

Abstract
Mutations in the human Shwachman-Bodian-Diamond syndrome (SBDS) gene cause defective ribosome assembly and are associated with exocrine pancreatic insufficiency, chronic neutropenia and skeletal defects. However, the mechanism underlying these phenotypes remains unclear. Here we show that knockdown of the zebrafish sbds ortholog fully recapitulates the spectrum of developmental abnormalities observed in the human syndrome, and further implicate impaired proliferation of ptf1a-expressing pancreatic progenitor cells as the basis for the observed pancreatic phenotype. It is thought that diseases of ribosome assembly share a p53-dependent mechanism. However, loss of p53 did not rescue the developmental defects associated with loss of zebrafish sbds. To clarify the molecular mechanisms underlying the observed organogenesis defects, we performed transcriptional profiling to identify candidate downstream mediators of the sbds phenotype. Among transcripts displaying differential expression, functional group analysis revealed marked enrichment of genes related to ribosome biogenesis, rRNA processing and translational initiation. Among these, ribosomal protein L3 (rpl3) and pescadillo (pes) were selected for additional analysis. Similar to knockdown of sbds, knockdown or mutation of either rpl3 or pes resulted in impaired expansion of pancreatic progenitor cells. The pancreatic phenotypes observed in rpl3- and pes-deficient embryos were also independent of p53. Together, these data suggest novel p53-independent roles for ribosomal biogenesis genes in zebrafish pancreas development.
Keywords: Shwachman-Diamond syndrome, Ribosome, Zebrafish, p53 (tp53), rpl3, pescadillo, sbds
INTRODUCTION
Shwachman-Diamond syndrome (SDS) is an autosomal recessive, multi-system pediatric disorder characterized by pancreatic insufficiency, skeletal abnormalities and chronic neutropenia. Infants present early with failure to thrive and require treatment for chronic infection and pancreatic enzyme deficiency. In 2002, the causal gene for SDS, SBDS, was identified (Boocock et al., 2003). SBDS is a highly conserved, essential gene that functions in late maturation of the large 60S ribosomal subunit. Studies in a variety of organisms and SDS patient lymphoblasts suggest that mutations in SBDS result in ribosomal subunit joining defects associated with altered subunit ratios (Zhang et al., 2006; Menne et al., 2007; Finch et al., 2011; Wong et al., 2011).
Ribosome biogenesis is normally accomplished in a tightly controlled, multi-step process. In addition to the 80 known ribosomal proteins (RPs), Sbds is one of ∼150 non-ribosomal proteins required to assemble a functional 80S ribosome (Deisenroth and Zhang, 2010). In addition to SDS, other diseases with causal mutations in genes related to ribosome assembly have been described and are collectively referred to as ribosomopathies (Luft, 2010; Narla and Ebert, 2010). One well-characterized ribosomopathy, Diamond-Blackfan anemia (DBA), is commonly associated with mutations in the RP gene RPS19 (Draptchinskaia et al., 1999). Models of DBA in mice and zebrafish have suggested a role for p53 (Tp53/Trp53) in mediating the disease phenotype (Danilova et al., 2008; McGowan et al., 2008; Uechi et al., 2008). Mice with mutations in Rps19 are phenotypically rescued by homozygous loss of p53 (McGowan et al., 2008), as are other mouse models of human ribosomopathies, including 5q-syndrome and Treacher Collins syndrome (Jones et al., 2008; Barlow et al., 2010). Thus, it is generally appreciated that ribosomopathies mediate their phenotype through p53-dependent mechanisms.
In order to determine whether the SDS phenotype exhibits similar p53 dependence, we used antisense morpholinos to recapitulate the composite SDS phenotype in zebrafish. Extending a previous report of the zebrafish sbds morphant phenotype (Venkatasubramani and Mayer, 2008), we found that sbds knockdown resulted in loss of neutrophils, skeletal defects and pancreatic hypoplasia, as well as changes in ribosomal subunit ratios. In addition, we have identified impaired proliferation but normal differentiation of ptf1a-expressing pancreatic progenitor cells as the primary defect underlying the pancreatic hypoplasia phenotype. Moreover, the observed organogenesis defects were unaffected by simultaneous inactivation of either p53 or its Δ113p53 variant, suggesting that SDS is a largely p53-independent ribosomopathy.
As a means to more fully understand the consequences of sbds loss, we used a discovery-based microarray platform to evaluate global transcriptional changes in sbds morphants. Functional group analysis suggested that loss of sbds results in widespread changes in the expression of genes related to ribosome biogenesis, rRNA processing and translational initiation. Among the genes displaying altered expression in sbds morphants, we performed additional functional analysis of ribosomal protein L3 (rpl3) and pescadillo (pes). Using antisense morpholinos or available mutants, we found that inactivation of either rpl3 or pes phenocopied the sbds pancreatic progenitor phenotype, also in a p53-independent manner. Together, these data identify novel developmental roles for genes related to ribosome biogenesis, independent of p53.
MATERIALS AND METHODS
Fish strains, genotyping and morpholinos
Fish were raised using standard husbandry procedures. The following strains were used: Tg BAC ptf1a:eGFPjh1 (herein ptf1a:eGFP) (Godinho et al., 2007), Tg Tol2 ins:mCherryjh2 (herein ins:mCherry) (Pisharath et al., 2007), Tg (gcga:GFP)ia1 (herein gcga:GFP) (Pauls et al., 2007), Tg (T2KTp1hglob:hmgb1-mCherry)jh11 (herein Tp1:hmgb1-mCherry) (Parsons et al., 2009), Tg (kdrl:GRCFP)zn1 (Cross et al., 2003), tp53zdf1 [M214K (Berghmans et al., 2005), obtained from the Zebrafish International Resource Center (ZIRC), Eugene, OR, USA], peshi2Tg (Allende et al., 1996) (ZIRC) and rpl3hi2347 (ZIRC). Tail fins of adult fish were digested in low TE (10 mM Tris-HCl pH 7.5, 1 mM EDTA) plus proteinase K. PCR genotyping was performed to confirm the genotype of tp53zdf1 homozygous embryos. peshi2Tg fish were functionally genotyped for clutches with 25% phenotypic embryos. Morpholinos (MOs) included: SbdsATG MO, SbdsSpl MO, p53ATG MO, p53Spl MO, Rpl3ATG MO and PesATG MO, all obtained from GeneTools (Philomath, OR, USA). MOs and genotyping primers are listed in supplementary material Table S1.
Polyribosome analysis
Zebrafish embryos were either mock injected or injected with SbdsATG MO 48 hours prior to polyribosome analysis. Approximately 150 embryos were washed and de-yolked in ice-cold PBS containing 150 μg/ml cycloheximide. Embryos were mechanically dissociated in ice-cold lysis buffer (300 mM NaCl, 15 mM Tris-HCl pH 7.5, 15 mM MgCl2, 0.75% Triton X-100, 100 μg/ml cycloheximide, 1 mg/ml heparin) and incubated on ice for 10 minutes. Lysates were cleared at 16,300 g at 4°C for 5 minutes. Cleared lysates were layered onto 10-50% sucrose gradients (in lysis buffer without the Triton X-100) with a 60% sucrose cushion. Gradients were spun in an SW 41 rotor (Beckman Coulter) at 40,000 rpm at 4°C for 3 hours and fractionated with an Isco Foxy R1/UA-6 detector system. Polyribosome profiles were analyzed using a custom macro in IGOR Pro (WaveMetrics).
Immunofluorescence
Whole-mount immunofluorescence was performed as previously described (Lin et al., 2004).
