry2 and Spry4 and, importantly, that Spry2 and Spry4 levels were reduced in the anterior end in K14-Eda;Fgf20βGal/βGal compared with K14-Eda molars. Although still speculative, a scenario can be envisioned in which Fgf20 would have a more prominent role in regulation of Spry2 and Spry4 expression compared with other Fgfs. Hence, loss of Fgf20 would in fact lead to increased Fgf signaling activity in the anterior end of the molar bud. In support of this hypothesis, expression of the Fgf target gene Etv5 was high in the anterior end epithelium of K14-Eda;Fgf20βGal/βGal molars. Whether these effects are directly caused by loss of Fgf20, or are partially due to alterations in the expression of mesenchymal Fgfs, is currently not known. We propose that the combined effect of high Eda and low Spry expression levels resulted in stabilization of the EM in K14-Eda;Fgf20βGal/βGal lower jaws. As Fgf20 activity appeared to inhibit the formation of the extra molar, other Eda targets must stimulate it. The reported Eda-induced genes include modulators of Bmp, Wnt and Hh pathways (Mikkola, 2009; Lefebvre et al., 2012), all of which have been linked to EM stabilization (Tummers and Thesleff, 2009). We suggest that the balance between the different Eda targets, i.e. those that promote and those that inhibit extra molar formation, determines the frequency of EM appearance.
Fgf20 and Eda play important roles in the activator-inhibitor balance regulating tooth number, size and shape
A characteristic feature of K14-Eda;Fgf20βGal/βGal molars was the increased reduction in size towards posterior molars and the relatively frequent lack of m3. We suggest that these two phenotypes are linked and can be explained by the inhibitory cascade model (Kavanagh et al., 2007). The increased reduction in size towards posterior molars indicates, according to this model, increased inhibition in K14-Eda;Fgf20βGal/βGal tooth development compared with K14-Eda. Moreover, 31% of K14-Eda;Fgf20βGal/βGal tooth rows lacked m3, although K14-Eda and Fgf20βGal/βGal mice do not lack m3 (Mustonen et al., 2003) (this study). The result follows the predictions of the inhibitory cascade model as a consequence of increased inhibition. Eda–/– mice lack m3 at a frequency of 17-55%, depending on the genetic background (Grüneberg, 1966; Pispa et al., 1999; Kristenová-Cermáková et al., 2002). The removal of Fgf20 in Eda gain-of-function mice caused the m3 phenotype to resemble the Eda loss-of-function phenotype. This highlights the importance of Fgf20 in the activator-inhibitor balance that modulates the initiation and size of posterior molars. The total effect of removal of Fgf20 in K14-Eda was ‘anteriorization’ of dentition: stabilization of the extra molar, and reduction or absence of m3. Thus, Fgf20 is likely to act as an activator during tooth development. The proportional reduction in tooth size towards posterior molars and reduced complexity are characteristic features of faunivorous rodent taxa (Kavanagh et al., 2007). Interestingly, both of these features were observed in K14-Eda;Fgf20βGal/βGal mice, which suggests that tooth characteristics regulated by Eda and Fgf20 might be, ultimately, under the control of ecology and that Eda- and Fgf20-induced variation might facilitate the microevolution of dentition.
Our work demonstrates how combinatorial genetic alteration within one pathway during tooth development can lead to a wide variety of phenotypes, causing subtle, but evolutionary important, dental variation. The reduction of Fgf20 expression in Fgf20+/βGal mice had no detectable effects on tooth development and the complete loss of Fgf20 signaling (Fgf20βGal/βGal) lead to phenotypic alteration of one characteristic (size), together with mild alteration in another (anteroconid cusps). When Fgf20 loss of function was combined with high Eda signaling, several tooth characteristics (size, number and cusp pattern) were changed, some more drastically than others, owing to imbalanced activation and inhibition. In nature, mutations in the regulatory regions of Eda and Fgf20 pathway genes could produce dental variation, the material for natural selection. Interestingly, Eda signaling activity is under natural selection in three-spined stickleback fish populations producing variation in armor plates (Colosimo et al., 2005), and, in humans, a specific allele of Edar is a major contributor to scalp hair thickness in Asian populations (Fujimoto et al., 2008). We demonstrated some mechanisms behind the Eda and Fgf20-induced variation. Maintenance of the embryonic Shh expression foci in the AE was supported by the loss of Fgf20 but completion of the formation of the EM required Eda upregulation. Epithelium-derived Fgf20 modulates mesenchymal cell proliferation and induces the expression of Fgf3, which reciprocally signals to the epithelium. By this mechanism at least, Fgf20 could regulate size of the molars and cusp development. Moreover, we connected Eda to this Fgf loop by demonstrating that Eda induces Fgf20 expression in the epithelium. Our results indicate that other epithelial Fgf ligands, Fgf4 and Fgf9 in particular, may compensate for the loss of Fgf20 explaining the relatively mild phenotype of Fgf20βGal/βGal dentition. The other targets of Eda probably include important inhibitors, as, according to the inhibitory cascade model, the characteristic molar proportions of K14-Eda;Fgf20βGal/βGal mice resulted from increased inhibition.
