

Abstract
The catalytic durability of an organic photocatalyst, 9-mesityl-10-methyl acridinium ion (Acr+–Mes), has been dramatically improved by the addition of [{tris(2-pyridylmethyl)amine}-CuII](ClO4)2 ([(tmpa)CuII]2+) in the photocatalytic oxygenation of p-xylene by molecular oxygen in acetonitrile. Such an improvement is not observed by the addition of Cu(ClO4)2 in the absence of organic ligands. The addition of [(tmpa)Cu]2+ in the reaction solution resulted in more than an 11 times higher turnover number (TON) compared with the TON obtained without [(tmpa)CuII]2+. In the photocatalytic oxygenation, a stoichiometric amount of H2O2 formation was observed in the absence of [(tmpa)CuII]2+, however, much less H2O2 formation was observed in the presence of [(tmpa)CuII]2+. The photocatalytic mechanism was investigated by laser flash photolysis measurements in order to detect intermediates. The reaction of O2˙− with [(tmpa)CuII]2+ monitored by UV-vis spectroscopy in propionitrile at 203 K suggested formation of [{(tmpa)CuII}2O2]2+, a transformation which is crucial for the overall 4-electron reduction of molecular O2 to water, and a key in the observed improvement in the catalytic durability of Acr+–Mes.
1. Introduction
A variety of organic reactions such as oxygenation, carbon–carbon bond formation and polymerization, have been accomplished by employing heavy transition metal catalysts,1,2 however, the increased attention on environmental problems demands a focus toward alternative and more environmentally benign methods.3,4 Recently, utilization of organic catalysts has been regarded as a promising method to avoid such problems because of their environmentally benign character. Especially, the employment of organic photocatalysts utilizing visible light has attracted many researchers expecting to produce products which are difficult to obtain via thermal reactions.5–7
Among such photocatalysts, a donor–acceptor linked dyad has been regarded as a prominent catalyst because of its already proven wide variety of applications.8–14 For example, oxidation of benzene to phenol where the oxygen atom derives from water as its source has been made possible without producing further oxygenated compounds.15,16 Bromination of various aromatic compounds has also been achieved without using toxic bromine.17 The donor–acceptor linked dyad can be also utilized in the oxygenation of aromatic compounds to produce aromatic aldehydes,18 which is a key chemical reaction for the production of precursors of a variety of fine or speciality chemicals such as pharmaceutics, organic dyes, pesticides and perfumes.19–24
9-Mesityl-10-methylacridinium ion (Acr+–Mes)9,10 and its derivatives have been reported to act as efficient photocatalysts for oxygenation of toluene derivatives to corresponding aldehydes in photocatalytic oxygenation under visible light irradiation.18 The electron-transfer state (Acr˙–Mes˙+), produced upon photoexcitation of Acr+–Mes, has both strong oxidizing and reducing ability, and this enables it to not only oxidize alkyl aromatic compounds but also to reduce O2.10,14 However, the accompanying formation of reactive oxygen species (ROS) may be problematic, because they may irreversibly oxidize the organic photocatalyst leading to its deactivation.
Organic catalyst durability is highly desirable for practical applications, especially in oxygenation reaction systems. In a previous report, the turnover number of an organic photocatalyst of Acr+–Mes was lower than 100 in the oxygenation of p-xylene by O2 using Acr+–Mes as a photocatalyst. Without a substrate, Acr+–Mes itself is oxidized by O2 under photoirradiation.25 The ROS O2˙− can be produced by electron transfer from the Acr˙ moiety of the photogenerated electron-transfer state (Acr˙–Mes˙+) to O2, and this may be responsible for the oxidation of the photocatalyst. Yet O2˙− is known to be efficiently trapped by various metal ions including Cu(II) complexes; Cu–O2 complexes have recently been extensively investigated.26–29 Thus, our thinking was that the presence of a copper complex in oxygenation reaction systems might improve organic catalyst durability and the reaction mechanism might be clarified by spectroscopic detection of a Cu–O2 intermediate.
We herein report a significant improvement of the photocatalytic durability of Acr+–Mes in the selective oxygenation of p-xylene with O2 by the addition of [{tris(2-pyridylmethyl)-amine}CuII](ClO4)2 ([(tmpa)CuII]2+).26–29 This Cu complex is expected to efficiently react with O2˙−,28,29 the latter is likely a significant product of O2-reduction. For comparison, Cu(ClO4)2 was added to the reaction solution but found to not be useful. Spectroscopic studies using nanosecond laser flash photolysis and UV-vis absorption at low temperature have enabled us to clarify the role of [(tmpa)CuII]2+ and to propose a mechanism for the significantly improved photocatalytic durability of the Acr+–Mes catalyst.
2. Experimental section
9-Mesityl-10-methylacridinium perchlorate [(Acr+–Mes)(ClO4)] was purchased from Tokyo Chemical Industry and recrystallized from absolute ethanol before use. p-Xylene was obtained from Wako Pure Chemicals. Copper(II) perchlorate hexahydrate and tris(2-pyrydylmethyl)amine were purchased from Nacalai tesuque and Tokyo Chemical Industry, respectively. Purified water was provided by a Millipore MilliQ water purification system where the electronic conductance was 18.2 MΩ cm.