In situ hybridization
Whole-mount in situ hybridization was conducted as previously described (Lin et al., 2004). To perform in situ hybridization on paraffin sections, 12-μm sections were deparaffinized in Histoclear (Fisher Scientific) and rehydrated. Sections were prehybridized (50% formamide, 5× SSC pH 4.5 with citric acid, 50 μg/ml yeast tRNA, 1% SDS, 50 μg/ml heparin) at 55°C for 2 hours. Primers used to generate in situ probes are listed in supplementary material Table S1. Antisense in situ probes were boiled in prehybridization buffer (10 μl probe/ml), added to tissue sections, covered with a coverslip and hybridized at 68°C in a humidity chamber overnight. Tissue sections were washed in 5× SSC at 68°C, followed by 0.2× SSC at 68°C and maelic acid buffer (MAB) at room temperature. Sections were blocked in 10% sheep serum diluted in MAB for 1 hour and incubated with anti-digoxigenin alkaline phosphatase antibody at 1:5000 overnight at 4°C. Slides were washed with MAB containing 0.1% Tween 20 and signal detected using BM Purple (Roche) at room temperature. Reactions were stopped with 1 mM EDTA pH 8.0, dehydrated, mounted and imaged.
Alcian Blue and Alizarin Red staining
Embryos were stained as described previously (Walker and Kimmel, 2007).
cDNA microarray analysis
Embryos were carefully staged at 24 hpf and RNA was extracted from ∼50 embryos per condition in Trizol (Invitrogen), followed by purification using RNA Miniprep columns (Qiagen). Four experimental replicates were analyzed. cDNA microarrays were performed at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins Microarray Core Facility using 4×44K zebrafish gene expression microarray slides (Agilent, Santa Clara, CA, USA). Differential expression was calculated using a moderated eBayes t-test as implemented in the Limma Bioconductor package (Smyth, 2004). The resulting P-values were false discovery rate (FDR)-adjusted across the whole microarray dataset to correct for multiple testing. Statistically significant changes in gene expression were determined at three different levels of stringency (P<0.001, P<0.01, P<0.05). Gene set enrichment analysis (GSEA) was performed with the preranked moderated t statistics obtained from Limma analysis (Smyth, 2004), according to the GO functional categories for the indicated microarray comparison. Additional analysis was carried out using Partek Genomics Suite (St Louis, MO, USA). Microarray data are available at Gene Expression Omnibus under accession number GSE39399.
RESULTS
Knockdown of zebrafish Sbds recapitulates the developmental defects observed in SDS
The sbds ortholog in zebrafish is highly homologous to the mouse and human Sbds/SBDS genes. Previously, it was shown that zebrafish sbds is expressed in the area of the pancreas, with highest expression in peri-pancreatic mesenchymal tissues (Venkatasubramani and Mayer, 2008). We confirmed this expression pattern at 5 days postfertilization (dpf) (Fig. 1A), and further identified expression in the acinar epithelia of adult zebrafish (Fig. 1B). In addition, analysis of publicly available mouse ChIP-Seq datasets (http://www.betacell.org/resources/data/studies/view.php?study_id=3994) revealed overlapping binding of Ptf1a and Rbpj1 (two members of the larger pancreas-specific PTF1 transcriptional complex) in the 5′ promoter region of the murine Sbds locus (supplementary material Fig. S1A).
Fig. 1.

Zebrafish sbds is an essential developmental gene with high-level expression in pancreas and surrounding tissues. (A) Expression of sbds examined by in situ hybridization in wild-type zebrafish embryos at 120 hpf. The boxed pancreatic region is shown at higher magnification to illustrate pancreatic and peri-pancreatic expression (arrowheads) of sbds. (B) Adult zebrafish pancreas (boxed) expresses sbds. (C) SbdsSpl MO results in durable loss of sbds transcript as assessed by RT-PCR. (D) Survival curve for recipients of SbdsATG MO illustrates near complete embryonic lethality by 7 dpf, as compared with mock injected embryos. (E) Ribosome profile of lysate prepared from 48-hpf control and SbdsATG MO-injected embryos. (F) Quantification of ribosomal subunit ratios, monosomes and polysomes in control and SbdsATG MO-injected embryos. Error bars indicate mean ± s.e.m. i, intestine; L, liver; P, pancreas.
To evaluate the role of Sbds during development we designed two morpholinos (MOs), one targeting the start codon (SbdsATG MO) and one targeting the splice junction between exon 2 and exon 3 (SbdsSpl MO) of zebrafish sbds. Injection of the SbdsSpl MO resulted in a loss of sbds transcript, as assessed by RT-PCR, that persisted beyond 72 hours postfertilization (hpf) (Fig. 1C). Although gene inactivation following MO injection is typically transient, embryos injected with either MO did not recover from the early loss of sbds. Embryos injected with the SbdsATG MO developed significant edema at later stages of development and did not survive past 7 dpf (Fig. 1D).
Work in yeast (Menne et al., 2007), mouse liver (Finch et al., 2011), Dictyostelium and SDS patient lymphoblasts (Wong et al., 2011) has suggested a role for Sbds in late ribosome assembly, with loss of Sbds function resulting in a subunit joining defect. To identify whether our SbdsATG MO-injected embryos exhibited similar ribosome assembly defects, we generated ribosome profiles at 48 hpf under native polysome assembly conditions. We identified altered ratios of the 40S subunit, 60S subunit and 80S monosome, as well as a decrease in the number of heavy polysomes in the SbdsATG MO-injected embryos (Fig. 1E,F, supplementary material Fig. S1B,C). This reduction in the number of heavy polysomes suggests an overall loss of fully functional ribosomes in SbdsATG MO-injected embryos.
Using the SbdsATG MO we evaluated three components of the developmental phenotype observed in SDS: the number of neutrophils, the size of the exocrine and endocrine pancreas, and the structure of the developing cartilage and bone. As in other vertebrates, myeloperoxidase (mpo; mpx – Zebrafish Information Network) is an early marker of neutrophils in zebrafish (Chen and Zon, 2009). We therefore evaluated neutrophil numbers in control and SbdsATG MO-injected embryos using an antisense mpo in situ probe. In mock injected embryos at 24 hpf, mpo-positive neutrophils were observed in the anterior lateral mesoderm (Fig. 2A, top arrow) and the intermediate cell mass (Fig. 2A, bottom arrow). By contrast, we observed a profound loss of mpo-positive neutrophils in SbdsATG MO-injected embryos (Fig. 2A). Other aspects of hematopoietic development were unaltered in SbdsATG MO-injected embryos, including normal numbers of L-plastin (Lcp1)-positive macrophages, normal vasculature, normal expression of gata1 in erythroid progenitors and normal expression of the neutrophil/macrophage progenitor marker spi1 (pu.1) (supplementary material Fig. S2A-C,E,F). As in other species, zebrafish neutrophils are dynamically recruited to sites of injury (Mathias et al., 2006; Renshaw et al., 2006). Even when SbdsATG MO-injected embryos were subjected to epithelial injury, no recruitment of mpo-positive neutrophils was observed (supplementary material Fig. S2D). Thus, we conclude that loss of Sbds results in a specific defect in the neutrophil lineage at a point beyond the formation of common neutrophil/macrophage progenitors.
Fig. 2.