Supplementary Material
Acknowledgments
We thank Nobuyki Itoh for support during the initiation of the project; Gail Martin and Xin Sun for the generation of Fgf4 conditional mutant mouse; and Raija Savolainen, Riikka Santalahti, Merja Mäkinen and Tuomas Kankaanpää for technical assistance.
Footnotes
Funding
The project was supported financially by Viikki Doctoral Programme in Molecular Biosciences, Academy of Finland, and Sigrid Juselius Foundation. Generation of Fgf20βGal mice was aided by National Institutes of Health support grants [P30DC04665, P30DK052574 and P30AR057235] at Washington University, and a generous contribution from Edward and Linda Ornitz. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.079558/-/DC1
References
- Åberg T., Wang X. P., Kim J. H., Yamashiro T., Bei M., Rice R., Ryoo H. M., Thesleff I. (2004). Runx2 mediates FGF signaling from epithelium to mesenchyme during tooth morphogenesis. Dev. Biol. 270, 76–93 [DOI] [PubMed] [Google Scholar]
- Bateson W. (1894). Materials for the study of variation, treated with special regard to discontinuity in the Origin of Species. London: Macmillan; [Google Scholar]
- Bitgood M. J., McMahon A. P. (1995). Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev. Biol. 172, 126–138 [DOI] [PubMed] [Google Scholar]
- Chamorro M. N., Schwartz D. R., Vonica A., Brivanlou A. H., Cho K. R., Varmus H. E. (2005). FGF-20 and DKK1 are transcriptional targets of β-catenin and FGF-20 is implicated in cancer and development. EMBO J. 24, 73–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles C., Pantalacci S., Tafforeau P., Headon D., Laudet V., Viriot L. (2009). Distinct impacts of Eda and Edar loss of function on the mouse dentition. PLoS ONE 4, e4985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo P. F., Hosemann K. E., Balabhadra S., Villarreal G., Jr, Dickson M., Grimwood J., Schmutz J., Myers R. M., Schluter D., Kingsley D. M. (2005). Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science 307, 1928–1933 [DOI] [PubMed] [Google Scholar]
- D’Souza R. N., Åberg T., Gaikwad J., Cavender A., Owen M., Karsenty G., Thesleff I. (1999). Cbfa1 is required for epithelial-mesenchymal interactions regulating tooth development in mice. Development 126, 2911–2920 [DOI] [PubMed] [Google Scholar]
- Dassule H. R., Lewis P., Bei M., Maas R., McMahon A. P. (2000). Sonic hedgehog regulates growth and morphogenesis of the tooth. Development 127, 4775–4785 [DOI] [PubMed] [Google Scholar]
- De Moerlooze L., Spencer-Dene B., Revest J. M., Hajihosseini M., Rosewell I., Dickson C. (2000). An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development 127, 483–492 [DOI] [PubMed] [Google Scholar]
- Evans A. R., Wilson G. P., Fortelius M., Jernvall J. (2007). High-level similarity of dentitions in carnivorans and rodents. Nature 445, 78–81 [DOI] [PubMed] [Google Scholar]
- Fliniaux I., Mikkola M. L., Lefebvre S., Thesleff I. (2008). Identification of dkk4 as a target of Eda-A1/Edar pathway reveals an unexpected role of ectodysplasin as inhibitor of Wnt signalling in ectodermal placodes. Dev. Biol. 320, 60–71 [DOI] [PubMed] [Google Scholar]
- Fujimoto A., Ohashi J., Nishida N., Miyagawa T., Morishita Y., Tsunoda T., Kimura R., Tokunaga K. (2008). A replication study confirmed the EDAR gene to be a major contributor to population differentiation regarding head hair thickness in Asia. Hum. Genet. 124, 179–185 [DOI] [PubMed] [Google Scholar]
- Gaide O., Schneider P. (2003). Permanent correction of an inherited ectodermal dysplasia with recombinant EDA. Nat. Med. 9, 614–618 [DOI] [PubMed] [Google Scholar]