2.1. Synthesis of [CuII(tmpa)](ClO4)2
[CuII(tmpa)](ClO4)2 was synthesized by the reported method.30 Copper(II) perchlorate hexahydrate (131 mg, 0.035 mmol) was slowly added to an acetonitrile (MeCN) solution (4.0 mL) of tris(2-pyridylmethyl)amine (100 mg, 0.035 mmol). The deep blue solution obtained was stirred for 30 min at room temperature and then ether (10 mL) was slowly added to precipitate blue powder which was collected by filtration and recrystallized from MeCN and diethyl ether (Yield 155 mg, 75%). Anal. Calcd. for C20H21Cl2CuN5O: C, 40.45; H, 3.56; N, 11.79. Found; C, 40.36; H, 3.32; N, 11.68.
2.2. Photocatalytic p-xylene oxygenation
The photocatalytic oxygenation of p-xylene was carried out by the following procedure. Typically, an MeCN solution (0.6 mL) containing (Acr+–Mes)(ClO4) (2.0 × 10−4 M), p-xylene (3.0 × 10−2 M) and [(tmpa)CuII](ClO4)2 in a quartz glass tube (i.d. 5 mm) with a rubber septum was saturated with oxygen by bubbling pure oxygen through a stainless steel needle for 5 min. The solution was then irradiated with a 500 W xenon lamp (Ushio Optical ModelX SX-UID 500XAMQ) through a colour filter glass (Asahi Techno Glass, λ > 390 nm) at 298 K. After photoirradiation, the solution was analysed periodically by 1H NMR spectroscopy (JEOL JNM-AL300 spectrometer) to identify and quantify the products. The amount of H2O2 formed was determined by titration by iodide ion; the diluted reaction mixture was treated with an excess amount of NaI in MeCN. The concentration of I3− formed was then determined from the UV-vis spectrum (λmax = 361 nm, ε = 2.50 × 104 M−1 cm−1 in MeCN).31 The yield of H2O2 was also determined by 1H NMR spectroscopy. H2O2: 1H NMR (CD3CN): 8.67 (s, 2H).
2.3. Time-resolved absorption spectral measurements
For nanosecond laser flash photolysis experiments, an MeCN solution (2.0 mL) containing (Acr+–Mes)(ClO4) and [(tmpa)CuII](ClO4)2 was deaerated by bubbling N2 for 10 min. Then, a certain amount of O2-saturated MeCN was added to the deaerated reaction solution. Then the mixed solution was photoirradiated by an Nd:YAG laser (Continuum, SLII-10, 4–6 ns fwhm) at λ = 430 nm with the power of 3.6 mJ per pulse. The transient absorption measurements were performed using a continuous xenon lamp (150 W) as a probe light and a photomultiplier (Hamamatsu R2949; 350–800 nm) as a detector, respectively.
2.4. UV-vis absorption spectroscopy under photoirradiation at low temperature
An O2-saturated propionitrile (EtCN) solution containing 1-benzyl–1,4-dihydronicotinamide dimer (0.25 mM), (Acr+–Mes)(ClO4) (0.050 mM) and [CuII(tmpa)](ClO4)2 (0.50 mM) was cooled in a quartz cuvette (light path length: 1.0 cm) to 203 K. UV-vis spectra were taken after photoirradiation (λ = 430 nm).
3. Results and discussion
3.1. p-Xylene oxygenation catalysed by Acr+–Mes in the presence of [(tmpa)CuII]2+
The photocatalytic p-xylene oxygenation using molecular oxygen was performed by photoirradiation (λ > 390 nm) of a deuterated MeCN solution using Acr+–Mes as a photocatalyst in the absence and presence of [(tmpa)CuII]2+. The oxygenation of p-xylene resulted in conversion to p-tolualdehyde and H2O2 (eqn (1)). Fig. 1 shows a typical 1H NMR
 |
(1) |
spectrum of the solution during the reaction. The 1H NMR spectroscopic signals assigned to p-xylene appeared around 2.6 ppm (CH3 group) and 7.0 ppm (phenyl ring) and those assigned to p-tolualdehyde appeared around 2.7 ppm (CH3 group), 7.3 ppm and 7.8 ppm (phenyl group) and 9.9 ppm (aldehyde). No overlapping of peaks of the reactant and product assures that the quantification by 1H NMR is reliable in this reaction system. The 1H NMR peaks assignable to Acr+–Mes were negligible due to its relatively low concentration (2.0 × 10−4 M). Formation of p-methylbenzyl alcohol is possible during the oxygenation as an intermediate, however, none of this product was observed in any significant amount when in the presence of [(tmpa)CuII]2+.
Fig. 1.
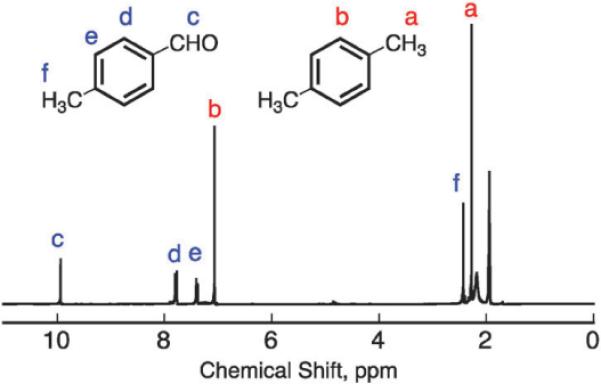
A 1H NMR spectrum of oxygen-saturated CD3CN solution containing Acr+–Mes (2.0 × 10−4 M), [(tmpa)CuII]2+ (1.0 × 10−3 M) and p-xylene (3.0 × 10−2 M) after photoirradiation for 3.5 h. The peaks labelled a and b were assigned to p-xylene and those labelled c–f were due to p-tolualdehyde.