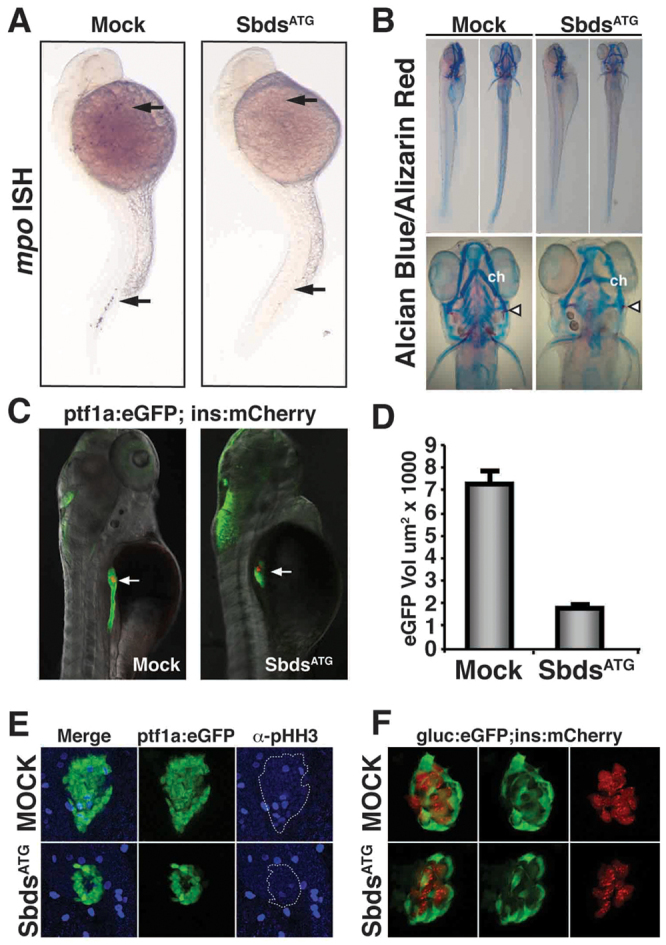
sbds knockdown in zebrafish embryos recapitulates human SDS organogenesis phenotypes. (A) At 24 hpf, loss of neutrophils in SbdsATG MO-injected embryos is revealed by in situ hybridization for the neutrophil marker mpo. mpo-positive cells were absent from both the anterior lateral mesoderm and the intermediate cell mass (top and bottom arrows, respectively). (B) At 120 hpf, abnormal cartilage of the gill arches and cerotohyal (ch) cartilage and bone (arrowheads) were observed in SbdsATG MO-injected embryos. (C) Visualization of ptf1-expressing pancreatic progenitor cells and differentiated beta cells in 72-hpf ptf1a:eGFP;ins:mCherry embryos (arrows). SbdsATG MO-injected embryos showed defects in ptf1-expressing pancreatic progenitors (green), whereas beta cells in the principal islet appeared normal (red). (D) Quantification of the volume of ptf1a:eGFP-expressing cells reveals an almost 75% reduction of pancreatic volume in SbdsATG MO-injected embryos. Error bars indicate s.e.m. (E) Immunofluorescence of phospho-histone H3 (pHH3, blue) marks proliferation in the exocrine pancreatic mass. Proliferation was decreased in the SbdsATG MO-injected embryos at 72 hpf. (F) High-resolution images of the pancreatic islet using gcga:eGFPia1;ins:mCherry double-transgenic zebrafish reveal normal islet architecture with peripheral alpha cells (green) and central beta cells (red) in SbdsATG MO-injected embryos at 72 hpf.
When we evaluated the skeletal phenotype of SbdsATG MO-injected embryos versus controls at 5 dpf by staining with Alcian Blue (cartilage) and Alizarin Red (bone), we detected defects in both the cartilage and bone. Both the cerotohyal cartilage of the lower jaw and the ceratobranchial cartilage of the gill arches (Kimmel et al., 2001) were malformed (Fig. 2B). At 5 dpf, the opercal, a bone of the inner ear, was misshapen and thinner in SbdsATG MO-injected embryos than in control embryos (Fig. 2B, arrowheads).
Finally, we evaluated the effect of SbdsATG MO injection on the developing pancreas using a ptf1a:eGFPjh1 transgenic line to visualize early pancreatic progenitor cells and later differentiating acinar cells (Park et al., 2008), in combination with an ins:mCherryjh2 transgenic line (Pisharath et al., 2007) to visualize beta cells in the principal islet. At 72 hpf, embryos injected with the SbdsATG MO exhibited dramatic pancreatic hypoplasia, initially manifested by a marked decrease in the number of undifferentiated ptf1a-expressing pancreatic progenitor cells (Fig. 2C). Quantification of the volume of eGFP-positive cells in ptf1a:eGFP larvae at 72 hpf revealed that the SbdsATG MO-injected embryos had an almost 75% reduction in the volume of ptf1a-expressing pancreatic progenitor cells compared with control embryos (Fig. 2D). Despite their dramatic reduction in number, eGFP-positive pancreatic progenitor cells were able to appropriately migrate away from the gut tube and associate with the preserved principal islet at 48 hpf (supplementary material Fig. S3A). To further document the pancreatic phenotype in the SbdsATG MO-injected embryos, we observed a dose-dependent reduction in pancreatic volume in ptf1a:eGFP larvae over multiple doses of both the SbdsATG MO (supplementary material Fig. S3B) and an additional splice-blocking MO, SbdsSpl (supplementary material Fig. S3C,D).
Loss of Sbds results in defective proliferation of pancreatic progenitors
Although the human SDS pancreatic phenotype is well characterized, its developmental basis has not been elucidated. To further explore the mechanism underlying the hypoplastic pancreas phenotype of SbdsATG MO-injected embryos, we analyzed phospho-histone H3 (pHH3) labeling of ptf1a-expressing pancreatic progenitor cells at 48 hpf. Similar to previous reports suggesting that Sbds might influence cellular proliferation (Zhang et al., 2006; Orelio et al., 2009), we observed a decrease in the number of pHH3-positive mitotic cells within the ptf1a:eGFP-defined pancreatic progenitor population at 48 hpf (Fig. 2E), with several sbds morphant embryos showing a complete absence of pHH3 labeling within the ptf1a:eGFP expression domain. When the total volume of pHH3-positive nuclei was normalized to the total volume of eGFP-expressing pancreatic progenitor cells, sbds morphants displayed a 39% reduction in pHH3 labeling. In contrast to this defect in the proliferation of early pancreatic progenitor cells, we observed no defects in the ability of progenitor cells to undergo normal exocrine differentiation. At 72 hpf, the digestive enzymes carboxypeptidase and trypsin were detected even in the reduced pancreatic mass of SbdsATG MO-injected embryos (supplementary material Fig. S3E,F).
In contrast to the abnormal expansion of ptf1a-expressing pancreatic progenitor cells, ptf1a-independent endocrine progenitors were able to appropriately expand and form the principal islet in SbdsATG MO-injected embryos (Fig. 2F). Using the transgenic zebrafish line gcga:GFPia1, which expresses GFP from the glucagon a promoter, we were able to visualize the alpha cells of the endocrine pancreas in conjunction with ins:mCherryjh2-expressing beta cells. In both control and SbdsATG MO-injected embryos, alpha cells appropriately localized to the periphery of the islet surrounding a central core of beta cells (Fig. 2F). Despite normal islet architecture, we observed a tendency toward diminished islet size in SbdsATG MO-injected embryos, although not at a level of statistical significance (P=0.072) (supplementary material Fig. S3G). This result mirrors the recently reported conditional knockout of sbds, where the adult islet structure is normal but islet volume is modestly reduced (Tourlakis et al., 2012).
Islet development in zebrafish involves early specification (prior to 24 hpf) of the principal islet, followed by the formation of secondary islets (after 5 dpf). These secondary islets arise from Notch-responsive progenitor cells that reside in the main pancreatic duct (Parsons et al., 2009; Wang et al., 2011). Although our SbdsATG MO-injected embryos die before secondary islet formation occurs, we were able to determine the influence of Sbds knockdown on this Notch-responsive islet progenitor population using Tp1:hmgb1-mCherry transgenic Notch reporter fish. Following Sbds knockdown, Tp1:hmgb1-mCherry-expressing cells failed to form a main pancreatic duct, thus corresponding to the failure of ptf1a-expressing progenitor cells to undergo normal morphogenesis. However, there was no change in the ratio of eGFP to mCherry volume in SbdsATG MO-treated ptf1a:eGFP;Tp1;hmgb1-mCherry double-transgenic embryos (supplementary material Fig. S3H,I). Although the early lethality of Sbds knockdown precludes direct observation of later-occurring secondary islet formation, these observations suggest that the relative number of secondary islet progenitor cells is retained, even in the absence of Sbds.
Loss of p53 does not rescue the sbds organogenesis phenotypes
When normal ribosome biogenesis is disrupted, a resulting p53 response is commonly accepted to play a role in the associated disease phenotypes (Lohrum et al., 2003; Dai and Lu, 2004; Jin et al., 2004; Danilova et al., 2008; Jones et al., 2008; Barlow et al., 2010; Duan et al., 2011; Sasaki et al., 2011). To determine whether SDS disease phenotypes are similarly p53 dependent, we evaluated the influence of p53 inactivation on the organogenesis defects induced by SbdsATG MO injection, using both MO-mediated p53 knockdown and available p53 mutant alleles.