- Grüneberg H. (1966). The molars of the tabby mouse, and a test of the ‘single-active X-chromosome’ hypothesis. J. Embryol. Exp. Morphol. 15, 223–244 [PubMed] [Google Scholar]
- Häärä O., Fujimori S., Schmidt-Ullrich R., Hartmann C., Thesleff I., Mikkola M. L. (2011). Ectodysplasin and Wnt pathways are required for salivary gland branching morphogenesis. Development 138, 2681–2691 [DOI] [PubMed] [Google Scholar]
- Harjunmaa E., Kallonen A., Voutilainen M., Hämäläinen K., Mikkola M. L., Jernvall J. (2012). On the difficulty of increasing dental complexity. Nature 483, 324–327 [DOI] [PubMed] [Google Scholar]
- Harris M. P., Rohner N., Schwarz H., Perathoner S., Konstantinidis P., Nüsslein-Volhard C. (2008). Zebrafish eda and edar mutants reveal conserved and ancestral roles of ectodysplasin signaling in vertebrates. PLoS Genet. 4, e1000206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh S. H., Jones J., Warchol M. E., Ornitz D. M. (2012). Differentiation of the lateral compartment of the cochlea requires a temporally restricted FGF20 signal. PLoS Biol. 10, e1001231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järvinen E., Salazar-Ciudad I., Birchmeier W., Taketo M. M., Jernvall J., Thesleff I. (2006). Continuous tooth generation in mouse is induced by activated epithelial Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 103, 18627–18632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernvall J., Kettunen P., Karavanova I., Martin L. B., Thesleff I. (1994). Evidence for the role of the enamel knot as a control center in mammalian tooth cusp formation: non-dividing cells express growth stimulating Fgf-4 gene. Int. J. Dev. Biol. 38, 463–469 [PubMed] [Google Scholar]
- Kangas A. T., Evans A. R., Thesleff I., Jernvall J. (2004). Nonindependence of mammalian dental characters. Nature 432, 211–214 [DOI] [PubMed] [Google Scholar]
- Kassai Y., Munne P., Hotta Y., Penttilä E., Kavanagh K., Ohbayashi N., Takada S., Thesleff I., Jernvall J., Itoh N. (2005). Regulation of mammalian tooth cusp patterning by ectodin. Science 309, 2067–2070 [DOI] [PubMed] [Google Scholar]
- Kavanagh K. D., Evans A. R., Jernvall J. (2007). Predicting evolutionary patterns of mammalian teeth from development. Nature 449, 427–432 [DOI] [PubMed] [Google Scholar]
- Kettunen P., Thesleff I. (1998). Expression and function of FGFs-4, -8, and -9 suggest functional redundancy and repetitive use as epithelial signals during tooth morphogenesis. Dev. Dyn. 211, 256–268 [DOI] [PubMed] [Google Scholar]
- Kettunen P., Karavanova I., Thesleff I. (1998). Responsiveness of developing dental tissues to fibroblast growth factors: expression of splicing alternatives of FGFR1, -2, -3, and of FGFR4; and stimulation of cell proliferation by FGF-2, -4, -8, and -9. Dev. Genet. 22, 374–385 [DOI] [PubMed] [Google Scholar]
- Kettunen P., Laurikkala J., Itäranta P., Vainio S., Itoh N., Thesleff I. (2000). Associations of FGF-3 and FGF-10 with signaling networks regulating tooth morphogenesis. Dev. Dyn. 219, 322–332 [DOI] [PubMed] [Google Scholar]