The time profiles of p-tolualdehyde and H2O2 yields in this photooxygenation with different concentrations of [(tmpa)CuII]2+ are shown in Fig. 2. In the absence of [(tmpa)CuII]2+, the yields of p-tolualdehyde and H2O2 reached 30% and 26% with photoirradiation for 3.5 h, respectively. The similarity of yields assures that the oxygenation proceeded following eqn (1). The yield of p-tolualdehyde increased with increasing concentrations of [(tmpa)CuII]2+ up to 2.0 mM to reach a constant value of 57% in 3.5 h, which is nearly double the yield achieved in the absence of [(tmpa)CuII]2+. On the other hand, the H2O2 yield decreased from 26% achieved in the absence of [(tmpa)CuII]2+ to 5% in the presence of 2.0 mM [(tmpa)CuII]2+. A separate experiment involving reaction of [(tmpa)CuII]2+ with H2O2 was conducted to confirm whether or not [(tmpa)CuII]2+ catalyzed the decomposition of H2O2 produced. No significant change in H2O2 concentration was detected by iodometry, as shown in Fig. S1 in the ESI.† Thus, the reaction mechanism of the O2 reduction appears to be altered in the presence of [(tmpa)CuII]2+. On the other hand, the quantum yield for the photocatalytic oxidation of p-xylene in the presence of [(tmpa)CuII]2+ (1.0 mM), Acr+–Mes (2.0 × 10−4 mM) and p-xylene (1.0 × 10−2 M) excited at 420 nm was determined to be 0.13 using ferrioxalate actinometer. The quantum yield was similar to that determined for the reaction system in the absence of [(tmpa)CuII]2+.18
Fig. 2.
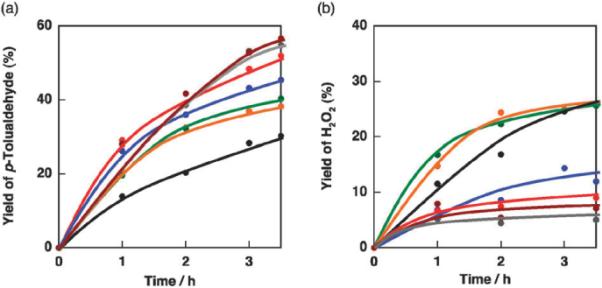
(a) Time profiles of the yield of p-tolualdehyde in the photo-oxygenation of p-xylene (3.0 × 10−2 M) in oxygen-saturated CD3CN containing Acr+–Mes (2.0 × 10−4 mM) and [(tmpa)CuII]2+ (0–3.0 mM). (b) Time profiles of the yield of H2O2 in the photooxygenation of p-xylene (3.0 × 10−2 M) in oxygen-saturated CD3CN containing Acr+–Mes (2.0 × 10−4 M) and [(tmpa)CuII]2+ (0 mM, black; 0.05 mM, orange; 0.10 mM, green; 0.5 mM, blue; 1.0 mM, red; 2.0 mM, grey and 3.0 mM, brown).
Fig. 3a shows the UV-vis absorption spectra of the reaction solutions with different concentrations of [(tmpa)CuII]2+ after photoirradiation for 3.5 h alongside a solution not undergoing photoirradiation. The characteristic bands of Acr+–Mes around 360 nm and 423 nm decreased after photoirradiation, indicating the decomposition of Acr+–Mes during the reaction. Fig. 3b shows the survival ratio of Acr+–Mes estimated from the relative absorbance at 423 nm depending on the reaction solution concentration of [(tmpa)CuII]2+. The survival ratio of Acr+–Mes increased in proportion to the concentration of [(tmpa)CuII]2+ up to 0.5 mM reaching a high of 78%.
Fig. 3.
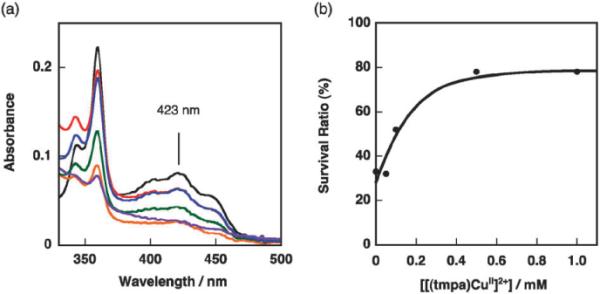
(a) UV-vis spectra of reaction solutions after photoirradiation for 3.5 h. ([Acr–Mes]: 2.0 × 10−4 M, [(tmpa)CuII]2+: 0 mM, black; 0.05 mM, orange; 0.10 mM, green; 0.5 mM, blue; 1.0 mM, red; 2.0 mM). (b) The survival ratio of Acr+–Mes after photoirradiation for 3.5 h as a function of the concentration of [(tmpa)CuII]2+.
The high survival ratio of Acr+–Mes in the presence of [(tmpa)CuII]2+ also benefits the turnover number (TON) in the photooxygenation of p-xylene. Fig. 4 shows the time profiles of the TON for the photocatalytic p-xylene oxidation in MeCN. These experiments were performed by photoirradiation of an MeCN solution containing p-xylene (6.0 × 10−2 M) and Acr+–Mes (2.0 × 10−4 M) in the absence or presence of [(tmpa)CuII]2+ (2.0 × 10−3 M). In the absence of [(tmpa)CuII]2+, the TON of Acr+–Mes reached ~100 although the ratio of p-xylene to Acr+–Mes was 300, indicating the deactivation of Acr+–Mes during the reaction. In the presence of [(tmpa)CuII]2+, p-xylene in the reaction solution was completely converted to p-tolualdehyde. When p-xylene (3.6 × 10−5 mol) was further added to the reaction solution after this photoirradiation, it was also completely converted to p-tolualdehyde, in this second round. For three additions of p-xylene, its complete conversion was observed. Thus, the TON of Acr+–Mes in the presence of [(tmpa)CuII]2+ became higher than 1200, which is more than 11 times higher than the TON obtained without [(tmpa)CuII]2+. In contrast to the case of [(tmpa)CuII]2+, a significant deceleration was observed in the p-xylene oxygenation reaction with the addition of CuII(ClO4)2.