As previously reported (Robu et al., 2007), injection of 1 ng of a p53ATG MO had no adverse effect on developing zebrafish embryos. Overall, co-injection of the p53ATG MO in combination with the SbdsATG MO resulted in healthier-appearing embryos than following injection of SbdsATG MO alone. At 48 hpf we observed a twisted tail phenotype in SbdsATG MO-injected embryos, and co-injection of p53ATG MO relieved this axis defect (Fig. 3A). To further validate the efficacy of the p53 knockdown, we performed semi-quantitative PCR for the p53 target genes Δ113p53 and p21 (cdkn1a – Zebrafish Information Network). Injection of the SbdsATG MO increased both Δ113p53 and p21 expression, but this expression was attenuated when the SbdsATG MO and p53ATG MO were co-injected (Fig. 3B).
Fig. 3.

Loss of p53 does not rescue sbds organogenesis defects. (A) The body curvature observed in SbdsATG MO-injected zebrafish embryos is attenuated in SbdsATG/p53ATG MO-injected embryos at 48 hpf. (B) The p53 transcriptional targets Δ113p53 and p21 are induced upon injection of SbdsATG MO, as assessed by semi-quantitative PCR; co-injection of p53ATG MO abrogates this response. Error bars indicate s.e.m. (C) The 72-hpf pancreatic progenitor phenotype induced by the SbdsATG MO is not rescued by co-injection of p53ATG MO (arrows). (D) The absence of mpo-expressing neutrophils in the intermediate cell mass of SbdsATG/p53ATG MO-injected embryos was detected at 24 hpf (arrowheads). (E) Co-injection of p53Spl MO and SbdsATG MO into ptf1a:eGFP;ins:mCherry embryos does not rescue the defective expansion of pancreatic progenitor cells (arrows). (F) Genetic loss of p53 in p53zdf1/zdf1 embryos does not rescue the SbdsATG MO-induced neutrophil phenotype, as assessed by in situ hybridization for mpo at 24 hpf (arrowheads). (G) The SbdsATG MO-induced pancreatic progenitor defect is not rescued in p53zdf1/zdf1;ptf1a:eGFP;ins:mCherry embryos at 72 hpf.
Next we evaluated whether knockdown of p53 could rescue SbdsATG MO-induced organogenesis phenotypes. Despite the improved gross appearance of the embryos, co-injection of SbdsATG MO and p53ATG MO did not rescue the pancreatic progenitor defect (Fig. 3C, supplementary material Fig. S4A). Likewise, mpo-positive neutrophils were not rescued by concomitant p53 knockdown (Fig. 3D, supplementary material Fig. S4B). Finally, the cartilage phenotype was not rescued in embryos co-injected with SbdsATG and p53ATG MOs, as assessed by Alcian Blue staining at 72 hpf (supplementary material Fig. S4C,D). We conclude that even though p53 knockdown effectively minimizes the pleiotropic effects associated with Sbds knockdown, it is unable to rescue SbdsATG MO-induced organogenesis phenotypes.
Δ113p53 (human) is both a shortened isoform and target gene of full-length p53 (Chen et al., 2005; Chen et al., 2009). Although full-length p53 is dispensable for normal development, work in the zebrafish mutant def (digestive organ expansion factor, or diexf) has demonstrated that Δ113p53 plays an important role in endoderm development. def mutants have properly differentiated but severely hypoplastic digestive organs, which are partially rescued by knockdown of Δ113p53 (Chen et al., 2005).
To evaluate the role of Δ113p53 in the zebrafish sbds phenotype, we used a splice-blocking p53 MO targeting the exon 5-intron 5 boundary (Chen et al., 2005). This p53Spl MO effectively depletes both full-length p53 and Δ113p53. As we observed no rescue associated with loss of full-length p53, any ability of the p53Spl MO to rescue normal pancreas development was expected to be the result of Δ113p53 knockdown. Co-injection of the SbdsATG and p53Spl MOs into ptf1a:eGFP;ins:mCherry embryos did not rescue defective pancreatic organogenesis (Fig. 3E, supplementary material Fig. S4E). RT-PCR of p53Spl MO-injected embryos confirmed the near complete loss of the Δ113p53 transcript, its absence persisting through 72 hpf (supplementary material Fig. S4F). Thus, we conclude that knockdown of p53 and/or Δ113p53 is unable to rescue the pancreas progenitor defect observed in SbdsATG MO-injected embryos.
In these p53 knockdown experiments, we noted that co-injection of SbdsATG and p53ATG MOs reduced, but did not eliminate, transcription of the p53 target genes Δ113p53 and p21 (Fig. 3B). To be certain that the persistent SbdsATG MO-induced phenotype observed following p53ATG MO injection was not due to residual p53 activity, we extended our p53 loss-of-function analysis using an available p53 null mutation. The zebrafish tp53zdf1 allele, which was generated by ENU mutagenesis, harbors an M216K missense mutation in the p53 DNA-binding domain, preventing activation of p53 target genes (Berghmans et al., 2005). Because the Δ113p53 variant is dependent upon full-length p53 for its expression, Δ113p53 is also lost in the tp53zdf1 (p53zdf1) mutant (Chen et al., 2009). Consistent with our previous results, mpo-positive neutrophils were absent at 24 hpf in SbdsATG MO-injected p53zdf1/zdf1 embryos (Fig. 3F). Likewise, there was no rescue of the pancreatic phenotype in SbdsATG MO-injected p53zdf1/zdf1 embryos (Fig. 3G). In conclusion, neither p53 knockdown using two different MOs nor genetic loss of p53 is able to rescue defective organogenesis in SbdsATG MO-injected embryos. Thus, we conclude that the organogenesis defects observed following Sbds knockdown are p53 independent.
Loss of Sbds is associated with a broad change in ribosome-related gene expression
To begin to understand additional cellular consequences of Sbds knockdown in developing zebrafish embryos, we analyzed global transcriptional profiles in SbdsATG MO-injected embryos with and without co-injection of p53ATG MO. Clutches produced from ptf1a:eGFP outcrosses were injected with the SbdsATG and/or the p53ATG MO, and embryos were carefully staged to 24 hpf. Non-transgenic embryos were then harvested for isolation of RNA. We chose 24 hpf as our earliest phenotypic time point because embryos can be more precisely staged early in development, allowing correction for any developmental delay. The remaining ptf1a:eGFP transgenic embryos were evaluated at 72 hpf to confirm defective pancreas organogenesis as a measure of successful SbdsATG MO injection. Four experimental replicates were run on the Agilent zebrafish oligo microarray 4×44k platform. Using FDR-adjusted P-value cut-offs of 0.05, 0.01 and 0.001 to identify differentially expressed genes at increasing levels of statistical stringency, we identified genes differentially expressed in SbdsATG MO-injected versus control embryos, as well as in SbdsATG plus p53ATG (SbdsATG/p53ATG) MO-injected versus p53ATG MO-injected embryos. The numbers of differentially expressed genes at each level of statistical significance are presented in supplementary material Table S2, and the list of individual differentially expressed genes including fold changes is presented in supplementary material Table S3. Among 24,278 annotated zebrafish genes in the platform, we identified 4251 to be differentially expressed (P<0.01) in SbdsATG MO-injected versus control embryos. By comparison, 931 genes were found to be differentially expressed in SbdsATG/p53ATG MO-injected versus p53ATG MO-injected embryos, with 556 genes differentially expressed in both comparisons (Fig. 4A).
Fig. 4.