- Klein O. D., Minowada G., Peterkova R., Kangas A., Yu B. D., Lesot H., Peterka M., Jernvall J., Martin G. R. (2006). Sprouty genes control diastema tooth development via bidirectional antagonism of epithelial-mesenchymal FGF signaling. Dev. Cell 11, 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein O. D., Lyons D. B., Balooch G., Marshall G. W., Basson M. A., Peterka M., Boran T., Peterkova R., Martin G. R. (2008). An FGF signaling loop sustains the generation of differentiated progeny from stem cells in mouse incisors. Development 135, 377–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppinen P., Pispa J., Laurikkala J., Thesleff I., Mikkola M. L. (2001). Signaling and subcellular localization of the TNF receptor Edar. Exp. Cell Res. 269, 180–192 [DOI] [PubMed] [Google Scholar]
- Kristenová-Cermáková P., Peterka M., Lisi S., Lesot H., Peterková R. (2002). Postnatal lower jaw dentition in different phenotypes of tabby mice. Connect. Tissue Res. 43, 283–288 [DOI] [PubMed] [Google Scholar]
- Laurikkala J., Mikkola M., Mustonen T., Åberg T., Koppinen P., Pispa J., Nieminen P., Galceran J., Grosschedl R., Thesleff I. (2001). TNF signaling via the ligand-receptor pair ectodysplasin and edar controls the function of epithelial signaling centers and is regulated by Wnt and activin during tooth organogenesis. Dev. Biol. 229, 443–455 [DOI] [PubMed] [Google Scholar]
- Laurikkala J., Pispa J., Jung H. S., Nieminen P., Mikkola M., Wang X., Saarialho-Kere U., Galceran J., Grosschedl R., Thesleff I. (2002). Regulation of hair follicle development by the TNF signal ectodysplasin and its receptor Edar. Development 129, 2541–2553 [DOI] [PubMed] [Google Scholar]
- Lefebvre S., Fliniaux I., Schneider P., Mikkola M. L. (2012). Identification of ectodysplasin target genes reveals the involvement of chemokines in hair development. J. Invest. Dermatol. 132, 1094–1102 [DOI] [PubMed] [Google Scholar]
- Line S. R. (2003). Variation of tooth number in mammalian dentition: connecting genetics, development, and evolution. Evol. Dev. 5, 295–304 [DOI] [PubMed] [Google Scholar]
- Liu F., Chu E. Y., Watt B., Zhang Y., Gallant N. M., Andl T., Yang S. H., Lu M. M., Piccolo S., Schmidt-Ullrich R., et al. (2008). Wnt/beta-catenin signaling directs multiple stages of tooth morphogenesis. Dev. Biol. 313, 210–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkola M. L. (2009). Molecular aspects of hypohidrotic ectodermal dysplasia. Am. J. Med. Genet. A 149, 2031–2036 [DOI] [PubMed] [Google Scholar]
- Mou C., Jackson B., Schneider P., Overbeek P. A., Headon D. J. (2006). Generation of the primary hair follicle pattern. Proc. Natl. Acad. Sci. USA 103, 9075–9080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustonen T., Pispa J., Mikkola M. L., Pummila M., Kangas A. T., Pakkasjärvi L., Jaatinen R., Thesleff I. (2003). Stimulation of ectodermal organ development by Ectodysplasin-A1. Dev. Biol. 259, 123–136 [DOI] [PubMed] [Google Scholar]
- Mustonen T., Ilmonen M., Pummila M., Kangas A. T., Laurikkala J., Jaatinen R., Pispa J., Gaide O., Schneider P., Thesleff I., et al. (2004). Ectodysplasin A1 promotes placodal cell fate during early morphogenesis of ectodermal appendages. Development 131, 4907–4919 [DOI] [PubMed] [Google Scholar]
- Närhi K., Thesleff I. (2010). Explant culture of embryonic craniofacial tissues: analyzing effects of signaling molecules on gene expression. Methods Mol. Biol. 666, 253–267 [DOI] [PubMed] [Google Scholar]
- Niswander L., Martin G. R. (1992). Fgf-4 expression during gastrulation, myogenesis, limb and tooth development in the mouse. Development 114, 755–768 [DOI] [PubMed] [Google Scholar]
- Ornitz D. M., Yin Y. (2012). Signaling networks regulating development of the lower respiratory tract. Cold Spring Harb. Perspect. Biol. 4, a008318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn H. F. (1888). The evolution of mammalian molars to and from the tritubercular type. Am. Nat. 22, 1067–1079 [Google Scholar]