Fig. 4.
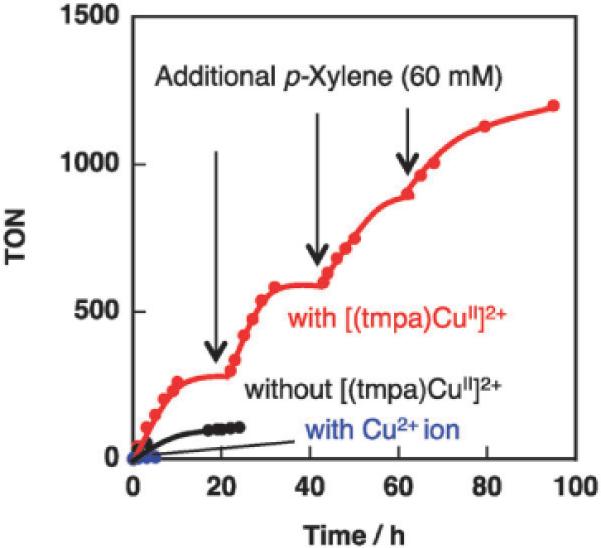
Time profiles of the turnover number for p-xylene oxygenation (6.0 × 10−2 M) under photoirradiation (λ > 390 nm) of an O2-saturated MeCN solution containing Acr+–Mes (2.0 × 10−4 M) in the absence or presence of [(tmpa)CuII]2+ or Cu(ClO4)2 (2.0 × 10−3 M). TON = [oxygenated products]/[Acr+–Mes].
3.2. Kinetic studies of electron transfer from photogenerated Acr˙–Mes˙+ to O2 and [(tmpa)CuII]2+
Nanosecond laser flash photolysis measurements on Acr+–Mes were conducted to detect the electron-transfer state (Acr˙–Mes˙+) in the absence and presence of O2 and [(tmpa)CuII]2+. Laser excitation at 430 nm of an MeCN solution containing Acr+–Mes (1.0 × 10−4 M) results in the formation of the electron-transfer state (Acr˙–Mes˙+).7,32 Fig. 5a shows the decay curves of absorption at 500 nm due to the Acr˙ moiety of Acr˙–Mes˙+. The absorption decay became faster by increasing the concentration of O2 in the solution. The decay obeyed pseudo-first-order kinetics and the pseudo-first-order rate constant (kobs) increased linearly with increasing concentration of O2, as shown in Fig. 5b. The second-order rate constant (kO2) of electron transfer from the Acr˙ moiety of Acr˙–Mes˙+ to O2 was determined from the slope of a linear plot in Fig. 5b to be 6.5 × 108 M−1 s−1. The presence of [(tmpa)CuII]2+ also increased the decay rate of the Acr˙ moiety of Acr˙–Mes˙+ generated by photoirradiation. The decay rate also obeyed pseudo-first-order kinetics and the pseudo-first-order rate constant (kobs) increased linearly with increasing concentration of [(tmpa)CuII]2+, as shown in Fig. 5d. The second-order rate constant (kCu) of electron transfer from Acr˙–Mes˙+ to [(tmpa)CuII]2+ was determined to be 1.5 × 108 M−1 s−1 from the slope of the plot shown in Fig. 5d. The smaller second-order rate constants of kCu compared with kO2 confirms that the electron transfer from the Acr˙ moiety of Acr˙–Mes˙+ to [(tmpa)CuII]2+ is much slower than that to O2 under the present reaction conditions, because the concentration of O2 is 13 mM, which is more than 5 times higher than the concentration of [(tmpa)CuII]2+.
Fig. 5.
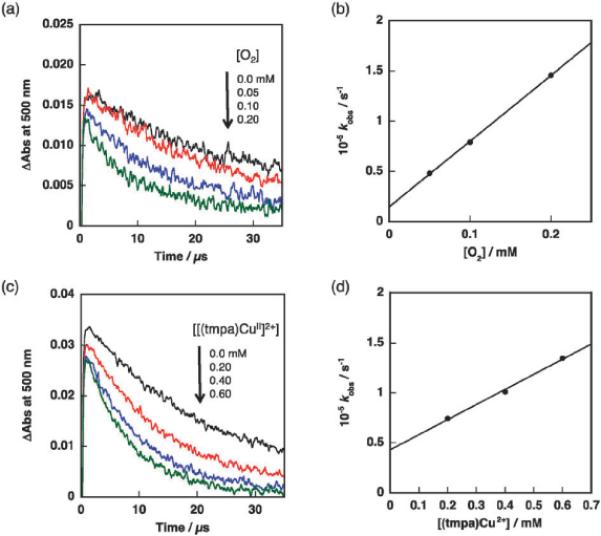
(a) Transient absorbance time profiles at 500 nm due to Acr˙–-Mes˙ in CH3CN containing Acr+–Mes (1.0 × 10−4 M) and oxygen (0 to 2.0 × 10−4 mM) after laser irradiation (λ = 430 nm). (b) A plot of the pseudo-first-order rate constant (kobs) for electron transfer from the Acr˙ moiety of Acr˙–Mes˙+ to O2 vs. [O2]. (c) Time profiles of absorbance at 500 nm due to Acr˙–Mes˙+ in CH3CN containing Acr+–Mes (1.0 × 10−4 M) and [(tmpa)CuII]2+ (0 to 6.0 × 10−4 mM) after photoirradiation (λ = 430 nm). (d) A plot of the pseudo-first-order rate constant (kobs) for electron transfer from the Acr˙ moiety of Acr˙–Mes˙+ to [(tmpa)CuII]2+ vs. [[(tmpa)CuII]2+].