Gene set enrichment analysis of genes differentially expressed in SbdsATG MO-injected versus control and in SbdsATG/p53ATG MO-injected versus p53ATG MO-injected embryos. (A) Venn diagram depicting differentially expressed genes with a P<0.01 statistical cut-off. The red circle indicates genes differentially expressed in SbdsATG MO-injected versus control embryos, whereas the blue circle represents genes differentially expressed in SbdsATG/p53ATG MO-injected versus p53ATG MO-injected embryos. (B) The most highly enriched GO categories for genes upregulated in SbdsATG/p53ATG MO-injected versus p53ATG MO-injected embryos. (C) The most highly enriched GO categories for genes upregulated in SbdsATG MO-injected versus control embryos. For B and C, enriched GO categories are ranked based on the negative logarithm of their P-value. Note the high-level enrichment of genes related to ribosome biogenesis and rRNA processing in both comparisons.
Gene set enrichment analysis using Gene Ontology (GO) functional group categories demonstrated that transcripts displaying altered expression following sbds knockdown were markedly enriched for genes assigned to the ‘ribosome biogenesis’, ‘rRNA processing’, ‘nucleolus’ and ‘translational initiation’ categories. These trends were especially robust among genes that were upregulated in the absence of Sbds, where we observed enrichment for ribosome-related GO functional groups in both the presence and absence of p53 (Fig. 4B,C, supplementary material Fig. S5A).
Loss of rpl3 phenocopies the p53-independent sbds pancreatic phenotype
Significant changes in the expression of ribosome-related genes were previously noted in the analysis of human SDS patient bone marrow samples (Rujkijyanont et al., 2009). Similar to that report, we identified ribosomal protein L3 (rpl3) as a gene downregulated in the absence of Sbds. Rpl3 is an RP that is associated with the large 60S subunit. In 24-hpf zebrafish embryos, expression of rpl3 is initially broad, but becomes increasingly restricted to the endoderm as development proceeds (Fig. 5A). Our microarray studies indicated that embryos injected with SbdsATG MO or SbdsATG/p53ATG MOs had a significant reduction in rpl3 expression (Fig. 5B). We confirmed this reduction in rpl3 expression in Sbds knockdown embryos by in situ hybridization for rpl3 in SbdsATG MO-injected p53zdf1/zdf1 embryos. At 24 hpf, expression of rpl3 was reduced in the somites and nervous system of SbdsATG MO-injected embryos (Fig. 5C). This reduction in rpl3 expression became more pronounced at 72 hpf, particularly in the pharyngeal arches and intestine (Fig. 5C).
Fig. 5.

Knockdown of rpl3 phenocopies SbdsATG MO-induced defects in pancreas development. (A) Expression of rpl3 is widespread at 24 hpf and becomes progressively restricted to the endoderm, with high-level pancreatic expression at 120 hpf. The principal islet (I) is labeled by in situ hybridization for insulin (red). (B) rpl3 expression is decreased in SbdsATG MO-injected embryos, as quantified by microarray analysis of transcript abundance. Error bars indicate s.e.m. (C) In situ hybridization for rpl3 in SbdsATG MO-injected p53zdf1/zdf1 embryos confirms loss of rpl3 expression in somites (black arrows), central nervous system (white arrows), pharyngeal arches (red arrows) and intestine (yellow arrows) of developing embryos. (D) An MO dose-dependent defect in pancreas progenitor expansion was observed in Rpl3ATG MO-injected ptf1a:eGFP;ins:mCherry;p53zdf1/zdf1 embryos. (E) The small pancreas (arrow) of p53zdf1/zdf1 embryos injected with 0.5 ng Rpl3ATG MO is positive for trypsin expression by in situ hybridization. (F) p53zdf1/zdf1 embryos injected with 0.5 ng Rpl3ATG MO at 72 hpf exhibit small heads and hydroencephaly (yellow arrowhead). (G) rpl3hi2437/+;ptf1a:eGFP;ins:mCherry heterozygotes bearing a mutagenic rpl3 transgenic insertion were incrossed. rpl3hi2437/hi2437 homozygous embryos displayed disrupted pancreas progenitor expansion at 72 hpf. Injection of rpl3hi2437;ptf1a:eGFP;ins:mCherry with the p53ATG MO did not rescue the pancreatic progenitor phenotype of homozygous rpl3hi2437hi/2437 embryos. L, liver; P, pancreas; I, islet; Ex, exocrine pancreas.
In order to determine whether loss of Rpl3 phenocopies loss of Sbds, we designed an ATG MO to rpl3 and evaluated its role during development. We identified an MO dose-dependent defect in pancreatic volume in Rpl3ATG MO-injected p53zdf1/zdf1 embryos (Fig. 5D, supplementary material Fig. S5A). Similar to SbdsATG MO-injected embryos, the rpl3 pancreatic phenotype originated in a failure of progenitor expansion, rather than differentiation, as cells in the hypoplastic pancreas showed normal activation of trypsin expression (Fig. 5E). As with our SbdsATG MO, Rpl3ATG MO-injected embryos appeared healthier on a p53zdf1/zdf1 background. In addition to the pancreatic organogenesis defect, we also observed swelling in the brain and reduced eye size at a phenotypic dose of Rpl3ATG MO (Fig. 5F).
A retroviral mutagenesis screen in zebrafish had identified a mutagenic insertion in the rpl3 locus, rpl3hi2347 (Gaiano et al., 1996; Lai et al., 2009). We therefore examined pancreatic development at 72 hpf in progeny of an incross of rpl3hi2347/+;ptf1a:eGFP;ins:mCherry adults. Similar to the effects of the Rpl3ATG MO, embryos homozygous for the rpl3hi2347 allele developed a defect in pancreatic progenitor expansion, while the primary islet appeared unaffected. This effect was not rescued by injection of the p53ATG MO (Fig. 5G).
We conclude that Rpl3 is essential for the normal expansion of pancreatic progenitors. Furthermore, loss of Rpl3 phenocopies the hypoplastic pancreas phenotype observed in SbdsATG MO-injected embryos and does so in a p53-independent manner.
Loss of pescadillo also phenocopies the p53-independent sbds pancreatic phenotype
In yeast, Rpl3 is an early-assembling RP that is essential for nucleolar ribosome biogenesis. Additionally, Rpl3 is a component of the multi-protein Pescadillo complex involved in both ribosome biogenesis and cell cycle control (Iouk et al., 2001; Schaper et al., 2001; Du and Stillman, 2002; Killian et al., 2004). A gene encoding another member of this complex, pescadillo (pes), was among those displaying altered expression in sbds morphants. The pes gene was originally identified in a zebrafish mutant characterized by major axis defects, small eyes and early embryonic lethality (Allende et al., 1996). Both in vitro and in vivo, disruption of pes has been reported to induce a p53 response (Gessert et al., 2007; Rohrmoser et al., 2007). We therefore undertook an analysis of pancreatic development in pes mutant and morphant embryos.
At 24 hpf, zebrafish pes is highly expressed in the embryonic eye and midbrain. At 72 hpf, pes expression is detected broadly in the endoderm, including the developing pancreas (Fig. 6A). An incross of peshi2Tg/wt fish results in 25% of the clutch exhibiting a profoundly curved axis, a small degenerating head and small eyes. Heterozygous and wild-type siblings develop normally (Fig. 6B).
Fig. 6.

The pes mutant phenocopies SbdsATG MO-induced p53-independent defects in pancreas development. (A) In situ hybridization at 24 hpf demonstrates pes expression in the eye and central nervous system. At 72 hpf, pes expression is detected in endoderm, including the developing pancreas. (B) The peshi2Tg/hi2Tg phenotype includes axis defects (tail curvature), a small dark head and a small eye. (C) Semi-quantitative RT-PCR of peshi2Tg/hi2Tg embryos reveals activation of the p53 target genes Δ113p53 (24-fold, P<0.0001) and p21 (2.4-fold, P<0.01). Error bars indicate s.e.m. (D) Loss of p53 does not rescue axis defects in peshi2Tg/hi2Tg embryos. Representative images of siblings (sib) generated either from a peshi2Tg/WT incross and injected with or without (Mock) p53ATG MO or p53Spl MO, or from a peshi2Tg/WT;p53zdf1/zdf1 incross. Asterisks indicate siblings displaying the pes phenotype. See supplementary material Fig. S6 for quantification. (E) peshi2Tg/hi2Tg embryos exhibit a p53-independent defect in expansion of ptf1-expressing pancreatic progenitor cells. The pancreatic progenitor defect in 72-hpf peshi2Tg/hi2Tg;ptf1a:eGFP;ins:mCherry embryos is not rescued by p53 MO-mediated knockdown, nor by genetic inactivation of p53 as assessed by incross of peshi2Tg/WT;p53zdf1/zdf1;ptf1a:eGFP;ins:mCherry fish.