- Pispa J., Jung H. S., Jernvall J., Kettunen P., Mustonen T., Tabata M. J., Kere J., Thesleff I. (1999). Cusp patterning defect in Tabby mouse teeth and its partial rescue by FGF. Dev. Biol. 216, 521–534 [DOI] [PubMed] [Google Scholar]
- Porntaveetus T., Otsuka-Tanaka Y., Basson M. A., Moon A. M., Sharpe P. T., Ohazama A. (2011). Expression of fibroblast growth factors (Fgfs) in murine tooth development. J. Anat. 218, 534–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochazka J., Pantalacci S., Churava S., Rothova M., Lambert A., Lesot H., Klein O., Peterka M., Laudet V., Peterkova R. (2010). Patterning by heritage in mouse molar row development. Proc. Natl. Acad. Sci. USA 107, 15497–15502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pummila M., Fliniaux I., Jaatinen R., James M. J., Laurikkala J., Schneider P., Thesleff I., Mikkola M. L. (2007). Ectodysplasin has a dual role in ectodermal organogenesis: inhibition of Bmp activity and induction of Shh expression. Development 134, 117–125 [DOI] [PubMed] [Google Scholar]
- Roehl H., Nüsslein-Volhard C. (2001). Zebrafish pea3 and erm are general targets of FGF8 signaling. Curr. Biol. 11, 503–507 [DOI] [PubMed] [Google Scholar]
- Salazar-Ciudad I., Jernvall J. (2002). A gene network model accounting for development and evolution of mammalian teeth. Proc. Natl. Acad. Sci. USA 99, 8116–8120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Ciudad I., Jernvall J. (2010). A computational model of teeth and the developmental origins of morphological variation. Nature 464, 583–586 [DOI] [PubMed] [Google Scholar]
- Sarkar L., Sharpe P. T. (1999). Expression of Wnt signalling pathway genes during tooth development. Mech. Dev. 85, 197–200 [DOI] [PubMed] [Google Scholar]
- Schmidt-Ullrich R., Aebischer T., Hülsken J., Birchmeier W., Klemm U., Scheidereit C. (2001). Requirement of NF-kappaB/Rel for the development of hair follicles and other epidermal appendices. Development 128, 3843–3853 [DOI] [PubMed] [Google Scholar]
- Sun X., Lewandoski M., Meyers E. N., Liu Y.-H., Maxson R. E., Martin G. R. (2000). Conditional inactivation of Fgf4 reveals complexity of signalling during limb bud development. Nat. Genet. 25, 83–86 [DOI] [PubMed] [Google Scholar]
- Tucker A. S., Headon D. J., Schneider P., Ferguson B. M., Overbeek P., Tschopp J., Sharpe P. T. (2000). Edar/Eda interactions regulate enamel knot formation in tooth morphogenesis. Development 127, 4691–4700 [DOI] [PubMed] [Google Scholar]
- Tummers M., Thesleff I. (2009). The importance of signal pathway modulation in all aspects of tooth development. J. Exp. Zoolog. B Mol. Dev. Evol. 312, 309–319 [DOI] [PubMed] [Google Scholar]
- Wang X. P., Suomalainen M., Jorgez C. J., Matzuk M. M., Wankell M., Werner S., Thesleff I. (2004). Modulation of activin/bone morphogenetic protein signaling by follistatin is required for the morphogenesis of mouse molar teeth. Dev. Dyn. 231, 98–108 [DOI] [PubMed] [Google Scholar]
- Wang X. P., Suomalainen M., Felszeghy S., Zelarayan L. C., Alonso M. T., Plikus M. V., Maas R. L., Chuong C. M., Schimmang T., Thesleff I. (2007). An integrated gene regulatory network controls stem cell proliferation in teeth. PLoS Biol. 5, e159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Lin Y., Itäranta P., Yagi A., Vainio S. (2001). Expression of Sprouty genes 1, 2 and 4 during mouse organogenesis. Mech. Dev. 109, 367–370 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}