3.3. Detection of reaction intermediate at low temperature
The kinetic measurements suggest that the one-electron reduction of O2 by the Acr˙ moiety occurred in both the absence and presence of [(tmpa)CuII]2+. In the absence of [(tmpa)CuII]2+, H2O2 was formed by disproportionation of two molecules of O2˙−, which is formed by the one-electron reduction of O2 with the Acr˙ moiety. Thus, the lesser formation of H2O2 in the presence of [(tmpa)CuII]2+ suggests that O2˙− may react with [(tmpa)CuII]2+ before the disproportionation. The intermediate species can be detected in the presence of 1-benzyl-1,4-dihydronicotinamide dimer [(BNA)2], which acts as a two-electron donor to be able to reduce O2 to two equivalents of O2˙−.33,34
The UV-vis spectral change was recorded for an EtCN solution containing (BNA)2 (2.5 × 10−4 M), Acr+–Mes (5.0 × 10−5 M) and [(tmpa)CuII]2+ (5.0 × 10−4 M) by photoirradiation (λ = 420 nm) at 203 K as shown in Fig. 6a. Characteristic absorption bands around 525 nm and 610 nm assignable to [{(tmpa)CuII}2O2]2+ complex26–29 increased while the absorption around 410 nm owing to (BNA)2 decreased with photoirradiation. Time profiles of the absorbance at 410 nm and 525 nm are indicated in Fig. 6b. These clearly showed the oxidation of (BNA)2 and formation of [{(tmpa)CuII}2O2]2+ in the same time regime.
Fig. 6.
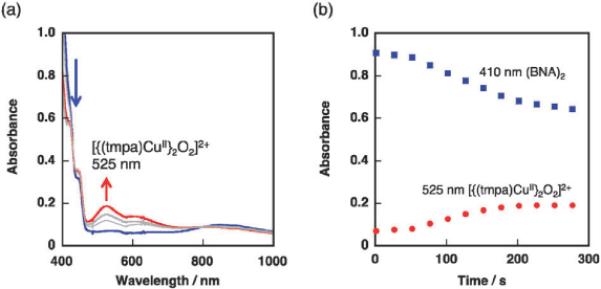
(a) The UV-vis spectral change of an EtCN solution containing (BNA)2 (2.5 × 10−4 M), Acr+–Mes (5.0 × 10−5 M) and [(tmpa)CuII]2+ (5.0 × 10−4 M) before (blue line) and after (red line) photoirradiation (λ = 420 nm) at 203 K. (b) Time profiles of absorbance at 410 nm due to (BNA)2 (blue square) and 525 nm due to [{(tmpa)CuII}2O2]2+ complex (red circle).
When Cu(ClO4)2 was used instead of [(tmpa)CuII]2+, neither the appearance of absorption around 520 nm nor the decay of absorbance at 410 nm was observed (Fig. 7). The photoirradiation of Acr+–Mes results in the formation of Acr˙–Mes˙+ and the Acr˙ moiety of Acr˙–Mes˙+ reduces O2 to produce O2˙− and Acr+–Mes˙+.8 O2˙− may reduce a Cu2+ ion to produce a Cu+ ion and O2, and the Cu+ ion may be immediately oxidized by the Mes˙+ moiety of Acr+–Mes˙+ to regenerate Acr+–Mes and a Cu2+ ion. Thus, the back electron transfer from Cu+ to the Mes˙+ moiety of Acr+–Mes˙+ prohibits the oxidation of (BNA)2 in EtCN as well as the p-xylene substrate in the original reaction solution.
Fig. 7.
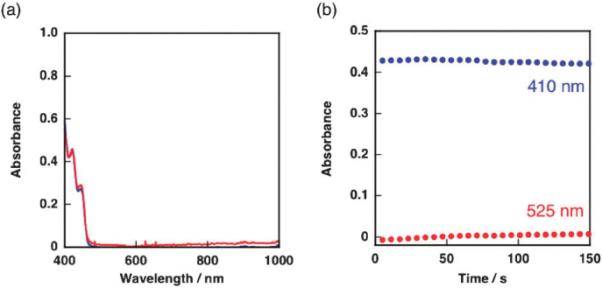
(a) The UV-vis spectral change of an EtCN solution containing (BNA)2 (2.5 × 10−4 M), Acr+–Mes (5.0 × 10−5 M) and CuII(ClO4)2 (5.0 × 10−4 M) before (blue line) and after (red line) photoirradiation (λ = 420 nm) at 203 K. (b) Time profiles of absorbance at 410 nm due to (BNA)2 (blue circle) and 525 nm due to dimeric copper species (red circle).