Based on previous work with pes and our observation that peshi2Tg/hi2Tg embryos display head degeneration characteristic of a p53 apoptotic response (Fig. 6B), we analyzed Δ113p53 and p21 expression by quantitative PCR. Both were strongly upregulated in peshi2Tg/hi2Tg embryos compared with their phenotypically normal siblings (24-fold increase for Δ113p53, P<0.0001; 2.4-fold increase for p21, P<0.01) (Fig. 6C). We next tested whether knockdown of p53 could rescue the peshi2Tg/hi2Tg phenotype. When incrossed peshi2Tg/wt embryos were injected with p53ATG MO or p53Spl MO, neither was able to rescue the peshi2Tg/hi2Tg phenotype (Fig. 6D, supplementary material Fig. S5B). We confirmed these results genetically by incrossing peshi2Tg/wt;p53zdf1/zdf1 fish. Again, we were unable to rescue the peshi2Tg/hi2Tg phenotype, even on a p53 null background (Fig. 6D, supplementary material Fig. S5C).
It has been suggested that peshi2Tg/hi2Tg mutants have disrupted endoderm development (Allende et al., 1996). Imaging of peshi2Tg/hi2Tg embryos in combination with ptf1a:eGFP and ins:mCherry reporters revealed a profoundly hypoplastic pancreas (Fig. 6E). As with the gross phenotype of peshi2Tg/hi2Tg fish, neither p53 knockdown nor genetic inactivation of p53 in p53zdf1/zdf1 embryos was able to rescue the observed defect in pancreatic organogenesis (Fig. 6E). Thus, we conclude that loss of p53 is insufficient to rescue multiple components of the peshi2Tg/hi2Tg phenotype, including the axis defects, small eyes and abnormal pancreas development.
Because the peshi2Tg/hi2Tg phenotype is severe, we reasoned that abnormal pancreas development might occur due to a more general disruption of endodermal patterning. To address this, we titrated increasing doses of PesATG MO in p53zdf1/zdf1;ptf1a:eGFP;ins:mCherry embryos. Embryos injected with the PesATG MO showed an MO dose-dependent pancreatic development defect (Fig. 7A,B). PesATG MO-injected embryos also exhibited swelling in the brain and an underdeveloped eye (Fig. 7C), but overall appeared much healthier than peshi2Tg/hi2Tg embryos. Because we detected a persistent pancreatic progenitor defect even in embryos with otherwise grossly normal development, we suggest that pes is specifically required for normal pancreas development.
Fig. 7.

MO knockdown of pes phenocopies SbdsATG MO-induced p53-independent defects in pancreas development. (A) Injection of PesATG MO into p53zdf1/zdf1 embryos results in a dose-dependent defect in pancreatic progenitor expansion, as assessed in 72-hpf ptf1a:eGFP;ins:mCherry embryos. (B) Quantification of three independent experiments scored for the percentage of embryos with wild-type pancreas following injection of PesATG MO into a p53zdf1/zdf1;ptf1a:eGFPjh1;ins:mCherryjh2 background. (C) The gross appearance of PesATG MO-injected embryos at 72 hpf includes a small head and eye but normal body axis. (D) Microarray quantification of pes transcript levels indicates an increase in pes expression in SbdsATG MO-injected p53zdf1/zdf1 embryos. (E) Increased abundance of pes transcripts in SbdsATG MO-injected p53zdf1/zdf1 embryos is confirmed by in situ hybridization. Error bars indicate s.e.m.
In our microarray studies, pes transcripts were significantly increased in SbdsATG MO-injected and SbdsATG/p53ATG MO-injected embryos (Fig. 7D). We confirmed this by in situ hybridization. At 24 hpf, pes expression is expanded in the somites of p53zdf1/zdf1 SbdsATG MO-injected embryos. This difference becomes more striking at 48 hpf, when expression of pes is stronger and more widespread in the SbdsATG MO-injected embryos (Fig. 7E). Based on the convergent phenotypes observed following knockdown of Sbds, Rpl3 and Pes, these results suggest that disruptions in pathways crucial for ribosome biogenesis result in the failure of normal pancreas development, mediated by defective expansion of ptf1a-expressing pancreatic progenitors.
DISCUSSION
Our work demonstrates that loss of sbds function in zebrafish effectively recapitulates the organogenesis defects observed in human SDS: loss of neutrophils, abnormal skeletal architecture and pancreatic hypoplasia. These results confirm and extend a previous analysis of the zebrafish sbds morphant phenotype (Venkatasubramani and Mayer, 2008). They also complement a recent analysis of pancreas-specific deletion of murine Sbds (Tourlakis et al., 2012), and implicate defective proliferation of ptf1a-expressing pancreatic progenitor cells as a basis for the observed pancreatic phenotype. The zebrafish Sbds knockdown phenotype was not rescued by loss of p53, suggesting that, in contrast to Treacher Collins syndrome and 5q-syndrome (Jones et al., 2008; Barlow et al., 2010), SDS is a largely p53-independent ribosomopathy. Furthermore, by unbiased screening of genes exhibiting altered expression following Sbds knockdown, we have identified a previously unappreciated requirement for additional genes related to ribosome assembly in early pancreas development. Importantly, the exquisite sensitivity of pancreatic progenitor cells to the altered expression of ribosome assembly genes is developmentally apparent well before the elaboration of extensive rough endoplasmic reticulum associated with subsequent exocrine differentiation. This suggests that a later requirement for high-level synthesis of digestive zymogens is not the sole basis for the resulting pancreatic hypoplasia.
Although loss of p53 failed to rescue the organogenesis phenotypes and the changes in expression of multiple ribosome-related genes observed following Sbds knockdown, it did improve the overall health of the embryos. The twisted tail phenotype and CNS degeneration we observed in SbdsATG MO-injected embryos were relieved by loss of p53 (Fig. 3A; data not shown). Similarly, the majority of genes displaying altered expression following Sbds knockdown were normalized by concomitant loss of p53. Using our present MO-based model, we cannot rule out the possibility that the p53 dependence of these events reflects non-specific MO toxicity (Robu et al., 2007). However, it remains a formal possibility that the composite SDS phenotype includes both p53-dependent and p53-independent aspects. Support for p53-dependent and p53-independent aspects of ribosome biogenesis disorders is also provided by studies of rps19, the gene responsible for DBA. Initial analyses of the zebrafish rps19 morphant phenotype described a loss of red blood cells (RBCs) and other gross developmental defects, and further reported that concomitant knockdown of p53 improved the health and survival of the embryos (Danilova et al., 2008; Uechi et al., 2008). However, more recent work found that only gross developmental defects were rescued by p53 knockdown, whereas loss of RBCs persisted in a p53-independent manner (Torihara et al., 2011).
It is fascinating to contemplate how mutations in genes responsible for an essential cellular process, such as ribosome assembly, result in largely tissue-restricted phenotypes. One possibility is that individual assembly factors are expressed in a cell type-specific or tissue-restricted manner. A recent report using an Rpl38 mutant mouse described a previously unappreciated enrichment of Rpl38 expression during development in tissues that are affected by its loss (Kondrashov et al., 2011). This also appears to be the case for pes, where tissues with high-level pes expression, including brain, eye and pancreas, are those most severely affected in the pes mutant phenotype (Fig. 6A,B). However, unlike pes, sbds appears to be expressed in all tissues except muscle (Boocock et al., 2003). Despite this, the sbds loss-of-function phenotype disproportionately affects the pancreas, the neutrophil lineage and the developing skeleton. It is still unclear why different tissues display differential sensitivity to sbds loss. However, the convergence of the sbds, rpl3 and pes phenotypes, together with the observed changes in ribosome profiles and the p53 independence of the sbds organogenesis phenotypes, all argue that these developmental defects represent the direct consequence of altered ribosome biogenesis.
As an additional component of the sbds loss-of-function phenotype, we identified widespread changes in the expression of genes related to ribosome biogenesis, rRNA processing and translational initiation. We functionally characterized two additional genes related to ribosome biogenesis, rpl3 and pes, with loss-of-function phenotypes that converge upon a similar p53-independent defect in pancreatic progenitor expansion. The fact that the rpl3 and pes loss-of-function phenotypes are similar even though rpl3 is downregulated and pes is upregulated in sbds morphants emphasizes that directional changes in gene expression do not necessarily correlate with gain- or loss-of-function phenotypes. Indeed, we observed increased expression of many ribosome-related genes in sbds MO-injected embryos, even though ribosome biogenesis is clearly disrupted in these embryos. These data suggest that the observed increased expression of pes and other ribosome-related genes is likely to represent a compensatory mechanism by which cells attempt to overcome disruptions in ribosome assembly. Rather than using our microarray analysis as a means of inferring function based on the directionality of observed changes in gene expression, these data serve primarily as a discovery tool to identify additional genes and pathways altered by sbds knockdown. Using this approach, we were able to identify a previously unappreciated role for multiple genes involved in ribosome biogenesis as early essential genes in pancreas development.
Because of the general understanding that genes encoding RPs are transcribed in excess (Lam et al., 2007), it was surprising to find that loss of Sbds was associated with profound changes in the expression of multiple RP genes. However, this view of RP transcription is likely to be an oversimplification, and evidence exists for uncoupling of individual RPs from otherwise coordinated expression. For example, both Rpl3 and Pes interact with another member of the yeast Pescadillo complex, Rrb1p (GRWD1 in human) (Iouk et al., 2001; Killian et al., 2004). The function of Rrb1p in ribosome biogenesis is linked to its physical interaction with Rpl3. Overexpression of Rrb1p results in an increase in the expression of rpl3, whereas the expression of other RP genes is unaffected. Likewise, loss of Rrb1p expression results in increased expression of other RP genes, but not rpl3 (Iouk et al., 2001). In addition to nuanced transcriptional control of RPs, post-translational modification of RPs and non-ribosome-related roles for RPs have been described (Bhavsar et al., 2010). This suggests that our previous understanding of the role of ribosome assembly genes during normal development and in disease states is incomplete, with additional tissue- and cell type-specific roles likely to emerge.
Supplementary Material
Acknowledgments
We thank Danielle Blake and Seneca Bessling for expert technical support and Dr Raymond MacDonald and the Beta Cell Biology Consortium for publicly available ChIP-Seq datasets.
Footnotes
Funding
This work was supported by grants from the National Institutes of Health (NIH) [DK077480 to E.P., DK080730 and DK082060 to M.J.P., DK61215 and DK56211 to S.D.L.]; an NIH National Institute of General Medical Sciences (NIGMS) RISE Grant [5R25GM059244-11 to Barry University]; and the Johns Hopkins Brain Science Institute (to K.A.W.). R.G. is an HHMI Investigator. S.D.L. is further supported by the Paul K. Neumann Professorship in Pancreatic Cancer at Johns Hopkins University. Deposited in PMC for release after 6 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.077107/-/DC1
References
- Allende M. L., Amsterdam A., Becker T., Kawakami K., Gaiano N., Hopkins N. (1996). Insertional mutagenesis in zebrafish identifies two novel genes, pescadillo and dead eye, essential for embryonic development. Genes Dev. 10, 3141–3155 [DOI] [PubMed] [Google Scholar]
- Barlow J. L., Drynan L. F., Hewett D. R., Holmes L. R., Lorenzo-Abalde S., Lane A. L., Jolin H. E., Pannell R., Middleton A. J., Wong S. H., et al. (2010). A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q–syndrome. Nat. Med. 16, 59–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghmans S., Murphey R. D., Wienholds E., Neuberg D., Kutok J. L., Fletcher C. D., Morris J. P., Liu T. X., Schulte-Merker S., Kanki J. P., et al. (2005). tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc. Natl. Acad. Sci. USA 102, 407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhavsar R. B., Makley L. N., Tsonis P. A. (2010). The other lives of ribosomal proteins. Hum. Genomics 4, 327–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boocock G. R., Morrison J. A., Popovic M., Richards N., Ellis L., Durie P. R., Rommens J. M. (2003). Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat. Genet. 33, 97–101 [DOI] [PubMed] [Google Scholar]
- Chen A. T., Zon L. I. (2009). Zebrafish blood stem cells. J. Cell. Biochem. 108, 35–42 [DOI] [PubMed] [Google Scholar]
- Chen J., Ruan H., Ng S. M., Gao C., Soo H. M., Wu W., Zhang Z., Wen Z., Lane D. P., Peng J. (2005). Loss of function of def selectively up-regulates Delta113p53 expression to arrest expansion growth of digestive organs in zebrafish. Genes Dev. 19, 2900–2911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Ng S. M., Chang C., Zhang Z., Bourdon J. C., Lane D. P., Peng J. (2009). p53 isoform delta113p53 is a p53 target gene that antagonizes p53 apoptotic activity via BclxL activation in zebrafish. Genes Dev. 23, 278–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross L. M., Cook M. A., Lin S., Chen J. N., Rubinstein A. L. (2003). Rapid analysis of angiogenesis drugs in a live fluorescent zebrafish assay. Arterioscler. Thromb. Vasc. Biol. 23, 911–912 [DOI] [PubMed] [Google Scholar]
- Dai M. S., Lu H. (2004). Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 279, 44475–44482 [DOI] [PubMed] [Google Scholar]
- Danilova N., Sakamoto K. M., Lin S. (2008). Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood 112, 5228–5237 [DOI] [PubMed] [Google Scholar]
- Deisenroth C., Zhang Y. (2010). Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene 29, 4253–4260 [DOI] [PubMed] [Google Scholar]
- Draptchinskaia N., Gustavsson P., Andersson B., Pettersson M., Willig T. N., Dianzani I., Ball S., Tchernia G., Klar J., Matsson H., et al. (1999). The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 21, 169–175 [DOI] [PubMed] [Google Scholar]
- Du Y. C., Stillman B. (2002). Yph1p, an ORC-interacting protein: potential links between cell proliferation control, DNA replication, and ribosome biogenesis. Cell 109, 835–848 [DOI] [PubMed] [Google Scholar]
- Duan J., Ba Q., Wang Z., Hao M., Li X., Hu P., Zhang D., Zhang R., Wang H. (2011). Knockdown of ribosomal protein S7 causes developmental abnormalities via p53 dependent and independent pathways in zebrafish. Int. J. Biochem. Cell Biol. 43, 1218–1227 [DOI] [PubMed] [Google Scholar]
- Finch A. J., Hilcenko C., Basse N., Drynan L. F., Goyenechea B., Menne T. F., González Fernández A., Simpson P., D’Santos C. S., Arends M. J., et al. (2011). Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 25, 917–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiano N., Amsterdam A., Kawakami K., Allende M., Becker T., Hopkins N. (1996). Insertional mutagenesis and rapid cloning of essential genes in zebrafish. Nature 383, 829–832 [DOI] [PubMed] [Google Scholar]
- Gessert S., Maurus D., Rössner A., Kühl M. (2007). Pescadillo is required for Xenopus laevis eye development and neural crest migration. Dev. Biol. 310, 99–112 [DOI] [PubMed] [Google Scholar]
- Godinho L., Williams P. R., Claassen Y., Provost E., Leach S. D., Kamermans M., Wong R. O. (2007). Nonapical symmetric divisions underlie horizontal cell layer formation in the developing retina in vivo. Neuron 56, 597–603 [DOI] [PubMed] [Google Scholar]