Scheme 1 depicts the derived photocatalytic reaction mechanism with [(tmpa)CuII]2+. The photocatalyst Acr+–Mes forms the long-lived electron-transfer state Acr˙–Mes˙+ following photoirradiation.7,32 The Mes˙+ moiety of Acr˙–Mes˙+ oxidizes p-xylene to a p-xylene radical cation, because the oxidation potential of p-xylene (1.93 V vs. SCE) is lower than the reduction potential of Acr˙−–Mes˙+ (2.06 V vs. SCE).18 Because the one-electron oxidation potential of the product (p-tolualdehyde) is also higher than the reduction potential of Acr˙–Mes˙+, no further oxidation of p-tolualdehyde occurred, making the present photocatalytic oxygenation of p-xylene highly selective.18 Then, the p-xylene radical cation formed is immediately deprotonated to produce a p-methylbenzyl radical. This carbon-centered radical reacts with molecular oxygen to give the p-methylbenzylperoxyl radical, which disproportionates to yield p-methylbenzyl alcohol and p-tolualdehyde.16p-Methylbenzyl alcohol is further oxidized by the same catalytic cycle, even more efficiently, to produce p-tolualdehyde as the final oxidized product of p-xylene.14,18 On the other hand, the Acr˙ moiety can react with O2 as evidenced by nanosecond laser flash photolysis (Fig. 5a). The O2˙− produced reacts with [(tmpa)CuII]2+ to form [(tmpa)CuII(O2˙−)]+, which is known to be in equilibrium with [(tmpa)CuI]+ and O2.28 The Cu(II)-superoxo complex [(tmpa)CuII(O2˙−)]+ is further reduced by [(tmpa)CuI]+ to form the dinuclear Cu(II)-peroxo complex [{(tmpa)CuII}2O2]2+ as shown in Fig. 6a.32 The fast trap of O2˙− by [(tmpa)CuII]2+ prevents back electron transfer from O2˙− to the Mes˙+ moiety of Acr˙–Mes˙+, resulting in acceleration of the photocatalytic oxygenation of p-xylene with O2 as shown in Fig. 2a. It was reported that [{(tmpa)CuII}2O2]2+ was further reduced in the presence of protons to produce H2O accompanied by regeneration of [(tmpa)CuII]2+.35 Thus, the formation of H2O2 is avoided by the presence of [(tmpa)CuII]2+, which can catalyze the four-electron reduction of O2 (Fig. 2b).35 The removal of potential oxidants (O2˙− and H2O2) by [(tmpa)CuII]2+ led to improvement of the durability of the photocatalyst (Acr+–Mes) as shown in Figs 3 and 4.
Scheme 1.
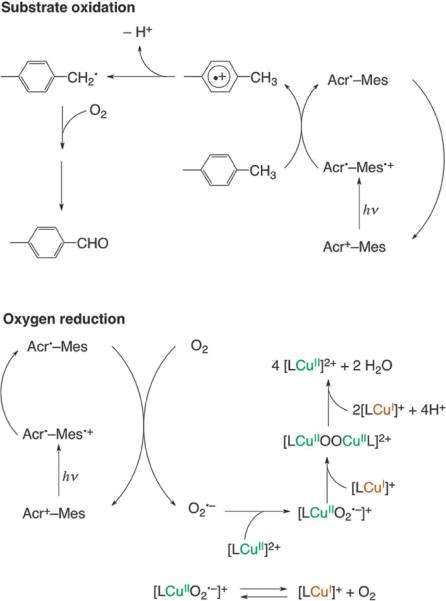
The reaction mechanism of the photocatalytic oxygenation of p-xylene with O2 in the presence of [(tmpa)CuII]2+. (L = tmpa).
Conclusions
The photocatalytic durability of Acr+–Mes in the selective photocatalytic oxygenation of p-xylene with O2 to yield p-tolualdehyde was significantly improved by addition of [(tmpa)CuII]2+; the latter can trap O2˙− produced by the one-electron reduction of O2 by the Acr˙ moiety of Acr˙–Mes˙+, which is the photoproduct of the organic Acr+–Mes catalyst. Overall, molecular oxygen is reduced to water via the dinuclear CuII-peroxo complex, [{(tmpa)CuII}2O2]2+. In contrast to the case of [(tmpa)CuII]2+, CuII(ClO4)2 prohibits the photocatalytic oxygenation of p-xylene with O2 because of the fast back electron transfer from the Cu+ ion to the Mes˙+ moiety of Acr˙–Mes˙+. In conclusion, the efficient trapping of O2˙− by [(tmpa)CuII]2+ and its further reduction to water with [(tmpa)CuI]+ prevents degradation of Acr+–Mes by its oxidation with reactive oxygen species.
Supplementary Material
Acknowledgements
This work was partially supported by a Grant-in-Aid (Nos. 20108010 and 23750014) and a Global COE program, “the Global Education and Research Centre for Bio-Environmental Chemistry” (to S.F.) from the MEXT, Japan, and NRF/MEST of Korea through WCU (R31-2008-000-10010-0) and GRL (2010-00353) Programs (to S.F.). K.D.K. acknowledges support from the USA National Institutes of Health (GM28962).
Footnotes
Electronic supplementary information (ESI) available: UV-vis spectra of titration of H2O2 with NaI in MeCN (Fig. S1).