- Iouk T. L., Aitchison J. D., Maguire S., Wozniak R. W. (2001). Rrb1p, a yeast nuclear WD-repeat protein involved in the regulation of ribosome biosynthesis. Mol. Cell. Biol. 21, 1260–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin A., Itahana K., O’Keefe K., Zhang Y. (2004). Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol. Cell. Biol. 24, 7669–7680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones N. C., Lynn M. L., Gaudenz K., Sakai D., Aoto K., Rey J. P., Glynn E. F., Ellington L., Du C., Dixon J., et al. (2008). Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 14, 125–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killian A., Le Meur N., Sesboüé R., Bourguignon J., Bougeard G., Gautherot J., Bastard C., Frébourg T., Flaman J.-M. (2004). Inactivation of the RRB1-Pescadillo pathway involved in ribosome biogenesis induces chromosomal instability. Oncogene 23, 8597–8602 [DOI] [PubMed] [Google Scholar]
- Kimmel C. B., Miller C. T., Moens C. B. (2001). Specification and morphogenesis of the zebrafish larval head skeleton. Dev. Biol. 233, 239–257 [DOI] [PubMed] [Google Scholar]
- Kondrashov N., Pusic A., Stumpf C. R., Shimizu K., Hsieh A. C., Xue S., Ishijima J., Shiroishi T., Barna M. (2011). Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 145, 383–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K., Amsterdam A., Farrington S., Bronson R. T., Hopkins N., Lees J. A. (2009). Many ribosomal protein mutations are associated with growth impairment and tumor predisposition in zebrafish. Dev. Dyn. 238, 76–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam Y. W., Lamond A. I., Mann M., Andersen J. S. (2007). Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr. Biol. 17, 749–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. W., Biankin A. V., Horb M. E., Ghosh B., Prasad N. B., Yee N. S., Pack M. A., Leach S. D. (2004). Differential requirement for ptf1a in endocrine and exocrine lineages of developing zebrafish pancreas. Dev. Biol. 270, 474–486 [DOI] [PubMed] [Google Scholar]
- Lohrum M. A., Ludwig R. L., Kubbutat M. H., Hanlon M., Vousden K. H. (2003). Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3, 577–587 [DOI] [PubMed] [Google Scholar]
- Luft F. (2010). The rise of a ribosomopathy and increased cancer risk. J. Mol. Med. 88, 1–3 [DOI] [PubMed] [Google Scholar]
- Mathias J. R., Perrin B. J., Liu T. X., Kanki J., Look A. T., Huttenlocher A. (2006). Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J. Leukoc. Biol. 80, 1281–1288 [DOI] [PubMed] [Google Scholar]
- McGowan K. A., Li J. Z., Park C. Y., Beaudry V., Tabor H. K., Sabnis A. J., Zhang W., Fuchs H., de Angelis M. H., Myers R. M., et al. (2008). Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat. Genet. 40, 963–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menne T. F., Goyenechea B., Sánchez-Puig N., Wong C. C., Tonkin L. M., Ancliff P. J., Brost R. L., Costanzo M., Boone C., Warren A. J. (2007). The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat. Genet. 39, 486–495 [DOI] [PubMed] [Google Scholar]
- Narla A., Ebert B. L. (2010). Ribosomopathies: human disorders of ribosome dysfunction. Blood 115, 3196–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelio C., Verkuijlen P., Geissler J., van den Berg T. K., Kuijpers T. W. (2009). SBDS expression and localization at the mitotic spindle in human myeloid progenitors. PLoS ONE 4, e7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. W., Davison J. M., Rhee J., Hruban R. H., Maitra A., Leach S. D. (2008). Oncogenic KRAS induces progenitor cell expansion and malignant transformation in zebrafish exocrine pancreas. Gastroenterology 134, 2080–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M. J., Pisharath H., Yusuff S., Moore J. C., Siekmann A. F., Lawson N., Leach S. D. (2009). Notch-responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech. Dev. 126, 898–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauls S., Zecchin E., Tiso N., Bortolussi M., Argenton F. (2007). Function and regulation of zebrafish nkx2.2a during development of pancreatic islet and ducts. Dev. Biol. 304, 875–890 [DOI] [PubMed] [Google Scholar]
- Pisharath H., Rhee J. M., Swanson M. A., Leach S. D., Parsons M. J. (2007). Targeted ablation of beta cells in the embryonic zebrafish pancreas using E. coli nitroreductase. Mech. Dev. 124, 218–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw S. A., Loynes C. A., Trushell D. M., Elworthy S., Ingham P. W., Whyte M. K. (2006). A transgenic zebrafish model of neutrophilic inflammation. Blood 108, 3976–3978 [DOI] [PubMed] [Google Scholar]
- Robu M. E., Larson J. D., Nasevicius A., Beiraghi S., Brenner C., Farber S. A., Ekker S. C. (2007). p53 activation by knockdown technologies. PLoS Genet. 3, e78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrmoser M., Hölzel M., Grimm T., Malamoussi A., Harasim T., Orban M., Pfisterer I., Gruber-Eber A., Kremmer E., Eick D. (2007). Interdependence of Pes1, Bop1, and WDR12 controls nucleolar localization and assembly of the PeBoW complex required for maturation of the 60S ribosomal subunit. Mol. Cell. Biol. 27, 3682–3694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rujkijyanont P., Adams S. L., Beyene J., Dror Y. (2009). Bone marrow cells from patients with Shwachman-Diamond syndrome abnormally express genes involved in ribosome biogenesis and RNA processing. Br. J. Haematol. 145, 806–815 [DOI] [PubMed] [Google Scholar]
- Sasaki M., Kawahara K., Nishio M., Mimori K., Kogo R., Hamada K., Itoh B., Wang J., Komatsu Y., Yang Y. R., et al. (2011). Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat. Med. 17, 944–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper S., Fromont-Racine M., Linder P., de la Cruz J., Namane A., Yaniv M. (2001). A yeast homolog of chromatin assembly factor 1 is involved in early ribosome assembly. Curr. Biol. 11, 1885–1890 [DOI] [PubMed] [Google Scholar]
- Smyth G. K. (2004). Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3 [DOI] [PubMed] [Google Scholar]
- Torihara H., Uechi T., Chakraborty A., Shinya M., Sakai N., Kenmochi N. (2011). Erythropoiesis failure due to RPS19 deficiency is independent of an activated Tp53 response in a zebrafish model of Diamond-Blackfan anaemia. Br. J. Haematol. 152, 648–654 [DOI] [PubMed] [Google Scholar]
- Tourlakis M. E., Zhong J., Gandhi R., Zhang S., Chen L., Durie P. R., Rommens J. M. (2012). Deficiency of SBDS in the mouse pancreas leads to features of Shwachman-Diamond syndrome, with loss of zymogen granules. Gastroenterology (in press). [DOI] [PubMed] [Google Scholar]
- Uechi T., Nakajima Y., Chakraborty A., Torihara H., Higa S., Kenmochi N. (2008). Deficiency of ribosomal protein S19 during early embryogenesis leads to reduction of erythrocytes in a zebrafish model of Diamond-Blackfan anemia. Hum. Mol. Genet. 17, 3204–3211 [DOI] [PubMed] [Google Scholar]
- Venkatasubramani N., Mayer A. N. (2008). A zebrafish model for the Shwachman-Diamond syndrome (SDS). Pediatr. Res. 63, 348–352 [DOI] [PubMed] [Google Scholar]
- Walker M. B., Kimmel C. B. (2007). A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotech. Histochem. 82, 23–28 [DOI] [PubMed] [Google Scholar]
- Wang Y., Rovira M., Yusuff S., Parsons M. J. (2011). Genetic inducible fate mapping in larval zebrafish reveals origins of adult insulin-producing β-cells. Development 138, 609–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C. C., Traynor D., Basse N., Kay R. R., Warren A. J. (2011). Defective ribosome assembly in Shwachman-Diamond syndrome. Blood 118, 4305–4312 [DOI] [PubMed] [Google Scholar]
- Zhang S., Shi M., Hui C. C., Rommens J. M. (2006). Loss of the mouse ortholog of the shwachman-diamond syndrome gene (Sbds) results in early embryonic lethality. Mol. Cell. Biol. 26, 6656–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}