Notes and references
- 1.Tsuji J. Transition Metal Reagents and Catalysts: Innovations in Organic Synthesis. Wiley; Chichester, U.K.: 2000. [Google Scholar]
- 2.(a) Dobereiner GE, Crabtree RH. Chem. Rev. 2010;110:681. doi: 10.1021/cr900202j. [DOI] [PubMed] [Google Scholar]; (b) Fairlamb IJS. Tetrahedron. 2005;61:9661. [Google Scholar]
- 3.(a) Fagnoni M, Dondi D, Ravelli D, Albini A. Chem. Rev. 2007;107:2725. doi: 10.1021/cr068352x. [DOI] [PubMed] [Google Scholar]; (b) Oelgemöller M, Jung C, Ortner J, Mattay J, Zimmermann E. Green Chem. 2005;7:35. [Google Scholar]
- 4.(a) Marin ML, Santos-Juanes L, Arques A, Amat AM, Miranda MA. Chem. Rev. 2012;112:1710. doi: 10.1021/cr2000543. [DOI] [PubMed] [Google Scholar]; (b) Ravelli D, Fagnoni M. ChemCatChem. 2012;4:169. [Google Scholar]; (c) Ravelli D, Protti S, Neri P, Fagnoni M, Albini A. Green Chem. 2011;13:1876. [Google Scholar]
- 5.(a) Galian RE, Perez-Prieto J. Energy Environ. Sci. 2010;3:1488. [Google Scholar]; (b) Lewis FD, Bedell AM, Dykstra RE, Elbert JE, Gould IR, Farid S. J. Am. Chem. Soc. 1990;112:8055. [Google Scholar]; (c) Fox MA, Chanon M. In: Photoinduced Electron Transfer. Fox MA, editor. Elsevier; Amsterdam: 1988. p. 1. [Google Scholar]; (d) Julliard M, Legris C, Chanon M. J. Photochem. Photobiol., A. 1991;61:137. [Google Scholar]
- 6.(a) Lechner R, Kümmel S, König B. Photochem. Photobiol. Sci. 2010;9:1367. doi: 10.1039/c0pp00202j. [DOI] [PubMed] [Google Scholar]; (b) Megerle U, Wenninger M, Kutta R-J, Lechner R, König B, Dickb B, Riedle E. Phys. Chem. Chem. Phys. 2011;13:8869. doi: 10.1039/c1cp20190e. [DOI] [PubMed] [Google Scholar]; (c) Svoboda J, Schmaderer H, König B. Chem.–Eur. J. 2008;14:1854. doi: 10.1002/chem.200701319. [DOI] [PubMed] [Google Scholar]; (d) Fukuzumi S, Yasui K, Suenobu T, Ohkubo K, Fujitsuka M, Ito O. J. Phys. Chem. A. 2001;105:10501. [Google Scholar]; (e) Fukuzumi S, Tanii K, Tanaka T. J. Chem. Soc., Chem. Commun. 1989:816. [Google Scholar]; (f) Fukuzumi S, Kuroda S, Tanaka T. J. Am. Chem. Soc. 1985;107:3020. [Google Scholar]
- 7.(a) Xu H-J, Lin Y-C, Wan X, Yang C-Y, Feng Y-S. Tetrahedron. 2010;66:8823. [Google Scholar]; (b) Suga K, Ohkubo K, Fukuzumi S. J. Phys. Chem. A. 2003;107:4339. [Google Scholar]; (c) Ohkubo K, Suga K, Fukuzumi S. Chem. Commun. 2006:2018. doi: 10.1039/b518127e. [DOI] [PubMed] [Google Scholar]; (d) Suga K, Ohkubo K, Fukuzumi S. J. Phys. Chem. A. 2005;109:10168. doi: 10.1021/jp053465q. [DOI] [PubMed] [Google Scholar]; (e) Fukuzumi S, Kuroda S, Tanaka T. J. Chem. Soc., Chem. Commun. 1986:1553. [Google Scholar]
- 8.(a) Fukuzumi S. Org. Biomol. Chem. 2003;1:609. doi: 10.1039/b300053b. [DOI] [PubMed] [Google Scholar]; (b) Fukuzumi S. Bull. Chem. Soc. Jpn. 2006;79:177. [Google Scholar]; (c) Fukuzumi S. Phys. Chem. Chem. Phys. 2008;10:2283. doi: 10.1039/b801198m. [DOI] [PubMed] [Google Scholar]; (d) Fukuzumi S, Kojima T. J. Mater. Chem. 2008;18:1427. [Google Scholar]; (e) Fukuzumi S, Ohkubo K. J. Mater. Chem. 2012;22:4575. [Google Scholar]
- 9.(a) Fukuzumi S, Kotani H, Ohkubo K, Ogo S, Tkachenko NV, Lemmetyinen H. J. Am. Chem. Soc. 2004;126:1600. doi: 10.1021/ja038656q. [DOI] [PubMed] [Google Scholar]; (b) Fukuzumi S, Hanazaki R, Kotani H, Ohkubo K. J. Am. Chem. Soc. 2010;132:11002. doi: 10.1021/ja105314x. [DOI] [PubMed] [Google Scholar]; (c) Hoshino M, Uekusa H, Tomita A, Koshihara S, Sato T, Nozawa S, Adachi S, Ohkubo K, Kotani H, Fukuzumi S. J. Am. Chem. Soc. 2012;134:4569. doi: 10.1021/ja300602h. [DOI] [PubMed] [Google Scholar]
- 10.Kotani H, Ohkubo K, Fukuzumi S. J. Am. Chem. Soc. 2004;126:15999. doi: 10.1021/ja048353b. [DOI] [PubMed] [Google Scholar]
- 11.Yamada Y, Miyahigashi T, Kotani H, Ohkubo K, Fukuzumi S. J. Am. Chem. Soc. 2011;133:16136. doi: 10.1021/ja206079e. [DOI] [PubMed] [Google Scholar]
- 12.Yamada Y, Miyahigashi T, Kotani H, Ohkubo K, Fukuzumi S. Energy Environ. Sci. 2012;5:6111. [Google Scholar]
- 13.(a) Ohkubo K, Fukuzumi S. Bull. Chem. Soc. Jpn. 2009;82:303. [Google Scholar]; (b) Kotani H, Ohkubo K, Fukuzumi S. Appl. Catal., B. 2008;77:317. [Google Scholar]; (c) Ohkubo K, Nanjo T, Fukuzumi S. Catal. Today. 2006;117:356. [Google Scholar]; (d) Ohkubo K, Yukimoto K, Fukuzumi S. Chem. Commun. 2006:2504. doi: 10.1039/b601418f. [DOI] [PubMed] [Google Scholar]; (e) Ohkubo K, Iwata R, Miyazaki S, Kojima T, Fukuzumi S. Org. Lett. 2006;8:6079. doi: 10.1021/ol062554y. [DOI] [PubMed] [Google Scholar]
- 14.Fukuzumi S, Doi K, Suenobu T, Ohkubo K, Yamada Y, Karlin KD. Proc. Natl. Acad. Sci. USA. 2012 doi: 10.1073/pnas.1119994109. DOI: 10.1073/pnas.1119994109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohkubo K, Kobayashi T, Fukuzumi S. Angew. Chem., Int. Ed. 2011;50:8652. doi: 10.1002/anie.201102931. [DOI] [PubMed] [Google Scholar]
- 16.Ohkubo K, Kobayashi T, Fukuzumi S. Opt. Express. 2012;20:A360. doi: 10.1364/OE.20.00A360. [DOI] [PubMed] [Google Scholar]
- 17.Ohkubo K, Mizushima K, Fukuzumi Shunichi. Chem. Sci. 2011;2:715. [Google Scholar]
- 18.Ohkubo K, Iwata R, Mizushima K, Souma K, Yamamoto Y, Suzuki N, Fukuzumi S. Chem. Commun. 2010;46:601. doi: 10.1039/b920606j. [DOI] [PubMed] [Google Scholar]
- 19.Franz G, Sheldon RA. Ullmann's Encyclopedia of Industrial Chemistry. 5th edn VCH, Weinheim; Germany: 1991. [Google Scholar]
- 20.Sheldon RA, Kochi JK. Metal Catalyzed Oxidation of Organic Compounds. Academic Press; New York: 1981. ch. 10. [Google Scholar]
- 21.Ballard RE, McKillop A. US. Pat., 4482, 438. 1984
- 22.Clarke R, Kuhn A, Okoh E. Chem. Brit. 1975;11:59. [Google Scholar]
- 23.Syper L. Tetrahedron Lett. 1966;7:4493. [Google Scholar]
- 24.Ho T-L. Synthesis. 1973:347. [Google Scholar]
- 25.Benniston AC, Elliott KJ, Harrington RW, Clegg W. Eur. J. Org. Chem. 2009;253 [Google Scholar]
- 26.(a) Karlin KD, Wei N, Jung B, Kaderli S, Zuberbühler AD. J. Am. Chem. Soc. 1991;113:5868. [Google Scholar]; (b) Karlin KD, Wei N, Jung B, Kaderli S, Niklaus P, Zuberbühler AD. J. Am. Chem. Soc. 1993;115:9506. [Google Scholar]; (c) Tyekiar Z, Karlin KD. Acc. Chem. Res. 1989;22:241. [Google Scholar]; (d) Karlin KD, Kaderli S, Zuberbühler D. Acc. Chem. Res. 1997;30:139. [Google Scholar]
- 27.Weitzer M, Schindler S, Brehm G, Schneider S, Hormann E, Jung B, Kaderli S, Zuberbühler AD. Inorg. Chem. 2003;42:1800. doi: 10.1021/ic025941m. [DOI] [PubMed] [Google Scholar]
- 28.Zhang C, Kaderli S, Costas M, Kim E, Neuhold Y, Karlin KD, Zuberbühler AD. Inorg. Chem. 2003;42:18073. doi: 10.1021/ic0205684. [DOI] [PubMed] [Google Scholar]
- 29.(a) Smirnov VV, Roth JP. J. Am. Chem. Soc. 2006;128:3683. doi: 10.1021/ja056741n. [DOI] [PubMed] [Google Scholar]; (b) Jitsukawa K, Harata M, Arii H, Sakurai H, Masuda H. Inorg. Chim. Acta. 2001;324:108. [Google Scholar]; (c) Harata M, Jitsukawa K, Masuda H, einaga H. J. Am. Chem. Soc. 1994;116:10817. [Google Scholar]
- 30.Wang J, Schopfer MP, Puiu SC, Sarjeant AAN, Karlin KD. Inorg. Chem. 2010;49:1404. doi: 10.1021/ic901431r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mair RD, Graupner AJ. Anal. Chem. 1964;36:194. [Google Scholar]
- 32.(a) Ohkubo K, Kotani H, Fukuzumi S. Chem. Commun. 2005:4520. doi: 10.1039/b506479a. [DOI] [PubMed] [Google Scholar]; (b) Fukuzumi S, Kotani H, Ohkubo K. Phys. Chem. Chem. Phys. 2008;10:5159. doi: 10.1039/b809264h. [DOI] [PubMed] [Google Scholar]
- 33.Fukuzumi S, Suenobu T, Patz M, Hirasaka T, Itoh S, Fujitsuka M, Ito O. J. Am. Chem. Soc. 1998;120:8060. [Google Scholar]
- 34.(a) Fukuzumi S, Patz M, Suenobu T, Kuwahara Y, Itoh S. J. Am. Chem. Soc. 1999;121:1605. [Google Scholar]; (b) Kawashima T, Ohkubo K, Fukuzumi S. Phys. Chem. Chem. Phys. 2011;13:3344. doi: 10.1039/c0cp00916d. [DOI] [PubMed] [Google Scholar]
- 35.Fukuzumi S, Kotani H, Lucas HR, Doi K, Suenobu T, Peterson R, Karlin KD. J. Am. Chem. Soc. 2010;132:6874. doi: 10.1021/ja100538x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.